

Abstract
Lipid oxidation products, including lysophosphatidylcholine (lysoPC), activate canonical transient receptor potential 6 (TRPC6) channels, and the subsequent increase in intracellular Ca2+ leads to TRPC5 activation. The goal of this study is to elucidate the steps in the pathway between TRPC6 activation and TRPC5 externalization. Following TRPC6 activation by lysoPC, extracellular regulated kinase (ERK) is phosphorylated. This leads to phosphorylation of p47phox and subsequent NADPH oxidase activation with increased production of reactive oxygen species. ERK activation requires TRPC6 opening and influx of Ca2+ as evidenced by the failure of lysoPC to induce ERK phosphorylation in TRPC6−/− endothelial cells. ERK siRNA blocks the lysoPC-induced activation of NADPH oxidase, demonstrating that ERK activation is upstream of NADPH oxidase. The reactive oxygen species produced by NADPH oxidase promote myosin light chain kinase (MLCK) activation with phosphorylation of MLC and TRPC5 externalization. Downregulation of ERK, NADPH oxidase, or MLCK with the relevant siRNA prevents TRPC5 externalization. Blocking MLCK activation prevents the prolonged rise in intracellular calcium levels and preserves endothelial migration in the presence of lysoPC.
Keywords: TRPC6, TRPC5, NADPH oxidase, calcium, MLCK, endothelial
endothelial cell (EC) migration is essential for the healing of arterial injuries such as those that accompany angioplasty. Rapid reendothelialization of an area of EC denudation limits the proliferation of smooth muscle cells and the subsequent development of intimal hyperplasia (2, 15, 23, 47, 48). Unfortunately, EC migration in vitro is inhibited by oxidized LDL and lipid oxidation products, including lysophosphatidylcholine (lysoPC) (25, 26), that are abundant in plasma and lesions of patients with atherosclerosis (9, 19, 31). Hypercholesterolemia inhibits reendothelialization of arterial injuries in vivo, and the delay in healing correlates with the elevation of plasma lysoPC (32, 34).
Normal cell migration requires exquisite control of calcium transients to allow detachment of focal adhesions, cytoskeletal reorganization to move the cell forward, and formation of new attachments to the substratum. EC monolayer disruption is followed by influx of Ca2+ from the extracellular milieu and a transient rise in intracellular Ca2+ concentration ([Ca2+]i) that is required for detachment of focal adhesions and initiation of movement (41). Following the initial rise in [Ca2+]i, caveolae and the machinery for calcium wave initiation relocate to the trailing edge of the cell (20), allowing new focal adhesions to assemble in the leading portion of the cell. A generalized, sustained increase in [Ca2+]i, such as that induced by lipid oxidation products, disrupts the time- and site-specific changes in focal adhesions and cytoskeleton that are required for cell movement and thus inhibits EC migration required to repair monolayer disruption (6).
We have shown that oxidized LDL and lysoPC, the primary lysophospholipid in oxidized LDL, inhibit EC migration, in part, by causing Ca2+ influx through canonical transient receptor potential (TRPC) channels (5). Specifically, lysoPC activates TRPC6 and the subsequent brief increase in [Ca2+]i leads to externalization of TRPC5 and a prolonged increase in [Ca2+]i. The increase in [Ca2+]i activates calpains causing breakdown of cytoskeletal proteins and inhibition of EC migration (6). The importance of this pathway in vivo is demonstrated by the finding that a high-cholesterol diet dramatically inhibits endothelial healing of a carotid injury in wild-type (WT) mice but has no effect in TRPC6-deficient (TRPC6−/−) mice (33). The mechanism of TRPC channel activation by lipid oxidation products is not completely elucidated.
Ca2+-dependent regulation of Ca2+-permeable cation channels is common, and all TRPC proteins have binding regions for calmodulin (CaM), a ubiquitous Ca2+-binding protein that can regulate TRPC activation (40). We have shown that lysoPC induces phosphorylation of CaM at Tyr99, and this phosphorylation is required for the dissociation of CaM and TRPC6 and externalization of TRPC6 in the cell membrane (8). Interesting, lysoPC does not cause dissociation of CaM and TRPC5, but lysoPC-induced TRPC5 externalization does require TRPC6 activation and influx of calcium (5, 8). The mechanism by which TRPC6 activation leads to TRPC5 externalization is the topic of this study.
TRPC5 channels can be activated by a multiplicity of signals including G protein-coupled receptor activation, extracellular gadolinium or Ca2+, modest elevation of [Ca2+]i, or depletion of intracellular Ca2+ stores (50), but TRPC5 channel activation is inhibited by high [Ca2+]i (28). We have shown that lysoPC-induced activation of TRPC5 requires the presence of TRPC6 channels and Ca2+ in the medium (5). To determine the mechanism by which an increase in [Ca2+]i through TRPC6 channels leads to activation of TRPC5, we assessed the contribution of extracellular regulated kinase (ERK) activation, activation of NADPH oxidase, increased reactive oxygen species (ROS) production, and myosin light chain kinase (MLCK) activation in the externalization of TRPC5. Determination of the mechanism by which lysoPC activates TRPC5 and identifying components of the activation cascade from TRPC6 to TRPC5 provide opportunities to interrupt these events, restore normal endothelial migration, and promote healing after interventions in atherosclerotic arteries.
MATERIALS AND METHODS
EC isolation and culture.
Bovine aortic ECs were isolated from fresh adult bovine aortas and cultured as previously described (6). ECs in passage 4 to 10 were used for experiments. Under a protocol approved by the Cleveland Clinic Institutional Animal Care and Use Committee, mouse aortic ECs (MAECs) were harvested from 129Sv/C57BL/6J WT, TRPC6−/−, or TRPC5−/− mice (14, 30), as described previously (5). The genetic deletion of the specific TRPC channel was confirmed by PCR using DNA extracted from tail or toe biopsies. MAECs in passage 3 or 4 were used for experiments.
Immunoprecipitation and immunoblot analysis of target protein.
Confluent ECs were used after being made quiescent in serum-free Dulbecco's modified Eagle's medium for 3 h before experimental conditions were initiated. Immunoprecipitation of target protein was performed as described previously (7).
Immunoblot analysis was performed after proteins (60–80 µg/lane) were loaded in 4–20% gradient SDS-PAGE as previously described (5, 7) and detected by antibodies specific for TRPC6 (1:200; catalog no. 600-401-FQ5; Rockland, St. Louis, MO), TRPC5 (1:250; catalog no. 73-104; UC Davis/NIH NeuroMab Facility), ERK1/2 (1:1,000; catalog no. 4695; Cell Signaling Technology, Danvers, MA), phospho-ERK1/2 (Thr202/Tyr 204; 1:1,000; catalog no. SC-16982; Santa Cruz Biotechnology, Santa Cruz, CA), p47phox (1:500; catalog no. 43125; Cell Signaling Technology), phospho-p47phox (Ser345; 1:200; catalog no. A8391; Assay Biotechnology, Sunnyvale, CA), MLCK (1:500; catalog no. MABT194; Millipore), phospho-MLCK (Tyr471; 1:500; catalog no. SC-17183; Santa Cruz Biotechnology), MLC (1:250; catalog no. M4401; Sigma), and phospho-MLC (Thr18/Ser19; 1:500; catalog no.3474; Cell Signaling Technology). After being washed, the horseradish peroxidase-conjugated secondary antibody was applied and the signal developed using a chemiluminescent reagent (Perkin Elmer-Cetus, Boston, MA) for 1 min, and the image was acquired on HyBlot CL autoradiography film (Denville Scientific). The blot was then stripped and reprobed using an anti-actin antibody (1:2,000; catalog no. MA1-744; Invitrogen). For immunoblot analysis of phosphorylated proteins, samples were divided into two aliquots, one used for the phorphorylated protein and the other used for total ERK, p47phox, MLCK, or MLC, as indicated. Protein band density on the HyBlot CL autoradiography film was quantititated using ImageJ software (NIH, Bethesda, MD). The film was then scanned with an HP Scanjet G4050 using HP Photosmart Premier Software.
Detection of TRPC externalization by biotinylation assay.
Externalization of TRPC6 or TRPC5 was determined by biotinylation assay as previously described (5). Briefly, cell surface proteins were biotinylated and complexed with streptavidin-agarose beads, beads were collected, and precipitated proteins were released, resolved by SDS-PAGE, and identified by immunoblot analysis. Total TRPC6 or TRPC5 protein was determined by immunoblot analysis of an aliquot of cell lysate removed before incubation with streptavidin-agarose beads.
Overexpression of mutant CaM.
The full-length human CaM cDNA (MGC-7) was obtained from ATCC. PCR-based site-directed mutagenesis was used to generate a cDNA for a mutant CaM in which Tyr99 was replaced with Phe (Phe99-CaM; ExonBio, San Diego, CA), as previously described (8). ECs at 60% confluence were transiently transfected with 2 μg of plasmids containing pcDNA3.1-myc-His-human CaM (WT-CaM) or pcDNA3.1-myc-His-Phe99-CaM using Effectene (Qiagen, Chatsworth, CA) according to the manufacturer's protocol. The effectiveness of transfection was verified after 48 h by fluorescence microscopy and immunoblot analysis of CaM as previously descibed (8).
Downregulation of target protein by small interfering RNA.
Using a transfection kit (RNAiFect; Qiagen), ECs at 80% confluence were transiently transfected with the small interfering (si)RNA duplex of CaM isoform1 (20 nM; Santa Cruz Biotechnology), ERK1 (20 nM; Santa Cruz Biotechnology), ERK2 (20 nM; Santa Cruz Biotechnology), p47phox subunit of NADPH oxidase (20 nM; Santa Cruz Biotechnology), or MLCK (30 nM; Santa Cruz Biotechnology) for 24 h. Downregulation of endogenous target protein level was examined after 48 h by immunoblot analysis. A negative control siRNA (NsiRNA; 20–40 nM; Ambion, Austin, TX) without homology to any known gene sequence was used as a negative control.
Detection of ROS production.
Intracellular ROS production was measured by oxidation of di(acetoxymethyl ester)(6-carboxy-2'-7'-dichlorodihydrofluorescein diacetate (H2DCF-DA; Life Technologies, Grand Island, NY). ECs at 70% confluence were incubated with H2DCF-DA (10 µM) for 20 min and washed twice before addition of lysoPC for 15 min. Fluorescence was monitored using Leica fluorescence microscope (Heidelberg, Germany) with FITC filter. The fluorescence intensity was quantititated using NIH ImageJ software.
Measurement of [Ca2+]i.
[Ca2+]i was measured using the Ca2+-binding fluorophore fura 2-AM (Life Technologies), as previously described (5, 6). ECs were cultured in 35-mm dishes designed for fluorescence microscopy (Bioptechs, Butler, PA), incubated with fura 2-AM (1 µM) for 30 min, and washed with Krebs-Ringer (KR) buffer to remove excess fura 2-AM. Fluorescence of a group of 10–12 cells inside the light path was continuously examined with an Olympus 1X-70 inverted fluorescence microscope (Melville, NY). The relative change of [Ca2+]i was estimated by the ratio of fura 2 excitation at wavelengths of 340 nm and 380 nm (340/380 ratio).
EC migration assay.
ECs were made quiescent in serum-free Dulbecco's modified Eagle's medium for 8–12 h before the migration assay. EC migration was assessed by the razor-scrape assay as described in our previous study (6). Migrating cells were quantitated using NIH Image 1.63 software by an observer blinded to the experimental conditions.
Statistics.
Results are presented as means ± SD. Experiments were performed in triplicate with ECs isolated from at least three different animals. Statistical analysis was performed using Student's t-test or ANOVA, and differences between means were considered statistically significant at P < 0.05.
RESULTS
LysoPC induced Ca2+-CaM dependent ERK phosphorylation.
Previously, we showed that lysoPC (1-palmitol-2-hydroxi-sn-glycerol-3-phosphocholine; Avanti Polar Lipids, Alabaster, AL) increased [Ca2+]i through a unique TRPC6 channel to TRPC5 channel activation cascade (5). An increase in [Ca2+]i through TRPC6 channels can lead to ERK activation (18), and ERK can activate MLCK (22), which is essential in TRPC5 externalization (21, 37). Therefore, a possible role of ERK in the lysoPC-induced TRPC activation cascade was studied. In ECs incubated with lysoPC (12.5 µM), but not phosphatidylcholine (PC; 12.5 µM), for 15 min, ERK1/2 phosphorylation at Thr202/Tyr204 increased to 280 ± 30% of control (n = 4; P < 0.01; Fig. 1A). Total ERK levels did not change.
Fig. 1.

LysoPC induces Ca2+-, calmodulin (CaM)-, and TRPC6-dependent ERK phosphorylation. A: ECs were incubated with medium alone (Control), lysoPC (12.5 µM), or PC (12.5 µM) for 15 min. ERK and phospho-ERK1/2(Thr202/Tyr204) were detected by immunoblot (IB) analysis. Actin served as loading control (n = 4). IB, immunoblot. B: ECs were incubated with calcium-free or calcium-containing KR buffer for 10 min or ECs were incubated with BAPTA/AM (25 µM) for 30 min before addition of lysoPC. Phospho-ERK1/2(Thr202/Tyr204) and total ERK1/2 were detected by immunoblot analysis. Actin served as loading control (n = 4). C and D: ECs were transiently transfected with control siRNA (NsiRNA) or CaM siRNA (20 nM) for 24 h. The siRNA was then removed. C: at 24 h after siRNA removal, cells were lysed and CaM was identified by immunoblot analysis. Actin served as loading control (n = 3). D: after siRNA removal, ECs were incubated with lysoPC (12.5 µM) for 15 min. Phospho-ERK1/2(Thr202/Tyr204) and total ERK1/2 were detected by immunoblot analysis. Actin served as loading control (n = 4). E and F: ECs were transiently transfected with pcDNA3.1-myc-His-WT-CaM or pcDNA3.1-myc-His- Phe99-CaM for 24 h. E: overexpression of CaM was confirmed after 48 h by immunoblot analysis with anti-Myc Tag antibody. Actin served as loading control (n = 3). F: after transfection, ECs were incubated with lysoPC (12.5 µM) for 15 min. Phospho-ERK1/2(Thr202/Tyr204) and total ERK were detected by immunoblot analysis. Actin served as loading control (n = 4). G and H: WT, TRPC6−/−, or TRPC5−/− MAECs were incubated with lysoPC (10 µM) for min. G: phospho-ERK1/2(Thr202/Tyr204) and total ERK were detected by immunoblot analysis. Actin served as loading control (n = 3). H: densitometry measurements of phospho-ERK(Thr202/Tyr204) are represented in graphic form: ●, medium control with WT ECs; ■, lysoPC with WT ECs; ▲, medium control with TRPC6−/− ECs; ▼, lysoPC with TRPC6−/− ECs; ◆, medium control with TRPC5−/− ECs; ○, lysoPC with TRPC5−/− ECs. (In A–G, lines indicate lanes rearranged from the same gel. All bands are from the same gel.)
To determine if lysoPC-induced ERK phosphorylation was Ca2+ dependent, ECs were incubated in Ca2+-free or Ca2+-containing KR buffer or with the intracellular Ca2+-chelator BAPTA/AM (25 µM) before the addition of lysoPC (12.5 µM). LysoPC did not induce ERK1/2 phosphorylation in the absence of extracellular Ca2+ or presence of BAPTA/AM, suggesting that ERK1/2 phosphorylation was Ca2+ dependent (n = 4; P < 0.01 compared with incubation with lysoPC in Ca2+-containing KR buffer; Fig. 1B).
ERK activation can be regulated by CaM (1), and lysoPC induces CaM phosphorylation at Tyr99 that is required for TRPC6 externalization (8). Transient transfection of ECs with CaM siRNA significantly reduced the level of CaM (Fig. 1C) and the lysoPC-induced ERK1/2 phosphorylation [n = 4; P < 0.01, compared with ECs transiently transfected with negative control siRNA (NsiRNA) that has no homology to a known gene sequence; Fig. 1D]. When ECs were transiently transfected with WT-CaM or mutant CaM in which the Tyr99 site was replaced with Phe (Phe99-CaM) that cannot be phosphorylated (Fig. 1E), lysoPC increased ERK1/2 phosphorylation in ECs transfected with WT-CaM but not in ECs overexpressing Phe99-CaM (n = 4; P < 0.01, compared with WT-CaM; Fig. 1F). Taken together, these findings suggested that lysoPC-induced ERK activation in ECs was Ca2+-CaM dependent and required CaM phosphorylation at Tyr99.
ERK activation by lysoPC was downstream of TRPC6 externalization but not TRPC5 externalization.
CaM phosphorylation is required for TRPC6 externalization (8); therefore, the relationship between lysoPC-induced ERK activation and TRPC activation was explored using TRPC6−/− and TRPC5−/− MAECs. The basal level of ERK1/2 phosphorylation was similar in WT, TRPC6−/−, and TRPC5−/− MAECs under control conditions (Fig. 1G). Incubation with lysoPC (10 µM) for 15 min caused an increase in ERK1/2 phosphorylation to 301 ± 10% of control in WT MAECs (n = 3; P < 0.01; Fig. 1, G and H), but no change in ERK1/2 phosphorylation in TRPC6−/− MAECs (n = 3; Fig. 1, G and H). Similar to WT MAECs, lysoPC induced an increase in ERK1/2 phosphorylation to 300 ± 10% of control in TRPC5−/− MAECs (n = 3; P < 0.03; Fig. 1, G and H). These data indicated that lysoPC-induced ERK activation required TRPC6 but not TRPC5, suggesting that ERK activation is downstream from TRPC6 but upstream from TRPC5 activation.
To further assess the role of ERK in TRPC externalization, ERK was downregulated with siRNA. Transient transfection of ECs with ERK siRNA resulted in a significant decrease in ERK at 48 h (n = 3; P < 0.01, compared with NsiRNA; Fig. 2A). The effect of ERK downregulation on TRPC6 and TRPC5 externalization was assessed by biotinylation assay (Fig. 2, B–G). Transfection with ERK siRNA had no effect on lysoPC-induced TRPC6 externalization (n = 4; Fig. 2, D and E); however, TRPC5 externalization was significantly inhibited (n = 4; P < 0.04, compared with NsiRNA transfected ECs incubated with lysoPC; Fig. 2, F and G). These results suggested that lysoPC increased TRPC5 externalization through an ERK-dependent mechanism.
Fig. 2.

LysoPC-induced ERK activation regulates TRPC5 externalization. A: ECs were transiently transfected with NsiRNA (40 nM) or ERK1 siRNA and ERK2 siRNA (20 nM of each) for 24 h. The siRNA was removed, and 24 h later the cells were lysed. ERK was detected by immunoblot analysis. Actin served as loading control (n = 3). B and C: TRPC externalization was determined by biotinylation of proteins on the cell surface, precipitation of biotin-streptavidin complexes, resolution of proteins by SDS-PAGE, and immunoblot analysis. To verify the specificity of the assay, controls included omission of the primary antibody for TRPC6 (B) or TRPC5 (C) but inclusion of the horseradish peroxidase-conjugated secondary antibody. Total TRPC6 or TRPC5 was determined by immunoblot analysis of an aliquot of the same sample (n = 3). Actin served as loading control (n = 3). D–G: ECs were transiently transfected with NsiRNA or ERK1 siRNA and ERK2 siRNA for 24 h before incubation with lysoPC (12.5 µM) for 15 min. Externalization of TRPC6 (D) or TRPC5 (F) was detected by biotinylation assay and total TRPC6 (D) or TRPC5 (F) by immunoblot analysis. Actin served as loading control (n = 4). The increase in externalized TRPC6 (E) or TRPC5 (G) is represented in graphic form: ●, medium control; ■, medium + lysoPC; ▲, NsiRNA + lysoPC; ▼, ERK 1 and 2 siRNA + lysoPC; ◆, NsiRNA medium control; ○, ERK 1 and 2 siRNA medium control. (In D and F, lines indicate lanes rearranged from the same gel. All bands are from the same gel.)
LysoPC induced ERK-dependent NADPH activation.
LysoPC and ERK can activate NADPH oxidase (35, 43), and TRPC5 can be activated by ROS (27). Therefore, we explored the role of NADPH oxidase in lysoPC-induced TRPC5 externalization. We previously showed that lysoPC caused the p47phox and p67phox subunits of NADPH oxidase to translocate from the cytosol to the membrane (32), characteristic of NADPH oxidase activation. After EC incubation in lysoPC (12.5 µM), but not PC (12.5 µM), for 15 min, p47phox phosphorylation at Ser345 increased to 340 ± 20% of control (n = 4; P < 0.02; Fig. 3A), again suggesting NADPH oxidase activation.
Fig. 3.

LysoPC-induced NADPH oxidase activation is Ca2+- and ERK dependent. A: ECs were incubated with PC (12.5 µM) or lysoPC (12.5 µM) for 15 min. Phospho-p47phox(Ser345) was detected by immunoblot analysis, and actin served as loading control (n = 4). B: ECs were incubated in calcium-free or calcium-containing KR buffer for 10 min or ECs were incubated with BAPTA/AM (25 µM) for 30 min before adding lysoPC. Phospho-p47phox(Ser345) was detected by immunoblot analysis. Actin served as loading control (n = 4). C: ECs were incubated with calcium-free or calcium-containing KR buffer for 10 min before adding lysoPC. Cytosolic and membrane fraction proteins were separated, and p47phox was detected by immunoblot analysis (n = 4). D and E: ECs were incubated with lysoPC (12.5 µM) for 15 min. Phospho-ERK1/2(Thr202/Tyr 204) was immunoprecipitated, and associated p47phox (D) or p67phox (E) was detected by immunoblot analysis (n = 3). Total p47phox or p67phox was measured by immunoblot analysis of the lysate before immunoprecipitation (n = 3). F: ECs were transiently transfected with NsiRNA (40 nM) or ERK1 siRNA and ERK2 siRNA (20 nM of each) for 24 h before incubation with lysoPC. Phospho-p47phox(Ser345) was detected by immunoblot analysis, and actin served as loading control (n = 5). G: ECs were transfected with NsiRNA or p47phox siRNA (20 nM) for 24 h, then incubated with lysoPC (12.5 µM) for 15 min. ERK phosphorylation (n = 4) was detected by immunoblot analysis. Actin served as loading control (n = 4). H and I: ECs were transfected with NsiRNA or p22phox siRNA (30 nM) for 24 h. The siRNA was then removed. H: At 24 h after siRNA removal, the cells were lysed, and p22phox was identified by immunoblot analysis. Actin served as loading control (n = 3). (I) After siRNA removal, ECs were incubated with lysoPC (12.5 µM) for 15 min. Phospho-ERK1/2(Thr202/Tyr204) and ERK1/2 were detected by immunoblot analysis. Actin served as loading control (n = 3). (In A and B, F and G, lines indicate lanes rearranged from the same gel. All bands are from the same gel.)
To determine if lysoPC-induced NADPH oxidase activation was Ca2+ dependent, ECs were incubated in Ca2+-free or Ca2+-containing KR buffer, or with BAPTA/AM (25 µM), before addition of lysoPC (12.5 µM). LysoPC did not induce p47phox phosphorylation at Ser345 in absence of extracellular Ca2+ (n = 4; P < 0.01, compared with Ca2+-containing KR buffer; Fig. 3B) or in the presence of BAPTA/AM (n = 4; P < 0.02, compared with incubation with lysoPC in Ca2+-containing KR buffer; Fig. 3B). In addition, lysoPC-induced translocation of p47phox to the membrane from the cytosol was reduced in the absence of Ca2+ (n = 4; Fig. 3C), suggesting that NADPH oxidase activation was Ca2+ dependent.
The role of ERK activation in NADPH oxidase activation was explored. LysoPC induced ERK phosphorylation, and this was accompanied by increased association of p47phox and phospho-ERK (Fig. 3D; n = 3) as well as p67phox and phospho-ERK (Fig. 3E; n = 3). To determine if NADPH oxidase activation by lysoPC required ERK, ERK was downregulated with siRNA for 24 h and then, lysoPC (12.5 µM) was added for 15 min. ERK downregulation caused a significant reduction in lysoPC-induced p47phox phosphorylation (n = 5; P < 0.02, compared with NsiRNA; Fig. 3F), indicating that lysoPC-induced NADPH oxidase activation was ERK dependent. Conversely, downregulation of p47phox did not alter lysoPC-induced ERK phosphorylation (n = 4; Fig. 3G). Similarly, downregulation of p22phox (n = 3; Fig. 3H) did not alter lysoPC-induced ERK phosphorylation (n = 3; Fig. 3I).
To confirm that p47phox phosphorylation induced by lysoPC required TRPC6 activation, p47phox phosphorylation was assessed in TRPC6−/− or TRPC5−/− MAECs. LysoPC induced p47phox phosphorylation in WT MAECs but not in TRPC6−/− MAECs (n = 4; P < 0.01, compared with WT MAECs with lysoPC; Fig. 4, A and B). LysoPC-induced p47phox phosphorylation in TRPC5−/− MAECs was similar to that in WT MAECs (n = 4; Fig. 4, A and B). These results suggested that lysoPC-induced NADPH oxidase activation was TRPC6 dependent. To determine if TRPC6 was required for NADPH oxidase activation by lysoPC in human ECs, TRPC6 was downregulated using siRNA (n = 3; Fig. 4C). This blocked lysoPC-induced p47phox phosphorylaton (n = 3; Fig. 4D).
Fig. 4.
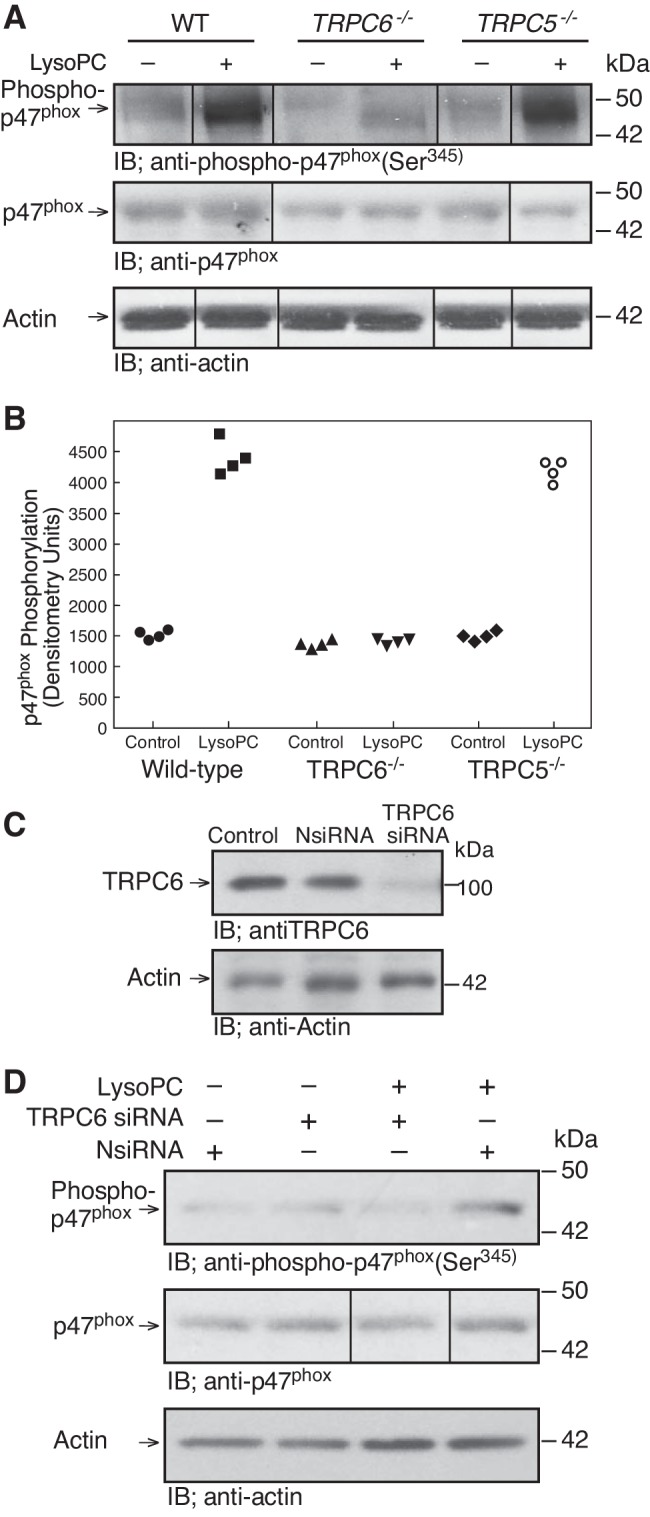
LysoPC-induced NADPH oxidase activation requires TRPC6. A and B: WT, TRPC6−/−, or TRPC5−/− MAECs were incubated with lysoPC (10 µM) for 15 min. A: phospho-p47phox(Ser345) and total p47phox were detected by immunoblot analysis. Actin served as loading control (n = 4). B: densitometry measurements of and phospho-p47phox(Ser345) are represented in graphic form: ●, medium control with WT ECs; ■, lysoPC with WT ECs; ▲, medium control with TRPC6−/− ECs; ▼, lysoPC with TRPC6−/− ECs, ◆, medium control with TRPC5−/− ECs; ○, lysoPC with TRPC5−/− ECs. C and D: human ECs (EA.hy926) were transiently transfected with NsiRNA (20 nM) or TRPC6 siRNA (20 nM) for 24 h. siRNA was removed. C: at 24 h after siRNA removal, cells were lysed and TRPC6 was detected by immunoblot analysis. Actin served as loading control (n = 3). D: after siRNA removal, EA.hy926 cells were incubated with lysoPC (12.5 µM) for 15 min. Phosphorylation of p47phox and total p47phox were detected by immunoblot analysis. Actin served as loading control (n = 3). (In A and D, lines indicate lanes rearranged from the same gel. All bands are from the same gel.)
Superoxide and NADPH oxidase activation were required for lysoPC-induced TRPC5 externalization.
Because ROS can activate TRPC5 (27), the role of superoxide in lysoPC-induced TRPC6 and TRPC5 externalization was assessed by biotinylation assay. ECs were pretreated with superoxide dismutase (SOD; 50 units/ml; Sigma) or PEG-catalase (5,000 units/ml) for 30 min before incubation with lysoPC (12.5 µM) for 15 min. LysoPC-induced TRPC6 externalization was not altered by either agent (n = 3; Fig. 5A). TRPC5 externalization, however, was blocked by SOD, suggesting that superoxide production was required for lysoPC-induced TRPC5 externalization (n = 3; Fig. 5B). Preincubation of ECs with PEG-catalase did not block lysoPC-induced TRPC5 externalization (Fig. 5B).
Fig. 5.

NADPH oxidase activation is required for TRPC5 externalization. A and B: ECs were made serum-free for 3 h, then pretreated with superoxide dismutase (SOD; 50 units/ml) or PEG catalase (5,000 units/ml) for 30 min before incubation with lysoPC (12.5 µM) for 15 min. TRPC6 or TRPC5 externalization was detected by biotinylation assay and total TRPC6 or TRPC5 was determined by immunoblot analysis of an aliquot of the same sample (n = 3). Actin served as loading control (n = 3). C: ECs were transiently transfected with NsiRNA or p47phox siRNA (20 nM) for 24 h. The siRNA was removed, and 24 h later cells were lysed. Total p47phox was detected by immunoblot analysis. Actin served as loading control (n = 5). D and E: ECs were transiently transfected with NsiRNA or p47phox siRNA for 24 h and then incubated with H2DCF-DA for 20 min before incubation with lysoPC (n = 5). D: ROS production was detected by DCF assay using fluorescence microscopy. E: fluorescence intensity was quantitated using image analysis software and graphed. Results are represented as means ± SD (n = 5; *P < 0.01, compared with NsiRNA-transfected EC control; **P < 0.02, compared with NsiRNA-transfected ECs incubated with lysoPC). F and G: ECs were transiently transfected with NsiRNA or p47phox siRNA for 24 h before incubation with lysoPC. Externalization of TRPC6 or TRPC5 was detected by biotinylation assay and total TRPC6 or TRPC5 by immunoblot analysis. Actin served as loading control (F: n = 4; G: n = 5). H and I: ECs were incubated with PC (12.5 µM) for 15 min, TNF-α (500 U/ml) for 30 min, or lysoPC (12.5 µM) for 15 min. Externalization of TRPC6 or TRPC5 was detected by biotinylation assay and total TRPC6 or TRPC5 by immunoblot analysis. Actin served as loading control (n = 3). (In A–C and G, lines indicate lanes rearranged from the same gel. All bands are from the same gel.)
To verify the role NADPH oxidase activity in the lysoPC-induced superoxide production, p47phox was downregulated with siRNA. In ECs transiently transfected with p47phox siRNA for 48 h, p47phox protein levels were significantly reduced (n = 5; P < 0.01, compared with NsiRNA; Fig. 5C). Next, ROS production was determined using H2DCF-DA. Addition of lysoPC (12.5 µM) for 15 min induced an increase in ROS production to 780 ± 40% of control in NsiRNA-transfected ECs as detected by H2DCF-DA assay (n = 5; P < 0.01; Fig. 5, D and E). In ECs transiently transfected with p47phox siRNA, incubation with lysoPC caused an increase in ROS production to only 201 ± 40% of control (n = 5; P < 0.02 compared with NsiRNA-transfected ECs incubated with lysoPC; Fig. 5, D and E). Thus downregulation of p47phox decreased the lysoPC-induced ROS production by ~75%.
Next, the role of NADPH oxidase in TRPC6 and TRPC5 externalization was assessed. In ECs transiently transfected with p47phox siRNA, incubation with lysoPC induced TRPC6 externalization (n = 4; Fig. 5F), but TRPC5 externalization was blocked (n = 5; P < 0.02, compared with NsiRNA with lysoPC; Fig. 5G). In ECs incubated for 30 min with TNF-α (500 U/ml), an NADPH oxidase activator (17), TRPC6 was not externalized (n = 3; Fig. 5H), but TRPC5 externalization was increased to 202 ± 40% of control (n = 3; P < 0.01; Fig. 5I). These results suggested that NADPH oxidase activation could promote TRPC5 externalization but not TRPC6 externalization.
LysoPC-induced MLCK activation required Ca2+ influx and TRPC6 channels.
ROS can activate MLCK (51), a Ca2+-CaM-dependent kinase that phosphorylates MLC, and MLC can modulate the insertion of TRPC5 containing vesicles into the plasma membrane, resulting in activation of TRPC5 (21, 37). We assessed MLCK phosphorylation at Tyr471 as this can contribute to increased enzymatic activity (3). In ECs incubated with lysoPC (12.5 µM), but not PC (12.5 µM), for 15 min, MLCK phosphorylation at Tyr471 increased to 230 ± 40% of medium alone (n = 3; P < 0.01; Fig. 6A). LysoPC-induced MLCK phosphorylation was inhibited in the absence of extracellular Ca2+ (n = 4; P < 0.03, compared with Ca2+-containing KR buffer; Fig. 6B) or the presence of BAPTA/AM (n = 4; P < 0.01, compared with Ca2+-containing KR buffer; Fig. 6B), suggesting that lysoPC-induced MLCK phosphorylation was Ca2+ dependent.
Fig. 6.

LysoPC-induced MLCK phosphorylation is dependent on Ca2+ and TRPC6 channels. A: ECs were incubated with PC (12.5 µM) or lysoPC (12.5 µM) for 15 min. Phospho-MLCK(Tyr471) was detected by immunoblot analysis, and actin served as loading control (n = 3). B: ECs were incubated in calcium-free or calcium-containing KR buffer for 10 min or ECs were incubated with BAPTA/AM (25 µM) for 30 min before adding lysoPC. Phospho-MLCK(Tyr471) was detected by immunoblot analysis, and actin served as loading control (n = 4). C–F: WT, TRPC5−/−, or TRPC6−/− MAECs were incubated with lysoPC (10 µM) for 15 min. Phospho-MLCK(Tyr471) and total MLCK (C) or phospho-MLC(Thr18/Ser19) and total MLC (E) were detected by immunoblot analysis. Actin served as loading control (n = 4). Densitometry measurements of phospho-MLCK(Tyr471) (D) or phospho-MLC(Thr18/Ser19 (F) are represented in graphic form: ●, medium control with WT ECs; ■, lysoPC with WT ECs; ▲, medium control with TRPC6−/− ECs; ▼, lysoPC with TRPC6−/− ECs; ◆, medium control with TRPC5−/− ECs; ○, lysoPC with TRPC5−/− ECs. (In A–C and E, lines indicate lanes rearranged from the same gel. All bands are from the same gel.)
To determine if MLCK activation was dependent on TRPC activation, we assessed MLCK phosphorylation in WT, TRPC6−/−, or TRPC5−/− MAECs. LysoPC induced a robust increase in MLCK phosphorylation to 220 ± 30% of control in WT MAECs (n = 4; P < 0.02) and an increase in MLCK phosphorylation to 220 ± 26% of control in TRPC5−/− MAECs (n = 4, P < 0.02; Fig. 6, C and D). No increase was noted when TRPC6−/− MAECs were incubated with lysoPC (n = 4; P = 0.7, compared with WT control; Fig. 6, C and D), confirming that lysoPC-induced MLCK phosphorylation was TRPC6 dependent. The MLCK phosphorylation was accompanied by an increase in MLC phosphorylation. Incubation with lysoPC increased MLC phosphorylation to 230 ± 40% of control in WT MAECs (n = 4; P < 0.02; Fig. 6, E and F). LysoPC also increased MLC phosphorylation to 220 ± 30% of control in TRPC5−/− MAECs (n = 4; P < 0.02; Fig. 6, E and F). No lysoPC-induced increase in MLC phosphorylation was noted in TRPC6−/− MAECs (n = 4; P = 0.8, compared with TRPC6−/− control; Fig. 6, E and F). The endogenous levels of MLC were not changed in the presence of lysoPC (n = 3; Fig. 6E). These results supported the TRPC6 dependence of MLCK activation by lysoPC.
ERK and NADPH oxidase activation were required for lysoPC-induced MLCK activation and MLC phosphorylation.
The role of ERK and NADPH oxidase in MLCK phosphorylation was assessed. In ECs incubated with the NADPH oxidase activator TNF-α (500 U/ml) for 30 min, MLCK phosphorylation was increased significantly (n = 3; P < 0.03, compared with control; Fig. 7A). The roles of ERK and NADPH oxidase in MLCK phosphorylation were confirmed by downregulating ERK or p47phox. ERK siRNA significantly reduced lysoPC-induced MLCK phosphorylation (n = 4; P < 0.01, compared with NsiRNA; Fig. 7B). Similarly, p47phox siRNA inhibited lysoPC-induced MLCK phosphorylation (n = 4; P < 0.02, compared with NsiRNA; Fig. 7B), suggesting the involvement of both ERK and NADPH oxidase in lysoPC-induced MLCK phosphorylation. The lysoPC-induced MLCK phosphorylation was accompanied by an increase in MLC phosphorylation. In the medium control, lysoPC increased MLC phosphorylation to 240 ± 20% of control (n = 3; P < 0.01; Fig. 7C). In ECs transfected with NsiRNA, lysoPC also increased MLC phosphorylation to 240 ± 20% of control (n = 3; P < 0.01; Fig. 7C). Downregulation of ERK using siRNA blocked lysoPC-induced MLC phosphorylation (n = 3; P < 0.02, compared with NsiRNA with lysoPC; P < 0.01, compared with medium control; Fig. 7C). Similarly, downregulation of p47phox using siRNA significantly reduced lysoPC-induced MLC phosphorylation (n = 3; P < 0.01, compared with NsiRNA with lysoPC; Fig. 7D). These results suggested that lysoPC-induced MLCK phosphorylation and MLC phosphorylation were dependent on the activation of ERK and NADPH oxidase.
Fig. 7.

LysoPC-induced MLCK and MLC phosphorylation are dependent on ERK and NADPH oxidase. A: ECs were incubated with PC (12.5 µM) or TNF-α (500 U/ml) for 30 min. Phospho-MLCK(Tyr471) was detected by immunoblot analysis, and actin served as loading control (n = 3). B–D: ECs were transiently transfected with NsiRNA (40 nM), ERK1 siRNA and ERK2 siRNA (20 nM of each), or p47phox siRNA (20 nM) for 24 h before incubation with lysoPC or TNF-α. B: phospho-MLCK(Tyr471) and total MLCK were detected by immunoblot analysis, and actin served as loading control (n = 4). C and D: phospho-MLC(Thr18/Ser19) and total MLC were detected by immunoblot analysis, and actin served as loading control (n = 3). E and F: ECs were incubated with superoxide dismutase (50 U/ml) for 30 min, then lysoPC (12.5 µM) was added for 15 min. E: phospho-MLCK(Tyr471) and total MLCK were a detected by immunoblot analysis. Actin served as loading control (n = 3). F: phospho-MLC(Thr18/Ser19) and total MLC were detected by immunoblot analysis. Actin served as loading control (n = 3). (In A and B, E and F, lines indicate lanes rearranged from the same gel. All bands are from the same gel.)
MLCK activation by lysoPC required superoxide.
The results with p47phox siRNA suggested that superoxide production is required for lysoPC-induced MLCK activation. To confirm the role of ROS, MLCK phosphorylation was measured in the presence of SOD. ECs were preincubated with SOD for 30 min and then lysoPC (12.5 µM) was added for 15 min. Incubation with lysoPC increased MLCK phosphorylation to 260 ± 20% of control (n = 3; P < 0.01; Fig. 7E), but after SOD pretreatment lysoPC did not cause a significant increase (n = 3; P = 0.8, compared with SOD control; Fig. 7E). Similarly, lysoPC increased MLC phosphorylation to 260 ± 10% of medium control (n = 3; P < 0.01; Fig. 7F), but after SOD pretreatment lysoPC did not cause a significant increase (n = 3; P = 0.8, compared with SOD control; Fig. 7F). Endogenous levels of MLCK and MLC were not changed by SOD pretreatment (n = 3; Fig. 7, E and F). These results suggested that lysoPC-induced MLCK activation required superoxide.
LysoPC-induced TRPC5 externalization required MLCK phosphorylation.
The role of MLCK in TRPC5 externalization was analyzed by inhibiting or downregulating MLCK. ECs were preincubated with ML-9 (10 µM), a relatively selective inhibitor that impairs MLCK phosphorylation by competing for the ATP binding site. After 30 min, lysoPC (12.5 µM) was added for 15 min. ML-9 had no effect on total TRPC6 or TRPC5 level and did not inhibit the lysoPC-induced TRPC6 translocation to the cell membrane suggesting that TRPC6 activation was MLCK independent (n = 4; Fig. 8A). However, ML-9 blocked lysoPC-induced TRPC5 translocation (n = 4; P < 0.02, compared with lysoPC control; Fig. 8B). In ECs transiently transfected with MLCK siRNA for 48 h, MLCK was downregulated significantly (n = 4; P < 0.02, compared with NsiRNA; Fig. 8C). When MLCK was downregulated, lysoPC-induced TRPC6 externalization was unchanged (n = 4, Fig. 8D), but TRPC5 externalization was markedly diminished (n = 4; P < 0.02, compared with NsiRNA with lysoPC; Fig. 8E). Similarly, when MLCK was downregulated with siRNA, incubation with TNF-α (500 U/ml) for 30 min did not increase TRPC5 externalization (n = 4; P = 0.8, compared with MLCK siRNA-transfected ECs without TNF-α; Fig. 8E). These results suggested that NADPH oxidase-mediated TRPC5 externalization, including that by lysoPC, was dependent on MLCK.
Fig. 8.

MLCK inhibition blocks lysoPC-induced TRPC5 externalization and increased [Ca2+]i, and partially preserves EC migration in lysoPC. A and B: ECs were pretreated with ML-9 (10 µM) for 30 min before incubation with lysoPC (12.5 µM). Externalization of TRPC6 or TRPC5 was detected by biotinylation assay and total TRPC6 or TRPC5 by immunoblot analysis. Actin served as loading control (n = 4). C: ECs were transiently transfected with NsiRNA or MLCK siRNA (30 nM) for 24 h. The siRNA was removed, and 24 h later the cells were lysed. MLCK was detected by immunoblot analysis. Actin served as loading control (n = 4). D and E: ECs were transiently transfected with NsiRNA or MLCK siRNA for 24 h before incubation with lysoPC or TNF-α. Externalization of TRPC6 or TRPC5 was detected by biotinylation assay and total TRPC6 or TRPC5 by immunoblot analysis. Actin served as loading control (n = 4). F: ECs were made quiescent for 18 h, then loaded with fura 2-AM. After the baseline [Ca2+]i was established, lysoPC (12.5 µM) was added to the cells (double arrow) and relative change of [Ca2+]i was measured. After [Ca2+]i plateaued, lysoPC was removed by washing with KR buffer. After readings returned to baseline (in 7–10 min), ML-9 (10 µM) was added and recordings made for 60 s. After 20 min, lysoPC (12.5 µM, arrow) was added, and the relative change of [Ca2+]i was measured. The tracing is representative of four separate experiments (//denotes the wash period and ML-9 incubation period during which times no data were collected). G: ECs were pretreated with ML-9 (10 µM) for 30 min, then the ML-9 was removed, and the migration assay initiated. Migration was assessed after 24 h in the presence or absence of lysoPC. Results are represented as means ± SD (n = 5; *P < 0.001, compared with control; **P < 0.001, compared with ML-9 control; †P < 0.001, compared with lysoPC). H: ECs were transiently transfected for 24 h with NsiRNA or MLCK siRNA (30 nM) and then made quiescent for 12 h. The migration assay initiated, and migration in the presence or absence of lysoPC was assessed after 24 h. Results were represented as means ± SD (n = 5; *P < 0.001, compared with control; **P < 0.001, compared with MLCK siRNA; †P < 0.001, compared with lysoPC with NsiRNA). (In A, D and E, lines indicate lanes rearranged from the same gel. All bands are from the same gel.)
MLCK inhibition reduced lysoPC-induced [Ca2+]i rise and inhibition of EC migration.
To determine if inhibition of MLCK blocked the lysoPC-mediated increase of [Ca2+]i, ECs were loaded with fura 2-AM for 30 min. After baseline [Ca2+]i readings were obtained, lysoPC (12.5 µM) was added and the increase of [Ca2+]i was assessed. LysoPC was removed and cells were washed. Then, ECs were incubated with ML-9 (10 µM) for 20 min and lysoPC again added. ML-9 did not alter the basal [Ca2+]i readings nor inhibit the initial transient increase in [Ca2+]i induced by lysoPC, presumably through TRPC6 channels but blocked the long plateau phase (n = 4; P < 0.02, compared with lysoPC control; Fig. 8F) that was attributed to TRPC5 activation (5).
To determine the role of MLCK in lysoPC-induced inhibition of EC migration, ECs were preincubated with ML-9 for 30 min. Basal migration of ECs with or without ML-9 was similar. LysoPC inhibited 24 h EC migration by 63%, but after pretreatment with ML-9, lysoPC inhibited EC migration by only 42% (n = 5; P < 0.001, compared with lysoPC without ML-9; Fig. 8G). Since ML-9 is only relatively specific for MLCK, the effect of MLCK downregulation using siRNA on EC migration was also assessed. Results using MLCK siRNA paralleled those with ML-9. Basal migration of control ECs and ECs transfected with NsiRNA or MLCK siRNA was similar. LysoPC inhibited 24-h migration of ECs transfected with NsiRNA by 64%, but in ECs transiently transfected with MLCK siRNA, lysoPC inhibited EC migration by only 43% (n = 3; P < 0.001, compared with ECs transfected with NsiRNA and incubated with lysoPC; Fig. 8H). This preservation of migration was similar to that seen in TRPC5−/− MAECs or when TRPC5 was downregulated using siRNA (5, 33). Taken together, these data suggested that MLCK is essential in the lysoPC-induced TRPC5 activation and inhibition of EC migration.
DISCUSSION
TRPC5 is activated by a variety of agonists and through multiple mechanisms (50). Ca2+-dependent regulation of Ca2+-permeable cation channels is a ubiquitous mechanism of regulation. We have shown that lysoPC, the major lysophospholipid in oxidized LDL, activates TRPC6 with a resultant increase in [Ca2+]i and this sets in motion a cascade of events resulting in TRPC5 externalization with a prolonged increase in [Ca2+]i that disrupts the coordinated calcium transients required for normal EC migration. The studies presented here define some of the steps in this cascade.
An increase in [Ca2+]i via TRPC6 channels can activate several pathways and kinases including ERK (18). Ca2+ and CaM can regulate the Ras/Raf/MEK/ERK pathway and positively modulate ERK1/2 activation (1, 24). We show that lysoPC-induced ERK activation is regulated by influx of Ca2+ specifically through TRPC6 channels. ERK and TRPC5 are not activated when ECs from TRPC6−/− mice are exposed to lysoPC. These findings are consistent with the work of other investigators showing increased ERK1/2 phosphorylation in 293T cells and podocytes that overexpress TRPC6 proteins with gain-of-function mutations (10).
We have shown that lysoPC increases EC production of ROS through activation of NADPH oxidase and this contributes to the inhibition of EC migration (43). Activation of NADPH oxidase in ECs involves phosphorylation and translocation of cytosolic components, including p47phox and p67phox, to the membrane where they assemble with Nox2 or Nox4 and p22phox (29). NADPH oxidase activation by specific agonists requires an increase in intracellular calcium (11), and lysophosphatidylcholines prime NADPH oxidase and stimulate multiple neutrophil functions through changes in cytosolic Ca2+ (38). We provide evidence that the lysoPC-induced activation of NADPH oxidase in ECs depends on Ca2+ influx through TRPC6-containing channels because lysoPC does not activate NADPH oxidase in TRPC6−/− MAECs. TRPC6 can be forming a complex with other TRPC proteins to permit Ca2+ influx to activate NADPH oxidase as has been shown in granulocytes (4). Furthermore, the resultant Ca2+ influx can activate NADPH oxidase directly or indirectly through activation of a kinase.
ERK can activate NADPH oxidase (13, 35). When ECs are exposed to transient hypoxia, ERK2 is activated, and this in turn activates NADPH oxidase causing an increase in ROS production (35). Our finding that downregulation of ERK blocks lysoPC-induced NADPH activation also indicates that ERK activation is upstream from NADPH oxidase activation. We find that downregulation of p47phox or p22phox does not alter lysoPC-induced ERK phosphorylation. Similarly, angiotensin II-induced ROS formation is blocked by p22phox antisense, but this has no effect on angiotensin II-induced ERK activation (44). Our results suggest that Ca2+ influx through TRPC6 channels activates ERK that in turn activates NADPH oxidase.
The importance of NADPH oxidase activation lies, in part, in the ability of superoxide to function as an intracellular signaling molecule. Although ROS can activate MLCK (51), and MLCK can activate TRPC5 channels (21, 37), there is conflicting evidence in the literature on the ability of ROS to activate TRPC5. One report indicates that TRPC5 can be activated by ROS (27). Another study suggests that ROS do not activate TRPC5, but only one inhibitor was used and its effectiveness was not assessed (16). Our findings support a major role for ROS in the lysoPC-induced externalization of TRPC5.
ERK can phosphorylate MLCK and thereby increase its activity (3, 22), and MLCK is essential for TRPC5 channel externalization (21, 37). MLCK phosphorylates MLC, and MLC phosphorylation/dephosphorylation regulates the insertion of TRPC5 containing vesicles into the plasma membrane (21, 37). Intracellular Ca2+-calmodulin may constitutively activate MLCK maintaining a basal distribution of TRPC5 channels at plasma membrane (37). Our data suggest that lysoPC increases MLCK activity through ERK activation and subsequent MLCK phosphorylation.
Inhibition of MLCK impairs TRPC5 translocation to the plasma membrane and TRPC5 activity (21, 37). We find that ML-9 can inhibit the lysoPC-induced externalization of TRPC5 channels in ECs, complementing the findings in HEK293 cells overexpressing TRPC5 where ML-9 decreased the cell surface localization of TRPC5 channels (37). In HEK293 cells overexpressing TRPC6, ML-9 has been reported to reduce cationic currents elicited by carbacol by an MLCK-independent mechanism (36). Our data do not show a reduction in basal [Ca2+]i in ECs pretreated with ML-9 nor do the results reported by other investigators studying ECs (39, 45, 46). The ability of ML-9 to reduce [Ca2+]i may be dependent on concentration, cell type, and native expression of calcium channels.
The pattern of lysoPC-induced increase in [Ca2+]i seen in ECs pretreated with ML-9 is very similar to that seen after blocking of TRPC5 activity with siRNA (5). In both cases, lysoPC induces only an initial transient increase in [Ca2+]i that we attribute to TRPC6 channels, but the prolonged elevation of [Ca2+]i is absent. ML-9 inhibits the lysoPC-induced externalization of TRPC5 but not TRPC6, supporting the role of MLCK in TRPC5, but not TRPC6, insertion into the cell membrane. LysoPC-induced phosphorylation of MLCK is blocked in ECs from TRPC6−/− mice but not in those from TRPC5−/− mice, suggesting that Ca2+ influx through TRPC6 is critical for lysoPC-induced MLCK activation.
Our findings should not be taken to suggest that there is a single linear pathway between TRPC6 channel opening and TRPC5 channel externalization. Many alternative pathways, redundancies, and regulatory influences exist. ROS production by NADPH oxidase can activate ERK (12, 49). MLCK can facilitate recruitment of cytoskeletal proteins and NADPH oxidase to the plasma membrane with subsequent activation of NADPH oxidase and increased ROS production (42). Pathologic feed-forward activities could occur with cycles of increasing ROS production and ERK and MLCK activation, culminating in elevated TRPC5 externalization.
The in vivo relevance of these studies is seen in comparing endothelial healing of arterial injuries in mice on a high-cholesterol diet compared with those on a chow diet (34). The high-cholesterol diet increases plasma lysoPC levels and inhibits endothelial healing in WT mice. Although a high-cholesterol diet causes the same increase of plasma cholesterol and a greater increase of plasma lysoPC in TRPC6−/− and TRPC5−/− mice, endothelial healing is not impaired in TRPC6−/− mice and the impairment is significantly reduced in TRPC5−/− mice (33). These findings suggest that the presence or absence of TRPC6 and TRPC5 channels has a significant influence on the cellular response to environmental factors.
Based on our results, we propose that lysoPC induces CaM-dependent TRPC6 activation allowing an initial transient rise in [Ca2+]i. This triggers ERK activation, which leads to the activation of NADPH oxidase. The increase in ROS activates MLCK, which in turn facilitates TRPC5 channel insertion into the plasma membrane and causes a prolonged influx of Ca2+ that disrupts the finely coordinated local signals that regulate calcium transients required for normal EC migration (see proposed model in Fig. 9). Although studies were conducted using animal ECs, TRPC6 and TRPC5 are expressed in human ECs, and downregulation of TRPC6 blocks lysoPC-induced p47phox phosphorylation (Fig. 4D), suggesting that the pathway outlined in this paper is functional in human ECs. Knowledge of the signaling pathway provides opportunites for its regulation and the restoration of endothelial healing under conditions of elevated lipid oxidation products or oxidative stress leading to TRPC6 or TRPC5 activation.
Fig. 9.

Proposed signaling pathway between TRPC6 activation and TRPC5 externalization. Ca2+ entry through TRPC6 activates a kinase, leading to ERK phosphorylation. ERK then phosphorylates p47phox, leading to NADPH oxidase activation and production of ROS, which contribute to the phosphorylation of MLCK that is required for externalization of TRPC5. The components of this process may be localized in a “signalsome” of the plasma membrane (PM).
GRANTS
This work was supported by National Institutes of Health National Heart, Lung, and Blood Institute (Grant R01-HL-064357 to L. M. Graham) and by the Intramural Research Program of the National Institutes of Health (National Institute of Environmental Health Sciences Project Z01-ES-101684 to L. Birnbaumer). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The contents of this article do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
P.C., L.B., and L.M.G. conceived and designed research; P.C. and M.A.R. performed experiments; P.C., M.A.R., and L.M.G. analyzed data; P.C., M.A.R., and L.M.G. interpreted results of experiments; P.C., M.A.R., and L.M.G. prepared figures; P.C. and L.M.G. drafted manuscript; P.C., M.A.R., L.B., and L.M.G. edited and revised manuscript; P.C., M.A.R., L.B., and L.M.G. approved final version of manuscript.
REFERENCES
- 1.Agell N, Bachs O, Rocamora N, Villalonga P. Modulation of the Ras/Raf/MEK/ERK pathway by Ca(2+), and calmodulin. Cell Signal 14: 649–654, 2002. doi: 10.1016/S0898-6568(02)00007-4. [DOI] [PubMed] [Google Scholar]
- 2.Bauters C, Isner JM. The biology of restenosis. Prog Cardiovasc Dis 40: 107–116, 1997. doi: 10.1016/S0033-0620(97)80003-5. [DOI] [PubMed] [Google Scholar]
- 3.Birukov KG, Csortos C, Marzilli L, Dudek S, Ma SF, Bresnick AR, Verin AD, Cotter RJ, Garcia JG. Differential regulation of alternatively spliced endothelial cell myosin light chain kinase isoforms by p60(Src). J Biol Chem 276: 8567–8573, 2001. doi: 10.1074/jbc.M005270200. [DOI] [PubMed] [Google Scholar]
- 4.Bréchard S, Melchior C, Plançon S, Schenten V, Tschirhart EJ. Store-operated Ca2+ channels formed by TRPC1, TRPC6 and Orai1 and non-store-operated channels formed by TRPC3 are involved in the regulation of NADPH oxidase in HL-60 granulocytes. Cell Calcium 44: 492–506, 2008. doi: 10.1016/j.ceca.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 5.Chaudhuri P, Colles SM, Bhat M, Van Wagoner DR, Birnbaumer L, Graham LM. Elucidation of a TRPC6-TRPC5 channel cascade that restricts endothelial cell movement. Mol Biol Cell 19: 3203–3211, 2008. doi: 10.1091/mbc.E07-08-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chaudhuri P, Colles SM, Damron DS, Graham LM. Lysophosphatidylcholine inhibits endothelial cell migration by increasing intracellular calcium and activating calpain. Arterioscler Thromb Vasc Biol 23: 218–223, 2003. doi: 10.1161/01.ATV.0000052673.77316.01. [DOI] [PubMed] [Google Scholar]
- 7.Chaudhuri P, Colles SM, Fox PL, Graham LM. Protein kinase Cdelta-dependent phosphorylation of syndecan-4 regulates cell migration. Circ Res 97: 674–681, 2005. doi: 10.1161/01.RES.0000184667.82354.b1. [DOI] [PubMed] [Google Scholar]
- 8.Chaudhuri P, Rosenbaum MA, Sinharoy P, Damron DS, Birnbaumer L, Graham LM. Membrane translocation of TRPC6 channels and endothelial migration are regulated by calmodulin and PI3 kinase activation. Proc Natl Acad Sci USA 113: 2110–2115, 2016. doi: 10.1073/pnas.1600371113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen L, Liang B, Froese DE, Liu S, Wong JT, Tran K, Hatch GM, Mymin D, Kroeger EA, Man RY, Choy PC. Oxidative modification of low density lipoprotein in normal and hyperlipidemic patients: effect of lysophosphatidylcholine composition on vascular relaxation. J Lipid Res 38: 546–553, 1997. [PubMed] [Google Scholar]
- 10.Chiluiza D, Krishna S, Schumacher VA, Schlöndorff J. Gain-of-function mutations in transient receptor potential C6 (TRPC6) activate extracellular signal-regulated kinases 1/2 (ERK1/2). J Biol Chem 288: 18407–18420, 2013. doi: 10.1074/jbc.M113.463059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cook-Mills JM, Johnson JD, Deem TL, Ochi A, Wang L, Zheng Y. Calcium mobilization and Rac1 activation are required for VCAM-1 (vascular cell adhesion molecule-1) stimulation of NADPH oxidase activity. Biochem J 378: 539–547, 2004. doi: 10.1042/bj20030794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Datla SR, Peshavariya H, Dusting GJ, Mahadev K, Goldstein BJ, Jiang F. Important role of Nox4 type NADPH oxidase in angiogenic responses in human microvascular endothelial cells in vitro. Arterioscler Thromb Vasc Biol 27: 2319–2324, 2007. doi: 10.1161/ATVBAHA.107.149450. [DOI] [PubMed] [Google Scholar]
- 13.Dewas C, Fay M, Gougerot-Pocidalo MA, El-Benna J. The mitogen-activated protein kinase extracellular signal-regulated kinase 1/2 pathway is involved in formyl-methionyl-leucyl-phenylalanine-induced p47phox phosphorylation in human neutrophils. J Immunol 165: 5238–5244, 2000. doi: 10.4049/jimmunol.165.9.5238. [DOI] [PubMed] [Google Scholar]
- 14.Dietrich A, Mederos Y Schnitzler M, Gollasch M, Gross V, Storch U, Dubrovska G, Obst M, Yildirim E, Salanova B, Kalwa H, Essin K, Pinkenburg O, Luft FC, Gudermann T, Birnbaumer L. Increased vascular smooth muscle contractility in TRPC6−/− mice. Mol Cell Biol 25: 6980–6989, 2005. doi: 10.1128/MCB.25.16.6980-6989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fishman JA, Ryan GB, Karnovsky MJ. Endothelial regeneration in the rat carotid artery and the significance of endothelial denudation in the pathogenesis of myointimal thickening. Lab Invest 32: 339–351, 1975. [PubMed] [Google Scholar]
- 16.Flemming PK, Dedman AM, Xu S-Z, Li J, Zeng F, Naylor J, Benham CD, Bateson AN, Muraki K, Beech DJ. Sensing of lysophospholipids by TRPC5 calcium channel. J Biol Chem 281: 4977–4982, 2006. doi: 10.1074/jbc.M510301200. [DOI] [PubMed] [Google Scholar]
- 17.Frey RS, Rahman A, Kefer JC, Minshall RD, Malik AB. PKCzeta regulates TNF-alpha-induced activation of NADPH oxidase in endothelial cells. Circ Res 90: 1012–1019, 2002. doi: 10.1161/01.RES.0000017631.28815.8E. [DOI] [PubMed] [Google Scholar]
- 18.Heiser JH, Schuwald AM, Sillani G, Ye L, Müller WE, Leuner K. TRPC6 channel-mediated neurite outgrowth in PC12 cells and hippocampal neurons involves activation of RAS/MEK/ERK, PI3K, and CAMKIV signaling. J Neurochem 127: 303–313, 2013. doi: 10.1111/jnc.12376. [DOI] [PubMed] [Google Scholar]
- 19.Ishii I, Fukushima N, Ye X, Chun J. Lysophospholipid receptors: signaling and biology. Annu Rev Biochem 73: 321–354, 2004. doi: 10.1146/annurev.biochem.73.011303.073731. [DOI] [PubMed] [Google Scholar]
- 20.Isshiki M, Ando J, Yamamoto K, Fujita T, Ying Y, Anderson RGW. Sites of Ca(2+) wave initiation move with caveolae to the trailing edge of migrating cells. J Cell Sci 115: 475–484, 2002. [DOI] [PubMed] [Google Scholar]
- 21.Kim MT, Kim BJ, Lee JH, Kwon SC, Yeon DS, Yang DK, So I, Kim KW. Involvement of calmodulin and myosin light chain kinase in activation of mTRPC5 expressed in HEK cells. Am J Physiol Cell Physiol 290: C1031–C1040, 2006. doi: 10.1152/ajpcell.00602.2004. [DOI] [PubMed] [Google Scholar]
- 22.Klemke RL, Cai S, Giannini AL, Gallagher PJ, de Lanerolle P, Cheresh DA. Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol 137: 481–492, 1997. doi: 10.1083/jcb.137.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Libby P, Schwartz D, Brogi E, Tanaka H, Clinton SK. A cascade model for restenosis. A special case of atherosclerosis progression. Circulation 86, Suppl: III47–III52, 1992. [PubMed] [Google Scholar]
- 24.Mottet D, Michel G, Renard P, Ninane N, Raes M, Michiels C. Role of ERK and calcium in the hypoxia-induced activation of HIF-1. J Cell Physiol 194: 30–44, 2003. doi: 10.1002/jcp.10176. [DOI] [PubMed] [Google Scholar]
- 25.Murugesan G, Chisolm GM, Fox PL. Oxidized low density lipoprotein inhibits the migration of aortic endothelial cells in vitro. J Cell Biol 120: 1011–1019, 1993. doi: 10.1083/jcb.120.4.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murugesan G, Fox PL. Role of lysophosphatidylcholine in the inhibition of endothelial cell motility by oxidized low density lipoprotein. J Clin Invest 97: 2736–2744, 1996. doi: 10.1172/JCI118728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naylor J, Al-Shawaf E, McKeown L, Manna PT, Porter KE, O’Regan D, Muraki K, Beech DJ. TRPC5 channel sensitivities to antioxidants and hydroxylated stilbenes. J Biol Chem 286: 5078–5086, 2011. doi: 10.1074/jbc.M110.196956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ordaz B, Tang J, Xiao R, Salgado A, Sampieri A, Zhu MX, Vaca L. Calmodulin and calcium interplay in the modulation of TRPC5 channel activity. Identification of a novel C-terminal domain for calcium/calmodulin-mediated facilitation. J Biol Chem 280: 30788–30796, 2005. doi: 10.1074/jbc.M504745200. [DOI] [PubMed] [Google Scholar]
- 29.Pendyala S, Usatyuk PV, Gorshkova IA, Garcia JG, Natarajan V. Regulation of NADPH oxidase in vascular endothelium: the role of phospholipases, protein kinases, and cytoskeletal proteins. Antioxid Redox Signal 11: 841–860, 2009. doi: 10.1089/ars.2008.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phelan KD, Shwe UT, Abramowitz J, Wu H, Rhee SW, Howell MD, Gottschall PE, Freichel M, Flockerzi V, Birnbaumer L, Zheng F. Canonical transient receptor channel 5 (TRPC5) and TRPC1/4 contribute to seizure and excitotoxicity by distinct cellular mechanisms. Mol Pharmacol 83: 429–438, 2013. doi: 10.1124/mol.112.082271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Portman OW, Alexander M. Lysophosphatidylcholine concentrations and metabolism in aortic intima plus inner media: effect of nutritionally induced atherosclerosis. J Lipid Res 10: 158–165, 1969. [PubMed] [Google Scholar]
- 32.Rosenbaum MA, Chaudhuri P, Abelson B, Cross BN, Graham LM. Apolipoprotein A-I mimetic peptide reverses impaired arterial healing after injury by reducing oxidative stress. Atherosclerosis 241: 709–715, 2015. doi: 10.1016/j.atherosclerosis.2015.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenbaum MA, Chaudhuri P, Graham LM. Hypercholesterolemia inhibits re-endothelialization of arterial injuries by TRPC channel activation. J Vasc Surg 62: 1040–1047.e2, 2015. doi: 10.1016/j.jvs.2014.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosenbaum MA, Miyazaki K, Graham LM. Hypercholesterolemia and oxidative stress inhibit endothelial cell healing after arterial injury. J Vasc Surg 55: 489–496, 2012. doi: 10.1016/j.jvs.2011.07.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schäfer M, Schäfer C, Ewald N, Piper HM, Noll T. Role of redox signaling in the autonomous proliferative response of endothelial cells to hypoxia. Circ Res 92: 1010–1015, 2003. doi: 10.1161/01.RES.0000070882.81508.FC. [DOI] [PubMed] [Google Scholar]
- 36.Shi J, Takahashi S, Jin XH, Li YQ, Ito Y, Mori Y, Inoue R. Myosin light chain kinase-independent inhibition by ML-9 of murine TRPC6 channels expressed in HEK293 cells. Br J Pharmacol 152: 122–131, 2007. doi: 10.1038/sj.bjp.0707368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shimizu S, Yoshida T, Wakamori M, Ishii M, Okada T, Takahashi M, Seto M, Sakurada K, Kiuchi Y, Mori Y. Ca2+-calmodulin-dependent myosin light chain kinase is essential for activation of TRPC5 channels expressed in HEK293 cells. J Physiol 570: 219–235, 2006. doi: 10.1113/jphysiol.2005.097998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Silliman CC, Elzi DJ, Ambruso DR, Musters RJ, Hamiel C, Harbeck RJ, Paterson AJ, Bjornsen AJ, Wyman TH, Kelher M, England KM, McLaughlin-Malaxecheberria N, Barnett CC, Aiboshi J, Bannerjee A. Lysophosphatidylcholines prime the NADPH oxidase and stimulate multiple neutrophil functions through changes in cytosolic calcium. J Leukoc Biol 73: 511–524, 2003. doi: 10.1189/jlb.0402179. [DOI] [PubMed] [Google Scholar]
- 39.Takahashi R, Watanabe H, Zhang XX, Kakizawa H, Hayashi H, Ohno R. Roles of inhibitors of myosin light chain kinase and tyrosine kinase on cation influx in agonist-stimulated endothelial cells. Biochem Biophys Res Commun 235: 657–662, 1997. doi: 10.1006/bbrc.1997.6856. [DOI] [PubMed] [Google Scholar]
- 40.Tang J, Lin Y, Zhang Z, Tikunova S, Birnbaumer L, Zhu MX. Identification of common binding sites for calmodulin and inositol 1,4,5-trisphosphate receptors on the carboxyl termini of trp channels. J Biol Chem 276: 21303–21310, 2001. doi: 10.1074/jbc.M102316200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tran POT, Hinman LE, Unger GM, Sammak PJ. A wound-induced [Ca2+]i increase and its transcriptional activation of immediate early genes is important in the regulation of motility. Exp Cell Res 246: 319–326, 1999. doi: 10.1006/excr.1998.4239. [DOI] [PubMed] [Google Scholar]
- 42.Usatyuk PV, Singleton PA, Pendyala S, Kalari SK, He D, Gorshkova IA, Camp SM, Moitra J, Dudek SM, Garcia JG, Natarajan V. Novel role for non-muscle myosin light chain kinase (MLCK) in hyperoxia-induced recruitment of cytoskeletal proteins, NADPH oxidase activation, and reactive oxygen species generation in lung endothelium. J Biol Chem 287: 9360–9375, 2012. doi: 10.1074/jbc.M111.294546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Aalst JA, Zhang DM, Miyazaki K, Colles SM, Fox PL, Graham LM. Role of reactive oxygen species in inhibition of endothelial cell migration by oxidized low-density lipoprotein. J Vasc Surg 40: 1208–1215, 2004. doi: 10.1016/j.jvs.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 44.Viedt C, Soto U, Krieger-Brauer HI, Fei J, Elsing C, Kübler W, Kreuzer J. Differential activation of mitogen-activated protein kinases in smooth muscle cells by angiotensin II: involvement of p22phox and reactive oxygen species. Arterioscler Thromb Vasc Biol 20: 940–948, 2000. doi: 10.1161/01.ATV.20.4.940. [DOI] [PubMed] [Google Scholar]
- 45.Watanabe H, Takahashi R, Zhang XX, Goto Y, Hayashi H, Ando J, Isshiki M, Seto M, Hidaka H, Niki I, Ohno R. An essential role of myosin light-chain kinase in the regulation of agonist- and fluid flow-stimulated Ca2+ influx in endothelial cells. FASEB J 12: 341–348, 1998. [DOI] [PubMed] [Google Scholar]
- 46.Watanabe H, Takahashi R, Zhang XX, Kakizawa H, Hayashi H, Ohno R. Inhibition of agonist-induced Ca2+ entry in endothelial cells by myosin light-chain kinase inhibitor. Biochem Biophys Res Commun 225: 777–784, 1996. doi: 10.1006/bbrc.1996.1250. [DOI] [PubMed] [Google Scholar]
- 47.Wegiel B, Gallo DJ, Raman KG, Karlsson JM, Ozanich B, Chin BY, Tzeng E, Ahmad S, Ahmed A, Baty CJ, Otterbein LE. Nitric oxide-dependent bone marrow progenitor mobilization by carbon monoxide enhances endothelial repair after vascular injury. Circulation 121: 537–548, 2010. doi: 10.1161/CIRCULATIONAHA.109.887695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Werner N, Junk S, Laufs U, Link A, Walenta K, Böhm M, Nickenig G. Intravenous transfusion of endothelial progenitor cells reduces neointima formation after vascular injury. Circ Res 93: e17–e24, 2003. doi: 10.1161/01.RES.0000083812.30141.74. [DOI] [PubMed] [Google Scholar]
- 49.Yamakawa T, Tanaka S, Yamakawa Y, Kamei J, Numaguchi K, Motley ED, Inagami T, Eguchi S. Lysophosphatidylcholine activates extracellular signal-regulated kinases 1/2 through reactive oxygen species in rat vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 22: 752–758, 2002. doi: 10.1161/01.ATV.0000015903.02749.71. [DOI] [PubMed] [Google Scholar]
- 50.Zeng F, Xu SZ, Jackson PK, McHugh D, Kumar B, Fountain SJ, Beech DJ. Human TRPC5 channel activated by a multiplicity of signals in a single cell. J Physiol 559: 739–750, 2004. doi: 10.1113/jphysiol.2004.065391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao Y, Davis HW. Hydrogen peroxide-induced cytoskeletal rearrangement in cultured pulmonary endothelial cells. J Cell Physiol 174: 370–379, 1998. doi:. [DOI] [PubMed] [Google Scholar]