

Abstract
As an α-chemokine receptor specific for stromal-derived-factor-1 (SDF-1, also called CXCL12), C-X-C chemokine receptor type 4 (CXCR4) plays a vital role in chemotactically attracting lymphocytes during inflammation. CXCR4 also regulates HIV infection due to its role as one of the chemokine coreceptors for HIV entry into CD4+ T cells. Chemokine receptors and their signaling pathways have been shown to be regulated by the process of ubiquitination, a posttranslational modification, guided by ubiquitin E3 ligases, which covalently links ubiquitin chains to lysine residues within target substrates. Here we describe a novel mechanism regulating CXCR4 protein levels and subsequent CXCR4/CXCL12 signaling pathway through the ubiquitination and degradation of the receptor in response to ligand stimulation. We identify that an uncharacterized really interesting new gene (RING) finger ubiquitin E3 ligase, RING finger protein 113A (RNF113A), directly ubiquitinates CXCR4 in cells, leading to CXCR4 degradation, and therefore disrupts the signaling cascade. We determined that the K331 residue within CXCR4 is essential for RNF113A-mediated ubiquitin conjugation. Overexpression of RNF113A significantly reduces CXCL12-induced kinase activation in HeLa cells, whereas RNF113A knockdown enhances CXCL12-induced downstream signaling. Further, RNF113A expression and silencing directly affect cell motility in a wound healing assay. These results suggest that RNF113A plays an important role in CXCR4 signaling through the ubiquitination and degradation of CXCR4. This mechanistic study might provide new understanding of HIV immunity and neutrophil activation and motility regulated by CXCR4.
Keywords: ubiquitination, cell migration, RING E3 ligase, CXCR4
c-x-c chemokine receptor type 4 (CXCR4) is a G protein-coupled receptor on the cellular membrane and is expressed across many tissue types. It serves as the main receptor for the chemokine stromal cell-derived factor 1 (CXCL12). The binding of CXCL12 leads to signaling through several G protein-dependent pathways (4). Specifically, this activates G protein subunits and phosphorylates SRK kinases, leading to signaling through the ERK pathway (43). This signaling pathway is critical for cellular processes such as chemotaxis and proliferation. Concurrently, CXCR4 signaling activates phosphatidylinositide 3-kinases, initiating a signaling cascade of second messengers resulting in downstream activation of RAC-α serine/threonine-protein kinase (Akt) and mitogen-associated protein kinase (35, 48). As with ERK, these kinases are important regulators of cell adhesion, migration, and survival. Following ligand binding, CXCR4 is extensively phosphorylated by G protein-coupled receptor kinases, causing desensitization, and its internalization (3, 42). Following internalization, CXCR4 can be recycled back to the membrane or sorted to endosomes for degradation.
Dysregulated CXCR4 activity exists in human pathologies. CXCR4 was initially characterized as one of two coreceptors aiding HIV-1 viral entry into cells (25). Classically, HIV-1 uses the CD4-lymphocyte receptor as the main coordinating anchor, with CCR5 or CXCR4 receptors as a secondary receptor. Use of CXCR4 as a coreceptor is primarily associated with the later stages of infection, for reasons that remain unclear (15). Genetic deletion of CXCR4 in CD4+ T cells in vitro confers resistance to HIV-1 infectivity, underscoring the importance of the receptor (22). However, direct antagonist inhibitors of CXCR4 have not proven clinically meaningful, despite its importance as a coreceptor (45). This suggests other avenues of inhibition need to be explored. CXCR4 plays a role in patients with comorbidities as well. Macrophages from patients with tuberculosis show higher pulmonary expression of CXCR4, which leads to an acceleration of HIV infection (21). While CXCR4 functions as a coreceptor most notably for T-cell invasion, pulmonary epithelia are susceptible to CXCR4-mediated HIV infection (8). Due to the chemotactic consequences of its signaling, CXCR4 is implicated in cancer progression (6). Li et al. (28) have demonstrated that CXCR4 is linked to HER2-mediated breast cancer metastasis. Specifically, they observed that HER2 prevents ligand-induced CXCR4 degradation and increases the protein expression of CXCR4, thus facilitating cell migration. Moreover, depletion of CXCR4 through siRNA can block the ability of breast and liver cancer cells to migrate in vitro (29, 41). Overall, CXCR4 protein level is known to be elevated in cancerous tissue and to be associated with worsened patient outcomes (49). Thus, CXCR4 protein expression, regulation, and stability are directly tied to disease progression.
There are multiple mechanisms regulating the expression and stability of CXCR4 (4). CXCR4 is regulated by factors such as nuclear respiratory factor-1 (NRF-1), SP-1, and negatively regulated by YY1 (39). This has disease implications, as HIV hijacks NRF-1 to increase CXCR4 expression, and thus infectivity. Posttranslational modifications also regulate CXCR4 stability. The extensive phosphorylation following ligand binding serves as phospho-degrons for subsequent ubiquitination and degradation (3). In fact, ligand binding promotes ubiquitination, leading to internalization and sorting prior to recycling (31). It has been reported that a HECT-domain ubiquitin E3 ligase, ITCH (E3 ubiquitin-protein ligase Itchy homolog), mediates ubiquitination of CXCR4 (32). ITCH specifically interacts with arrestins in the endosomal sorting complex required for transport (ESCRT) machinery, leading to CXCR4 ubiquitination and endosomal transport to the lysosome (2). However, researchers have observed other E3 ligases to manipulate CXCR4, specifically E3 ubiquitin-protein ligase Deltex-3L (DTX3L), which antagonizes ITCH activity and maintains CXCR4 stability (20). Regulation of CXCR4 degradation is a highly regulated and complex process (24). These recent discoveries suggest that multiple proteins and multiple ubiquitin E3 ligases work to regulate CXCR4 stability.
Ubiquitination is the main mechanism of targeting cellular proteins for degradation through the lysosome or proteasome (19). A series of protein complexes facilitate the conjugation of ubiquitin to target proteins in an enzymatic cascade. Initially, the E1 ubiquitin activating enzyme transmits a ubiquitin moiety to one of several dozen types of ubiquitin E2 conjugating enzymes via a high-energy thioester bond. The charged ubiquitin~E2 complex is then guided by a ubiquitin E3 ligase protein or complex to the target substrate, and ubiquitin is delivered onto the target lysine residue. There are >600 E3 ligases known and estimated to exist in the human proteome, and they are organized into several families (36, 40). Specifically, the largest family of E3 ligases, really interesting new gene (RING) E3 ligases remains poorly characterized. RING E3 ligases contain a RING domain, which is a sequence of cysteine and histidine residues that function to chelate two zinc ions (37). This charged domain serves as a binding interface for E2 enzymes and orients the coordinated E2 into a position to facilitate ubiquitination of substrate proteins (13).
We screened a library of RING E3 ligases to assay their activity on CXCR4 stability and observed a previously uncharacterized E3 ligase, RING finger protein 113A (RNF113A), mediates CXCR4 degradation. Here we report RNF113A is a bona-fide RING E3 ligase that directly binds CXCR4, shortens CXCR4 protein half-life, downregulates CXCR4 signaling, and impairs cellular motility. This study represents a new means in regulating CXCR4 stability.
MATERIALS AND METHODS
Antibodies.
Anti-CXCR4 antibody (UMB2) (ab124824) was from Abcam. Horseradish peroxidase-conjugated secondary antibodies (170-515/6) were from Bio-Rad. Antibodies against ERK1/2 (137F5; 4695), pERK1/2 (Thr202/Tyr204) (20G11; 4376), pAkt (Ser473) (D9E; 4060), and Akt (40D4; 2920) were from Cell Signaling Technologies. Anti-ubiquitin antibody (VU101) was from Life Sensors. Antibodies against RNF113A (V-25, sc-133965) were from Santa Cruz Biotechnology. Anti-actin antibody (A5441) was from Sigma Aldrich. Anti-V5 Tag (R960-25) was from Thermo Fisher Scientific.
Materials.
QuikChange II XL Site-Directed Mutagenesis Kit (200522) was from Aglient Technologies. Eagle's minimum essential medium (EMEM) (30-2003) and HeLa cells (CCL-2) were from ATCC. Thermal Cycler Life ECO (BYQ6078) was from BIOER Technology. Cytation5 Imager was from BioTek. Cycloheximide (BML-GR310) was from Enzo. Plasmids (pLKO.1) encoding shRNA against RNF113A were from GE Dharmacon. FBS (100-106) was from Gemini. DNA sequencing was performed at Genewiz. Phusion High-Fidelity DNA Polymerase (M0530) was from NEB. TnT T7 Quick Coupled Transcription/Translation System (L1170) was from Promega. Recombinant hCXCL12 (350-NS-010) was from R&D Systems. Agar (A5306) and XtremeGene HP (XTGHP-RO ROCHE) were from Sigma Aldrich. HisPur Ni-NTA Magnetic Beads (88831), PureLink Quick Plasmid Miniprep Kit (K210010), Pierce Protein A/G Magnetic Beads (88802), and pcDNA3.1D V5/HIS/TOPO kit (K490040) were from Thermo Fisher Scientific.
Cell culture.
HeLa cells were cultured in Eagle's minimum essential medium (ATCC) supplemented with 10% fetal bovine serum (EMEM-10). Cell line morphology was monitored via microscopy and immunoblotted for multiple markers. Mycoplasma contamination was checked using the MycoAlert Mycoplasma Detection Kit (Lonza, Switzerland). For plasmid overexpression in HeLa cells, plasmids were combined with XtremeGene HP kit following manufacturer’s protocol. After 24 h, cells were treated with CXCL12 (6 nM) or cycloheximide (CHX; 50 µg/ml) for the indicated times. For RNF113A silencing studies in HeLa cells, scrambled shRNA control and RNF113A shRNA were transfected into cells for 48 h using XtremeGene HP kit following the manufacturer’s protocol. Exposed cells were collected and processed for immunoblotting.
Cloning and mutagenesis.
All wild-type (WT) and mutant CXCR4, RNF113A, and RING E3 ligase plasmid constructs were generated using PCR-based approaches using appropriate primers and were then subcloned into a pcDNA3.1D/V5-His vector (9, 34). Point mutants were generated using the QuikChange II XL kit (Aglient). All plasmid constructs were sequence-confirmed against NCBI reference sequences before experimentation, (e.g., NM_001008540 for CXCR4, NM_006978 for RNF113A).
Western blotting.
Cell sample lysates were collected and digested in buffer A (150 mM NaCl, 50 mM Tris, 1.0 mM EDTA, 2 mM dithiothreitol, 0.025% sodium azide, and 1 mM phenylmethylsulfonyl fluoride) on ice. Lysates were prepared by brief sonication at 4°C. Insoluble cellular debris was precipitated through centrifugation at 15,000 g for 10 min at 4°C. Lysate supernatant was normalized for protein concentration and diluted in denaturing loading buffer, with a final 1× formulation of: 50 mM Tris·HCl (pH 6.8), 2% SDS, 10% glycerol, and 100 mM DTT. Samples were resolved via SDS-PAGE prior to immunoblotting. Signal was detected via chemiluminescence on a Kodak Imaging Station. Densitometry was calculated via ImageJ (National Institutes of Health, Bethesda, MD).
HIS-pull down.
Full-length CXCR4-HIS-V5 plasmid was overexpressed in HeLa cells without and with coexpression of tagless RNF113A using the above protocol. Following 18 h of expression, cells were exposed to vehicle or CXCL12 (6 nM) for 1 h, before collection and lysis of cells. Clarified cell lysate was incubated with HisPur Ni-NTA agarose resin for 1 h at 25°C. Resin pull-downs were washed before elution at 70°C in 1× denaturing loading buffer and subsequent immunoblot analysis.
In vitro protein-binding assays.
CXCR4 protein was immunoprecipitated from 1 mg HeLa cell lysate using CXCR4 antibody (rabbit) and coupled to protein A/G agarose resin. CXCR4 beads were then incubated with in vitro synthesized products (50 μl) expressing RNF113A-V5 deletion mutants. After washing, the proteins were eluted and processed for V5 immunoblotting. Similarly, RNF113A protein was immunoprecipitated from HeLa cell lysate using RNF113A antibody (rabbit), and subjected to binding against V5-tagged CXCR4 mutants.
RNF113A shRNA knockdown.
pLKO.1 plasmids encoding nontargeting control (RHS6848) and shRNA against RNF113A were derived from the RNAi Consortium (TRC-Hs1.0, human) and purchased from GE Dharmacon. Mature antisense sequences are as follows: shRNA no. 1 (TRCN0000033729): TTTCCGGTCGAACCACAGTGC; shRNA no. 2 (TRCN0000033730): TAGCAATCAATTCTTTCGCTG; shRNA no. 3 (TRCN0000033731): TATCATTGGATTGTGGGTCAC; shRNA no. 4 (TRCN0000033732): AAAGATGGCTTGTGCATCGCG; and shRNA no. 5 (TRCN0000033733): ATTGAAGACGCCATTGGTCTG. Plasmids encoding shRNA were delivered to cells using transfection protocols described above.
Cellular migration assay.
HeLa cells were transfected with empty plasmid, RNF113A-V5, control shRNA, or shRNA against RNF113A as described above. Transfected cells were seeded in six-well plates to 80–90% of confluence and starved for 24 h before scratching the monolayer with a pipette tip. CXCL12 (12 nM) was added to wounded monolayer immediately following injury and replaced every 24 h. Images were collected at 48 and 72 h post scratch. Images were collected with ×4 objective using BioTek Cytation5 Imager.
Statistical analysis.
Statistical comparisons were performed with means ± SE for continuous variables. All data were statistically analyzed by the indicated statistical tests with P < 0.05 indicative of statistical significance. All analyses were performed using GraphPad Prism 6.
RESULTS
RNF113A affects CXCR4 stability.
We prepared a library of over 200 E3 ligases and expressed them in HeLa cells to assess their effect on the protein abundance of CXCR4 (9, 27). A representative panel of E3 ligase overexpression is shown in Fig. 1A. We observed that RNF113A expression decreases protein abundance of CXCR4 (Fig. 1A). Ligand binding is known to stimulate CXCR4 ubiquitination and degradation (31). To examine the role of RNF113A and CXCL12 on CXCR4 ubiquitination, we coexpressed HIS-tagged CXCR4 with RNF113A before treatment with CXCL12 (6 nM) for 1 h. Lysates were subjected to HIS pull-down, washed, and the eluate was processed for ubiquitin immunoblotting (Fig. 1B). We recapitulated that CXCR4 protein is degraded upon CXCL12 exposure, as well as increased ubiquitin signal with pulled-down CXCR4 following CXCL12 treatment. Furthermore, we observed an even greater degradation and greater ubiquitin signal upon coexpression of RNF113A, suggesting that RNF113A aids in CXCR4 ubiquitination and degradation in response to CXCL12 stimulant (Fig. 1B). Next, we observed that the overexpression of RNF113A resulted in the decrease in CXCR4 protein level in a dose-dependent manner, but not by its paralogue RNF113B (Fig. 1C). RING E3 ligases have a canonical RING domain containing critical Cys and His residues for zinc coordination (37). To validate the activity of RNF113A, we mutated two critical residues within the RING domain of RNF113A: C262 and C277. We observed that C262A and C277A mutants were unable to reduce CXCR4 protein levels as compared with wild-type RNF113A (Fig. 1D). Upon the blockage of protein synthesis by exposure to cycloheximide (CHX), the overexpression of RNF113A led to an accelerated degradation of CXCR4, compared with the empty vector control group (Fig. 2A). Silencing of RNF113A via several different shRNA all led to the increased CXCR4 protein abundance (Fig. 2B), and combination of shRNA silencing of RNF113A and CHX treatment significantly slowed down CXCR4 protein turnover (Fig. 2C).
Fig. 1.
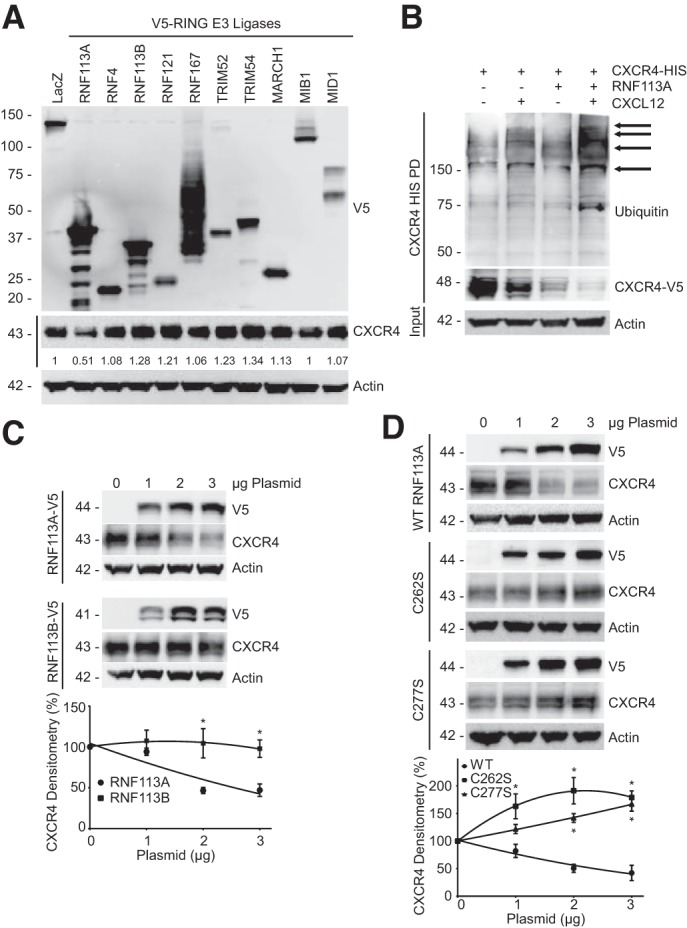
RING ubiquitin E3 ligase RNF113A facilitates C-X-C chemokine receptor type 4 (CXCR4) degradation. A: RING E3 ligase screening. A library of RING E3 ligases were transfected in HeLa cells before CXCR4 immunoblotting. CXCR4 densitometry relative to LacZ is shown beneath immunoblot. B: HeLa cells were cotransfected with CXCR4-HIS and RNF113A before CXCL12 treatment (6 nM, 1 h.), HIS pull-down, and immunoblotting. C, top: V5-tagged RNF113A and its paralogue RNF113B were expressed in HeLa cells in a dose course before CXCR4 immunoblotting. Bottom: densitometry on CXCR4 protein signal normalized to 0 µg for each treatment. Data represent mean values ± SE (n = 4, *P < 0.05, significant compared with RNF113A, two-way ANOVA, Bonferroni multiple comparisons). D, top: point mutants of RNF113A RING domain (V5-tagged) were expressed in HeLa cells before CXCR4 immunoblotting. Bottom: densitometry on CXCR4 protein signal normalized to 0 µg for each treatment. Data represent mean values ± SE (n = 4, *P < 0.05, significant compared with RNF113A WT signal at indicated dose, two-way ANOVA, Bonferroni multiple comparisons).
Fig. 2.

RNF113A decreases CXCR4 stability and half-life. A, left: HeLa cells were transfected with RN113A-V5 plasmid before cycloheximide (CHX) chase (50 μg/ml), and CXCR4 immunoblotting. Right: densitometry on CXCR4 protein signal normalized to time 0 for each treatment. Data represent mean values ± SE (n = 4, *P < 0.05, significant compared with Empty at indicated time, two-way ANOVA, Bonferroni multiple comparisons). B: screening of shRNA against RNF113A. HeLa cells were transfected with shRNA plasmids targeted against RNF113A before CXCR4 immunoblotting. Densitometry of RNF113A and CXCR4 signal relative to control (Ctrl) shRNA is shown beneath immunoblots. C, left: HeLa cells were transfected with shRNA scramble or shRNA plasmids targeted against RNF113A before CHX chase (50 μg/ml). Cells were then subjected to immunoblotting. Right: densitometry on CXCR4 protein signal normalized to time 0 for each treatment. Data represent mean values ± SE (n = 4, *P < 0.05, significant compared with Ctrl at indicated time, two-way ANOVA, Bonferroni multiple comparisons). D, top: HeLa cells were cotransfected with RNF113A and CXCR4 K-R lysine mutants before immunoblotting. Bottom: densitometry on CXCR4-V5 protein signal normalized to (−) RNF113A for each treatment. Data represent mean values ± SE (n = 3, NS, P > 0.05, not significant, *P < 0.05, significant compared with CXCR4 WT + RNF113A, one-way ANOVA, Bonferroni multiple comparisons). E, left: candidate lysine point mutants were transfected into HeLa cells before CHX chase (50 μg/ml). Right: densitometry on CXCR4-V5 protein signal normalized to time 0 for each treatment. Data represent mean values ± SE (n = 3, *P < 0.05, significant compared with WT at indicated time, two-way ANOVA, Bonferroni multiple comparisons).
CXCR4 K331 is critical for RNF113A-mediated degradation.
We next sought to identify a potential ubiquitin lysine site within CXCR4. The intracellular carboxy-terminal tail of CXCR4 contains several lysines. Previous reports have shown that the CXCR4 triple mutant of lysine to arginine (lysines 327, 331, and 333) is resistant to ubiquitin-mediated degradation (31, 38). We further constructed single CXCR4 K-R point mutants and observed K331R to be resistant to RNF113A-mediated degradation (Fig. 2D). As a negative control, K310R remained susceptible to RNF113A-dependent degradation. Further, K331R CXCR4 exhibited a prolonged half-life during CHX chase, compared with CXCR4 wild-type (WT) and K310R mutant (Fig. 2E).
RNF113A binds CXCR4 through a positively charged region.
E3 ligases are known to target substrates through specific motifs on the E3 ligase and the target substrate (27, 46). To further characterize the interaction of RNF113A and CXCR4, we employed a reductionist mapping approach to elucidate the putative binding regions within RNF113A and CXCR4. A series of amino-terminal and carboxy-terminal truncation mutants of RNF113A were prepared, in vitro expressed, and subjected to CXCR4 binding (Fig. 3A). We observed a loss of association between CXCR4 immunoprecipitate and RNF113A when the 15-residue region between 50 and 65 was deleted (Fig. 3B). Finer mapping experiments narrowed this region down to 10 residues (Fig. 3C). The RNF113A construct with an internal deletion of these 10 residues largely decreased the interaction between RNF113A and CXCR4 (Fig. 3D), and its overexpression in HeLa cells failed to induce CXCR4 degradation (Fig. 3, E and F). This led to the conclusion that this 10-residue region is the putative binding site. Conversely, we designed carboxy-terminal truncation mutants of CXCR4 and conducted binding assays with immunoprecipitated RNF113A (Fig. 3G). We observed a loss of association between RNF113A immunoprecipitate and CXCR4 when the region between residues 322 and 333 was deleted (Fig. 3H). As serines- 324 and 325 within this region have been described to be phosphorylated (3), we hypothesized that they are critical for RNF113A association. Mutation of these serines to alanines prevented the binding of CXCR4 to RNF113A (Fig. 3I). However, as a positive control, deletion of the final 19 residues of CXCR4 preserved its binding affinity to RNF113A (Fig. 3I).
Fig. 3.

RNF113A binds CXCR4 in a positively charged region. A: schematic of RNF113A deletion mutants constructed for the binding studies. B and C: binding assays between RNF113A and CXCR4. HeLa cell lysate (100 μg protein) was subjected to CXCR4 immunoprecipitation (IP). RNF113A mutants were synthesized via in vitro transcription and translation (TnT), and allowed to bind to overnight with immunoprecipitated resin. Eluates were subjected to immunoblotting (IB). Asterisk (*) indicates full-length RNF113A, which runs at the same size as IgG heavy chain (HC). D: V5-tagged RNF113A internal deletion mutant was expressed in HeLa cells before HIS pull-down (PD) and CXCR4 immunoblotting. E: V5-tagged RNF113A internal deletion mutant was expressed in HeLa cells in a dose course before CXCR4 immunoblotting. F: densitometry of E. Data represent mean values ± SE (n = 3, NS, P > 0.05, slope not significantly different from 0, F-test). G: schematic of CXCR4 deletion mutants constructed for the binding studies. H and I: binding assays between RNF113A and CXCR4. HeLa cell lysate (100 μg protein) was subjected to RNF113A immunoprecipitation. CXCR4 mutants were synthesized via in vitro transcription and translation (TnT) and allowed to bind to overnight with immunoprecipitated resin. Eluates were subjected to immunoblotting.
RNF113A regulates CXCR4 signaling.
Next, we investigated the effect of RNF113A on the CXCR4 signaling pathway. HeLa cells were transfected with RNF113A and treated with 6 nM CXCL12 in a time course before immunoblotting. RNF113A expression accelerates CXCL12-mediated degradation of CXCR4 (Fig. 4A). Further, RNF113A expression decreased the activation of downstream kinases in CXCR4 signaling, specifically the phosphorylation of Akt1 and ERK1/2, without affecting total kinase levels (Fig. 4A). Next, we silenced RNF113A in HeLa cells before CXCL12 treatment. We observed RNF113A depletion led to increased CXCR4 protein signal. Further, RNF113A depletion increases the activation of downstream kinases in CXCR4 signaling, specifically the phosphorylation of Akt1 and ERK1/2 (Fig. 4B).
Fig. 4.

RNF113A regulates CXCR4 signaling and cellular motility. A: HeLa cells were transfected with RNF113A-V5 before CXCL12 treatment and immunoblotting for phosphorylated kinases. Densitometry relative to Empty at time 0 is shown beneath immunoblots. B: HeLa cells were transfected with plasmid encoding shRNA targeting RNF113A before treatment with CXCL12 (6 nM) for the indicated times. Cells were collected and immunoblotted for activation of downstream CXCR4 signalers. Densitometry relative to Ctrl shRNA at time 0 is shown beneath immunoblots. C and D: wound healing assay. HeLa cells were transfected with empty or RNF113A-encoded plasmids, or with plasmids encoding shRNA against scramble or RNF113A and allowed to grow to 80–90% confluency before starvation and wounding with pipette tip. Extent of gap closure was measured at 48 or 72 h and in the absence or presence of CXCL12. Gap closure is quantified below. Data represent mean values ± SE (n = 3–4 per group; *P < 0.05, significant between indicated groups, NS, P > 0.05, not significant between indicated groups, two-sided t-test). “KD” refers to shRNA.
RNF113A affects cell motility.
Finally, we investigated the functional role of RNF113A on cellular motility through its regulation of CXCR4. The CXCR4 signaling cascade mediates cellular motility, especially in the presence of CXCL12. HeLa cells were transfected with RNF113A-V5 plasmid or shRNA against RNF113A and were seeded to a density of 80–90%. Confluent cells were starved and wounded using 200 µl pipette tip to create a 400 μm gap in the monolayer. Additionally, cells were exposed to CXCL12 (12 nM) during course of healing. The monolayer was imaged and gap closure was quantified (n = 3–4). RNF113A overexpression significantly precluded gap closure, compared with empty plasmid (Fig. 4C). Moreover, treatment with CXCL12 resulted in a significantly increased gap closure with empty plasmid cells. However, compared with CXCL12 treatment, additional overexpression of RNF113A only slightly increased gap closure, and only closely within significance (P = 0.0458). From these observations, we conclude that RNF113A expression affects cell motility through regulating CXCR4. As a complementary assay, we used shRNA to silence RNF113A before wound healing assay (Fig. 4D). We further confirmed that CXCL12 enhanced gap closure among the control shRNA plasmid group. Furthermore, additional depletion of RNF113A significantly increased gap closure, compared with CXCL12 treatment alone.
These data demonstrate that RNF113A is a negative regulator of CXCR4 protein stability, signaling, and CXCR4-mediated cellular motility.
DISCUSSION
Here we report a new mechanism in the regulation of CXCR4 protein stability. We observed that the RING ubiquitin E3 ligase RNF113A promotes CXCR4 degradation, impairs downstream signaling, and affects cell motility. Depletion of RNF113A through shRNA preserves CXCR4 protein level, extends CXCR4 protein half-life, and enhances CXCR4 signaling. CXCR4 has been shown to be monoubiquitinated, specifically by ubiquitin E3 ligase ITCH, leading to its endosomal sorting and degradation (4, 32). However, polyubiquitination of CXCR4 has also been observed and is suggested to play a role in immune cell aging (5, 26). We demonstrated that RNF113A expression and CXCL12 treatment can increase high-molecular-weight ubiquitin signal of CXCR4 compared with control (Fig. 1B). This suggests that RNF113A facilitates the polyubiquitination of CXCR4.
The ubiquitin system is a critical mediator of the dynamics of cellular motility and migration (44). Specifically, RING E3 ligases, such as inhibitors of apoptosis, can both positively or negatively regulate cell migration through the ubiquitination of substrates including plasma membrane surface proteins or members of the signaling cascade (14). We observed that RNF113A overexpression attenuates CXCL12-CXCR4-dependent activation of Akt and ERK, kinases classically associated with cellular movement (Fig. 4A). Conversely, silencing of RNF113A enhanced Akt and ERK activation (Fig. 4B). RNF113A mediation of CXCR4 degradation also has functional consequences. Cells with overexpressed RNF113A were less able to close the gap in a wound healing assay relative to empty vector plasmid (Fig. 4C). However, among cells with RNF113A overexpression, the difference in gap closure between untreated and CXCL12-treatment trends toward insignificance (P = 0.0458). This suggests that RNF113A-expressing cells are less sensitive to CXCL12-stimulated motility, possibly due to RNF113A-mediated degradation of the primary CXCL12 receptor, CXCR4. Manipulation of CXCR4 signaling through antagonist inhibition or siRNA silencing has been shown to affect cellular motility in a wound healing assay (17, 41). Further, CXCL12 treatment can accelerate wound healing (17). When RNF113A is silenced, which leads to the increased CXCR4 stability and abundance, cells exhibit a stronger response to CXCL12, with enhanced gap closure relative to control shRNA treatment (Fig. 4D).
G protein-coupled receptors and chemokine receptor signaling are heavily regulated by the stability of the receptor, specifically through posttranslational modification. Phosphorylation by G protein-specific kinases has been known to be a main regulator through desensitization. However, ubiquitination is increasingly understood to be a potent regulator of stability and downstream function (33). Indeed, several ubiquitin E3 ligases are known to regulate the function and signaling of CXCR4.
Substrates fated for ubiquitination are often characterized by specific targeting sequences that aid ubiquitin E3 ligases in identification and association. Specifically, protein binding motifs involving phosphorylation have proven critical for E3-substrate interaction (27, 46). Protein binding motifs have been shown to be critical in engaging CXCR4. Previous research has shown that ITCH binds CXCR4 with WW-motifs, which are binding regions with proline-rich affinity (1). We observed RNF113A to have a positively charged 10-residue region necessary for binding to CXCR4 (Fig. 3A). Further, mutation of critical serine residues of CXCR4, known to be phosphorylated and thus negatively charged, led to decreased association with RNF113A (Fig. 3I). These mechanistic studies suggest a complementary charge-dependent interaction facilitating the association between E3 ligase and substrate. Understanding of the mechanistic underpinnings of association will serve as the basis for future structure-based small molecule drug development. As a coreceptor for HIV-1, depletion of CXCR4 protein would be beneficial to prevent viral entry, similar to drug development efforts in antagonizing CCR5 (18). Previous efforts to chemically target CXCR4 with direct antagonism suffered from significant off-targeting effects (45). An alternative pathway of inhibition may avoid such off-targeting issues. Further, inhibitors of CXCR4 have shown promise in slowing cancer progression (12). Enhancing RNF113A-CXCR4 interaction or prolonging RNF113A half-life may show efficacy in several CXCR4-related pathologies and potentially have therapeutic benefits.
RNF113A may be working in concert with other proteins or E3 ligases. Arrestin-2 has been shown to cooperate with ITCH in promoting ubiquitination of CXCR4, leading to its degradation (2, 30). Conversely, Holleman and Marchese (20) described antagonism between the RING E3 ligase Deltex-3L (DTX3L) and ITCH, as DTX3L inhibits ITCH ubiquitin ligase activity toward CXCR4. Similarly, the downstream kinase CISK has been shown to inhibit ITCH activity in degrading CXCR4 (47). Interestingly, we observed that RNF113A protein signal also decays during cyclohexamide chase (Fig. 2A), suggesting that RNF113A protein stability is subject to regulation. We have observed a similar multiple E3 ligase relationship, as F-box only protein 3 (FBXO3) regulates F-box/LRR-repeat protein 2 (FBXL2), leading to pleiotropic cellular and pathological consequences (7). Substrates have been shown to be regulated by a variety of ubiquitin E3 ligases depending on the specific spatiotemporal environment, and in response to discrete stimuli, a classic example being the ubiquitin-mediated regulation of p53 (23). It is possible that the regulation of CXCR4 by RNF113A works within a comparable regulatory framework.
Aside from this study, the function of RNF113A remains unclear. Bioinformatics studies predict a RING domain in RNF113A (16). We observed that mutation of critical RING residues rescues substrate CXCR4 from degradation (Fig. 1D). As the RING domain aids in interfacing and orientating the ubiquitin E2 enzyme (13), we believe mutation within this domain would abolish E2-binding, yet still bind CXCR4, thus sterically preventing access to the substrate, and functioning as a dominant-negative. We have previously observed this phenotype with the E3 ligase FIEL1, in which a dominant-negative mutant unable to bind substrate not only prevented substrate degradation, but enhanced overall substrate abundance (27). Genomic studies have shown that nonsense mutations within the catalytic RING domain of RNF113A occur in patients with the autosomal recessive disease trichothiodystrophy (10). While trichothiodystrophy is a pathologically heterogeneous disease, malfunction of DNA-repair mechanisms has been associated with patients (11). It could be that RNF113A is involved in regulation of these potentially disease-causing repair mechanisms. Further investigation is needed for determining the regulation of RNF113A on biologic processes.
We believe that this study is a stepping stone for further investigations into a novel regulator of CXCR4 stability and signaling.
GRANTS
This work was supported in whole or part by American Heart Association Scientist Development Grant 16SDG27650008 (to Y. Liu), National Institutes of Health National Heart, Lung, and Blood Institute R01 Grants HL-116472 and HL-132862 (to B. B. Chen), and a University of Pittsburgh Vascular Medicine Institute seed fund (to B. B. Chen).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
T.L., S.R.D., A.C.M., A.M., J.E., and B.B.C. performed experiments; T.L., S.R.D., A.C.M., A.M., B.B.C., and Y.L. analyzed data; T.L., S.R.D., J.E., B.B.C., and Y.L. interpreted results of experiments; T.L., S.R.D., A.C.M., and B.B.C. prepared figures; T.L. drafted manuscript; T.L., B.B.C., and Y.L. edited and revised manuscript; T.L., S.R.D., A.C.M., A.M., J.E., B.B.C., and Y.L. approved final version of manuscript; B.B.C. and Y.L. conceived and designed research.
ACKNOWLEDGMENTS
We thank Shristi Rajbhandari for technical support to initiate this project.
REFERENCES
- 1.Bhandari D, Robia SL, Marchese A. The E3 ubiquitin ligase atrophin interacting protein 4 binds directly to the chemokine receptor CXCR4 via a novel WW domain-mediated interaction. Mol Biol Cell 20: 1324–1339, 2009. doi: 10.1091/mbc.E08-03-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhandari D, Trejo J, Benovic JL, Marchese A. Arrestin-2 interacts with the ubiquitin-protein isopeptide ligase atrophin-interacting protein 4 and mediates endosomal sorting of the chemokine receptor CXCR4. J Biol Chem 282: 36971–36979, 2007. doi: 10.1074/jbc.M705085200. [DOI] [PubMed] [Google Scholar]
- 3.Busillo JM, Armando S, Sengupta R, Meucci O, Bouvier M, Benovic JL. Site-specific phosphorylation of CXCR4 is dynamically regulated by multiple kinases and results in differential modulation of CXCR4 signaling. J Biol Chem 285: 7805–7817, 2010. doi: 10.1074/jbc.M109.091173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Busillo JM, Benovic JL. Regulation of CXCR4 signaling. Biochim Biophys Acta 1768: 952–963, 2007. doi: 10.1016/j.bbamem.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cané S, Ponnappan S, Ponnappan U. Altered regulation of CXCR4 expression during aging contributes to increased CXCL12-dependent chemotactic migration of CD4(+) T cells. Aging Cell 11: 651–658, 2012. doi: 10.1111/j.1474-9726.2012.00830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chatterjee S, Behnam Azad B, Nimmagadda S. The intricate role of CXCR4 in cancer. Adv Cancer Res 124: 31–82, 2014. doi: 10.1016/B978-0-12-411638-2.00002-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen BB, Coon TA, Glasser JR, McVerry BJ, Zhao J, Zhao Y, Zou C, Ellis B, Sciurba FC, Zhang Y, Mallampalli RK. A combinatorial F box protein directed pathway controls TRAF adaptor stability to regulate inflammation. Nat Immunol 14: 470–479, 2013. doi: 10.1038/ni.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chinnapaiyan S, Parira T, Dutta R, Agudelo M, Morris A, Nair M, Unwalla HJ. HIV infects bronchial epithelium and suppresses components of the mucociliary clearance apparatus. PLoS One 12: e0169161, 2017. doi: 10.1371/journal.pone.0169161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coon TA, McKelvey AC, Lear T, Rajbhandari S, Dunn SR, Connelly W, Zhao JY, Han S, Liu Y, Weathington NM, McVerry BJ, Zhang Y, Chen BB. The proinflammatory role of HECTD2 in innate immunity and experimental lung injury. Sci Transl Med 7: 295ra109–295ra109, 2015. doi: 10.1126/scitranslmed.aab3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corbett MA, Dudding-Byth T, Crock PA, Botta E, Christie LM, Nardo T, Caligiuri G, Hobson L, Boyle J, Mansour A, Friend KL, Crawford J, Jackson G, Vandeleur L, Hackett A, Tarpey P, Stratton MR, Turner G, Gécz J, Field M. A novel X-linked trichothiodystrophy associated with a nonsense mutation in RNF113A. J Med Genet 52: 269–274, 2015. doi: 10.1136/jmedgenet-2014-102418. [DOI] [PubMed] [Google Scholar]
- 11.Czugala M, Karolak JA, Nowak DM, Polakowski P, Pitarque J, Molinari A, Rydzanicz M, Bejjani BA, Yue BY, Szaflik JP, Gajecka M. Novel mutation and three other sequence variants segregating with phenotype at keratoconus 13q32 susceptibility locus. Eur J Hum Genet 20: 389–397, 2012. doi: 10.1038/ejhg.2011.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Clercq E. Recent advances on the use of the CXCR4 antagonist plerixafor (AMD3100, Mozobil™) and potential of other CXCR4 antagonists as stem cell mobilizers. Pharmacol Ther 128: 509–518, 2010. doi: 10.1016/j.pharmthera.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 13.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem 78: 399–434, 2009. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 14.Dubrez L, Rajalingam K. IAPs and cell migration. Semin Cell Dev Biol 39: 124–131, 2015. doi: 10.1016/j.semcdb.2015.02.015. [DOI] [PubMed] [Google Scholar]
- 15.Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272: 872–877, 1996. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 16.Frattini A, Faranda S, Bagnasco L, Patrosso C, Nulli P, Zucchi I, Vezzoni P. Identification of a new member (ZNF183) of the Ring finger gene family in Xq24-25. Gene 192: 291–298, 1997. doi: 10.1016/S0378-1119(97)00108-X. [DOI] [PubMed] [Google Scholar]
- 17.Ghosh MC, Makena PS, Gorantla V, Sinclair SE, Waters CM. CXCR4 regulates migration of lung alveolar epithelial cells through activation of Rac1 and matrix metalloproteinase-2. Am J Physiol Lung Cell Mol Physiol 302: L846–L856, 2012. doi: 10.1152/ajplung.00321.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henrich TJ, Kuritzkes DR. HIV-1 entry inhibitors: recent development and clinical use. Curr Opin Virol 3: 51–57, 2013. doi: 10.1016/j.coviro.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem 67: 425–479, 1998. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 20.Holleman J, Marchese A. The ubiquitin ligase deltex-3l regulates endosomal sorting of the G protein-coupled receptor CXCR4. Mol Biol Cell 25: 1892–1904, 2014. doi: 10.1091/mbc.E13-10-0612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoshino Y, Tse DB, Rochford G, Prabhakar S, Hoshino S, Chitkara N, Kuwabara K, Ching E, Raju B, Gold JA, Borkowsky W, Rom WN, Pine R, Weiden M. Mycobacterium tuberculosis-induced CXCR4 and chemokine expression leads to preferential X4 HIV-1 replication in human macrophages. J Immunol 172: 6251–6258, 2004. doi: 10.4049/jimmunol.172.10.6251. [DOI] [PubMed] [Google Scholar]
- 22.Hou P, Chen S, Wang S, Yu X, Chen Y, Jiang M, Zhuang K, Ho W, Hou W, Huang J, Guo D. Genome editing of CXCR4 by CRISPR/cas9 confers cells resistant to HIV-1 infection. Sci Rep 5: 15577, 2015. doi: 10.1038/srep15577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jain AK, Barton MC. Regulation of p53: TRIM24 enters the RING. Cell Cycle 8: 3668–3674, 2009. doi: 10.4161/cc.8.22.9979. [DOI] [PubMed] [Google Scholar]
- 24.Kennedy JE, Marchese A. Regulation of GPCR trafficking by ubiquitin. Prog Mol Biol Transl Sci 132: 15–38, 2015. doi: 10.1016/bs.pmbts.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klasse PJ. The molecular basis of HIV entry. Cell Microbiol 14: 1183–1192, 2012. doi: 10.1111/j.1462-5822.2012.01812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lapham CK, Romantseva T, Petricoin E, King LR, Manischewitz J, Zaitseva MB, Golding H. CXCR4 heterogeneity in primary cells: possible role of ubiquitination. J Leukoc Biol 72: 1206–1214, 2002. [PubMed] [Google Scholar]
- 27.Lear T, McKelvey AC, Rajbhandari S, Dunn SR, Coon TA, Connelly W, Zhao JY, Kass DJ, Zhang Y, Liu Y, Chen BB. Ubiquitin E3 ligase FIEL1 regulates fibrotic lung injury through SUMO-E3 ligase PIAS4. J Exp Med 213: 1029–1046, 2016. doi: 10.1084/jem.20151229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li YM, Pan Y, Wei Y, Cheng X, Zhou BP, Tan M, Zhou X, Xia W, Hortobagyi GN, Yu D, Hung M-C. Upregulation of CXCR4 is essential for HER2-mediated tumor metastasis. Cancer Cell 6: 459–469, 2004. doi: 10.1016/j.ccr.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 29.Liang Z, Yoon Y, Votaw J, Goodman MM, Williams L, Shim H. Silencing of CXCR4 blocks breast cancer metastasis. Cancer Res 65: 967–971, 2005. [PMC free article] [PubMed] [Google Scholar]
- 30.Malik R, Marchese A. Arrestin-2 interacts with the endosomal sorting complex required for transport machinery to modulate endosomal sorting of CXCR4. Mol Biol Cell 21: 2529–2541, 2010. doi: 10.1091/mbc.E10-02-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marchese A, Benovic JL. Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR4 mediates lysosomal sorting. J Biol Chem 276: 45509–45512, 2001. doi: 10.1074/jbc.C100527200. [DOI] [PubMed] [Google Scholar]
- 32.Marchese A, Raiborg C, Santini F, Keen JH, Stenmark H, Benovic JL. The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein-coupled receptor CXCR4. Dev Cell 5: 709–722, 2003. doi: 10.1016/S1534-5807(03)00321-6. [DOI] [PubMed] [Google Scholar]
- 33.Marchese A, Trejo J. Ubiquitin-dependent regulation of G protein-coupled receptor trafficking and signaling. Cell Signal 25: 707–716, 2013. doi: 10.1016/j.cellsig.2012.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McKelvey AC, Lear TB, Dunn SR, Evankovich J, Londino JD, Bednash JS, Zhang Y, McVerry BJ, Liu Y, Chen BB. RING finger E3 ligase PPP1R11 regulates TLR2 signaling and innate immunity. eLife 5: 5, 2016. doi: 10.7554/eLife.18496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mellado M, Rodríguez-Frade JM, Vila-Coro AJ, Fernández S, Martín de Ana A, Jones DR, Torán JL, Martínez-A C. Chemokine receptor homo- or heterodimerization activates distinct signaling pathways. EMBO J 20: 2497–2507, 2001. doi: 10.1093/emboj/20.10.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Metzger MB, Hristova VA, Weissman AM. HECT and RING finger families of E3 ubiquitin ligases at a glance. J Cell Sci 125: 531–537, 2012. doi: 10.1242/jcs.091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Metzger MB, Pruneda JN, Klevit RE, Weissman AM. RING-type E3 ligases: master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim Biophys Acta 1843: 47–60, 2014. doi: 10.1016/j.bbamcr.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mines MA, Goodwin JS, Limbird LE, Cui FF, Fan GH. Deubiquitination of CXCR4 by USP14 is critical for both CXCL12-induced CXCR4 degradation and chemotaxis but not ERK ativation. J Biol Chem 284: 5742–5752, 2009. doi: 10.1074/jbc.M808507200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moriuchi M, Moriuchi H, Margolis DM, Fauci AS. USF/c-Myc enhances, while Yin-Yang 1 suppresses, the promoter activity of CXCR4, a coreceptor for HIV-1 entry. J Immunol 162: 5986–5992, 1999. [PubMed] [Google Scholar]
- 40.Morreale FE, Walden H. Types of ubiquitin ligases. Cell 165: 248–248.e1, 2016. doi: 10.1016/j.cell.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 41.Niu J, Huang Y, Zhang L. CXCR4 silencing inhibits invasion and migration of human laryngeal cancer Hep-2 cells. Int J Clin Exp Pathol 8: 6255–6261, 2015. [PMC free article] [PubMed] [Google Scholar]
- 42.Orsini MJ, Parent JL, Mundell SJ, Marchese A, Benovic JL. Trafficking of the HIV coreceptor CXCR4. Role of arrestins and identification of residues in the c-terminal tail that mediate receptor internalization. J Biol Chem 274: 31076–31086, 1999. [Erratum. J Biol Chem 275: 25876, 2000.] doi: 10.1074/jbc.274.43.31076. [DOI] [PubMed] [Google Scholar]
- 43.Pozzobon T, Goldoni G, Viola A, Molon B. CXCR4 signaling in health and disease. Immunol Lett 177: 6–15, 2016. doi: 10.1016/j.imlet.2016.06.006. [DOI] [PubMed] [Google Scholar]
- 44.Schaefer A, Nethe M, Hordijk PL. Ubiquitin links to cytoskeletal dynamics, cell adhesion and migration. Biochem J 442: 13–25, 2012. doi: 10.1042/BJ20111815. [DOI] [PubMed] [Google Scholar]
- 45.Scholten DJ, Canals M, Maussang D, Roumen L, Smit MJ, Wijtmans M, de Graaf C, Vischer HF, Leurs R. Pharmacological modulation of chemokine receptor function. Br J Pharmacol 165: 1617–1643, 2012. doi: 10.1111/j.1476-5381.2011.01551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Skaar JR, Pagan JK, Pagano M. Mechanisms and function of substrate recruitment by F-box proteins. Nat Rev Mol Cell Biol 14: 369–381, 2013. doi: 10.1038/nrm3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Slagsvold T, Marchese A, Brech A, Stenmark H. CISK attenuates degradation of the chemokine receptor CXCR4 via the ubiquitin ligase AIP4. EMBO J 25: 3738–3749, 2006. doi: 10.1038/sj.emboj.7601267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun Y, Cheng Z, Ma L, Pei G. Beta-arrestin2 is critically involved in CXCR4-mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J Biol Chem 277: 49212–49219, 2002. doi: 10.1074/jbc.M207294200. [DOI] [PubMed] [Google Scholar]
- 49.Zhao H, Guo L, Zhao H, Zhao J, Weng H, Zhao B. CXCR4 over-expression and survival in cancer: a system review and meta-analysis. Oncotarget 6: 5022–5040, 2015. doi: 10.18632/oncotarget.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]