

SUMMARY
How cancer cells respond to nutrient deprivation remains poorly understood. In certain cancer cells, deprivation of cystine induces a non-apoptotic, iron-dependent form of cell death termed ferroptosis. Recent evidence suggests that ferroptosis sensitivity may be modulated by the stress-responsive transcription factor and canonical tumor suppressor protein p53. Using CRISPR/Cas9 genome editing, small-molecule probes, and high-resolution, time-lapse imaging, we find that stabilization of wild-type p53 delays the onset of ferroptosis in response to cystine deprivation. This delay requires the p53 transcriptional target CDKN1A (encoding p21) and is associated with both slower depletion of intracellular glutathione and a reduced accumulation of toxic lipid-reactive oxygen species (ROS). Thus, the p53-p21 axis may help cancer cells cope with metabolic stress induced by cystine deprivation by delaying the onset of non-apoptotic cell death.
In Brief
Tarangelo et al. show that stabilization of wild-type p53, leading to expression of the downstream target CDKN1A (encoding p21CIP1/WAF1), delays the onset of ferroptosis in response to subsequent cystine deprivation in human and in mouse cancer cells.
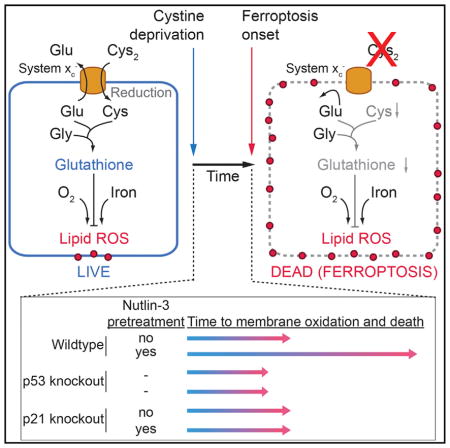
INTRODUCTION
The p53 tumor suppressor is mutated or functionally inactivated in many tumors (Bieging et al., 2014; Pfister and Prives, 2017). How wild-type p53 suppresses tumor formation remains unclear despite decades of study. Recently, acetylation-defective p53 mutants were shown to promote ferroptosis, an iron-dependent, oxidative and non-apoptotic form of cell death (Jiang et al., 2015; Wang et al., 2016). These p53 mutants are unable to induce apoptosis, cell-cycle arrest, or senescence, yet retain the ability to prevent tumor formation in mice (Li et al., 2012), suggesting that p53-induced ferroptosis could be responsible for tumor suppression. However, other results suggest that p53 expression may inhibit ferroptosis in certain cells, either through a post-translational mechanism (Xie et al., 2017) or via effects on the transcription of metabolic genes (Jennis et al., 2016). Thus, the impact of p53 expression on ferroptosis sensitivity is poorly understood.
System xc− is a cystine/glutamate antiporter whose constitutive activity suppresses ferroptosis in many cell types (Dixon et al., 2012; Stockwell et al., 2017). System xc−-mediated cystine import is necessary for the synthesis of the antioxidant tripeptide (reduced) glutathione (GSH). Depletion of cystine and/or GSH results in the iron-dependent accumulation of lethal lipid-reactive oxygen species (ROS), a process that is suppressed by lipophilic antioxidants such as ferrostatin-1 (Dixon et al., 2012; Stockwell et al., 2017). Here, we examined the relationship between wild-type p53 expression and ferroptosis sensitivity in response to system xc− inhibition. We find that wild-type p53 stabilization can decrease system xc− activity yet simultaneously reduce ferroptosis sensitivity in a broad range of cell types. This protective effect requires expression of the p53 transcriptional target CDKN1A (encoding p21) and conservation of intracellular GSH. These results indicate that the p53-p21 transcriptional axis negatively regulates ferroptosis in cancer cells.
RESULTS
p53 Stabilization Suppresses Ferroptosis in Response to System xc− Inhibition
Human HT-1080 fibrosarcoma cells are a classic ferroptosis model that express wild-type p53 (Dixon et al., 2012; Stockwell et al., 2017; Tarunina and Jenkins, 1993). To characterize the effects of p53 expression on ferroptosis in these cells, we used CRISPR/Cas9 technology to isolate a clonal TP53 knockout (TP53KO) cell line. The small-molecule MDM2 inhibitor nutlin-3 (10 μM, 48 hr) increased p53 levels, upregulated the canonical p53 target genes CDKN1A and MDM2, and induced G1 cell-cycle arrest in parental (control) cells, but not in TP53KO cells, suggesting that TP53KO cells lacked p53 function (Figures 1A–1C). To determine how p53 expression affected ferroptosis sensitivity, we examined control and TP53KO cells pretreated for 48 hr with or without nutlin-3, then treated for a further 48 hr with or without the potent and specific system xc− inhibitor erastin2 (Dixon et al., 2014) (Figure 1D). Cell death was quantified during this second phase using scalable time-lapse analysis of cell death kinetics (STACK), an imaging-based method that measures cell death within a population over time (Forcina et al., 2017). We observed that control cells pretreated with nutlin-3 were less sensitive to erastin2-induced cell death, both across erastin2 concentrations and over time (Figures 1E and 1F). By contrast, TP53KO cells were equally sensitive to erastin2 regardless of nutlin-3 pretreatment (Figures 1E and 1F). The protective effect of nutlin-3 pretreatment on subsequent erastin2-induced cell death could also be detected in HT-1080 cells using an alternative measure of cell viability, the metabolic indicator Pres-toBlue (Figure S1A).
Figure 1. p53 Suppresses Ferroptosis.

(A) p53 levels determined by immunoblotting.
(B) Expression of p53 target genes ± nutlin-3 (10 μM, 48 hr).
(C) Cell-cycle profiles ± nutlin-3 (10 μM, 48 hr).
(D) Standard cell death experimental design, involving an unobserved 48-hr pretreatment phase and a 48-hr compound treatment phase, where cell death was measured using scalable time-lapse analysis of cell death kinetics (STACK).
(E) Cell death (lethal fraction) at 36 hr of erastin2 treatment. Erastin2 was tested in a 6-point, 2-fold dose-response series (indicated by black triangle), with a high concentration of 2 μM. On the y axis, 0 = all cells alive and 1 = all cells dead.
(F and G) Cell death over time in response to erastin2 (F) and cystine deprivation (G) following pretreatment with or without (10 μM, 48 hr).
(H) Representative images from (G). Scale bar, 25 μm.
(I) Cell death over time. Orange data points represent cells transiently (72 hr) deprived of cystine (Cys2) followed by Cys2 restoration. Orange arrows indicate the time point of Cys2 add back. An asterisk indicates cells maintained in cystine-containing medium; time courses were terminated at 96 hr due to high-population confluence.
Data represent mean ± SD from at least three independent biological replicates (B, C, E–G, and I).
We next tested the generalizability of the findings obtained in HT-1080 cells to other cell types. Similar to HT-1080 cells, nutlin-3 pretreatment increased the expression of canonical p53 target genes and transiently suppressed erastin2-induced ferroptosis in Trp53+/+, but not in Trp53−/−, primary mouse embryonic fibroblasts (Figures S1B and S1C). Nutlin-3 pretreatment also increased p53 protein levels in p53 wild-type U-2 OS, ACHN, Caki-1, and A549 cells without causing substantial cell death alone, and delayed the onset of erastin2-induced ferropto-sis (Figures S1D–S1F). SJSA-1 cells express wild-type p53 and are susceptible to nutlin-3-induced apoptosis (Vassilev et al., 2004). In these cells, a 24 hr nutlin-3 pretreatment elevated p53 levels and suppressed erastin2-induced cell death to the level of cell death observed with nutlin-3 alone, indicating that p53 stabilization may be capable of inducing apoptosis and suppressing ferroptosis simultaneously (Figures S2A–S2C). Nutlin-3 pretreatment had weaker effects on erastin2-induced ferroptosis in p53 null H1299 cells and p53 mutant T98G cells, consistent with a requirement for wild-type p53 expression and stabilization for suppression of erastin2-induced ferroptosis in cancer cells (Figures S1D–S1F). However, in non-cancerous IMR-90 human fetal lung fibroblasts that express wild-type p53, nutlin-3 pretreatment elevated p53 levels without altering the onset of erastin2-induced ferroptosis, indicating that the association of p53 stabilization with protection against ferroptosis is not universal (Figures S2D and S2E).
While erastin2 inhibits system xc− function (Dixon et al., 2014), it could potentially have other effects on the cell. To address this point, HT-1080 control and TP53KO cells were pretreated with nutlin-3 or DMSO then directly cultured in medium lacking cystine. Under these conditions, nutlin-3 pretreatment suppressed the death of control cells, but not TP53KO cells (Figures 1G and 1H; note that nutlin-3 treatment alone was not lethal to either cell line). Cell death in response to cystine deprivation was completely blocked by a potent and specific antioxidant inhibitor of ferroptosis, ferrostatin-1 (Fer-1, 2 μM) (Dixon et al., 2012) (Figure 1G). Erastin2-induced death was likewise completely suppressed by co-treatment with a structurally distinct nitroxide-based free radical scavenger (Figure S2F), indicating that nutlin-3 pretreatment does not alter the mode of cell death caused by cystine deprivation or erastin2 treatment.
Using the cystine deprivation paradigm, we examined the ability of p53 stabilization to promote long-term cell survival. We pretreated HT-1080 control and TP53KO cells with DMSO or nutlin-3, then deprived cells of cystine for 72 hr in the continued presence of DMSO or nutlin-3, at which point cystine was restored (without DMSO or nutlin-3) and cell viability examined over the following 96 hr. Nutlin-3 pretreatment delayed the onset of cell death over the 72-hr cystine deprivation period in control versus TP53KO cells, as expected (Figure 1I). Moreover, upon cystine restoration, only nutlin-3-pretreated control cells exhibited a steady decrease in the lethal fraction over time, which is best explained by an increase in the number of live cells within the population (Forcina et al., 2017) (Figure 1I, orange datapoints). Collectively, these results suggest that p53 stabilization promotes cell survival in response to cystine deprivation by blocking ferroptosis.
Delayed Ferroptosis Onset Requires p21
p53 may impact ferroptosis via transcriptional or posttranslational mechanisms (Jennis et al., 2016; Jiang et al., 2015; Xie et al., 2017). In HT-1080 cells, more than 12 hr of nutlin-3 pretreatment were required to delay the onset of erastin2-induced ferroptosis, which suggested a p53-dependent transcriptional effect (Figure S3A). To investigate this further, we isolated lung adenocarcinoma cell lines derived from KrasLA2/+; Trp53LSL-wt/LSL-wt (KPWT) and KrasLA2/+; Trp53LSL-25,26/LSL-25,26 (KP25,26) mice (Johnson et al., 2001, 2005). KPWT and KP25,26 cells express oncogenic KrasG12D. When infected with a virus directing the expression of Cre recombinase (Ad-Cre), but not a control adenovirus (Ad-empty), these cells re-expressed wild-type p53, upregulated the p53 target genes Mdm2 and Cdkn1a, and exhibited reduced sensitivity to erastin2-induced, Fer-1-sensitive cell death (Figures 2A, 2B, S3B, and S3C). Ad-Cre infected KP25,26 cells re-express a mutant p53 protein (i.e., p5325,26) that cannot robustly transactivate most canonical p53 target genes including Cdkn1a (Brady et al., 2011) (Figures S3D and S3E). In Ad-Cre-infected KP25,26 cells, erastin2-induced ferroptosis was not suppressed (Figures 2C). Thus, transcriptional upregulation of one or more p53 target genes appeared essential to suppress ferroptosis.
Figure 2. Effect of Genetic Manipulation of p53 Levels on Ferroptosis.

(A) p53 expression in KrasLA2/+;Trp53LSL-wt/LSL-wt mouse lung adenocarcinoma cells following infection with empty adenovirus (Ad-empty) or adenovirus directing the expression of Cre re-combinase (Ad-Cre).
(B and C) Normalized SYTOX Green (norm. SG+) dead cell counts over time in KPWT (B) and KP25,26 (C) cells infected with Ad-empty or Ad-Cre cells prior to treatment with erastin2 ± ferrostatin-1 (Fer-1, 2 μM).
(D) p53 and p21 levels determined by immuno-blotting.
(E and F) Cell-cycle profile (E) and MDM2 expression (F) in control and CDKN1AKO1/2 cell lines ± nutlin-3.
(G) Cell death following erastin2 treatment (36 hr). Erastin2 was tested in a 6-point, 2-fold dose-response series (black traingles) starting at 4 μM.
(H) Cell death over time in response to erastin2.
(I) Cell proliferation expressed as the fold change in live cell (mKate2+) counts over 48 hr in HT-1080 control (C) and TP53KO (KO) cells ± palbociclib (Palbo., 2 μM). The yellow dotted line indicates a ratio of 1 (e.g., no change).
(J) Cell-cycle profiles ± palbociclib (2 μM, 48 hr).
(K) Cell death over time in cells pretreated ± palbociclib and then treated with erastin2 ± ferrostatin-1 (Fer-1, 2 μM).
Data represent mean ± SD from at least three independent biological replicates (B, C, and E–K).
CDKN1A (encoding p21) is a key transcriptional target of p53 that can inhibit cell-cycle progression and impact glutathione metabolism, making it a logical candidate to examine in the context of ferroptosis (Abbas and Dutta, 2009; Maddocks et al., 2013). Nutlin-3 treatment resulted in upregulation of CDKN1A mRNA and p21 protein in control but not TP53KO HT-1080 cells, consistent with a possible role in ferroptosis regulation (Figures 1B and 2D). We therefore used CRISPR to generate two independent clonal HT-1080 CDKN1AKO cell lines that lacked p21 expression and failed to undergo nutlin-3-induced cell-cycle arrest despite normal stabilization of p53 and upregulation of the p53 target gene MDM2 (Figures 2D–2F). Strikingly, and in contrast to control cells, nutlin-3 pretreatment conferred essentially no protection against erastin2-induced cell death in CDKN1AKO1/2 cells, either across compound doses or over time (Figures 2G and 2H). Short hairpin RNA (shRNA)-mediated silencing of CDKN1A in Caki-1 cells likewise abrogated the ability of nutlin-3 pretreatment to delay subsequent erastin2-induced cell death (Figures S3F and S3G). These results suggest a requirement for p21, downstream of p53, to suppress ferroptosis in response to system xc− inhibition.
p21 causes cell-cycle arrest by inhibiting the activity of cyclin-dependent kinase (CDK) complexes that contain the G1/S kinases CDK2, CDK4 and CDK6 (Abbas and Dutta, 2009). We investigated whether cell-cycle arrest was sufficient to protect against ferroptosis. Lentiviral overexpression of CDKN1A in HT-1080 cells delayed the onset of erastin2-induced ferroptosis relative to cells transduced with an empty control virus (Figures S3H and S3I). To test whether cell-cycle arrest per se was sufficient to suppress ferroptosis, we inhibited cell-cycle progression downstream of p21 at the level of CDK4/6. Inhibition of CDK4/6 function using the specific inhibitor palbociclib (2 μM, 48 hr) arrested the proliferation of both control and TP53KO HT-1080 cells, predominantly in G1 (Figures 2I and 2J). However, pretreatment with palbociclib did not alter the sensitivity of either control or TP53KO cells to erastin2-induced ferroptosis (Figure 2J). Thus, cell-cycle arrest caused by CDK4/6 inhibition does not appear to be sufficient to inhibit ferroptosis.
p53 Stabilization Can Decrease System xc− Activity
Ferroptosis is inhibited by system xc− activity (Stockwell et al., 2017), and acetylation-defective p53 mutants sensitize to ferroptosis by repressing SLC7A11/xCT expression and system xc− function (Jiang et al., 2015). Indeed, direct small interfering RNA (siRNA)-mediated silencing of SLC7A11 expression sensitized HT-1080 cells to erastin2-induced cell death (Figures 3A and 3B). We therefore examined how stabilization of wild-type p53, which protects from cell death (e.g., Figures 1 and 2), altered xCT/SLC7A11 expression and system xc− function. In HT-1080 control cells basal system xc− activity (i.e., Na+-independent glutamate release [Dixon et al., 2014]), xCT protein levels and SLC7A11 mRNA expression were all reduced by nutlin-3 treatment (Figures 3C–3E). System xc− activity and SLC7A11/xCT expression were lower in TP53KO cells and insensitive to nutlin-3 (Figures 3C–3E). These effects may be due to reorganization of metabolic networks in mutant cells to compensate for the loss of p53. In both cell lines, system xc− activity was fully inhibited by treatment with erastin2 (1 μM, 2 hr), confirming the specificity of this assay (Figure 3C). A known inducer of xCT expression, tert-butylhydroquinone (tBHQ, 100 μM, 48 hr [Lewerenz et al., 2012]) increased xCT/ SLC7A11 levels, validating our reagents (Figures 3D and 3E).
Figure 3. p53 Stabilization affects System xc−.

(A) xCT levels following transfection (48 hr) with scrambled (SCR) siRNA or siRNA-targeting SLC7A11.
(B) Erastin2 dose-response at 48 hr, following 48 hr after transfection with siRNAs like in (A).
(C) Glutamate release, a measure of system xc− activity ± erastin2 (1 μM)following pretreatment ± nutlin-3. BD: below limit of detection.
(D) p53 and xCT protein levels following DMSO, tert-butylhydroquinone (tBHQ, 100 μM), or nutlin-3 (10 μM) treatment for 48 hr.
(E and F) SLC7A11 (E) and HMOX and GCLM (F) mRNA expression following DMSO (vehicle), nutlin-3 (10 μM), or tBHQ (100 μM) treatment for 48 hr.
Data represent mean ± SD from at least three independent biological replicates (B, C, E, and F).
We reasoned that the inhibitory effect of p53 stabilization on system xc− function must be counterbalanced by other alterations that ultimately reduce cellular ferroptosis sensitivity. The antioxidant master regulator transcription factor NFE 2-like 2 (NRF2) can be activated by p21 (Chen et al., 2009), and we wondered whether p21-stimulated NRF2 activity and antioxidant gene expression could be compromised by disruption of the p53-p21 axis. However, tBHQ, a bona fide inducer of NRF2 activity (Lewerenz et al., 2012), stimulated the expression of two canonical NRF2 target genes, HMOX1 and GCLM, equally in control, TP53KO and CDKN1AKO1/2 cells (Figure 3F). Conversely, nutlin-3 treatment did not consistently enhance HMOX1 and GCLM expression in these cells (Figure 3F). Thus, it appeared unlikely that the p53-p21 axis suppressed ferroptosis by enhancing NRF2 activity.
p53 Stabilization Leads to Conservation of Intracellular Glutathione
The toxic lipid ROS accumulation occurring during ferroptosis can be monitored using the lipid ROS probe C11 BODIPY 581/ 591 (Dixon et al., 2012). HT-1080 cells pretreated with DMSO and then treated with erastin2 (1 μM, 10 hr) exhibited robust probe oxidation, both on internal membranes and in a distinct ring around the periphery of the cell (Figure 4A). Pretreatment with nutlin-3 reduced probe oxidation upon subsequent erastin2 treatment in both locations (Figure 4A). Lipid ROS accumulation is opposed by GPX4, a (reduced) glutathione (GSH)-dependent lipid hydroperoxidase (Stockwell et al., 2017). In control HT-1080 cells, total glutathione levels (as determined using Ellman’s reagent) were not elevated by nutlin-3 pretreatment (Figure 4B). Notably, however, nutlin-3-pretreated control cells exhibited higher glutathione levels than control cells pretreated with DMSO, or TP53KO or CDKN1AKO1/2 cells pretreated with DMSO or nutlin-3, following erastin2 treatment (1 μM, 8 hr) (Figures 4B and S4A).
Figure 4. p53 Expression Alters GSH Metabolism.

(A) C11 BODIPY 581/591 labeling in HT-1080 cells. Scale bar, 15 μm.
(B) Total glutathione levels ± nutlin-3 pretreatment ± erastin2 (1 μM, 8 hr). BD, below the limit of detection.
(C) Outline of carbon flux in metabolic pathways leading from uniformly (U) labeled 13C-serine (yellow) to GSH. The dotted line indicates multiple steps (data not shown) in the pathway from serine to cystine via transsulfuration. Also not shown is the flux of (unlabeled) cysteine from system xc−-dependent cystine uptake.
(D) Relative metabolite abundance (all isotopologues) determined by mass spectrometry ± nutlin-3 pretreatment ± erastin2 (1 μM, 8 hr). U-13C-serine was added only during the 8-hr treatment phase.
(E) Isotopologues of GSH detected in the experiment outlined in (D).
(F) Effects of buthionine sulfoximine (BSO, 100 μM) inclusion during the pretreatment (left) or treatment (right) phases on erastin2-induced cell death. The timing of initial population cell death onset is extracted from curves of cell death over time for each condition (see the Supplemental Experimental Procedures).
Based on these results, we investigated whether p53 stabilization delayed ferroptosis by limiting GSH depletion or enhancing GSH synthesis using mass spectrometry and carbon-13 labeling. HT-1080 control and TP53KO cells were pretreated with either DMSO or nutlin-3 for 48 hr and then incubated in the presence or absence of erastin2 (1 μM) for 8 hr in medium containing uniformly (U) labeled-13C-serine. Within the cell, serine can be converted into glycine, which is then directly incorporated into GSH via de novo synthesis, yielding GSH M+2 (Figure 4C). Serine also can be utilized in the synthesis of cysteine via the transsulfuration pathway, ultimately resulting in GSH M+5 (via glycine and transsulfuration; Figure 4C). In control and TP53KO cells, most intracellular serine was labeled (M+3) at 8 hr, and this labeling was not grossly affected by nutlin-3 or erastin2, indicating that these treatments did not affect the uptake of the tracer (Figure S4B). In control cells, nutlin-3 pretreatment resulted in higher levels of total GSH (all isotopologues) following erastin2 treatment compared to cells pretreated with DMSO, or compared to TP53KO cells pretreated with DMSO or nutlin-3 (Figure 4D). GSSG represented a minor fraction of the total glutathione in control (< 10%) and TP53KO (< 20%) cells and was unaffected by nutlin-3 pretreatment, indicating that higher levels of GSH likely account for the increase in total glutathione.
Examining the distribution of GSH isotopologues, we observed that de novo GSH synthesis was inhibited in control cells pretreated with nutlin-3, as indicated by a reduction in GSH M+2 relative to control cells pretreated with DMSO (Figure 4E). This effect was not observed in TP53KO cells pretreated with either nutlin-3 or DMSO (Figure 4E). In control cells pre-treated with nutlin-3, most of the GSH retained upon erastin2 treatment was the unlabeled M+0 species, suggesting that it was derived from preexisting pools rather than de novo (i.e., stress-induced) synthesis. Under no conditions did we observe accumulation of GSH M+5, indicating that the transsulfuration pathway was not active in these cells (Figure 4E). Based on these results, we predicted that GSH synthesized during the pretreatment phase (i.e., detected as GSH M+0 in our experiments) was important for the protective effect of p53 stabilization in response to system xc− inhibition. Consistent with this prediction, addition of the GSH biosynthesis inhibitor buthionine sulfoximine (BSO, 100 μM, 24 hr) during the nutlin-3 pretreatment phase, but not the erastin2 treatment phase, sensitized control HT-1080 cells to ferroptosis, regardless of nutlin-3 pretreatment (Figure 4F).
DISCUSSION
Depending on the context, p53 can either promote or prevent cell death (Kruiswijk et al., 2015; Paek et al., 2016). We find that prolonged (>24 hr) stabilization of wild-type p53 renders many cancer cells less sensitive to ferroptosis induced by system xc− inhibition or direct cystine deprivation. This reduced sensitivity to ferroptosis requires p21 and the conservation of intracellular GSH (Figure S4C). How activation of the p53-p21 axis reduces cellular reliance on system xc−-mediated cystine import and ongoing de novo GSH synthesis is unclear. p21 inhibits cell-cycle progression (Abbas and Dutta, 2009), but experiments using the CDK4/6 inhibitor palbociclib tentatively suggest that inhibition of proliferation and cell-cycle arrest per se are insufficient to inhibit ferroptosis. It is possible that p21-dependent inhibition of additional or alternative CDKs (e.g., CDK1/2) is required to conserve GSH and inhibits ferroptosis through induction of a more complete cell-cycle arrest. Alternatively, p21 could potentially enhance GSH retention through effects on CDK-regulated metabolic enzymes (Ewald et al., 2016).
Our results are broadly consistent with the recent findings that ferroptosis is inhibited by basal p53 expression in colorectal cancer cells (Xie et al., 2017), but suggest a specific role for p53-mediated transcription and upregulation of p21 in the protective response conferred by p53 stabilization. Our results appear to contradict the findings that expression of acetylation-defective p53 mutants (i.e., 3KR) reduce system xc− function and sensitize cancer cells to ferroptosis (Jiang et al., 2015; Wang et al., 2016). However, acetylation-defective p53 mutants are unable to induce p21 (Jiang et al., 2015). We speculate that system xc− downregulation and p21 upregulation could be two branches of a coordinated p53-mediated response that normally decreases cystine import to match the lower metabolic demands of growth-arrested cells. One possibility is that such a coordinated response is modulated by acetylation and other p53 post-translational modifications (e.g., phosphorylation) (Jennis et al., 2016) to fine-tune ferroptosis sensitivity in specific contexts. Delaying the onset of ferroptosis by even a few hours could help a cancer cell survive transient exposure to harsh environmental conditions in vivo, such as the oxidizing conditions encountered in the bloodstream during metastasis (Piskounova et al., 2015). This could help explain why some cancers retain the ability to express wild-type p53 and p21.
Experimental Procedures
Further details and an outline of the resources used in this work can be found in the Supplemental Experimental Procedures.
Cell Lines
For cell viability studies, polyclonal Nuc::mKate2-expressing HT-1080 parental, TP53KO, CDKN1AKO1/2, U-2 OS, A549, T98G, H1299, ACHN, and CAKI-1 cell lines were generated by transducing parental cell lines with NucLight Red Lentivirus (Essen Bioscience) followed by selection with puromycin (1–2 μg/mL) for 4 days (Forcina et al., 2017). Murine lung tumor cell lines were generated by dissection of lung tumors from 11-week-old KrasLA2/+;Trp53LSL-wt/LSL-wt and KrasLA2/+;Trp53LSL-25,26/LSL-25,26 male mice. Cultures were established in N5 medium supplemented with epidermal growth factor (EGF) (20 ng/mL) and fibroblast growth factor (FGF) (20 ng/mL), sorted by fluorescence-activated cell sorting (FACS) (Stanford Shared FACS facility) for EpCam positivity (BioLegend) and then switched to DMEM Hi-glucose media. p53+/+ and p53−/− mouse embryonic fibroblasts (MEFs) were isolated from E13.5 embryos as previously described (Johnson et al., 2005) and cultured in DMEM Hi-glucose media. All media were supplemented with 10% fetal bovine serum and 1× Pen/Strep (Life Technologies). All cell lines were grown at 37°C with 5% CO2 in humidified tissue culture incubators (Thermo Scientific). All experiments involving cell isolation from tissues or embryos were performed in accordance with the Stanford APLAC #10382.
Data Analysis
Data were analyzed using Microsoft Excel and GraphPad Prism (v.6.0h) and are represented as mean ± SD or ± 95% confidence interval. Lethal fraction scoring was performed as using Microsoft Excel and GraphPad Prism, as previously described (Forcina et al., 2017).
Supplementary Material
Highlights.
p53 stabilization can delay the induction of ferroptosis in cancer cells
This effect requires the p53 transcriptional target gene CDKN1A (p21)
Cell-cycle arrest per se may not be sufficient to inhibit ferroptosis
Inhibition of ferroptosis correlates with conservation of glutathione
Acknowledgments
We thank P.-J. Ko and T. Stearns for help with confocal imaging; J.-Y. Cao, C. Poltorack, and M. Morris for help with experiments; and J. Sage, J. Carette, and A. Gitler for providing reagents. This work was supported by an NSF GRFP fellowship (to A.T.), awards from the NIH (R35 CA197591 to L.D.A.; 4R00CA166517 and 1R01GM122923 to S.J.D.), and a Damon Runyon-Rachleff innovation award (to S.J.D.).
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and four figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2017.12.077.
DECLARATION OF INTERESTS
All authors declare no competing interests.
AUTHOR CONTRIBUTIONS
Conceptualization, A.T., L.D.A., and S.J.D.; Methodology, A.T., L.M., K.T.B.-R., and Y.L.; Investigation, A.T., L.M., and Y.L.; Writing - Original Draft, A.T. and S.J.D. Writing - Review & Editing, L.M., K.T.B.-R. and L.D.A. Funding Acquisition and Supervision, J.Y., L.D.A. and S.J.D.
References
- Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400–414. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14:359–370. doi: 10.1038/nrc3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Kenzelmann Broz D, Basak S, Park EJ, McLaughlin ME, et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell. 2011;145:571–583. doi: 10.1016/j.cell.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Sun Z, Wang XJ, Jiang T, Huang Z, Fang D, Zhang DD. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol Cell. 2009;34:663–673. doi: 10.1016/j.molcel.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS, Stockwell BR. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife. 2014;3:e02523. doi: 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald JC, Kuehne A, Zamboni N, Skotheim JM. The yeast cyclin-dependent kinase routes carbon fluxes to fuel cell cycle progression. Mol Cell. 2016;62:532–545. doi: 10.1016/j.molcel.2016.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcina GC, Conlon M, Wells A, Cao JY, Dixon SJ. Systematic quantification of population cell death kinetics in mammalian cells. Cell Syst. 2017;4:600–610. e6. doi: 10.1016/j.cels.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennis M, Kung CP, Basu S, Budina-Kolomets A, Leu JIJ, Khaku S, Scott JP, Cai KQ, Campbell MR, Porter DK, et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev. 2016;30:918–930. doi: 10.1101/gad.275891.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, Jacks T. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- Johnson TM, Hammond EM, Giaccia A, Attardi LD. The p53QS transactivation-deficient mutant shows stress-specific apoptotic activity and induces embryonic lethality. Nat Genet. 2005;37:145–152. doi: 10.1038/ng1498. [DOI] [PubMed] [Google Scholar]
- Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. 2015;16:393–405. doi: 10.1038/nrm4007. [DOI] [PubMed] [Google Scholar]
- Lewerenz J, Sato H, Albrecht P, Henke N, Noack R, Methner A, Maher P. Mutation of ATF4 mediates resistance of neuronal cell lines against oxidative stress by inducing xCT expression. Cell Death Differ. 2012;19:847–858. doi: 10.1038/cdd.2011.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012;149:1269–1283. doi: 10.1016/j.cell.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddocks ODK, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, Vousden KH. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature. 2013;493:542–546. doi: 10.1038/nature11743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paek AL, Liu JC, Loewer A, Forrester WC, Lahav G. Cell-to-cell variation in p53 dynamics leads to fractional killing. Cell. 2016;165:631–642. doi: 10.1016/j.cell.2016.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister NT, Prives C. Transcriptional regulation by wild-type and cancer-related mutant forms of p53. Cold Spring Harb Perspect Med. 2017;7:a026054. doi: 10.1101/cshperspect.a026054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ, Morrison SJ. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527:186–191. doi: 10.1038/nature15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK, Kagan VE, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarunina M, Jenkins JR. Human p53 binds DNA as a protein homodimer but monomeric variants retain full transcription transactivation activity. Oncogene. 1993;8:3165–3173. [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Wang SJ, Li D, Ou Y, Jiang L, Chen Y, Zhao Y, Gu W. Acetylation is crucial for p53-mediated ferroptosis and tumor suppression. Cell Rep. 2016;17:366–373. doi: 10.1016/j.celrep.2016.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, Zhong M, Yuan H, Zhang L, Billiar TR, et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 2017;20:1692–1704. doi: 10.1016/j.celrep.2017.07.055. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.