

Abstract
Microtubules are recognized as crucial components of the mitotic spindle during cell division, and, for this reason, the microtubule system is an attractive target for the development of anticancer agents. Continuing our search strategy for novel tubulin targeting-compounds, a new series of 2-alkoxycarbonyl-3-(3′,4′,5′-trimethoxyanilino)-5-aryl/heteroarylthiophene derivatives was designed, synthesized and demonstrated to act as tubulin polymerization inhibitors at the colchicine site. A structure-activity relationship study on the phenyl at the 5-position of the thiophene ring was performed by introducing a variety of substituents containing electron-releasing and electron-withdrawing groups, with the 2-alkoxycarbonyl-3-(3′,4′,5′-trimethoxyanilino)thiophene scaffold being the minimum structural requirement for activity. Of the tested compounds, derivatives 4a, 4c, 4i and 4k possessed the highest overall potency and displayed high antiproliferative activities at submicromolar concentrations, with IC50 values ranging from 0.13 to 0.84 μM against four different cancer cell lines. Three agents (4a, 4c and 4i) in the present series had similar effects, and these were comparable to those of the reference compound combretastatin A-4 (CA-4) as inhibitors of tubulin assembly. The antitubulin effects correlated with the cytostatic activities and indicate that these compounds inhibit cell growth through inhibition of tubulin polymerization by binding at the colchicine site. Compound 4c, containing the 2′-thienyl ring at the 5-position of the 2-methoxycarbonyl-3-(3′,4′,5′-trimethoxyanilino)thiophene scaffold, exhibited substantial antiproliferative activity with a mean IC50 value of 140 nM, inhibited tubulin polymerization with an IC50 value of 1.2 μM, similar to that of CA-4 (IC50: 1.1 μM), and induced apoptosis in HeLa cells.
Keywords: Microtubule, Tubulin polymerization inhibitors, Antiproliferative agents, Colchicine site, Structure-activity relationship
1. Introduction
There has been in recent years an intense effort directed at the discovery and development of novel small molecules, many of which are natural products, able to interfere with tubulin polymerization because of their anti-cancer potential [1]. Microtubules represent a dynamic cellular compartment in both neoplastic and normal cells. The microtubule system of eukaryotic cells plays important roles in regulating cell architecture and has an essential role in cell division, since microtubules are a key component of the mitotic spindle [2]. This dynamicity is characterized by the continuous turnover of αβ-tubulin heterodimers in the polymeric microtubules. Because of their key roles in cell structure and cell division, they are involved in a variety of fundamental cellular functions, such as regulation of motility, cell signaling, formation and maintenance of cell shape, and bidirectional transport of material within the cell [3–5]. The disruption of microtubule dynamics increases the number of cells in metaphase arrest and mitotic catastrophe, and this interference with cell cycle progression has proven to be useful for designing anticancer agents, such as taxanes (paclitaxel, carbazitaxel and docetaxel) and vinca alkaloids (vinblastine, vinorelbine and vincristine), all of which are used clinically [6–8].
One of the most important microtubule depolymerizing agents is combretastatin A-4 (CA-4, 1; Chart 1). CA-4, isolated from the bark of the South African tree Combretum caffrum [9], affects microtubule dynamics by binding to the β-subunit of tubulin at the same site as colchicine and thus strongly inhibits tubulin polymerization [10]. This compound has been recognized to act as both a cytotoxic and a vascular disrupting agent (VDA), inducing the collapse of tumor vasculature via rapid microtubule depolymerisation [11,12]. The phosphate prodrug of CA-4, named CA-4P, with improved solubility with respect to CA-4, is in clinical trials as a VDA [13].
Chart 1.

Structures of CA-4 (1), benzo[b]thiophene and thieno[2,3-b]pyridine derivatives 2 and 3 previously published, and novel 2-alkoxycarbonyl-3-(3′,4′,5′-trimethoxyanilino)-5-aryl/heteroaryl thiophenes 4a–r.
Among the synthetic inhibitors of tubulin polymerization, we previously described the synthesis and biological characterization of two series of compounds based on the 2-alkoxycarbonyl-3-(3′,4′,5′-trimethoxyanilino)benzo[b]thiophene and thieno[2,3-b] pyridine molecular skeletons (compounds with general structure 2 and 3, respectively) that showed strong antiproliferative activity against a panel of cell lines and act as inhibitors of microtubule polymerization by interfering with the colchicine site of tubulin [14a]. To investigate the possible binding mode for this series of compounds, we performed a series of molecular docking simulations in the colchicine site. The results obtained showed that the trimethoxyphenyl unit of these compounds is placed in proximity of βCys241, and it is consistent with that previously reported for different tubulin polymerization inhibitors [14b]. Furthermore, the formation of an intramolecular hydrogen bonding interaction between the anilino and the carbonyl groups in these series of molecules allows the formation of a hydrogen bond between the ester itself and βAla250. This study indicated that important structural requirements playing a crucial role in enhancing anti-microtubule activity are the presence of an alkoxycarbonyl moiety and a 3′,4′,5′-trimethoxyanilino function at the 2- and 3-position, respectively, of the thiophene ring fused with benzene or pyridine.
Based on these observations, in this article a new series of 2-alkoxycarbonyl-3-(3′,4′,5′-trimethoxyanilino)thiophene derivatives with general formula 4 was designed to explore the role of the benzene and pyridine portion of the benzo[b]thiophene and thieno[2,3-b]pyridine nucleus, respectively, in binding in the colchicine site of tubulin. We examined the replacement of the benzo[b]thiophene and thieno[2,3-b]pyridine bicyclic systems by a thiophene ring substituted at its 5-position with an aryl or heteroaryl moiety, whereas the 2-methoxycarbonyl group and the 3-(3,4,5-trimethoxyanilino) function were kept unmodified.
To evaluate the influence of the new structural modifications on binding in the colchicine site of tubulin, preliminary docking studies were performed, following a previously reported method [15]. The new derivatives occupy the active site in a similar manner as the co-crystallized N-deacetyl-N-(2-mercaptoacetyl)-colchicine (DAMA-colchicine) (Fig. 1A: DAMA-colchicine and 2-methoxycarbonyl-3-(3′,4′,5′-trimethoxyanilino)-5-phenylthiophene derivative 4a), and their binding mode is consistent with that reported previously for the thieno[2,3-b]pyridine series (Fig. 1B: 4a and thieno[2,3-b]pyridine 3a). The trimethoxyphenyl ring is in proximity to Cys241, while the 5-phenyl ring sits deep in the small hydrophobic pocket and potentially interacts with hydrophobic amino acids Met259, Thr314, Val181 and others. These interactions are similar to those modeled with the heterocycle of the thieno[2,3-b]pyridine series (Fig. 2: compound 4a alone). Small substitutions on the 5-phenyl ring could be tolerated and did not affect the binding mode in a negative manner. As found previously for the benzo[b]thiophene and thieno [2,3-b]pyridine series, a molecular docking study revealed that the ortho-relationship between the alkoxycarbonyl group and the 3,4,5-trimethoxyanilino moiety plays an important role in activity. This allows the formation of an intramolecular hydrogen bond between the hydrogen of the anilino group and the carbonyl oxygen of the alkoxycarbonyl group, resulting in formation of a hydrogen bond between the ester itself and Ala250, further stabilizing these new compounds in the colchicine site. Encouraged by the activity obtained with compound 4a, we assessed the effects on biological activity of both the nature and position of electron-withdrawing (F, Cl, CF3 and NO2) and electron-releasing (CH3, OCH3 and OC2H5) substituents on the phenyl at the 5-position of the 2-alkoxycarbonyl-3-(3′,4′,5′-trimethoxyanilino)thiophene system as well as replacement of the phenyl with the bioisosteric thien-2′-yl moiety.
Fig. 1.

Proposed binding mode for compound 4a in comparison with DAMA-colchicine (A) and derivative 3a (B) in the colchicine site. Co-crystallized DAMA-colchicine is shown in green, the carbon atoms of compound 4a in magenta, and the carbon atoms of compound 3a in turquoise. The hydrophobic subpocket is highlighted with a turquoise-colored surface. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Fig. 2.
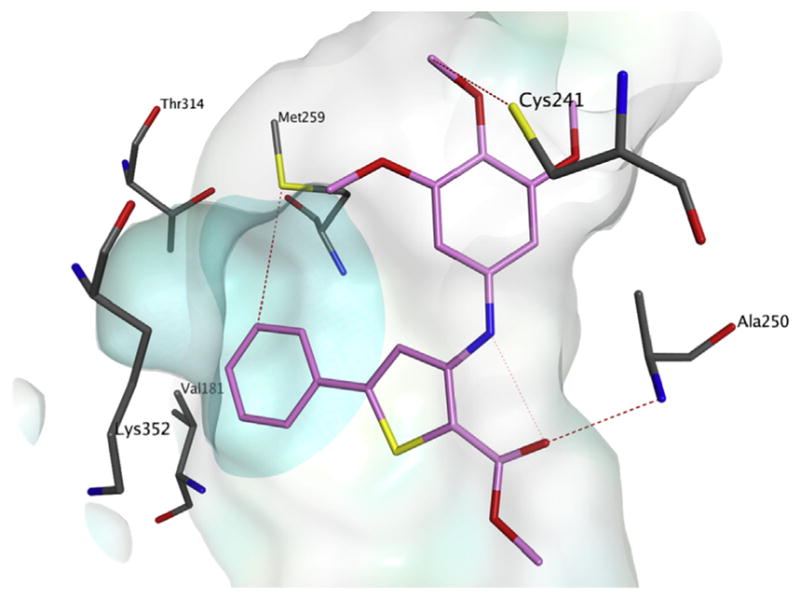
Proposed binding mode for compound 4a alone in the colchicine site. The hydrophobic subpocket is highlighted with a turquoise-colored surface. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
2. Chemistry
2-Alkoxycarbonyl-3-(3′,4′,5′-trimethoxyanilino)-5-aryl/heteroaryl thiophene derivatives 4a–r were synthesized by a four step procedure summarized in Scheme 1. β-Chloroaryl cinnamonitriles 6a–m were obtained by a modified Vilsmeier reaction of commercially available acetophenones 5a–m with phosphorus(V)oxy-chloride (POCl3) in dimethylformamide, followed by treatment with hydroxylamine hydrochloride (NH2OH·HCl) [16]. The subsequent condensation of compounds 6a–m with methyl or ethyl thioglycolate using sodium methoxide (MeONa) as base in a mixture of methanol/DMF furnished in mild conditions the corresponding 2-alkoxycarbonyl-3-amino-5-aryl/heteroaryl thiophene derivatives 7a–r in good yields through the nucleophilic displacement of chlorine, followed by base-induced ring closure in a single step. Subsequent deaminative bromination using modified Sandmeyer conditions of 3-aminothiophenes 7a–r using tert-butyl nitrite (t-BuONO) in acetonitrile in the presence of copper(II)bromide (CuBr2) gave the target bromothiophene 8a–r in excellent yield. Finally, the novel derivatives 4a–r were obtained using Buchwald-Hartwig conditions, by coupling of bromide 8a–r with 3,4,5-trimethoxyaniline in a palladium (II)acetate [Pd(OAc)2], 2,2-bis(diphenylphosphino)1,1′-binaphthyl (BINAP) catalytic system in the presence of cesium carbonate (Cs2CO3) as base in toluene at 100 °C for 18 h.
Scheme 1. Reagents.

a POCl3, DMF then NH2OH.HCl; b: HSCH2CO2CH3 or HSCH2CO2C2H5, MeONa, MeOH/DMF, 60 °C, 4 h; c: t-BuONO, CuBr2, CH3CN, 0 °C then room temperature for 2 h; d: 3,4,5-trimethoxyaniline, Pd(OAc)2, BINAP, Cs2CO3, PhMe, 100 °C, 18 h.
3. Biological results and discussion
3.1. In vitro antiproliferative activities
Table 1 summarizes the antiproliferative effects of the 2-alkoxycarbonyl-3-anilino-5-substituted thiophene derivatives 4a–r against the growth of murine leukemia (L1210), murine mammary carcinoma (FM3A/0), human T-lymphoblastoid leukemia (CEM) and human cervix carcinoma (HeLa) cells as compared with the reference compound CA-4. Comparing compounds which shared a common aryl moiety at the 5-position of the 2-alkoxycarbonyl-3-(3′,4′,5′-trimethoxyanilino)thiophene scaffold, the 2-methoxycarbonyl derivatives were more potent than the 2-ethoxycarbonyl counterparts (4a, 4d, 4f, 4i, 4k versus 4b, 4e, 4g, 4j, 4l, respectively). Ignoring compound 4p, all synthesized compounds possessed significant cell growth inhibitory activity, which was lower than 1 μM for compounds 4a, 4c–d, 4i–k, 4o and 4r in all cell lines.
Table 1.
In vitro inhibitory effects of compounds 4a–r and CA-4 (1) against the proliferation of murine leukemia (L1210), murine mammary carcinoma (FM3A), human T-lymphocyte leukemia (CEM) and human cervix carcinoma (HeLa) cells.
| Compound | IC50 (μM)a
|
|||
|---|---|---|---|---|
| L1210 | FM3A | CEM | HeLa | |
| 4a | 0.24 ± 0.01 | 0.21 ± 0.03 | 0.20 ± 0.08 | 0.67 ± 0.40 |
| 4b | 1.1 ± 0.0 | 0.98 ± 0.13 | 0.93 ± 0.08 | 1.4 ± 0.7 |
| 4c | 0.13 ± 0.07 | 0.16 ± 0.01 | 0.16 ± 0.08 | 0.16 ± 0.02 |
| 4d | 0.56 ± 0.38 | 0.72 ± 0.19 | 0.53 ± 0.38 | 0.88 ± 0.16 |
| 4e | 1.1 ± 0.0 | 1.0 ± 0.2 | 0.99 ± 0.24 | 2.1 ± 1.1 |
| 4f | 1.1 ± 0.1 | 0.88 ± 0.08 | 0.87 ± 0.18 | 0.87 ± 0.13 |
| 4g | 6.0 ± 0.0 | 5.1 ± 0.5 | 4.6 ± 0.6 | 9.8 ± 5.0 |
| 4h | 5.7 ± 0.1 | 4.6 ± 0.6 | 3.3 ± 1.7 | 5.0 ± 1.3 |
| 4i | 0.15 ± 0.08 | 0.18 ± 0.01 | 0.18 ± 0.04 | 0.26 ± 0.10 |
| 4j | 0.82 ± 0.31 | 0.82 ± 0.23 | 0.76 ± 0.38 | 0.78 ± 0.06 |
| 4k | 0.27 ± 0.03 | 0.25 ± 0.04 | 0.23 ± 0.10 | 0.84 ± 0.10 |
| 4l | 1.2 ± 0.1 | 1.2 ± 0.1 | 1.1 ± 0.3 | 2.3 ± 1.5 |
| 4m | 6.3 ± 0.0 | 5.7 ± 0.5 | 5.4 ± 1.3 | 4.4 ± 0.2 |
| 4n | 1.1 ± 0.1 | 0.99 ± 0.10 | 0.69 ± 0.46 | 0.80 ± 0.06 |
| 4o | 0.48 ± 0.27 | 0.43 ± 0.29 | 0.47 ± 0.35 | 0.89 ± 0.08 |
| 4p | >250 | >250 | >250 | 162 ± 25 |
| 4q | 1.2 ± 0.1 | 1.1 ± 0.1 | 0.81 ± 0.33 | 1.3 ± 0.2 |
| 4r | 0.99 ± 0.13 | 0.93 ± 0.08 | 0.76 ± 0.21 | 1.0 ± 0.3 |
| CA-4 (nM) | 3 ± 1 | 42 ± 6 | 2 ± 1 | 2 ± 1 |
IC50 = compound concentration required to inhibit tumor cell proliferation by 50%. Data are expressed as the mean ± SD from at least two to three independent experiments.
Hydrophobic moieties such as phenyl and thien-2′-yl were well tolerated at the 5-position of the 2-alkoxycarbonyl-3-(3′,4′,5′-trimethoxyanilino)thiophene scaffold, and variation of the phenyl substituents had variable effects on potency. Replacement of the phenyl ring of compound 4a by the bioisosteric thien-2′-yl ring, to yield derivative 4c, increased antiproliferative activity 2–4-fold against L1210 and HeLa cells, while the two compounds showed comparable potency against FM3A and CEM cells. Of all the tested compounds, the thien-2′-yl derivative 4c possessed the highest overall cytostatic potency and inhibited the growth of the four cancer cell lines with IC50 values ranging between 0.13 and 0.16 μM, being 10- to 100-fold less active than the reference compound CA-4. Compound 4i was virtually as active as 4c.
SAR was elucidated by substitution with electron-releasing and electron-withdrawing groups on the phenyl moiety at the 5-position of the 2-alkoxycarbonyl-3-(3′,4′,5′-trimethoxyanilino) thiophene system. In general, a single modification at the para-position of the phenyl ring was well tolerated, and para-substituted phenyl derivatives showed variable potencies, suggesting an opportunity for further synthetic exploration. In comparing the effect of electron-releasing (ERG's) or electron-witwdrawing groups (EWG's) at the para-position of the phenyl ring, in all cell lines compounds 4i and 4k with electron-donating methyl or methoxy groups, respectively, were generally more cytostatic than those with the electron-withdrawing fluoro or chloro moieties (derivatives 4d and 4f, respectively). The same effect was observed for the 2-ethoxycarbonyl derivatives (4j and 4l vs. 4e and 4g). Substituents at the para-position of the 5-phenyl ring showed anti-proliferative activity in the following order: Me > OMe > F > Cl=NO2>CF3≫OEt.
Potency was reduced from 4- to 7-fold after the weak ERG p-methyl (4i) was replaced with the EWG trifluoromethyl in compound 4q. Turning to the effect of ERG's on the phenyl moiety, with the exception of HeLa cells, we found that para-tolyl and para-methoxyphenyl groups (compounds 4i and 4k, respectively) caused only minor changes in antiproliferative activity relative to the unsubstituted phenyl analogue 4a.
Relative to the activity of the unsubstituted phenyl analogue 4a, the introduction of the EWG fluorine at the para-position of the phenyl ring (compound 4d) caused a 2–4-fold reduction of anti-proliferative activity in three of the four cancer cell lines. Increasing the size of the halogen from fluorine to chlorine (compounds 4d and 4f, respectively) reduced the anti-proliferative activity against L1210 and CEM cells, while the two compounds were equipotent against the FM3A and HeLa cell lines. While a single chlorine atom at the para-position of the phenyl group was tolerated for cytostatic activity (4f), double substitution by the introduction of a second chlorine atom to furnish the meta, para-dichlorophenyl derivative 4h caused a 4–5-fold reduction of potency relative to 4f in all tumor cell lines. Replacement of the chlorine atom with even stronger EWG's (trifluoromethyl or nitro; derivatives 4q and 4r, respectively) had generally little further effect on activity against the four cell lines.
Replacement of the fluorine atom of 4d with the weak ERG methyl, resulting in para-tolyl derivative 4i, increased anti-proliferative activity by 3–4-fold against all tumor cell lines, with a potency similar to that of the thien-2′-yl analogue 4c.
The number and location of methoxy substituents on the phenyl ring played a profound role in the antiproliferative activity. A comparison between the antiproliferative activities of compounds 4k and 4n–p, illustrated the antiproliferative SAR of the number of methoxy groups substituted on the phenyl ring (1 > 2»3). The introduction of a single methoxy group at the para-position (compound 4k) caused only minor changes in antiproliferative activity relative to the unsubstituted derivative 4a. Moving the methoxy group from the para-to the meta-position, to furnish 4n, reduced antiproliferative activity 3–4-fold on three of the cancer cell lines. Reduced activity (2-fold) on three cancer cell lines relative to 4k occurred with the insertion of a second methoxy group, to furnish the meta, para-dimethoxyphenyl analogue 4o, while activity was maintained against HeLa cells. This latter compound has antiproliferative activity similar to that of the meta-methoxy analogue 4n against HeLa cells and increased potency against the three other cell lines. These results suggest that the optimal position for mono-methoxy substitution is the para-position, as in compound 4k. Adding a third methoxy group, to yield derivative 4p, abolished antiproliferative activity.
These results suggested that the space for accepting substituents on the phenyl at the 5-position of 2-alkoxycarbonyl-3-(3′,4′,5′-trimethoxyanilino)thiophene system is highly limited and that the phenyl ring is specifically recognized by tubulin in a highly specific manner. In an effort to further understand the steric effect of the alkoxy substitution at the para-position of the phenyl ring, replacement of the para-methoxy with a para-ethoxy homologue (compound 4m) resulted in a 5–20-fold reduction of cytostatic activity in all cell lines.
3.2. Evaluation of cytotoxicity ofcompounds 4a, 4c and 4i in human non-cancer cells
To obtain a preliminary indication of the cytotoxic potential of these derivatives in normal human cells, three of the most active compounds (4a, 4c and 4i) were evaluated in vitro against peripheral blood lymphocytes (PBL) from healthy donors (Table 2). All compounds were practically devoid of significant cytotoxic activity in quiescent lymphocytes, with GI50's of 30–85 μM, while with the mitogenic stimulus phytohematoaglutinin (PHA), the GI50's were reduced to about 20–30 μM.
Table 2.
Cytotoxicity of compounds 4a, 4c and 4i for human peripheral blood lymphocytes (PBL).
| GI50 (μM)a
|
|||
|---|---|---|---|
| 4a | 4c | 4i | |
| PBLrestingb | 45.7 ± 10.9 | 85.7 ± 8.9 | 31.7 ± 2.0 |
| PBLPHAc | 19.0 ± 1.7 | 29.3 ± 1.2 | 20.0 ± 1.1 |
Values are the mean ± SEM from two separate experiments.
Compound concentration required to reduce cell growth inhibition by 50%.
PBL not stimulated with PHA.
PBL stimulated with PHA.
These values, even under proliferation conditions, were more than 100 times those found in the two lines of T lymphoblastic leukemia cells (L1210 and CEM) shown in Table 1. These results indicate that these compounds have little effect in rapidly proliferating normal cells and even less in quiescent cells, as previously observed for other antimitotic derivatives developed by our group [15,17,18].
3.3. Inhibition of tubulin polymerization and colchicine binding
To investigate whether the activities of these molecules were related to an interaction with the microtubule system, the more active compounds (4a, 4c–d, 4i, 4k and 4o) and reference compound CA-4 were evaluated for their in vitro tubulin polymerization inhibitory activity as well as for their inhibitory effects on the binding of [3H]colchicine to tubulin (in the latter assay, the compounds and colchicine were at 5 μM, and tubulin was at 1 μM) (Table 3). In the tubulin polymerization assay, these compounds showed IC50 values in a relatively narrow range (1.2–2.7 μM). Three compounds (4a, 4c and 4i) showed the best tubulin polymerization assembly inhibition ability (IC50: 1.2–1.3 μM), which is comparable to the IC50 of 1.1 μM obtained with CA-4, while derivatives 4d, 4k and 4o were about half as potent as CA-4. Derivatives 4a, 4c and 4i also displayed the most potent activities against the panel of four cancer cell lines. The results obtained demonstrated that antiproliferative activity correlated well with inhibition of tubulin polymerization.
Table 3.
Inhibition of tubulin polymerization and colchicine binding by compounds 4a, 4c–d, 4i, 4k, 4o and CA-4.
| Compound | Tubulin assemblya | Colchicine bindingb |
|---|---|---|
| IC50±SD (μM) | %±SD | |
| 4a | 1.3 ± 0.2 | 57 ± 5 |
| 4c | 1.2 ± 0.1 | 62 ± 1 |
| 4d | 2.2 ± 0.3 | 41 ± 5 |
| 4i | 1.2 ± 0.0 | 70 ± 3 |
| 4k | 2.0 ± 0.3 | 46 ± 4 |
| 4o | 2.7 ± 0.0 | 30 ± 3 |
| CA-4 (1) | 1.1 ± 0.1 | 99 ± 3 |
Inhibition of tubulin polymerization. Tubulin was at 10 μM.
Inhibition of [3H]colchicine binding. Tubulin, colchicine and tested compound were at 1, 5 and 5 μM, respectively.
In the colchicine binding studies, compounds 4a, 4c and 4i were also the best inhibitors of the binding of [3H]colchicine to tubulin. None, however, was quite as potent as CA-4, which in these experiments inhibited colchicine binding by 99%. The potent inhibition observed with these compounds indicated that 4a, 4c and 4i bind to tubulin at a site overlapping the colchicine site. This group of compounds were all highly potent in the biological assays (inhibition of cell growth, tubulin assembly and colchicine binding), and there was a good correlation between the three types of assays. We conclude that the antiproliferative activity of these derivatives derives from an interaction with the colchicine site of tubulin and interference with cellular microtubule assembly.
3.4. Effects of 4a, 4c and 4i on the cell cycle
The effects of compounds 4a, 4c and 4i on cell cycle progression was examined by flow cytometry in Hela cells. After a 24 h treatment (Fig. 3), the three compounds induced a G2/M arrest that became evident at the highest concentration used (500 nM). A concomitant reduction of cells in the G1 phase was also observed, while S phase cells remained essentially constant, although there was a slight increase in S phase cells that occurred at 500 nM with all compounds.
Fig. 3.

Percentage of cells in each phase of the cell cycle in HeLa cells, treated with compounds 4a (A), 4c (B) and 4i (C) at the indicated concentrations for 24 h. Cells were fixed and labeled with PI and analyzed by flow cytometry as described in the Experimental Section. Data are presented as the mean of two independent experiments ± SEM.
We also studied the association between 4c-induced G2/M arrest and alterations in G2/M regulatory protein expression in HeLa cells. As shown in Fig. 4, compound 4c caused, in a concentration-dependent manner, an increase in cyclin B1 expression after 24 h, followed by a marked reduction at 48 h, indicating an activation of the mitotic checkpoint following drug exposure [19]. This effect was confirmed by a marked reduction in the expression of phosphatase cdc25c after a 24 h incubation, even at the lowest concentration used (100 nM). In particular, it is worth noting in Fig. 4 the appearance of a slowly migrating form of cdc25c, indicating changes in its phosphorylation state, as previously observed for other antimitotics synthesized by our group [15,17,18]. The phosphorylation of cdc25c directly stimulates its phosphatase activity, and this is necessary to activate Cdk1/cyclin B on entry into mitosis [20]. Accordingly, we observed a decrease in the phosphorylated form of cyclin dependent kinase-1 (Cdk1) after 24 h and 48 h treatments at 250 nM.
Fig. 4.
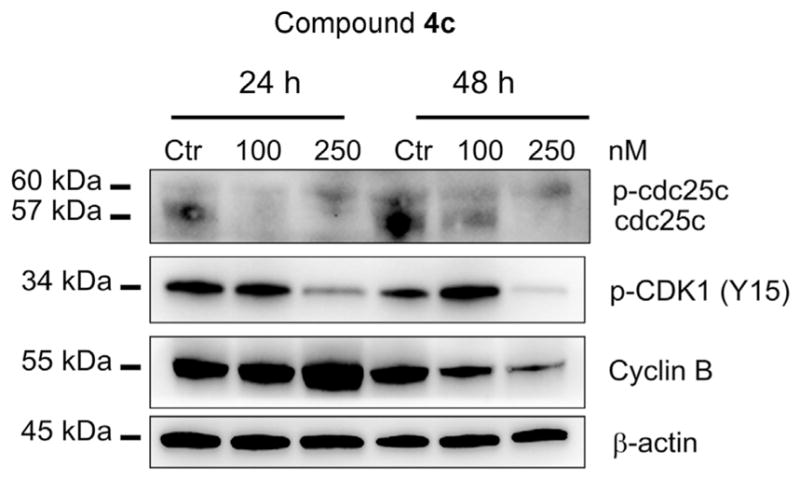
Effect of compound 4c on cell cycle checkpoint proteins. HeLa cells were treated for 24 or 48 h with the indicated concentrations of 4c. The cells were harvested and lysed for detection of the expression of the indicated proteins by western blot analysis. To confirm equal protein loading, each membrane was stripped and reprobed with anti-β-actin antibody.
3.5. Compounds 4a, 4c and 4i induce apoptosis
To evaluate the mode of cell death, we treated HeLa cells with compounds 4a, 4c and 4i, and, after 24 and 48 h incubations, we performed a biparametric cytofluorimetric analysis using propidium iodide (PI) and annexin-V-FITC, which stain DNA and phosphatidylserine (PS) residues, respectively. As shown in Fig. 5, the three compounds induced a significant proportion of apoptotic cells after the 24 h incubation period, and this proportion further increased at 48 h. The most active compounds appear to be 4c and 4i, in good agreement with the antiproliferative results presented in Table 1.
Fig. 5.

Flow cytometric analysis of apoptotic cells after treatment of HeLa cells with 4a, 4c and 4i at the indicated concentrations after incubation for 24 or 48 h. The cells were harvested and labeled with annexin-V-FITC and PI and analyzed by flow cytometry. Data are presented as mean ± SEM of three independent experiments.
3.6. Compound 4c induced mitochondrial depolarization and ROS production
It is well known that mitochondria are involved in the initiation of apoptosis, since, at an early stage, apoptotic stimuli alter the mitochondrial transmembrane potential (Δψmt) [21,22]. To determine whether the cells treated with compound 4c underwent mitochondrial depolarization, we assessed the changes in Δψmt in HeLa cells by flow cytometry using the fluorescent dye JC-1 [23]. HeLa cells treated with compound 4c (100–250 nM) showed a time-dependent increase in the percentage of cells with low Δψmt (Fig. 6, Panel A). The depolarization of the mitochondrial membrane is associated with the appearance of annexin-V positivity in the treated cells when they are in an early apoptotic stage [24]. One of the major consequences of the increase of mitochondrial membrane permeability is the release into the cytosol of pro-apoptotic molecules such as AIF, Smac/Diablo and, in particular, cytochrome c [21]. This release triggers ROS production at the mitochondrial level during the later stages of the cell death program [24–26]. We therefore investigated whether ROS production increased after treatment with compound 4c. We analyzed ROS production by flow cytometry, using the fluorescence indicator 2,7-dichlorodihydrofluorescein diacetate (H2-DCFDA). As shown in Fig. 6 (Panel B), compound 4c induced significant production of ROS starting after a treatment of 12–24 h at 250 nM, in good agreement with the mitochondrial depolarization described above.
Fig. 6.

Assessment of mitochondrial membrane potential (Δψmt) after treatment of HeLa cells (Panel A) with compound 4c. Cells were treated with the indicated concentration of compound for 6, 12, 24 or 48 h and then stained with the fluorescent probe JC-1 for analysis of mitochondrial potential. Cells were then analyzed by flow cytometry as described in the Experimental Section. Data are presented as mean ± SEM of three independent experiments. Assessment of ROS production after treatment of HeLa cells with compound 4c (Panel B). Cells were treated with the indicated concentration for 6, 12, 24 or 48 h and then stained with H2-DCFDA for evaluation of ROS levels. Cells were then analyzed by flow cytometry as described in the Experimental Section. Data are presented as mean ± SEM of three independent experiments.
3.7. Compound 4c induced PARP cleavage and decreased expression of anti-apoptotic proteins
To further investigate the mechanism of apoptosis induction by 4c, we analyzed the expression of poly (ADP-Ribose) polymerase (PARP), a protein involved in late stage apoptosis, and the expression of two anti-apoptotic proteins belonging to the Bcl-2 family. As shown in Fig. 7, compound 4c in HeLa cells caused a concentration-and time-dependent cleavage of PARP, confirming the pro-apoptotic properties of 4c.
Fig. 7.

Western blot analysis of Bcl-2, Mcl-1 and PARP after treatment of HeLa cells with 4c at the indicated concentrations and times. To confirm equal protein loading, each membrane was stripped and reprobed with anti-β-actin antibody.
We also investigated the expression of anti-apoptotic proteins such as Bcl-2 and Mcl-1. It is well-known that antimitotic agents can modulate both expression levels and activity of many proteins of the Bcl-2 family [27–29]. Our results (Fig. 7) showed that the expression of both anti-apoptotic proteins Bcl-2 and Mcl-1 were decreased starting after 24 h of treatment, even at the lowest 4c concentration used (0.1 μM). These studies underline the importance of Mcl-1 phosphorylation and its subsequent degradation in response to antimitotic agents and that this event potentiates cell death [30,31].
4. Conclusions
In conclusion, we have discovered a new class of simple synthetic inhibitors of tubulin polymerization based on the molecular skeleton of 2-alkoxycarbonyl-3-(3′,4′,5′-trimethoxyanilino)thiophene. These derivatives were designed and synthesized based on modification of benzo[b]thiophene and thieno[2,3-b]pyridine analogues previously published. The results demonstrated that the aryl or 2-thienyl moieties at the 5-position of the 2-alkoxycarbonyl-3-(3′,4′,5′-trimethoxyanilino)thiophene system could replace either the benzene or the pyridine portion of benzo[b]thiophene and thieno[2,3-b]pyridine derivatives with general structure 2 and 3, respectively. We explored SAR by examining various substitutions with EWGs and ERGs on the phenyl at the 5-position of the 2-alkoxycarbonyl-3-(3′,4′,5′-trimethoxyanilino)thiophene scaffold. The presence of ERGs such as methyl or methoxy was beneficial for antiproliferative activity, as these compounds proved to be more potent than the corresponding derivatives with the EWGs fluorine or chlorine. Generally, it was found that most substituents in the para-position resulted in lower activity as compared to the unsubstituted parent compound 4a, with the least deleterious being methyl and methoxy moieties (compounds 4i and 4k, respectively).
It is clear that the substitution pattern on the phenyl at the 5-position of the 2-methoxycarbonyl-3-(3′,4′,5′-trimethoxyanilino) thiophene system plays an important role for antitubulin and antiproliferative activities, and this was supported by the molecular docking studies. SAR studies showed that the 2-methoxycarbonyl-3-(3′,4′,5′-trimethoxyanilino)-5-phenylthiophene derivative 4a, its bioisosteric thien-2′-yl analogue 4c as well as the para-tolyl and para-methoxyphenyl analogues 4i and 4k, respectively, displayed high antiproliferative activities, with IC50 values ranging between 0.13 and 0.24, 0.16–0.25, 0.16–0.23 and 0.16–0.84 μM, respectively, against the L1210, FM3A, CEM and HeLa cell lines. Of all the tested compounds, derivative 4c possessed the highest overall cytostatic potency with IC50 values ranging from 0.13 to 0.16 μM against the panel of four cancer cell lines. The antiproliferative activity was considerably increased by replacing the EWG fluorine with the ERG methyl group (compounds 4d and 4i, respectively), with the latter compound being about 3–6-fold more active than the former. By comparing 4d and 4f, replacement of the para-fluoro group in 4d with the chloro (derivative 4f) led to little change in activity against FM3A and HeLa cells, while 4f was less potent than 4d in L1210 and CEM cells. Replacement of the EWG para-chlorine atom of 4f with the ERG para-methyl moiety, to furnish derivative 4i, resulted in 4–6-fold enhancement in antiproliferative activity against the four cancer cell lines. Replacement of the methyl with a methoxy group (derivative 4k) produced a 2- and 3-fold reduction in potency against L1210 and HeLa cells, respectively, while the difference between 4i and 4k were minimal for FM3A and CEM cells.
The antiproliferative activity of the thiophene derivatives 4k and 4n–p can be further characterized in terms of the substitution pattern and the number of methoxy groups on the phenyl ring. The introduction of a single methoxy group at the para-position caused only minor changes in antiproliferative activity relative to the unsubstituted derivative 4a. Reduced activity occurred in three of the four cancer cell lines when the methoxy substituent was moved from the para-to the meta-position (4n), with the exception of HeLa cells. As previously observed comparing the activities of para-chloro and meta, para-dichloro derivatives 4f and 4h, respectively, the introduction of a second methoxy group at the meta-position of para-methoxyphenyl derivative 4k, to furnish the meta, para-dimethoxyphenyl analogue 4o, decreased potency in three of the four cancer cell lines by 2-fold, with the exception of the HeLa cells. The 3′,4′,5′-trimethoxyphenyl derivative 4p was not active, and this result suggested the space for accepting substituents on the phenyl ring at the 5-position of the 2-alkoxycarbonyl-3-(3′,4′,5′-trime-thoxyanilino)thiophene scaffold is highly limited. This conclusion was supported by comparing the reduced activity (on average 20-fold) of the para-ethoxy homologue 4m relative to the para-methoxy analogue 4k.
The results we obtained indicated that compound 4c could induce tumor cell apoptosis through reducing the mitochondrial membrane potential and regulating the expression of apoptosis-related proteins in tumor cells.
5. Experimental protocols
5.1. Chemistry
5.1.1. Materials and methods
1H NMR experiments were recorded on either a Bruker AC 200 or a Varian 400 Mercury Plus spectrometer, while 13C NMR spectra were recorded on a Varian 400 Mercury Plus spectrometer. Chemical shifts (δ) are given in ppm upfield from tetramethylsilane as internal standard, and the spectra were recorded in appropriate deuterated solvents, as indicated. Positive-ion electrospray ionization (ESI) mass spectra were recorded on a double-focusing Finnigan MAT 95 instrument with BE geometry. Melting points (mp) were determined on a Buchi-Tottoli apparatus and are uncorrected. All products reported showed 1H and 13C NMR spectra in agreement with the assigned structures. The purity of tested compounds was determined by combustion elemental analyses conducted by the Microanalytical Laboratory of the Chemistry Department of the University of Ferrara with a Yanagimoto MT-5 CHN recorder elemental analyzer. All tested compounds yielded data consistent with a purity of at least 95% as compared with the theoretical values. Reaction courses and product mixtures were routinely monitored by TLC on silica gel (precoated F254 Merck plates), and compounds were visualized with aqueous KMnO4. Flash chromatography was performed using 230–400 mesh silica gel and the indicated solvent system. Organic solutions were dried over anhydrous Na2SO4.
5.1.2. General procedure A for the synthesis of compounds 7a–r
To a solution of methyl/ethyl thioglycolate (10 mmol) in methanol (5 mL) was added a solution of sodium methoxide (0.54 g, 10 mmol) in methanol (5 mL), and the mixture was stirred for 1 h. To the above mixture, a solution of the corresponding 3-chloroacrylonitrile 6a–m (7.5 mmol) in DMF (5 mL) was added dropwise for 10 min at room temperature and stirred at 60 °C for 2 h. Then, a solution of sodium methoxide (1.08 g, 20 mmol) in methanol (10 mL) was added dropwise at room temperature, and stirring was continued for 2 h at 60 °C. The mixture was poured into cold water and stirred for 10 min. The solution was extracted with CH2Cl2 (3 × 20 mL), and the combined CH2Cl2 layer was washed with water (20 mL), brine (20 mL), and dried. The solution was filtered, and, after concentration under reduced pressure, the residue was purified by silica gel column chromatography to furnish the corresponding thiophene derivatives 7a–r.
5.1.2.1. Methyl 3-amino-5-phenylthiophene-2-carboxylate (7a)
Following general procedure A, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 1:9 (v:v) as the eluting solution, to furnish 7a as a yellow solid (54% yield), mp 130–132 °C. 1H NMR (CDCl3) δ: 3.85 (s, 3H), 5.47 (bs, 2H), 6.77 (s, 1H), 7.38 (m, 3H), 7.57 (m, 2H). MS (ESI): [M+1]+ = 234.2.
5.1.2.2. Ethyl 3-amino-5-phenylthiophene-2-carboxylate (7b)
Following general procedure A, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as the eluting solution, to furnish 7b as a brown solid (95% yield), mp 90–91 °C. 1H NMR (CDCl3) δ: 1.33 (t, J = 7.0 Hz, 3H), 4.26 (q, J = 7.0 Hz, 2H), 5.46 (bs, 2H), 6.77 (s, 1H), 7.36 (m, 3H), 7.57 (m, 2H). MS (ESI): [M+1]+ = 248.3.
5.1.2.3. Methyl 4-amino-[2,2′-bithiophene]-5-carboxylate (7c)
Following general procedure A, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 3:7 (v:v) as the eluting solution, to furnish 7c as a brown solid (78% yield), mp 105–107 °C. 1H NMR (CDCl3) δ: 3.86 (s, 3H), 5.73 (bs, 2H), 7.38 (m, 2H), 7.45 (m, 1H), 7.86 (dd, J = 5.0 and 1.2 Hz, 1H). MS (ESI): [M+1]+ = 240.3.
5.1.2.4. Methyl 3-amino-5-(4-fluorophenyl)thiophene-2-carboxylate (7d)
Following general procedure A, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as the eluting solution, to furnish 7d as a white solid (84% yield), mp 164–166 °C. 1H NMR (DMSO-d6) δ: 3.71 (s, 3H), 6.59 (bs, 2H), 6.92 (s, 1H), 7.27 (t, J = 8.8 Hz, 2H), 7.63 (dd, J = 8.8 and 5.4 Hz, 2H). MS (ESI): [M+1]+ = 252.3.
5.1.2.5. Ethyl 3-amino-5-(4-fluorophenyl)thiophene-2-carboxylate (7e)
Following general procedure A, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as the eluting solution, to furnish 7e as a yellow solid (78% yield), mp 110–112 °C. 1H NMR (CDCl3) δ: 1.37 (t, J = 7.0 Hz, 3H), 4.29 (q, J = 7.0 Hz, 2H), 5.46 (bs, 2H), 6.70 (s, 1H), 7.03 (t, J = 8.8 Hz, 2H), 7.52 (dd, J = 8.8 and 5.4 Hz, 2H). MS (ESI): [M+1]+ = 266.3.
5.1.2.6. Methyl 3-amino-5-(4-chlorophenyl)thiophene-2-carboxylate (7f)
Following general procedure A, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as the eluting solution, to furnish 7f as a cream-colored solid (66% yield), mp 134–136 °C. 1H NMR (CDCl3) δ: 3.84 (s, 3H), 5,48 (bs, 2H), 6.74 (s, 1H), 7.33 (d, J = 8.8 Hz, 2H), 7.48 (d, J = 8.8 Hz, 2H). MS (ESI): [M+1]+ = 268.7.
5.1.2.7. Ethyl 3-amino-5-(4-chlorophenyl)thiophene-2-carboxylate (7g)
Following general procedure A, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 3:7 (v:v) as the eluting solution, to furnish 7g as a yellow solid (72% yield), mp 103–105 °C. 1H NMR (CDCl3) δ: 1.36 (t, J = 7.2 Hz, 3H), 4.29 (q, J = 7.2 Hz, 2H), 5.46 (bs, 2H), 6.74 (s, 1H), 7.33 (d, J = 8.8 Hz, 2H), 7.50 (d, J = 8.8 Hz, 2H). MS (ESI): [M+1]+ = 282.8.
5.1.2.8. Methyl 3-amino-5-(3,4-dichlorophenyl)thiophene-2-carboxylate (7h)
Following general procedure A, the crude residue was purified by flash chromatography, using ethyl acetate:-petroleum ether 3:7 (v:v) as the eluting solution, to furnish 7h as a grey solid (58% yield), mp 154–156 °C. 1H NMR (CDCl3) δ: 3.85 (s, 3H), 5.48 (bs, 2H), 6.75 (s, 1H), 7.40 (dd, J = 8.4 and 2.0 Hz, 1H), 7.44 (d, J = 8.4 Hz, 1H), 7.65 (d, J = 2.0 Hz, 1H). MS (ESI): [M]+ = 303.2.
5.1.2.9. Methyl 3-amino-5-(p-tolyl)thiophene-2-carboxylate (7i)
Following general procedure A, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 1:9 (v:v) as the eluting solution, to furnish 7i as an orange solid (54% yield), mp 140–142 °C. 1H NMR (CDCl3) δ: 2.37 (s, 3H), 3.84 (s, 3H), 5.46 (bs, 2H), 6.73 (s, 1H), 7.17 (d, J = 8.2 Hz, 2H), 7.46 (d, J = 8.2 Hz, 2H). MS (ESI): [M+1]+ = 248.2.
5.1.2.10. Ethyl 3-amino-5-(p-tolyl)thiophene-2-carboxylate (7j)
Following general procedure A, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 1:9 (v:v) as the eluting solution, to furnish 7j as a yellow solid (80% yield), mp 103–105 °C. 1H NMR (CDCl3) δ: 1.36 (t, J = 7.0 Hz, 3H), 2.37 (s, 3H), 4.29 (q, J = 7.0 Hz, 2H), 5.45 (bs, 2H), 6.73 (s,1H), 7.17 (d, J = 7.8 Hz, 2H), 7.46 (d, J = 7.8 Hz, 2H). MS (ESI): [M+1]+ = 262.3.
5.1.2.11. Methyl 3-amino-5-(4-methoxyphenyl)thiophene-2-carboxylate (7k)
Following general procedure A, the crude residue was purified by flash chromatography, using ethyl acetate:-petroleum ether 3:7 (v:v) as the eluting solution, to furnish 7k as a yellow solid (95% yield), mp 118–120 °C. 1H NMR (CDCl3) δ: 3.84 (s, 6H), 5.46 (bs, 2H), 6.67 (s, 1H), 6.89 (d, J = 9.2 Hz, 2H), 7.51 (d, J = 9.2 Hz, 2H). MS (ESI): [M+1]+ = 264.3.
5.1.2.12. Ethyl 3-amino-5-(4-methoxyphenyl)thiophene-2-carboxylate (7l)
Following general procedure A, the crude residue was purified by flash chromatography, using ethyl acetate:-petroleum ether 3:7 (v:v) as the eluting solution, to furnish 7l as a yellow solid (82% yield), mp 132–134 °C. 1H NMR (CDCl3) δ: 1.36 (t, J = 7.2 Hz, 3H), 3.83 (s, 3H), 4.28 (q, J = 7.2 Hz, 2H), 6.66 (s, 3H), 6.88 (d, J = 9.0 Hz, 2H), 7.51 (d, J = 9.0 Hz, 2H). MS (ESI): [M+1]+ = 278.3.
5.1.2.13. Methyl 3-amino-5-(4-ethoxyphenyl)thiophene-2-carboxylate (7m)
Following general procedure A, the crude residue was purified by flash chromatography, using ethyl acetate:-petroleum ether 3:7 (v:v) as the eluting solution, to furnish 7m as a pink solid (82% yield), mp 153–155 °C. 1H NMR (CDCl3) δ: 1.39 (t, J = 6.8 Hz, 3H), 3.83 (s, 3H), 4.04 (q, J = 6.8 Hz, 2H), 5.48 (bs, 2H), 6.66 (s,1H), 6.87 (d, J = 8.8 Hz, 2H), 7.48 (d, J = 8.8 Hz, 2H). MS (ESI): [M+1]+ = 278.3.
5.1.2.14. Methyl 3-amino-5-(3-methoxyphenyl)thiophene-2-carboxylate (7n)
Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:-petroleum ether 3:7 (v:v) as the eluting solution, to furnish 7n as a yellow oil (95% yield). 1H NMR (CDCl3) δ: 3.84 (s, 6H), 5.47 (bs, 2H), 6.76 (s, 1H), 6.93 (m, 1H), 7.09 (t, J = 1.8 Hz, 1H), 7.19 (m, 1H), 7.26 (t, J = 7.8 Hz, 1H). MS (ESI): [M+1]+ = 263.4.
5.1.2.15. Methyl 3-amino-5-(3,4-dimethoxyphenyl)thiophene-2-carboxylate (7o)
Following general procedure A, the crude residue was purified by flash chromatography, using ethyl acetate:-petroleum ether 3:7 (v:v) as the eluting solution, to furnish 7o as a brown solid (95% yield), mp 88–90 °C. 1H NMR (CDCl3) δ: 3.84 (s, 3H), 3.91 (s, 3H), 3.92 (s, 3H), 5.47 (bs, 2H), 6.68 (s, 1H), 6.85 (d, J = 8.6 Hz, 1H), 7.06 (d, J = 2.0 Hz, 1H), 7.15 (dd, J = 8.6 and 2.0 Hz, 1H). MS (ESI): [M+1]+ = 284.3.
5.1.2.16. Methyl 3-amino-5-(3,4,5-trimethoxyphenyl)thiophene-2-carboxylate (7p)
Following general procedure A, the crude residue was purified by flash chromatography, using ethyl acetate:-petroleum ether 4:6 (v:v) as the eluting solution, to furnish 7p as a brown solid (95% yield), mp 143–145 °C. 1H NMR (CDCl3) δ: 3.84 (s, 3H), 3.87 (s, 3H), 3.90 (s, 6H), 5.47 (bs, 2H), 6.7 (s,1H), 6.78 (s, 2H). MS (ESI): [M+1]+ = 324.4.
5.1.2.17. Methyl 3-amino-5-(4-(trifluoromethyl)phenyl)thiophene-2-carboxylate (7q)
Following general procedure A, the crude residue was purified by flash chromatography, using ethyl acetate:-petroleum ether 2:8 (v:v) as the eluting solution, to furnish 7q as a brown solid (63% yield), mp 139–140 °C. 1H NMR (CDCl3) δ: 3.86 (s, 3H), 5.49 (bs, 2H), 6.83 (s, 1H), 7.61 (d, J = 9.0 Hz, 2H), 7.68 (d, J = 9.0 Hz, 2H). MS (ESI): [M+1]+ = 302.3.
5.1.2.18. Methyl 3-amino-5-(4-nitrophenyl)thiophene-2-carboxylate (7r)
Following general procedure A, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 3:7 (v:v) as the eluting solution, to furnish 7r as a yellow solid (61% yield), mp 219–220 °C. 1H NMR (CDCl3) δ: 3.86 (s, 3H), 5.68 (bs, 2H), 6.90 (s, 1H), 7.69 (d, J = 9.2 Hz, 2H), 8.23 (d, J = 9.0 Hz, 2H). MS (ESI): [M+1]+ = 302.3.
5.1.3. General procedure B for the synthesis of compounds 8a–r
To a solution of tert-butyl nitrite (360 μL, 3 mmol) in anhydrous acetonitrile (10 mL) at 0 °C under an Ar atmosphere in a dry three-necked round-bottom flask, was added anhydrous CuBr2 (536 mg, 2.4 mmol). Derivative 7a–r (2 mmol) was then slowly added portion-wise to the mixture, which was stirred at 0 °C for 1 h. The dark mixture was allowed to reach room temperature, stirred for 2 h and then poured into an aqueous HCl solution (10%, 10 mL). The mixture was extracted with CH2Cl2 (3 × 15 mL). The organic phase was washed with brine (10 mL), dried and concentrated at reduced pressure to furnish a residue that was purified by flash chromatography on silica gel to give 8a–r.
5.1.3.1. Methyl 3-bromo-5-phenylthiophene-2-carboxylate (8a)
Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 1:9 (v:v) as the eluting solution, to furnish 8a as a yellow oil (69% yield). 1H NMR (CDCl3) δ: 3.91 (s, 3H), 7.29 (s, 1H), 7.42 (m, 3H), 7.58 (m, 2H). MS (ESI): [M+1]+ = 296.1 and 298.1.
5.1.3.2. Ethyl 3-bromo-5-phenylthiophene-2-carboxylate (8b)
Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 1:9 (v:v) as the eluting solution, to furnish 8b as a yellow solid (63% yield), mp 80–81 °C. 1H NMR (CDCl3) δ: 1.32 (t, J = 7.4 Hz, 3H), 4.30 (q, J = 7.6 Hz, 2H), 7.32 (m, 4H), 7.38 (s, 1H), 7.48 (m, 1H). MS (ESI): [M+1]+ = 327.1 and 329.1.
5.1.3.3. Methyl 4-bromo-[2,2′-bithiophene]-5-carboxylate (8c)
Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 1:9 (v:v) as the eluting solution, to furnish 8c as a yellow oil (55% yield). 1H NMR (CDCl3) δ: 3.89 (s, 3H), 7.18 (s, 1H), 7.30 (m, 1H), 7.41 (dd, J = 5.2 and 2.8 Hz, 1H), 7.51 (m, 1H). MS (ESI): [M+1]+ = 302.1 and 304.2.
5.1.3.4. Methyl 3-bromo-5-(4-fluorophenyl)thiophene-2-carboxylate (8d)
Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 1:9 (v:v) as the eluting solution, to furnish 8d as a yellow solid (58% yield), mp 126–128 °C. 1H NMR (CDCl3) δ: 3.91 (s, 3H), 7.08 (t, J = 8.8 Hz, 2H), 7.22 (s, 1H), 7.53 (dd, J = 8.8 and 5.2 Hz, 2H). MS (ESI): [M+1]+ = 314.9 and 316.9.
5.1.3.5. Ethyl 3-bromo-5-(4-fluorophenyl)thiophene-2-carboxylate (8e)
Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 1:9 (v:v) as the eluting solution, to furnish 8e as a yellow solid (67% yield), mp 90–92 °C. 1H NMR (CDCl3) δ: 1.36 (t, J = 7.0 Hz, 3H), 4.33 (q, J = 7.0 Hz, 2H), 7.07 (t, J = 8.8 Hz, 2H), 7.22 (s,1H), 7.53 (dd, J = 8.8 and 5.2 Hz, 2H). MS (ESI): [M+1]+ = 329.1 and 331.1.
5.1.3.6. Methyl 3-bromo-5-(4-chlorophenyl)thiophene-2-carboxylate (8f)
Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 0.5:9.5 (v:v) as the eluting solution, to furnish 8f as a brown solid (64% yield), mp 122–124 °C. 1H NMR (CDCl3) δ: 3.91 (s, 3H), 7.26 (s, 1H), 7.42 (d, J = 8.4 Hz, 2H), 7.52 (d, J = 8.4 Hz, 2H). MS (ESI): [M+1]+ = 330.9 and 332.9.
5.1.3.7. Ethyl 3-bromo-5-(4-chlorophenyl)thiophene-2-carboxylate (8g)
Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 1:9 (v:v) as the eluting solution, to furnish 8g as a yellow solid (58% yield), mp 98–100 °C. 1H NMR (CDCl3) δ: 1.36 (t, J = 7.0 Hz, 3H), 4.36 (q, J = 7.0 Hz, 2H), 7.232 (s, 1H), 7.37 (d, J = 8.8 Hz, 2H), 7.51 (d, J = 8.6 Hz, 2H). MS (ESI): [M+1]+ = 344.9 and 346.9.
5.1.3.8. Methyl 3-bromo-5-(3,4-dichlorophenyl)thiophene-2-carboxylate (8h)
Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:-petroleum ether 2:8 (v:v) as the eluting solution, to furnish 8h as a yellow solid (51% yield), mp 122–124 °C. 1H NMR (CDCl3) δ: 3.92 (s, 3H), 7.28 (s, 1H), 7.42 (dd, J = 8.4 and 2.0 Hz, 1H), 7.48 (d, J = 8.4 Hz, 1H), 7.68 (d, J = 2.0 Hz, 1H). MS (ESI): [M]+ = 364.9 and 366.9.
5.1.3.9. Methyl 3-bromo-5-(p-tolyl)thiophene-2-carboxylate (8i)
Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 1:9 (v:v) as the eluting solution, to furnish 8i as a yellow solid (75% yield), mp 90–92 °C. 1H NMR (CDCl3) δ: 2.38 (s, 3H), 3.89 (s, 3H), 7.20 (d, J = 8.2 Hz, 2H), 7.26 (s, 1H), 7.47 (d, J = 8.2 Hz, 2H). MS (ESI): [M+1]+ = 312.1 and 314.1.
5.1.3.10. Ethyl 3-bromo-5-(p-tolyl)thiophene-2-carboxylate (8j)
Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 1:9 (v:v) as the eluting solution, to furnish 8j as a yellow solid (54% yield), mp 98–100 °C. 1H NMR (CDCl3) δ: 1.36 (t, J = 7.2 Hz, 3H), 2.38 (s, 3H), 4.36 (q, J = 7.2 Hz, 2H), 7.20 (d, J = 8.2 Hz, 2H), 7.24 (s, 1H), 7.47 (d, J = 8.2 Hz, 2H). MS (ESI): [M+1]+ = 325.1 and 327.1.
5.1.3.11. Methyl 3-bromo-5-(4-methoxyphenyl)thiophene-2-carboxylate (8k)
Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:-petroleum ether 2:8 (v:v) as the eluting solution, to furnish 8k as a yellow solid (58% yield), mp 106–108 °C. 1H NMR (CDCl3) δ: 3.85 (s, 3H), 3.90 (s, 3H), 6.92 (d, J = 8.8 Hz, 2H), 7.18 (s, 1H), 7.51 (d, J = 8.8 Hz, 2H). MS (ESI): [M+1]+ = 327.1 and 329.1.
5.1.3.12. Ethyl 3-bromo-5-(4-methoxyphenyl)thiophene-2-carboxylate (8l)
Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as the eluting solution, to furnish 8l as a yellow solid (76% yield), mp 75–77 °C. 1H NMR (CDCl3) δ: 1.36 (t, J = 7.2 Hz, 3H), 3.82 (s, 3H), 4.36 (q, J = 7.2 Hz, 2H), 6.91 (d, J = 8.8 Hz, 2H), 7.17 (s, 1H), 7.52 (d, J = 8.8 Hz, 2H). MS (ESI): [M+1]+ = 341.1 and 343.1.
5.1.3.13. Methyl 3-bromo-5-(4-ethoxyphenyl)thiophene-2-carboxylate (8m)
Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:-petroleum ether 3:7 (v:v) as the eluting solution, to furnish 8m as a yellow solid (46% yield), mp 106–108 °C. 1H NMR (CDCl3) δ: 1.40 (t, J = 7.2 Hz, 3H), 3.90 (s, 3H), 4.05 (q, J = 7.2 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H), 7.18 (s, 1H), 7.49 (d, J = 8.8 Hz, 2H). MS (ESI): [M+1]+ = 341.1 and 343.1.
5.1.3.14. Methyl 3-bromo-5-(3-methoxyphenyl)thiophene-2-carboxylate (8n)
Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:-petroleum ether 2:8 (v:v) as the eluting solution, to furnish 8n as a yellow oil (84% yield). 1H NMR (CDCl3) δ: 3.82 (s, 3H), 3.86 (s, 3H), 6.96 (td, J = 8.0, 2.2 and 1.0 Hz, 1H), 7.11 (t, J = 2.2 Hz, 1H), 7.20 (m, 1H), 7.28 (s, 1H), 7.34 (d, J = 8.0 Hz, 1H). MS (ESI): [M+1]+ = 327.2 and 329.2.
5.1.3.15. Methyl 3-bromo-5-(3,4-dimethoxyphenyl)thiophene-2-carboxylate (8o)
Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:-petroleum ether 3:7 (v:v) as the eluting solution, to furnish 8o as an orange solid (77% yield), mp 105–107 °C. 1H NMR (CDCl3) δ: 3.89 (s, 3H), 3.90 (s, 3H), 3.92 (s, 3H), 6.87 (d, J = 8.6 Hz, 1H), 7.05 (d, J = 2.0 Hz, 1H), 7.19 (m, 2H). MS (ESI): [M+1]+ = 358.1 and 360.1.
5.1.3.16. Methyl 3-bromo-5-(3,4,5-trimethoxyphenyl)thiophene-2-carboxylate (8p)
Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:-petroleum ether 2:8 (v:v) as the eluting solution, to furnish 8p as a yellow solid (48% yield), mp 143–145 °C. 1H NMR (CDCl3) δ: 3.88 (s, 3H), 3.90 (s, 3H), 3.92 (s, 3H), 6.78 (s, 2H), 7.26 (s, 1H). MS (ESI): [M+1]+ = 389.1 and 391.1.
5.1.3.17. Methyl 3-bromo-5-(4-(trifluoromethyl)phenyl)thiophene-2-carboxylate (8q)
Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:-petroleum ether 1:9 (v:v) as the eluting solution, to furnish 8q as a brown solid (58% yield), mp 98–100 °C. 1H NMR (CDCl3) δ: 3.93 (s, 3H), 7.36 (s, 1H), 7.69 (s, 4H). MS (ESI): [M+1]+ = 364.1 and 366.2.
5.1.3.18. Methyl 3-bromo-5-(4-nitrophenyl)thiophene-2-carboxylate (8r)
Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as the eluting solution, to furnish 8r as a yellow solid (67% yield), mp 224–226 °C. 1H NMR (CDCl3) δ: 3.94 (s, 3H), 7.43 (s, 1H), 7.73 (d, J = 8.2 Hz, 2H), 8.27 (d, J = 8.8 Hz, 2H). MS (ESI): [M+1]+ = 364.1 and 366.1.
5.1.4. General procedure C for the preparation of compounds 4a–r
A dry Schlenk tube was charged with dry toluene (5 mL), bromo derivative 8a–r (0.5 mmol), Pd(OAc)2 (13 mol%, 15 mg), rac-BINAP (4 mol%, 15 mg), Cs2CO3 (230 mg, 0.7 mmol, 1.4 equiv.) and 3,4,5-trimethoxyaniline (137 mg, 0.75 mmol, 1.5 equiv.) under Ar, and the mixture was heated at 100 °C for 18 h. After cooling, the mixture was filtered through a pad of celite and the filtrate diluted with EtOAc (10 mL) and water (5 mL). The organic phase was washed with brine (5 mL), dried and concentrated under vacuum. The residue was purified by column chromatography on silica gel to furnish 4a–r.
5.1.4.1. Methyl 5-phenyl-3-[(3,4,5-trimethoxyphenyl)amino]thiophene-2-carboxylate (4a)
Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as the eluting solution, to furnish 4a as a yellow solid (52% yield), mp 120–121 °C. 1H NMR (CDCl3) δ: 3.85 (s, 9H), 3.89 (s, 3H), 6.47 (s, 2H), 7.37 (m, 4H), 7.61 (m, 2H), 8.67 (s, 1H). 13C NMR (CDCl3) δ: 56.30 (2x), 61.14, 65.96, 99.13 (2x), 113.70, 114.57, 126.08 (2C), 127.87, 129.16 (2C), 129.28, 133.46, 134.52, 137.47, 149.73, 152.32, 153.88 (2C). MS (ESI): [M+1]+ = 400.5. Anal. calcd for C21H21NO5S. C, 63.14; H, 5.30; N, 3.51; found: C, 63.01; H, 5.13; N, 3.36.
5.1.4.2. Ethyl 5-phenyl-3-[(3,4,5-trimethoxyphenyl)amino]thiophene-2-carboxylate (4b)
Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 3:7 (v:v) as the eluting solution, to furnish 4b as a yellow solid (81% yield), mp 124–125 °C. 1H NMR (CDCl3) δ: 1.36 (t, J = 7.6 Hz, 3H), 3.85 (s, 9H), 4.33 (q, J = 7.6 Hz, 2H), 6.46 (s, 2H), 7.37 (m, 4H), 7.56 (m, 2H), 8.70 (s, 1H). 13C NMR (CDCl3) δ: 14.63, 56.29 (2C), 60.47, 61.14, 99.02 (2C), 113.71, 114.52, 126.07 (2C), 127.78, 129.15 (2C), 129.22, 133.52, 134.48, 137.55, 149.51, 152.17, 153.88 (2C). MS (ESI): [M+1]+ = 414.4. Anal. calcd for C22H23NO5S: C, 63.90; H, 5.61; N, 3.39; found: C, 63.76; H, 5.45; N, 3.29.
5.1.4.3. Methyl 4-[(3,4,5-trimethoxyphenyl)amino]-[2,2′-bithiophene]-5-carboxylate (4c)
Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as the eluting solution, to furnish 4c as a yellow solid (58% yield), mp 167–169 °C. 1H NMR (CDCl3) δ: 3.85 (s, 9H), 3.88 (s, 3H), 6.45 (s, 2H), 7.13 (s, 1H), 7.26 (m, 1H), 7.35 (dd, J = 5.2 and 3.0 Hz, 1H), 7.48 (m, 1H), 8.67 (s, 1H). 13C NMR (CDCl3) δ: 51.53, 56.29 (2C), 61.14, 99.16 (2C), 113.65, 122.10, 125.82, 127.03, 134.54, 134.97, 137.43, 144.32, 152.27, 152.88, 153.86 (2C), 165.29. MS (ESI): [M+1]+ = 405.9. Anal. calcd for C19H19NO5S2: C, 56.28; H, 4.72; N, 3.45; found: C, 56.20; H, 4.59; N, 3.36.
5.1.4.4. Methyl 5-(4-fluorophenyl)-3-[(3,4,5-trimethoxyphenyl) amino]thiophene-2-carboxylate (4d)
Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as the eluting solution, to furnish 4d as a yellow solid (82% yield), mp 160–161 °C. 1H NMR (CDCl3) δ: 3.85 (s, 9H), 3.89 (s, 3H), 6.46 (s, 2H), 7.05 (t, J = 8.8 Hz, 2H), 7.17 (s, 1H), 7.52 (dd, J = 8.8 and 5.2 Hz, 2H), 8.66 (s, 1H). 13C NMR (CDCl3) δ: 51.59, 56.32 (2x), 61.14, 99.31 (2x), 113.69, 115.57, 116.12 (2C), 116.34 (2C), 127.87, 127.95, 135.32, 137.37, 148.53, 151.13, 152.42, 153.89 (2C). MS (ESI): [M+1]+ = 418.2. Anal. calcd for C21H20FNO5S: C, 60.42; H, 4.83; N, 3.36; found: C, 60.29; H, 4.69; N, 3.30.
5.1.4.5. Ethyl 5-(4-fluorophenyl)-3-[(3,4,5-trimethoxyphenyl)amino] thiophene-2-carboxylate (4e)
Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as the eluting solution, to furnish 4e as a yellow solid (77% yield), mp 150–151 °C. 1H NMR (CDCl3) δ: 1.39 (t, J = 7.2 Hz, 3H), 3.84 (s, 6H), 3.85 (s, 3H), 4.32 (q, J = 7.2 Hz, 2H), 6.45 (s, 2H), 7.06 (t, J = 8.8 Hz, 2H), 7.18 (s, 1H), 7.53 (dd, J = 8.8 and 5.2 Hz, 2H), 8.68 (s, 1H). 13C NMR (CDCl3) δ: 10.59, 52.28 (2C), 56.45, 57.10, 95.17 (2C), 109.66 (2C), 112.06 (2C), 112.27, 123.79 (J = 33.6 Hz), 125.78, 130.55, 133.42, 144.28, 148.21, 149.85, 158.03, 160.90 (2C). MS (ESI): [M+1]+ = 432.2. Anal. calcd for C22H22FNO5S: C, 61.24; H, 5.14; N, 3.25; found: C, 61.09; H, 5.02; N, 3.13.
5.1.4.6. Methyl 5-(4-chlorophenyl)-3-[(3,4,5-trimethoxyphenyl) amino]thiophene-2-carboxylate (4f)
Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as the eluting solution, to furnish 4f as a yellow solid (62% yield), mp 164–166 °C. 1H NMR (CDCl3) δ: 3.85 (s, 9H), 3.89 (s, 3H), 6.45 (s, 2H), 7.21 (s, 1H), 7.34 (d, J = 8.6 Hz, 2H), 7.48 (d, J = 8.6 Hz, 2H), 8.65 (s, 1H). 13C NMR (CDCl3) δ: 51.61, 56.32 (2C), 61.14, 99.35 (2C), 113.95, 127.27 (2C), 129.35 (2C), 131.98, 134.72, 135.18, 137.32, 148.19, 151.09, 152.40, 153.89 (2C), 165.20. MS (ESI): [M+1]+ = 434.2. Anal. calcd for C21H20ClN2O5S: C, 58.13; H, 4.65; N, 3.23; found: C, 58.01; H, 4.55; N, 3.12.
5.1.4.7. Ethyl 5-(4-chlorophenyl)-3-[(3,4,5-trimethoxyphenyl)amino] thiophene-2-carboxylate (4g)
Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 3:7 (v:v) as the eluting solution, to furnish 4g as a yellow solid (64% yield), mp 137–139 °C. 1H NMR (CDCl3) δ: 1.36 (t, J = 7.0 Hz, 3H), 3.84 (s, 6H), 3.85 (s, 3H), 4.30 (q, J = 7.0 Hz, 2H), 6.45 (s, 2H), 7.21 (s, 1H), 7.34 (d, J = 8.6 Hz, 2H), 7.49 (d, J = 8.6 Hz, 2H), 8.67 (s, 1H). 13C NMR (CDCl3) δ: 14.62, 56.32 (2C), 60.54, 61.13, 99.25 (2C), 113.97, 127.25 (2C), 129.35 (2C), 132.05, 134.70, 135.12, 137.41, 147.98, 151.00, 152.24, 153.89 (2C), 164.91. MS (ESI): [M+1]+ = 448.2. Anal. calcd for C22H22ClN2O5S: C, 58.99; H, 4.95; N, 3.13; found: C, 58.79; H, 4.82; N, 3.01.
5.1.4.8. Methyl 5-(3,4-dichlorophenyl)-3-[(3,4,5-trimethoxyphenyl) amino]thiophene-2-carboxylate (4h)
Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as the eluting solution, to furnish 4h as a yellow solid (78% yield), mp 167–168 °C. 1H NMR (DMSO-d6) δ: 3.64 (s, 3H), 3.78 (s, 6H), 3.82 (s, 3H), 6.60 (s, 2H), 7.69 (d, J = 8,2 Hz, 1H), 7.71 (m, 2H), 8.09 (d, J = 2.0 Hz, 1H), 8.70 (s, 1H). 13C NMR (CDCl3) δ: 51.53, 55.75 (2C), 59.97, 99.22 (2C), 101.78, 116.26, 125.99, 127.58, 131.14, 131.64, 131.96, 133.06, 133.31, 136.75, 145.28, 150.98, 153.22 (2C), 163.63. MS (ESI): [M]+ = 468.2. Anal. calcd for C21H19Cl2NO5S: C, 53.85; H, 4.09; N, 2.99; found: C, 53.68; H, 3.97; N, 2.79.
5.1.4.9. Methyl 5-(p-tolyl)-3-[(3,4,5-trimethoxyphenyl)amino]thiophene-2-carboxylate (4i)
Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 3:7 (v:v) as the eluting solution, to furnish 4i as a yellow solid (85% yield), mp 135–136 °C. 1H NMR (CDCl3) δ: 2.37 (s, 3H), 3.84 (s, 3H), 3.85 (s, 6H), 3.88 (s, 3H), 6.46 (s, 2H), 7.18 (d, J = 8.2 Hz, 2H), 7.26 (s, 1H), 7.48 (d, J = 8.2 Hz, 2H), 8.67 (s, 1H). 13C NMR (CDCl3) δ: 21.39, 51.51, 56.28 (2C), 61.14, 99.06 (2C), 113.17, 125.96 (2C), 129.82 (2C), 130.68, 134.46, 137.51, 139.50, 149.98, 152.36, 152.88, 153.86 (2C), 165.33. MS (ESI): [M+1]+ = 414.0, Anal. calcd for C22H23NO5S: C, 63.90; H, 5.61; N, 3.39; found: C, 63.78; H, 5.51; N, 3.27.
5.1.4.10. Ethyl 5-(p-tolyl)-3-[(3,4,5-trimethoxyphenyl)amino]thiophene-2-carboxylate (4j)
Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as the eluting solution, to furnish 4j as a yellow solid (80% yield), mp 141–143 °C. 1H NMR (CDCl3) δ: 1.36 (t, J = 7.2 Hz, 3H), 2.37 (s, 3H), 3.84 (s, 9H), 4.33 (q, J = 7.2 Hz, 2H), 6.45 (s, 2H), 7.17 (d, J = 8.4 Hz, 2H), 7.23 (s, 1H), 7.46 (d, J = 8.4 Hz, 2H), 8.70 (s, 1H). 13C NMR (CDCl3) δ: 14.62, 21.36, 56.26 (2C), 60.38, 61.11, 98.93 (2C), 113.17, 125.92 (2C), 129.79 (2C), 130.71, 134.37, 137.58, 139.41, 149.73, 152.16, 152.88, 153.83 (2C), 165.00. MS (ESI): [M+1]+ = 428.2. Anal. calcd for C23H25NO5S: C, 64.62; H, 5.89; N, 3.28; found: C, 64.51; H, 5.74; N, 3.16.
5.1.4.11. Methyl 5-(4-methoxyphenyl)-3-[(3,4,5-trimethoxyphenyl) amino]thiophene-2-carboxylate (4k)
Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 3:7 (v:v) as the eluting solution, to furnish 4k as a yellow solid (78% yield), mp 134–136 °C. 1H NMR (CDCl3) δ: 3.84 (s, 6H), 3.85 (s, 6H), 3.88 (s, 3H), 6.46 (s, 2H), 6.89 (d, J = 8.6 Hz, 2H), 7.16 (s, 1H), 7.47 (d, J = 8.6 Hz, 2H), 8.68 (s, 1H). 13C NMR (CDCl3) δ: 51.49, 55.51, 56.29 (2x), 61.14, 99.10 (2x), 112.60, 114.52, 126.18, 127.44 (2C), 136.32, 137.53 (2C), 148.34, 149.85, 151.13, 153.85 (2C),160.59. MS (ESI): [M+1]+ = 430.1. Anal. calcd for C22H23NO6S: C, 61.52; H, 5.40; N, 3.26; found: C, 61.43; H, 5.30; N, 3.15.
5.1.4.12. Ethyl 5-(4-methoxyphenyl)-3-[(3,4,5-trimethoxyphenyl) amino]thiophene-2-carboxylate (4l)
Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as the eluting solution, to furnish 4l as a yellow solid (76% yield), mp 150–151 °C. 1H NMR (CDCl3) δ: 1.37 (t, J = 7.2 Hz, 3H), 3.84 (s, 6H), 3.85 (s, 6H), 4.32 (q, J = 7.2 Hz, 2H), 6.46 (s, 2H), 6.90 (dd, J = 6.8 and 2.0 Hz, 2H), 7.16 (s, 1H), 7.51 (dd, J = 6.8 and 2.0 Hz, 2H), 8.71 (s, 1H). 13C NMR (CDCl3) δ: 14.66, 55.49, 56.29 (2C), 60.37, 61.14, 98.99 (2C), 112.61, 114.50 (2C), 126.26, 127.41 (2C), 134.40, 137.60, 146.24, 148.36, 149.62, 152.30, 153.85 (2C), 160.54, 165.03. MS (ESI): [M+1]+ = 444.3. Anal. calcd for C23H25NO6S: C, 62.29; H, 5.68; N, 3.16; found: C, 62.10; H, 5.54; N, 3.02.
5.1.4.13. Methyl 5-(4-ethoxyphenyl)-3-[(3,4,5-trimethoxyphenyl) amino]thiophene-2-carboxylate (4m)
Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 3:7 (v:v) as the eluting solution, to furnish 4m as a yellow solid (74% yield), mp 147–149 °C. 1H NMR (CDCl3) δ: 1.43 (t, J = 7.0 Hz, 3H), 3.82 (s, 3H), 3.85 (s, 6H), 3.87 (s, 3H), 4.04 (q, J = 7.0 Hz, 2H), 6.46 (s, 2H), 6.88 (d, J = 8.8 Hz, 2H), 7.15 (s,1H), 7.48 (d, J = 8.8 Hz, 2H), 8.68 (s,1H). 13C NMR (CDCl3) δ: 14.82, 51.46, 56.28 (2C), 61.12, 63.71, 99.10 (2C), 112.50, 114.99 (2C), 125.97, 127.39 (2C), 130.54, 134.46, 137.52, 149.43, 152.46, 153.83 (2C), 159.97, 165.31. MS (ESI): [M+1]+ = 444.2. Anal. calcd for C23H25NO6S: C, 62.29; H, 5.68; N, 3.16; found: C, 62.09; H, 5.51; N, 3.00.
5.1.4.14. Methyl 5-(3-methoxyphenyl)-3-[(3,4,5-trimethoxyphenyl) amino]thiophene-2-carboxylate (4n)
Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 3:7 (v:v) as the eluting solution, to furnish 4n as a yellow solid (65% yield), mp 127–129 °C. 1H NMR (CDCl3) δ: 3.84 (s, 6H), 3.85 (6H), 3.89 (s, 3H), 6.46 (s, 2H), 6.89 (td, J = 8.0, 2.2 and 1.0 Hz, 1H), 7.09 (t, J = 2.2 Hz, 1H), 7.15 (m, 1H), 7.24 (d, J = 5.8 Hz, 1H), 7.31 (d, J = 8.0 Hz, 1H), 8.64 (s, 1H). 13C NMR (CDCl3) δ: 51.57, 55.45, 56.28 (2x), 61.14, 99.09 (2x), 111.79, 113.91, 114.68, 118.60, 130.21, 134.52, 134.77, 137.44, 149.54, 152.24, 152.38, 152.78, 153.87 (2C), 160.07. MS (ESI): [M+1]+ = 430.1. Anal. calcd for C22H23NO6S: C, 61.52; H, 5.40; N, 3.26; found: C, 61.40; H, 5.26; N, 3.11.
5.1.4.15. Methyl 5-(3, 4-dimethoxyphenyl)-3-[(3, 4, 5-trimethoxyphenyl)amino]thiophene-2-carboxylate (4o)
Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 3:7 (v:v) as the eluting solution, to furnish 4o as a yellow solid (80% yield), mp 160–162 °C. 1H NMR (CDCl3) δ: 3.84 (s, 6H), 3.85 (s, 3H), 3.88 (s, 3H), 3.91 (s, 6H), 6.47 (s, 2H), 6.86 (d, J = 8.6 Hz, 1H), 7.05 (d, J = 2.2 Hz, 1H), 7.15 (m, 2H), 8.68 (s, 1H). 13C NMR (CDCl3) δ: 51.47, 56.03, 56.22 (2C), 61.10, 99.01 (2C), 112.60, 100.72, 109.18, 111.41, 112.76, 118.89, 126.40, 134.42, 137.47, 149.26, 149.89, 150.15, 152.31, 153.82 (2C), 165.21. MS (ESI): [M+1]+ = 460.1. Anal. calcd for C23H25NO7S: C, 60.12; H, 5.48; N, 3.05; found: C, 59.96; H, 5.38; N, 2.88.
5.1.4.16. Methyl 5-(3, 4, 5-trimethoxy ph enyl)-3-[(3,4,5-trimethoxyphenyl)amino]thiophene-2-carboxylate (4p)
Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 4:6 (v:v) as the eluting solution, to furnish 4p as a yellow solid (80% yield), mp 200–201 °C. 1H NMR (CDCl3) δ: 3.84 (s, 6H), 3.85 (s, 3H), 3.87 (s, 9H), 3.89 (s, 3H), 6.47 (s, 2H), 6.77 (s, 2H), 7.17 (s,1H), 8.67 (s, 1H). 13C NMR (CDCl3) δ: 51.54, 56.25 (2C), 56.31 (2C), 61.06, 61.12, 99.12 (2C), 103.56 (2C), 113.50, 129.15, 134.55, 136.26, 137.43, 139.26, 149.82, 152.18, 153.66 (2C), 153.89 (2C), 165.17. MS (ESI): [M+1]+ = 490.2. Anal. calcd for C24H27ClNO8S: C, 58.88; H, 5.56; N, 2.86; found: C, 58.76; H, 5.39; N, 2.72.
5.1.4.17. Methyl 5-(4-trifluorophenyl)-3-[(3,4,5-trimethoxyphenyl) amino]thiophene-2-carboxylate (4q)
Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 4:6 (v:v) as the eluting solution, to furnish 4q as a yellow solid (80% yield), mp 223–225 °C. 1H NMR (DMSO-d6) δ: 3.64 (s, 3H), 3.78 (s, 6H), 3.83 (s, 3H), 6.62 (s, 2H), 7.70 (s, 1H), 7.77 (d, J = 8.4 Hz, 2H), 7.97 (d, J = 8.4 Hz, 2H), 8.71 (s, 1H). 13C NMR (DMSO-d6) δ: 51.55, 55.78 (2C), 59.98, 98.26 (2C), 102.09, 116.26 (2C), 126.02, 126.57 (2C), 129.34, 133.35, 136.23, 136.79, 146.09, 147.23, 150,98, 153.25 (2C), 163.62. MS (ESI): [M+1]+ = 468.2. Anal. calcd for C22H20F3NO5S: C, 56.53; H, 4.31; N, 3.00; found: C, 56.42; H, 4.18; N, 2.87.
5.1.4.18. Methyl 5-(4-nitrophenyl)-3-[(3,4,5-trimethoxyphenyl) amino]thiophene-2-carboxylate (4r)
Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 3:7 (v:v) as the eluting solution, to furnish 4r as an orange solid (76% yield), mp 158–160 °C. 1H NMR (DMSO-d6) δ: 3.65 (s, 3H), 3.78 (s, 6H), 3.84 (s, 3H), 6.62 (s, 2H), 7.77 (s, 1H), 8.02 (d, J = 8.8 Hz, 2H), 8.23 (d, J = 8.8 Hz, 2H), 8.70 (s, 1H). 13C NMR (DMSO-d6) δ: 51.71, 55.85 (2C), 60.07, 98.49 (2C), 112.60, 117.28, 124.38 (2C), 127.00 (2C), 136.82, 138.57, 144.64. 145.22, 147.32, 151.09, 153.35 (2C), 163.63. MS (ESI): [M+1]+ = 445.1. Anal. calcd for C21H20N2O7S: C, 56.75; H, 4.54; N, 6.30; found: C, 56.67; H, 4.44; N, 6.13.
5.2. Biological assays and computational studies
5.2.1. Cell growth conditions and antiproliferative assay
Murine leukemia L1210, murine mammary carcinoma FM3A, human T-lymphocyte leukemia CEM and human cervix carcinoma (HeLa) cells were suspended at 300,000–500,000 cells/mL of culture medium, and 100 μL of a cell suspension was added to 100 μL of an appropriate dilution of the test compounds in wells of 96-well microtiter plates. After incubation at 37 °C for two (L1210 and FM3A), three (CEM) or 4 (HeLa) days, cell number was determined using a Coulter counter. The IC50 value was defined as the compound concentration required to inhibit cell proliferation by 50%.
Peripheral blood lymphocytes (PBL) from healthy donors were obtained by separation on Lymphoprep (Fresenius KABI Norge AS) gradient. After extensive washing, cells were resuspended (1.0 × 106 cells/mL) in RPMI-1640 with 10% fetal bovine serum and incubated overnight. For cytotoxicity evaluations in proliferating PBL cultures, non-adherent cells were resuspended at 5 × 105 cells/mL in growth medium, containing 2.5 μg/mL PHA (Irvine Scientific), whereas for cytotoxicity evaluations in resting PBL cultures, non-adherent cells were resuspended (5 × 105 cells/mL) in the same medium without PHA. Different concentrations of the test compounds were added, and viability was determined 72 h later by the MTT assay as described previously [32].
5.2.2. Effects on tubulin polymerization and on colchicine binding to tubulin
To evaluate the effect of the compounds on tubulin assembly in vitro [33], varying concentrations of compounds were pre-incubated with 10 μM bovine brain tubulin in 0.8 M monosodium glutamate (pH adjusted to 6.6 with HCl in a 2.0 M stock solution) at 30 °C and then cooled to 0 °C. After addition of 0.4 mM GTP, the mixtures were transferred to 0 °C cuvettes in a recording spectrophotometer and warmed to 30 °C. Tubulin assembly was followed turbidimetrically at 350 nm. The IC50 was defined as the compound concentration that inhibited the extent of assembly by 50% after a 20 min incubation. The capacity of the test compounds to inhibit colchicine binding to tubulin was measured as described [34]. The reaction mixtures contained 1 μM tubulin, 5 μM [3H]colchicine and 5 μM test compound.
5.2.3. Molecular modeling
All molecular modeling studies were performed on a MacPro dual 2.66 GHz Xeon running Ubuntu 14.04. The tubulin structure was downloaded from the PDB data bank (http://www.rcsb.org/; PDB code 1SA0) [35]. Hydrogen atoms were added to the protein, using the Protonate 3D routine of the Molecular Operating Environment (MOE) [36]. Ligand structures were built with MOE and minimized using the MMFF94x force field until a RMSD gradient of 0.05 kcal mol−1 Å−1 was reached. The docking simulations were performed using PLANTS [37]. A RMSD value of 1.5 Å was obtained when comparing the docked pose of DAMA-colchicine to its conformation present in the crystal structure.
5.2.4. Flow cytometric analysis of cell cycle distribution
5 × 105 HeLa cells were treated with different concentrations of the test compounds for 24 h. After the incubation period, the cells were collected, centrifuged, and fixed with ice-cold ethanol (70%). The cells were then treated with lysis buffer containing RNase A and 0.1% Triton X-100 and then stained with PI. Samples were analyzed on a Cytomic FC500 flow cytometer (Beckman Coulter). DNA histograms were analyzed using MultiCycle for Windows (Phoenix Flow Systems).
5.2.5. Apoptosis assay
Cell death was determined by flow cytometry of cells double stained with annexin V/FITC and PI. The Coulter Cytomics FC500 (Beckman Coulter) was used to measure the surface exposure of PS on apoptotic cells according to the manufacturer's instructions (Annexin-V Fluos, Roche Diagnostics).
5.2.6. Analysis of mitochondrial potential and reactive oxygen species (ROS)
The mitochondrial membrane potential was measured with the lipophilic cation JC-1 (Molecular Probes, Eugene, OR, USA), while the production of ROS was followed by flow cytometry using the fluorescent dye H2DCFDA (Molecular Probes), as previously described [35].
5.2.7. Western blot analysis
HeLa cells were incubated in the presence of 4c and, after different times, were collected, centrifuged, and washed two times with ice-cold phosphate buffered saline (PBS). The pellet was then resuspended in lysis buffer. After the cells were lysed on ice for 30 min, lysates were centrifuged at 15000 x g at 4 °C for 10 min. The protein concentration in the supernatant was determined using the BCA protein assay reagents (Pierce, Italy). Equal amounts of protein (10 μg) were resolved using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (Criterion Precast, BioRad, Italy) and transferred to a Immobilon-P membrane (Millipore). Membranes were blocked with a bovine serum albumin solution (3% in Tween PBS 1X) for at least 2 h at room temperature. Membranes were then incubated with primary antibodies against Bcl-2, PARP, cdc25c, cyclin B, p-cdc2Tyr15, Mcl-1 (all from Cell Signaling) and β-actin (Sigma-Aldrich) and gently rotated overnight at 4 °C. Membranes were next incubated with peroxidase labeled secondary antibodies for 60 min. All membranes were visualized using ECL Select (GE Healthcare), and images were acquired using an Uvitec-Alliance imaging system (Uvitec, Cambridge, UK). To ensure equal protein loading, each membrane was stripped and reprobed with anti-β-actin antibody.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge Alberto Casolari for excellent technical assistance. We also acknowledge the support of the Life Science Research Network Wales grant n° NRNPGSep14008, an initiative funded through the Welsh Government's Ser Cymru program. We acknowledge “Proyecto de Excelencia de la Consejería de Innovación y Ciencia de la Junta de Andalucía, Spain ref. P12- CTS-696” for its financial support.
Appendix A. Supplementary data
Supplementary data related to this article can be found at https://doi.org/10.1016/j.ejmech.2017.11.096.
Footnotes
Disclaimer
The content of this paper is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health.
References
- 1.van Vuuren RJ, Visagie MH, Theron AE, Joubert AM. Antimitotic drugs in the treatment of cancer. Cancer Chemother Pharmacol. 2015;76:1101–1112. doi: 10.1007/s00280-015-2903-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akhmanova A, Steinmetz MO. Control of microtubule organization and dynamics: two ends in the limelight. Nat Rev Mol Cell Biol. 2015;16:711–726. doi: 10.1038/nrm4084. [DOI] [PubMed] [Google Scholar]
- 3.Janke C. The tubulin code: molecular components, readout mechanisms, and functions. J Cell Biol. 2014;206:461–472. doi: 10.1083/jcb.201406055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kueh HY, Mitchison TJ. Structural plasticity in actin and tubulin polymer dynamics. Science. 2009;325:960–963. doi: 10.1126/science.1168823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brouhard GJ, Rice LM. The contribution of αβ-tubulin curvature to microtubule dynamics. J Cell Biol. 2014;207:323–334. doi: 10.1083/jcb.201407095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mukhtar E, Adhami VM, Mukhtar H. Targeting microtubules by natural agents for cancer therapy. Mol Cancer Ther. 2014;13:275–284. doi: 10.1158/1535-7163.MCT-13-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vindya NG, Sharma N, Yadav M, Ethiraj KR. Tubulins-the target for anti-cancer therapy. Curr Top Med Chem. 2015;125:73–82. doi: 10.2174/1568026615666150112115805. [DOI] [PubMed] [Google Scholar]
- 8.Nitika V, Kapil K. Microtubule targeting agents: a benchmark in cancer therapy. Curr Drug Ther. 2014;8:189–196. [Google Scholar]
- 9.Pettit GR, Singh SB, Hamel E, Lin CM, Alberts DS, Garcia-Kendall D. Isolation and structure of the strong cell growth and tubulin inhibitor combretastatin A-4. Experientia. 1989;45:209–211. doi: 10.1007/BF01954881. [DOI] [PubMed] [Google Scholar]
- 10.Lin CM, Ho HH, Pettit GR, Hamel E. Antimitotic natural products combretastatin A-4 and combretastatin A-2: studies on the mechanism of their inhibition of the binding of colchicine to tubulin. Biochemistry. 1989;28:6984–6991. doi: 10.1021/bi00443a031. [DOI] [PubMed] [Google Scholar]
- 11.Ji YT, Liu YN, Liu ZP. Tubulin colchicine binding site inhibitors as vascular disrupting agents in clinical developments. Curr Med Chem. 2015;22:1348–1360. doi: 10.2174/0929867322666150114163732. [DOI] [PubMed] [Google Scholar]
- 12.Greene LM, Meegan MJ, Zisterer DM. Combretastatins: more than just vascular targeting agents? J Pharmacol Exp Ther. 2015;355:212–227. doi: 10.1124/jpet.115.226225. [DOI] [PubMed] [Google Scholar]
- 13.Pérez-Pérez MJ, Priego EM, Bueno O, Martins MS, Canela MD, Liekens S. Blocking blood flow to solid tumors by destabilizing tubulin: an approach to targeting tumor growth. J Med Chem. 2016;59:8685–8711. doi: 10.1021/acs.jmedchem.6b00463. [DOI] [PubMed] [Google Scholar]
- 14.a) Romagnoli R, Baraldi PG, Kimatrai Salvador M, Preti D, Aghazadeh Tabrizi M, Bassetto M, Brancale A, Hamel E, Castagliuolo I, Bortolozzi R, Basso G, Viola G. Synthesis and biological evaluation of 2-alkoxycarbonyl-3-Anilino benzo[b]thiophenes and thieno[2,3-c]pyridines as new potent anti-cancer agents. J Med Chem. 2013;56:2606–2618. doi: 10.1021/jm400043d. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mangiatordi GF, Trisciuzzi D, Alberga D, Denora N, Iacobazzi RM, Gadaleta D, Catto M, Nicolotti O. Novel chemotypes targeting tubulin at the colchicine binding site and unbiasing P-glycoprotein. Eur J Med Chem. 2017;139:792–803. doi: 10.1016/j.ejmech.2017.07.037. [DOI] [PubMed] [Google Scholar]
- 15.Romagnoli R, Baraldi PG, Salvador MK, Camacho ME, Preti D, Aghazadeh Tabrizi M, Bassetto M, Brancale A, Hamel E, Bortolozzi R, Basso G, Viola G. Synthesis and biological evaluation of 2-substituted-4-(3′,4′,5′-trimethoxyphenyl)-5-aryl thiazoles as anticancer agents. Bioorg Med Chem. 2012;20:7083–7094. doi: 10.1016/j.bmc.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.For the characterization of compounds 6a–f see: Romagnoli R, Baraldi PG, Remusat V, Carrion MD, Lopez Cara C, Preti D, Fruttarolo F, Pavani MG, Aghazadeh Tabrizi M, Tolomeo M, Grimaudo S, Balzarini J, Jordan MA, Hamel E. Synthesis and biological evaluation of 2-(3′,4′,5′-trimethox-ybenzoyl)-3-amino 5-aryl thiophenes as a new class of tubulin inhibitors. J Med Chem. 2006;49:6425–6428. doi: 10.1021/jm060804a.
- 17.Romagnoli R, Baraldi PG, Brancale A, Ricci A, Hamel E, Bortolozzi R, Basso G, Viola G. Convergent synthesis and biological evaluation of 2-amino-4-(3′,4′,5′-trimethoxyphenyl)-5-arylthiazoles as microtubule targeting agents. J Med Chem. 2011;54:5144–5153. doi: 10.1021/jm200392p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Romagnoli R, Baraldi PG, Prencipe F, Oliva P, Baraldi S, Kimatrai Salvador M, Lopez Cara LC, Brancale A, Ferla S, Ronca R, Bortolozzi R, Mariotto E, Porcù E, Basso G, Viola G. Synthesis and biological evaluation of 2-methyl-4,5-disubstituted oxazoles as a novel class og highly potent tubulin polymerization inhibitors. derivatives as novel anticancer agents. Sci Rep. 2017;7:46356. doi: 10.1038/srep46356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weaver BAA, Cleveland DW. Decoding the links between mitosis, cancer, and chemotherapy: the mitotic checkpoint, adaptation, and cell death. Cancer Cell. 2005;8:7–12. doi: 10.1016/j.ccr.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 20.Clarke PR, Allan LA. Cell-cycle control in the face of damage- a matter of life or death. Trends Cell Biol. 2009;19:89–98. doi: 10.1016/j.tcb.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 21.Tsujimoto Y, Shimizu S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis. 2007;12:835–840. doi: 10.1007/s10495-006-0525-7. [DOI] [PubMed] [Google Scholar]
- 22.Xiong S, Mu T, Wang G, Jiang X. Mitochondria-mediated apoptosis in mammals. Protein Cell. 2014;5:737–749. doi: 10.1007/s13238-014-0089-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lugli E, Troiano L, Cossarizza A. Polychromatic analysis of mitochondrial membrane potential using JC-1. Curr Protoc Cytom. 2007 Jul;Chapter 7(Unit 7.32) doi: 10.1002/0471142956.cy0732s41. [DOI] [PubMed] [Google Scholar]
- 24.Zamzami N, Marchetti P, Castedo M, Decaudin D, Macho A, Hirsch T, Susin SA, Petit PX, Mignotte B, Kroemer G. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med. 1995;182:367–377. doi: 10.1084/jem.182.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cai J, Jones DP. Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J Biol Chem. 1998;273:11401–11404. doi: 10.1074/jbc.273.19.11401. [DOI] [PubMed] [Google Scholar]
- 26.Rovini A, Savry A, Braguer D, Carré M. Microtubule-targeted agents: when mitochondria become essential to chemotherapy. Biochim Biophys Acta-Bioenerg. 2011;6:679–688. doi: 10.1016/j.bbabio.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 27.Romagnoli R, Baraldi PG, Lopez-Cara C, Preti D, Aghazadeh Tabrizi M, Balzarini J, Bassetto M, Brancale A, Xian-Hua F, Gao Y, Li J, Zhang S-Z, Hamel E, Bortolozzi R, Basso G, Viola G. Concise synthesis and biological evaluation of 2-aroyl-5-amino benzo[b]thiophene derivatives as a novel class of potent antimitotic agents. J Med Chem. 2013;56:9296–9309. doi: 10.1021/jm4013938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Romagnoli R, Baraldi PG, Kimatrai Salvador M, Schiaffino Ortega S, Prencipe F, Brancale A, Hamel E, Castagliuolo I, Mitola S, Ronca R, Bortolozzi R, Porcù E, Basso G, Viola G. Design, synthesis, in vitro and in vivo anticancer and antiangiogenic activity of novel 3-arylamino benzofuran derivatives targeting the colchicine site on tubulin. J Med Chem. 2015;58:3209–3222. doi: 10.1021/acs.jmedchem.5b00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haschka MD, Soratroi C, Kirschnek S, Hacker G, Hilbe R, Geley S, Villunger A, Fava LL. The NOXA-MCL1-BIM axis defines lifespan on extended mitotic arrest. Nat Commun. 2015;6:6891. doi: 10.1038/ncomms7891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harley ME, Allan LA, Sanderson HS, Clarke PR. Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J. 2010;29:2407–2420. doi: 10.1038/emboj.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, Belmont LD, Kaminker JS, O'Rourke KM, Pujara K, Kohli PB, Johnson AR, Chiu ML, Lill JR, Jackson PK, Fairbrother WJ, Seshagiri S, Ludlam MJ, Leong KG, Dueber EC, Maecker H, Huang DC, Dixit VM. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471:110–114. doi: 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- 32.Ferlin MG, Bortolozzi R, Brun P, Castagliuolo I, Hamel E, Basso G, Viola G. Synthesis and in vitro evaluation of 3H-pyrrolo[3,2-f]-quinolin-9-one derivatives that show potent and selective anti-leukemic activity. Chem-MedChem. 2010;5:1373–1385. doi: 10.1002/cmdc.201000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamel E. Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin. Cell Biochem Biophys. 2003;38:1–21. doi: 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]
- 34.Verdier-Pinard P, Lai J-Y, Yoo H-D, Yu J, Marquez B, Nagle DG, Nambu M, White JD, Falck JR, Gerwick WH, Day BW, Hamel E. Structure-activity analysis of the interaction of curacin A, the potent colchicine site antimitotic agent, with tubulin and effects of analogs on the growth of MCF-7 breast cancer cells. Mol Pharmacol. 1998;53:62–67. doi: 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]
- 35.Ravelli RBG, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature. 2004;428:198–202. doi: 10.1038/nature02393. [DOI] [PubMed] [Google Scholar]
- 36.Molecular Operating Enviroment (MOE 2015.10) Chemical Computing Group, Inc; Montreal, Quebec, Canada: 2015. http://www.chemcomp.com. [Google Scholar]
- 37.Korb O, Stützle T, Exner TE. PLANTS: application of ant colony optimization to structure-based drug design. In: Dorigo M, Gambardella LM, Birattari M, Martinoli A, Poli R, Sttzle T, editors. Ant Colony Optimization and Swarm Intelligence; 5th International Workshop, ANTS 2006; Brussels, Belgium. Sep 4–7, 2006; Berlin: Springer; 2006. pp. 247–258. LNCS 4150. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.