

Abstract
Objective
To determine if a unique subtype of scleroderma muscle disease exists by comparing the clinical features of systemic sclerosis (SSc) patients with predominant fibrosis on muscle biopsy to those with inflammatory muscle histopathology.
Methods
This retrospective, cross-sectional study included SSc patients with muscle weakness and an available muscle biopsy. Biopsies with fibrosis but without inflammation/necrosis were designated as “fibrosing myopathy” and those with inflammation and/or necrosis were assigned a category of “inflammatory myopathy”. Clinical data including features of SSc, serum creatine kinase (CK) levels, electromyography (EMG), autoantibody profile, and survival were compared between the 2 groups.
Results
The study population consisted of 37 weak SSc patients, 8 with fibrosing myopathy, and 29 with inflammatory myopathy. Compared to those with inflammatory myopathy, patients with fibrosing myopathy were more likely to have diffuse SSc skin subtype (87% vs. 62%, p=0.18), African-American race (62.5% vs. 37.9%; p=0.20), and a lower FVC (55.5 ± 31.9 vs. 66.4 ± 17.6; p=0.23). They also had lower CK values (516 ± 391 vs. 2477 ± 3511, p=0.007) and lower aldolase values (13.8 ± 4.7 vs. 27.3 ± 4.7, p=0.01). Patients with fibrosing myopathy had a significantly higher mortality (5 of 8; 62.5% vs 4 of 29 (14.3%), p=0.005).
Conclusion
Fibrosing myopathy is a unique histological subtype of muscle disease among weak patients with SSc and is associated with significantly worse mortality compared to those with inflammation and/or necrosis on muscle biopsy.
Keywords: myopathy, scleroderma, fibrosis, mortality
INTRODUCTION
Myopathy in systemic sclerosis (SSc) is associated with poor outcomes, such as disability, even when controlling for potential confounders such as restrictive lung disease, disease subtype, and disease duration (1). The reported prevalence for myopathy is 25% (2,3)while the prevalence of muscle weakness ranges from 5–96% (4,5). This reported wide range is likely due both to the diverse etiologies accounting for muscle weakness and the lack of consensus for standard classification criteria.
We recently described the muscle biopsy characteristics of 42 SSc patients and found that the predominant individual features were inflammation, necrosis, and acute neurogenic atrophy (5). We also identified a small group of patients with fibrosis only on muscle histopathology. There is a lack of information on the clinical features and disease course in patients with a myopathy with fibrosis alone on biopsy. The purpose of our current investigation was to determine if patients with a fibrosing myopathy were unique by analyzing the clinical features and survival of these patients compared to those with inflammation and/or necrosis (an inflammatory myopathy) on muscle histopathology.
MATERIALS AND METHODS
Our study population was derived from a cohort that was previously described after reviewing data on all subjects enrolled in the Johns Hopkins Scleroderma Center database from May 1990 to June 2014 (6). Our inclusion criteria were as follows: 1) evidence of myopathy as defined by proximal weakness by an expert at our center, and 2) a muscle biopsy reviewed by a muscle pathologist at the Johns Hopkins Neuromuscular Laboratory.
Demographic data including age, sex, race, ethnicity, and SSc subtype were obtained from the Johns Hopkins Scleroderma Center Database. SSc disease severity measures were collected prospectively at 6 month intervals (Medsger severity scales) (7). Clinical data for this study included the lowest recorded percent predicted forced vital capacity (FVC) and diffusing capacity of the lungs for carbon monoxide (DLCO) on available pulmonary function tests (PFTs) standardized for gender and age (18,19), the lowest ejection fraction (EF (%)) and highest right ventricular systolic pressure (RVSP) on echocardiography, the peak recorded modified Rodnan skin score (mRSS), the highest Medsger muscle severity score, and any history of a renal crisis recorded during any of the subjects’ follow-up visits at the Center. These most extreme recorded values were selected to provide a composite picture of the severity of the clinical features or phenotype of the study subjects. The disease duration of SSc at the onset of muscle symptoms was calculated from the time of the first recorded non-Raynaud SSc symptom.
Autoantibody assays
For each patient, anti-nuclearantibody and U1-RNP antibody status was obtained from the clinical database; All other antibody specificities listed in Table 1 were assayed using a commercially available line immunoblot platform (EuroImmun; Systemic Sclerosis (Nucleoli) profile). Euroimmun results were considered positive when values were ≥ 26 (strong/very strong brand when compared to control, per the manufacturer’s protocol).
Table 1.
Clinical characteristics of patients with scleroderma with fibrosis only vs. inflammation and/or necrosis on muscle biopsy
| Fibrosis only (n=8) |
Inflammation and/or
necrosis (n=29) |
p-value | |
|---|---|---|---|
| Age of scleroderma diagnosis defined by 1st non-Raynaud’s symptom | 40.1 ± 12.1 years | 45.6 ± 2.5 years | 0.35 |
| Female | 6 (75%) | 20 (69.0%) | 0.78 |
| Diffuse skin subtype | 7 (87.5%) | 18 (62.0%) | 0.18 |
| Maximum Modified Rodnan Skin Score | 23 ± 12.7 | 15.5 ±13.8 | 0.17 |
| Renal crisis | 1 (12.5%) | 5 (17.2%) | 0.75 |
| African American race | 5 (62.5%) | 11 (37.9%) | 0.20 |
| Caucasian race | 3 (37.5%) | 18 (62.7%) | |
| Disease Duration (at time of First Visit) | |||
| 1st Non-Raynaud’s Symptom | 2.31 ± 1.91 years | 2.95 ± 3.35 years | 0.49 |
| Raynaud’s Symptom | 2.01 ± 1.37 years | 3.59 ± 7.44 years | 0.31 |
| Disease duration (From Date of 1st non-Raynaud’s symptom to Muscle symptom) | 1.64 ± 1.39 years | 3.0 ± 6.36 years | 0.30 |
| Disease duration (From date of 1st symptom to muscle biopsy) | 3.96 ± 2.45 years | 4.33 ± 4.59 years | 0.73 |
| Duration of follow-up | 1.86 ± 0.53 years | 5.93 ± 4.73 years | 0.0005 |
| Mean time to death after muscle biopsy | 0.28 ± 0.19 years | 2.50 ± 1.6 years | 0.034 |
| Mean time to death from 1st non-Raynaud’s symptom | 4.9 ± 2.3 years | 10.6 ± 9.8 years | 0.34 |
| Deceased | 5 (62.5%) | 4 (14.3%) | 0.005 |
| Maximum Muscle Severity Score | 0.20 | ||
| 0 | 0 | 0 | |
| 1 | 2 (25%) | 17 (58.6%) | |
| 2 | 2 (25%) | 8 (27.6%) | |
| 3 | 1 (12.5%) | 2 (6.9%) | |
| 4 | 3 (37.5%) | 2 (6.9%) | |
| Maximum Creatine Kinase (mean) | 516 ± 391 | 2477 ± 3511 | 0.007 |
| Maximum Aldolase (mean) | 13.8 ± 4.7 | 27.3 ± 4.7 | 0.01 |
| MRI edema | 4/5 (80%) | 20/22 (95.4%) | 0.24 |
| MRI fatty replacement | 1/5 (20%) | 6/22 (27.2%) | 0.74 |
| EMG- irritable myopathy | 3/7 (42.6%) | 15/24 (62.5%) | 0.31 |
| EMG- non-irritable myopathy | 4/7 (57.1%) | 9/24 (37.5%) | |
| Minimum ejection fraction on echocardiogram | 49.7 ± 15.8 | 55.6 ± 8.5 | 0.42 |
| Minimum RVSP on echocardiogram | 44 ± 12.1 | 36.8 ± 16.0 | 0.35 |
| Minimum FVC | 55.5 ± 31.9 | 66.4 ± 17.6 | 0.23 |
| Minimum DLCO | 59.5 ± 36 | 57.3 ± 23.5 | 0.83 |
| Anti-centromere | 0/8 (0%) | 3/29 (10.3%) | 0.35 |
| Scl-70 | 3/8 (37.5%) | 2/29 (6.9%) | 0.03 |
| U1-RNP | 0/8 (0%) | 3/29(10.3%) | 0.35 |
| RNA-Poly III | 0/8 (0%) | 4/29 (13.8%) | 0.30 |
| PM-Scl | 2/7 (28.6%) | 3/27 (11.1%) | 0.25 |
| U3-RNP | 2/7 (29%) | 0/27 (0%) | 0.004 |
| Anti-Ku | 0/7 (0%) | 2/27 (7.4%) | 0.46 |
| Anti-NOR90 | 1/7 (14.3%) | 1/27 (3.7%) | 0.30 |
| Anti-Ro 52 | 0/7 (0%) | 6/27 (22.2%) | 0.18 |
| Anti-Th/To | 0/7 (0%) | 2/27 (7.4%) | 0.46 |
| ANA-nucleolar pattern | 7/7 (100%) | 9/28 (32.1%) | 0.02 |
| Improvement at 3 months in muscle strength and/or CK 3 months after biopsy | 3/8 (37.5%) | 28/29 (96.5%) | 0.001 |
| Medications 3 months after biopsy* | |||
| Prednisone (Moderate to High dose) | 1/8 (12.5%) | 22/29 (75.9%) | 0.0013 |
| Methotrexate | 2/8 (25%) | 7/29 (24%) | 0.64 |
| Mycophenalate mofetil | 4/8 (50%) | 13/29 (40.7%) | 0.55 |
| Azathioprine | 0/8 (0%) | 6/29 (20.7%) | 0.20 |
| IVIG | 1/8 (12.5%) | 4/27 (14.8%) | 0.71 |
Not mutually exclusive; Autoantibody data in the Table were obtained from clinical lab results (ANA, U1-RNP) or by Euroimmune assay (all other tabulated specificities). Euroimmune assay results were scored as positive when the assay values were ≥26 (that is, strong to very strong band when compared to controls).
Muscle evaluation of study participants
Muscle weakness was determined by the provider at the time of evaluation and defined by the Medsger muscle severity score, which defines 5 categories as follows: a score of 0 is full motor strength; score of 1 is strength of 4/5; score of 2 is strength of 3/5; score of 3 is strength of 2/5, and score of 4 is strength that requires ambulation aids (7). Electromyography (EMG) with nerve conduction studies (NCS), muscle magnetic resonance imaging (MRI), and muscle biopsies that were obtained as part of routine clinical care were reviewed for this study. The presence of positive sharp waves and fibrillations on EMG in the context of myopathic motor units was defined as an irritable myopathy; myopathic motor units without spontaneous activity were defined as a non-irritable myopathy (10). Edema on STIR images and fatty replacement on T1 weighted sequences were read by a radiologist as part of routine clinical care (11). Highest ever recorded CK and aldolase levels were also obtained from comprehensive medical record review.
Muscle Biopsy Analysis
To ensure uniform assessment, we excluded samples that were not available to be read (by AC) at The Johns Hopkins Neuromuscular Pathology Lab. In only 2 instances, muscle biopsy slides were obtained from another institution and then read at Johns Hopkins. Muscle biopsy slides were read using a predesigned protocol for routine care and assessed for the presence or absence of the following individual histologic features: necrotic fibers, inflammation, perifasicular atrophy, fibrosis, angular atrophic esterase-positive fibers (a histopathologic feature of acute neurogenic atrophy), and fiber type grouping (a feature of chronic denervation with re-innervation).
Based on the individual histologic features, biopsies were assigned to one of 4 well-accepted histopathologic categories of myopathy: (i) polymyositis, (ii) dermatomyositis, (iii) necrotizing myopathy, or (iv) or non-specific myositis. These categories were adapted from the European Neuro Muscular Centre (ENMC) histopathologic criteria (12). If there were other findings such as numerous COX-negative fibers (indicating mitochondrial dysfunction), only neurogenic features, or non-specific myofiber atrophy in isolation, these were classified as “other” and excluded from further analysis. Patients with fibrosis as the only analyzed histopathologic feature were categorized as having “fibrosing myopathy”. For the purposes of this paper, those muscle biopsies that met the categories of either polymyositis, dermatomyositis, or necrotizing myopathy as adapted by the ENMC criteria were defined as having “inflammatory myopathy”.
Institutional review board approval and participants’ written informed consent were obtained from each participant.
Statistical Analysis
Statistical analyses were performed using the statistical software, Stata version 12.1 (Stata Corp, College Station, Texas). Continuous variables were reported as a mean value +/− standard deviation. Discrete variables were summarized as proportions. The Wilcoxon-Mann-Whitney test was used to compare between the two groups due to the small sample size. All reported p values are 2-sided with α=0.05. Kaplan-Meier cumulative survival curves were computed to compare patients with fibrosing and inflammatory myopathies.
RESULTS
Clinical characteristics of SSc patients with fibrosis only, compared to those with inflammatory muscle biopsies
Among the 2830 patients enrolled in the Johns Hopkins Scleroderma Center, 1711 had an available maximum muscle severity score; 405 (23.7%) of these were weak as defined by a muscle severity score of ≥ 1 (6). From among the identified weak patients, 65(16%) had a muscle biopsy and 47 of these were available for review. 8 of 47 (17%) had a fibrosing myopathy, 29 of 47 (62%) had an inflammatory myopathy (non-specific myositis=15, necrotizing=9, polymyositis=2, dermatomyositis=3), and 10 of 47 (21%) were excluded from further analysis because they had neuropathic features only (3 subjects) or only non-specific myopathic features such as type 2 fiber atrophy or evidence of mitochondrial dysfunction (7 subjects).
As shown in Figure 1, fibrosis in fibrosing myopathy patients was predominantly localized to the perimysium between muscle fascicles; but also extended into the endomysium. Despite the often extensive fibrosis, there was no accompanying inflammation or necrosis. Compared to those with inflammatory myopathy, SSc patients with fibrosing myopathy were more likely to have younger age of SSc onset (defined from the onset of first non-Raynaud’s phenomenon) (40.1 ± 12.1 years vs. 45.6 ± 2.5 years, p=0.35), diffuse cutaneous disease (87% vs. 62.5%, p=0.18), African-American race (62.5% vs. 37.9%; p=0.20), and lower FVC (55.5 ± 31.9% vs. 66.4 ± 17.6%; p=0.23) (Table 1).
Figure 1.
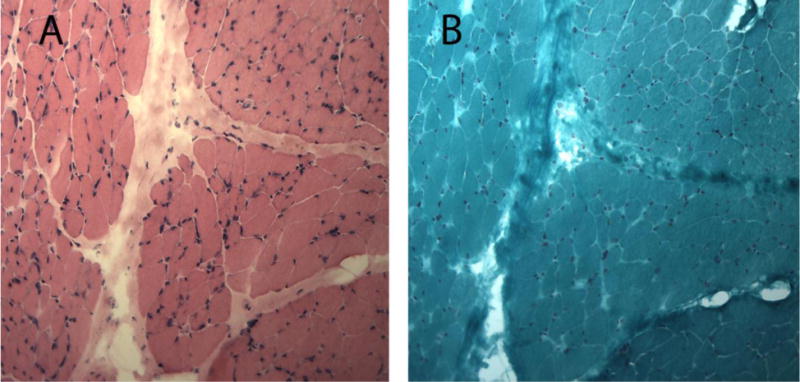
Fibrosing myopathy on Muscle Histopathology
Panel A: Hematoxylin and eosin (H&E) stain of fibrosing myopathy: Prominent perimysial and endomysial fibrosis. No inflammatory infiltrates. Panel B: Gomori trichome stain: prominent perimysial and endomysial fibrosis on 10X high power view.
When compared to the inflammatory group, patients with a fibrosing myopathy had a higher maximum Medsger Muscle severity score. Despite being weaker, these patients had strikingly lower CK levels (516 ± 391 vs. 2477 ± 3511 IU/L; p=0.007) and were less likely to have an irritable myopathy (42.6% vs. 62.5%; p=0.31) than those with inflammatory muscle biopsies. While muscle MRI suggested that the patients with predominant fibrosis had less edema (80% vs 95%) and fatty replacement (20% vs. 27%), this was not statistically significantly different. Complete EMG/NCS results are available in Supplemental Table 2.
A study of the well-defined antibody specificities showed that autoantibodies against Scl-70 and U3-RNP were detected more frequently in the fibrosing group compared to the inflammatory group (p=0.03, p=0.004 (Table 1). To address whether the fibrosing myopathy patients were defined by a common antibody specificity (other than those reported in Table 1), we tested these sera by immunoprecipitation from radiolabeled HeLa lysates as described (13) (Supplemental Figure 1). Seven sera in the fibrosing group were available for study, and 21 in the inflammatory group. Of the fibrosing myopathy set, 4/7 sera did not immunoprecipitate any bands and 2 sera immunoprecipitated bands of unknown specificity. Interestingly, 17/21 (81%) of the inflammatory group immunoprecipitated robust bands, compared to only 3/7 (43%) of the fibrosing myopathy set. To address the possibility that the fibrosing myopathy sera may have antibodies against muscle-specific proteins, we performed similar immunoprecipitations using lysates made from radiolabeled cultured myoblasts. This data was consistent with that generated using HeLa lysates, confirming that there were no antibodies against muscle-specific proteins in these sera.
Increased mortality in those with fibrosing myopathy
It is striking to report that the patients with a fibrosing myopathy had a significantly higher mortality compared to those in the inflammatory group (62.5% vs. 14.3%; p = 0.01), and a shorter follow-up duration (1.86 ± 0.53 years vs. 5.93 ± 4.73 years (p=0.02) (Table 1). These patients died shortly after muscle biopsy and early in their disease from cardiac causes. Consistent with this, the mean time to death after muscle biopsy was 0.28 ± 0.19 years in the fibrosing myopathy group and 2.50 ± 1.6 years in those with an inflammatory muscle biopsy (p=0.01). Kaplan Meier survival curves are shown in Figure 2 and demonstrate a statistically significant difference in survival (p=0.001). Of the 5 deceased fibrosing myopathy patients, 1 had myocardial fibrosis on endomyocardial biopsy without inflammation, and cardiac MRI revealed evidence of fibrosis; this patient died shortly after inpatient admission. Two other patients died from complications of heart failure with cardiomyopathy related to their scleroderma. One patient died of multisystem disease and the remaining patient died from complications of bowel obstruction and perforation. Thus, 3 of the 5 patients (60%) most likely had cardiac disease that primarily contributed to death. Of note, among 6 patients with a fibrosing myopathy who had a troponin-I checked at the time of biopsy, the troponin level was elevated in 5 (83.3%). In contrast, among those with an inflammatory myopathy, only 5 of 17 (29.4%) had elevated troponin at time of muscle biopsy (p=0.06).
Figure 2.
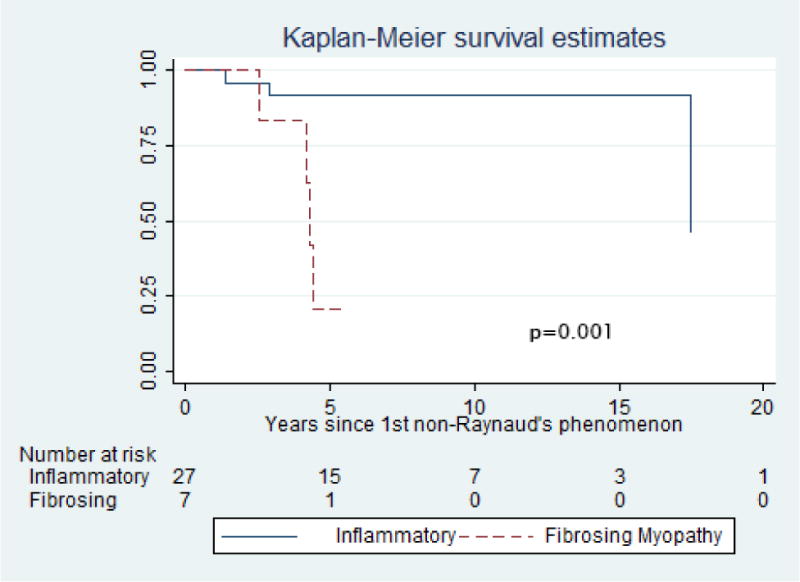
Kaplan-Meier survival estimates comparing SSc patients with fibrosing myopathy vs. inflammatory myopathy
DISCUSSION
This cross-sectional study was undertaken to characterize the clinical phenotype of SSc patients with muscle weakness and a muscle biopsy showing predominant muscle fibrosis in isolation (i.e., a fibrosing myopathy). These patients were then compared to a group of SSc patients with weakness and a muscle biopsy showing an inflammatory myopathy to determine if there were any clinical or serological measures that differed between these groups. While not statistically significant, we found that SSc patients with a fibrosing myopathy tended to be of the diffuse skin subtype, African-American race, have lower FVC, and have shorter disease duration (as defined by first non-Raynaud’s symptom). In addition, they had significantly lower CK levels and more prevalent non-irritable myopathy on EMG. Strikingly, the fibrosing group had a statistically significant higher risk of death compared to those with an inflammatory muscle biopsy. These fibrosing myopathy patients appeared to have died with cardiac involvement with documented myocardial fibrosis on biopsy in one case and complications of heart failure in 2 cases.
While predominant fibrosis in the absence of other histologic features has been described in a prior survey of weak SSc patients (4), it was reported in a study that included patients who had muscle tissue examined at autopsy. This predominantly autopsy study raised the question of whether fibrosis was exclusively a late stage feature of disease in which a previous inflammatory process was no longer seen. In contrast, the patients in our report had clinically relevant muscle weakness in real time that prompted a muscle biopsy. While we cannot exclude the possibility that predominant fibrosis occurred after some prior inflammatory process, given that the muscle biopsies were obtained close to the clinical onset of muscle weakness and at a comparable disease duration, we feel that fibrosis was most likely to be the primary ongoing pathological process.
The muscle evaluation of our patients included muscle strength testing, muscle enzymes, EMG, MRI of the muscles, and muscle biopsy. The fibrosing myopathy group tended to have higher maximum muscle severity scores, with 4 of the 8 (50%) subjects having a Maximum Muscle Severity Score ≥ 3. Despite the severe myopathic clinical phenotype of these patients, they tended to have lower CK levels and less frequent findings of membrane instability (i.e., spontaneous activity) on EMG. While practicing physicians often perform muscle biopsies only in the setting of elevated CK levels, our findings suggest that in SSc, obtaining muscle tissue should be considered when a patient exhibits prominent weakness even without markedly elevated CK levels or spontaneous activity on EMG. This may identify the muscle pathology causing the weakness and may have implications for therapeutic decisions.
The optimal treatment of a fibrosing myopathy is not defined. However, given the markedly increased mortality for patients who have this type of muscle histopathology, an aggressive treatment strategy should be considered to prevent the predicted steep decline in clinical trajectory. Our experience demonstrates that the fibrosing group did not respond to immunosuppressive treatment such as steroids, mycophenalate, or methotrexate. Two patients were biopsied immediately and started on MMF followed by IVIG; both had improvement in muscle strength, CK and aldolase and additional benefit to their skin disease. It is unclear if these medications had an immunomodulatory effect or it was the timing at which it was started. Long term follow-up of these patients will be helpful to identify the best medication for treatment. In contrast, patients in the inflammatory group did have a response to immunosuppression including moderate to high dose steroids which was statistically significant (Table 1). While it may be possible the fibrosing group died because of the lack of high dose steroids, given that these patients are at high risk for renal crisis these patients did not receive high dose steroids thereby explaining the difference between these two groups.
Given that a strong association between myopathy and sudden cardiac death has been previously reported (14), it may not be surprising that patients with a myopathy have a higher risk of cardiac involvement or a higher risk of morality over all (3). However, the distinction in our study is that a fibrosing myopathy was associated with cardiac complications which was not seen in the inflammatory myopathy group. Our study highlights that in the presence of a fibrosing myopathy, early cardiac work-up with troponin-I, cardiac MRI and EKG for conduction defects may be warranted.
SSc autoantibody profiles were determined for all patients in this study. Anti-Scl-70 and U3-RNP autoantibodies were more frequent in the fibrosing group. While U3-RNP has been associated with myopathy in SSc (15), Scl-70 has not been previously reported.
This study has several limitations. First, as not all weak patients in the cohort underwent a muscle biopsy, this could have introduced a selection bias. Second, our finding that they have increased mortality should be confirmed in a larger group who are identified early in disease and studied prospectively. Third, since this was a cross-sectional study, we cannot draw conclusions about causal relationships between the onset of muscle weakness and specific histopathologic diagnoses on muscle biopsies. The muscle biopsies may not reflect the biological process causing muscle fibrosis, but our data clearly demonstrates that a unique subtype of muscle disease exists. A better understanding of the exact disease course and opportunities for intervention is key for future investigations in this subtype of SSc muscle disease.
Supplementary Material
SIGNIFICANCE AND INNOVATIONS.
Fibrosis predominance on muscle biopsy, or fibrosing myopathy, represents a histopathological subset of myopathy in patients with systemic sclerosis.
Systemic sclerosis patients with fibrosing myopathy on muscle biopsy have higher mortality than patients who have inflammation or necrosis on muscle biopsies.
Patients with a fibrosing myopathy have lower muscle enzymes (CK, aldolase) levels requiring vigilance in identification and may benefit from more extensive muscle evaluation including a muscle biopsy.
Anti-U3-RNP and Scl-70 antibodies were more frequently detected in those with a fibrosing myopathy
Acknowledgments
We thank Adrianne Woods, Margaret Sampedro, and Gwendolyn Leatherman for their assistance with database management and coordination of study, and Dr Laura Gutierrez for her assistance in running the EUROIMMUN assays.
Supported in part by a John Staurulakis Endowed Scholar Award and Doris Duke Early Clinician Investigator Award (Dr. Paik), the Donald B. and Dorothy L. Stabler Foundation (Dr. Hummers), and the National Institutes of Health and Chresanthe Stauralakis Memorial Discovery Fund (NIAMS K23 AR061439 to Dr. Shah). This research was supported [in part] by the Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health (Dr. Mammen), Scleroderma Research Foundation and McCrory Professorship (Dr. Wigley). The Johns Hopkins Rheumatic Disease Research Core Center, where the immunoprecipitations were performed, is supported by NIH grant AR-053503.
References
- 1.Paik JJ, Wigley FM, Mejia AF, Hummers LK. Severity of muscle weakness independently associates with disability as measured by the Health Assessment Questionnaire-Disability Index (HAQ-DI) in scleroderma. Arthritis Care Res. 2016 doi: 10.1002/acr.22870. [DOI] [PubMed] [Google Scholar]
- 2.Muangchan C, Markland J, Robinson D, Jones N, Khalidi N, Docherty P, et al. The 15% rule in scleroderma: the frequency of severe organ complications in systemic sclerosis. A systematic review. J Rheumatol. 2013;40:1545–1556. doi: 10.3899/jrheum.121380. [DOI] [PubMed] [Google Scholar]
- 3.Jung M, Bonner A, Hudson M, Baron M, Pope J, on behalf of the Canadian Scleroder Myopathy is a poor prognostic feature in systemic sclerosis: results from the Canadian Scleroderma Research Group (CSRG) cohort. Scand J Rheumatol. 2014;43:217–220. doi: 10.3109/03009742.2013.868512. [DOI] [PubMed] [Google Scholar]
- 4.Medsger TA, Rodnan GP, Moossy J, Vester JW. Skeletal muscle involvement in progressive systemic sclerosis (scleroderma) Arthritis Rheum. 1968;11:554–568. doi: 10.1002/art.1780110405. [DOI] [PubMed] [Google Scholar]
- 5.Clements PJ, Furst DE, Campion DS, Bohan A, Harris R, Levy J, et al. Muscle disease in progressive systemic sclerosis: diagnostic and therapeutic considerations. Arthritis Rheum. 1978;21:62–71. doi: 10.1002/art.1780210111. [DOI] [PubMed] [Google Scholar]
- 6.Paik JJ, Wigley FM, Lloyd TE, Corse AM, Casciola-Rosen L, Shah AA, et al. Spectrum of Muscle Histopathologic Findings in Forty-Two Scleroderma Patients With Weakness. Arthritis Care Res. 2015;67:1416–1425. doi: 10.1002/acr.22620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Medsger TA, Silman AJ, Steen VD, Black CM, Akesson A, Bacon PA, et al. A disease severity scale for systemic sclerosis: development and testing. J Rheumatol. 1999;26:2159–2167. [PubMed] [Google Scholar]
- 8.Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general U.S. population. Am J Respir Crit Care Med. 1999;159:179–187. doi: 10.1164/ajrccm.159.1.9712108. [DOI] [PubMed] [Google Scholar]
- 9.Knudson RJ, Kaltenborn WT, Knudson DE, Burrows B. The single-breath carbon monoxide diffusing capacity. Reference equations derived from a healthy nonsmoking population and effects of hematocrit. Am Rev Respir Dis. 1987;135:805–811. doi: 10.1164/arrd.1987.135.4.805. [DOI] [PubMed] [Google Scholar]
- 10.Griggs R, Mendell J, Miller R. Evaluation of patients with myopathy. Philadelphia, PA: FA Davis; 1995. Evaluation and Treatment of Myopathies; pp. 17–78. [Google Scholar]
- 11.Napier N, Shortt C, Eustace S. Muscle edema: classification, mechanisms, and interpretation. Semin Musculoskelet Radiol. 2006;10:258–267. doi: 10.1055/s-2007-971997. [DOI] [PubMed] [Google Scholar]
- 12.Hoogendijk JE, Amato AA, Lecky BR, Choy EH, Lundberg IE, Rose MR, et al. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10–12 October 2003, Naarden, The Netherlands. Neuromuscul Disord NMD. 2004;14:337–345. doi: 10.1016/j.nmd.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 13.Hall JC, Casciola-Rosen L, Samedy L-A, Werner J, Owoyemi K, Danoff SK, et al. Anti-melanoma differentiation-associated protein 5-associated dermatomyositis: expanding the clinical spectrum. Arthritis Care Res. 2013;65:1307–1315. doi: 10.1002/acr.21992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Follansbee WP, Zerbe TR, Medsger TA. Cardiac and skeletal muscle disease in systemic sclerosis (scleroderma): a high risk association. Am Heart J. 1993;125:194–203. doi: 10.1016/0002-8703(93)90075-k. [DOI] [PubMed] [Google Scholar]
- 15.Aggarwal R, Lucas M, Fertig N, Oddis CV, Medsger TA. Anti-U3 RNP autoantibodies in systemic sclerosis. Arthritis Rheum. 2009;60:1112–1118. doi: 10.1002/art.24409. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.