

Abstract
A key physiological feature of acute respiratory distress syndrome (ARDS) is inflammation. Toll-like receptor (TLR) signaling is required to combat the infection that underlies many ARDS cases but also contributes to pathological inflammation. Several TLR signaling pathway genes encoding positive effectors of inflammation also produce alternatively spliced mRNAs encoding negative regulators of inflammation. An imbalance between these isoforms could contribute to pathological inflammation and disease severity. To determine whether splicing in TLR pathways is altered in patients with ARDS, we monitored alternative splicing of MyD88 and IRAK1, two genes that function in multiple TLR pathways. The MyD88 and IRAK1 genes produce long proinflammatory mRNAs (MyD88L and IRAK1) and shorter anti-inflammatory mRNAs (MyD88S and IRAK1c). We quantified mRNA encoding inflammatory cytokines and MyD88 and IRAK1 isoforms in peripheral blood mononuclear cells (PBMCs) from 104 patients with ARDS and 30 healthy control subjects. We found that MyD88 pre-mRNA splicing is altered in patients with ARDS in a proinflammatory direction. We also observed altered MyD88 isoform levels in a second critically ill patient cohort, suggesting that these changes may not be unique to ARDS. Early in ARDS, PBMC IRAK1c levels were associated with patient survival. Despite the similarities in MyD88 and IRAK1 alternative splicing observed in previous in vitro studies, there were differences in how MyD88 and IRAK1 alternative splicing was altered in patients with ARDS. We conclude that pre-mRNA splicing of TLR signaling genes is altered in patients with ARDS, and further investigation of altered splicing may lead to novel prognostic and therapeutic approaches.
Keywords: acute respiratory distress syndrome, inflammation, MyD88, IRAK1, alternative pre-mRNA splicing, peripheral blood mononuclear cell
acute respiratory distress syndrome (ARDS) affects roughly 190,000 people in the USA annually, with a mortality rate over 20% (13, 20, 29, 38, 53, 56, 74, 75, 77). Many of those who recover have lasting lung damage and increased mortality after hospital discharge (9, 13, 23, 73). ARDS, which is characterized by hypoxemia and pulmonary edema, is most commonly caused by pneumonia, sepsis, aspiration of gastric contents, or major trauma (13, 20, 29, 38, 53, 56, 74, 75, 77). One of the key physiological features of ARDS is significant inflammation, both in the lung and systemically (29, 38, 74). Inflammatory cytokines are increased in bronchoalveolar lavage and plasma from patients with ARDS, and the levels of these cytokines correlate with worsened disease status and mortality (3, 5, 6, 14, 39, 40, 52, 60, 66). Genetic association studies also implicate inflammatory cytokines and the signaling pathways involved in their production in ARDS pathogenesis (15, 37, 62). This suggests that limiting inflammation could be a useful therapeutic approach, although there have been mixed results in trials testing anti-inflammatory therapies for ARDS (13, 20, 29, 75).
Toll-like receptor (TLR) signaling is required to combat the infection that underlies many cases of ARDS but also contributes to systemic pathological inflammation (31, 67, 78). When activated, most TLRs initiate the formation of a large signaling complex that contains the signaling adaptor MyD88 and a family of IRAK kinases (30, 65). This signaling cascade continues, leading to the activation of the proinflammatory transcription factors NF-κB and activator protein 1 and inflammatory cytokine production (30, 65). Most proteins that function in TLR signaling pathways act as positive effectors of inflammation. However, many TLR pathway genes also encode alternative mRNA splice forms that produce negative regulators of TLR signaling and inflammation. For example, the genes that encode the LPS coreceptors TLR4 and MD-2, the TLR signaling adaptor MyD88, and the signaling kinases IRAK1 and IRAK2 all produce alternate mRNA splice forms that encode proteins that inhibit TLR signaling (11, 18, 21, 25–28, 32, 35, 50, 54, 64, 76). All these negatively acting splice forms are induced by stimulation with TLR agonists. Thus production of these alternative mRNA splice forms constitutes a negative feedback loop that limits inflammation. This allows the body to mount critical antipathogen responses when inflammatory stimuli are encountered and also limits the development of chronic inflammation.
Both MyD88 and IRAK1 have been implicated in the pathogenesis of ARDS or related conditions. MyD88 mediates mechanical ventilation-induced lung inflammation in mice (10). Genetic variants in IRAK1 that increase NF-κB activity associate with worsened patient outcome in sepsis (2, 34, 63, 68). MyD88 protein is encoded by a five-exon mRNA (long form or MyD88L). However, a shorter alternative pre-mRNA splice form (MyD88S) also has been identified that lacks the 135-bp second exon. MyD88S, like MyD88L, can bind to TLRs, but, unlike MyD88L, MyD88S cannot activate downstream signaling (7, 26, 41). MyD88S therefore acts as a dominant inhibitor of NF-κB activation. Similarly, an exon 11-skipping event in IRAK1 has been identified that converts the positive effector IRAK1 to a negative regulator called IRAK1c (54, 64). IRAK1c can also bind to the TLR signaling complex but cannot activate downstream signaling, instead acting as a dominant inhibitor of NF-κB activation (54).
New techniques using therapeutic antisense oligonucleotides have been developed to alter splicing of target genes in an effort to treat various diseases (59, 61, 69–71), including therapeutic oligonucleotides that alter pre-mRNA splicing in the TLR signaling pathway to limit inflammation (19, 72, 79, 80). Thus understanding the role that altered pre-mRNA splicing plays in diseases such as ARDS could lead to new therapeutic approaches. Although the importance of alternate splicing events in the TLR signaling pathway has been demonstrated in vitro (11, 18, 21, 25–28, 32, 35, 50, 54, 64, 76), little study of the effect of this alternate splicing on inflammatory disease in vivo has been performed. The expression level of some TLR pathway gene isoforms has been reported in septic patients (1, 57, 58); however, to our knowledge, no prior study has monitored alternative pre-mRNA splicing in any inflammatory disease, including ARDS. We therefore chose to monitor production of alternate mRNA splice forms of two key regulatory genes in the TLR signaling pathway, MyD88 and IRAK1, in peripheral blood mononuclear cells (PBMCs) from patients with ARDS. Our goal was to determine 1) whether pre-mRNA splicing in the TLR signaling pathway is altered in these patients, 2) whether this altered splicing is associated with clinical outcomes, and 3) whether this altered splicing offers a potential novel therapeutic avenue.
MATERIALS AND METHODS
Subject enrollment and sample collection.
The study protocol was approved by the Colorado Multiple Institutional Review Board (COMIRB) Committee and the National Jewish Health Institutional Review Board. All subjects, or an appropriate proxy, gave written, informed consent. The study was conducted in accordance with the Declaration of Helsinki. Patients enrolled were part of the Colorado ARDS Network site comprised of medical intensive care units at four urban hospitals.
PBMCs were isolated from patients after enrollment into one of three NHLBI ARDSnet studies at the University of Colorado affiliated hospitals, as described in detail previously (36, 48). Ninety-six percent of samples were collected within 4 days of enrollment into a study. The three parent studies were as follows: Drug Study of Albuterol to Treat ALI (ALTA, clinicaltrials.gov NCT00434993; n = 10) (46), Early Vs. Delayed Enteral Feeding and Omega-3 Fatty Acid/Antioxidant Supplementation for Treating People with Acute Lung Injury or Acute Respiratory Distress Syndrome (EDEN-Omega Study, NCT00609180; n = 80) (47, 55), or Statins for Acutely Injured Lungs From Sepsis (SAILS, NCT00979121; n = 14) (45). Peripheral blood samples were obtained 2.4 ± 1.1 (mean ± SD) days after ARDS criteria (4) were met (2.6 ± 1.2 days after intubation). PBMCs were isolated by the plasma Percoll method (22), at the 42% interface.
PBMCs also were obtained from healthy volunteers collected by the National Jewish Health Human Cell Core (48). These volunteers answered an 18-point questionnaire to verify that they were not experiencing acute symptoms of viral infection, did not have recently diagnosed medical conditions or vaccinations, did not have underlying chronic conditions, and did not have recent exposure to blood products. Additionally, the healthy volunteers were negative for hepatitis B and HIV. Vital signs monitored on the day of blood collection were normal.
Finally, to determine whether pre-mRNA splicing was altered in a second critically ill patient cohort, PBMCs were obtained from patients with fibrosing interstitial lung disease (ILD) that were undergoing an acute respiratory decline (24). Eligible patients had evidence of fibrosing interstitial lung disease, new or worsening respiratory symptoms present for ≤30 days that required hospitalization, and new or worsening infiltrates on chest imaging. These patients were prospectively enrolled during their hospitalization for acute respiratory decline. Blood was collected within 72 h of admission and processed as described above at the time of enrollment. Clinical characteristics for a subset of these patients have been previously reported (24).
Experimental measurements.
qPCR to monitor cytokine mRNA levels and MyD88 and IRAK1 isoform mRNA levels in PBMCs was performed on an ABI7900 Fast Real-Time PCR System using the Quantitect SYBR-Green RT-PCR assay kit (Qiagen). Data were normalized with the ddCt method using β-actin as a control. A subset of the data was also validated using Gapdh as a normalization control, which yielded similar results to β-actin normalization (not shown). Oligonucleotide sequences used for qPCR are listed in Table 1. MyD88L was assayed using a forward primer that annealed to exon 1 and a reverse primer that spanned the exon 2-exon 1 junction. MyD88S was assayed using a forward primer that annealed to exon 1 and a reverse primer that spanned the unique exon 3-exon 1 junction. IRAK1 was assayed using a forward primer that annealed to exon 10 and a reverse primer that spanned the exon 11-exon 10 junction. IRAK1c was assayed using a forward primer that annealed to exon 10 and a reverse primer that spanned the unique exon 12-exon 10 junction. A subset of the data was also validated using a second set of isoform-specific oligonucleotides (not shown). These approaches for measuring MyD88 and IRAK1 isoforms have been validated extensively by us (11, 12, 49).
Table 1.
Oligonucleotides used for qPCR analysis
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| β-Actin | CTGAACCCCAAGGCCAACCG | CCGTCACCGGAGTCCATCAC |
| TNF-α | CTTTGGAGTGATCGGCCCCCAGAGGGAAGA | CAGGCTTGTCACTCGGGGTTCGAGAAGATG |
| IL-6 | CTCCAGGAGCCCAGCTATGAACTCCTTCTC | GGGGGTACTGGGGCAGGGAAGGCAGC |
| MyD88L | GGCGCCTCTGTAGGCCGACTGCTCGAGCTG | GCTTCAAGATATACTTTTGGCAATCCTCCTCAATG |
| MyD88S | GGCGCCTCTGTAGGCCGACTGCTCGAGCTG | AACGCTCAGGCATATGCCCAATG |
| IRAK1 | TTCAGCTTTGGGGTGGTAGTGCTAGAGACC | CCTCAGCCTCCTCTTCCACCAGGTCTTTCAGATAC |
| IRAK1c | TTCAGCTTTGGGGTGGTAGTGCTAGAGACC | GCAGCTTCTCTAGCCTCTCGTACACCAGATAC |
Statistical methods.
Data analysis was conducted using R version 3.3.0 (2016-05-03). Summary statistics of demographic variables including age, sex, and race/ethnicity were calculated for the sample overall and separately for patients with ARDS and healthy subjects. Counts with corresponding percentages were calculated for categorical variables; means and standard deviations were calculated for continuous variables. We used an independent sample t-test to test for age differences between groups and a χ2 test of association to test for sex differences between groups. Factors found to be significantly different between groups were chosen as adjustment variables in future regression models. No statistical testing was performed on the distribution of race/ethnicity between healthy and patient groups because of the small number of subjects from several racial groups.
To compare expression of MyD88 isoforms between groups, boxplots showing the distribution of natural log-transformed MyD88S and MyD88L mRNA levels were reported in addition to testing for a mean difference between patients with ARDS and healthy controls via an independent sample t-test. Expression levels of IRAK1 isoforms (IRAK1c and IRAK1) in addition to inflammatory cytokines (IL-6 and TNF-α) for patients with ARDS were compared with healthy patients using the same approach. We compared expression of MyD88 and IRAK1 isoforms between patients with ARDS or ILD and healthy subjects via one-way ANOVA. The relationship between isoforms of MyD88 was examined via regression of MyD88L on MyD88S, case status and the product between MyD88S, and case status (interaction term) to allow for different linear relationships between MyD88L and MyD88S in the patient and control groups. A similar approach was performed for isoforms of IRAK1, in which, for both the MyD88 and IRAK1 models, interaction variables were removed if not significant (P > 0.05). All linear regression models examining these relationships were adjusted for age. Estimates for the change in log MyD88L mRNA expression for a one-unit increase in log MyD88S mRNA with corresponding 95% confidence intervals and P values were reported for patients with ARDS and healthy controls. The estimate for the change in log IRAK1 mRNA expression for a one-unit change in log IRAK1c mRNA was reported with a corresponding 95% confidence interval and P value.
To explore the association between expression of the various isoforms with clinical factors observed for patients with ARDS, logistic regression of status at day 28 and linear regression of APACHE III score on log transformed mRNA levels were used to test for association between these clinical factors and the isoform of interest; odds ratio estimates for status at day 28 and regression parameter estimates for APACHE III score with corresponding 95% confidence intervals and P values were reported.
The relationship between IL-6 mRNA and expression of isoforms of MyD88 was explored using linear regression of log-transformed IL-6 mRNA expression on the level of MyD88S, case status, and the product between MyD88S and case status (interaction term, as above). All linear regression models examining these relationships were adjusted for age. Estimates for the change in log IL-6 mRNA expression for a one-unit increase in log MyD88S mRNA expression with corresponding 95% confidence intervals and P values were reported for patients with ARDS and healthy controls. In addition, the estimate for the interaction is reported as the difference in the change in IL-6 expression corresponding to a one-unit change in MyD88S expression between patients with ARDS and healthy individuals. A similar approach to modeling the relationship between log IL-6 mRNA expression was performed with MyD88L, IRAK1, and IRAK1c. For a given model, if the interaction term for case status by isoform expression was not significant (P > 0.05), it was removed from the final model; subsequently, if the case status variable was not significant, it was removed from the model.
RESULTS
Characteristics of patients with ARDS and healthy control subjects.
We analyzed PBMCs from 104 patients with ARDS and 30 healthy control subjects. All subjects were adults, but the average age was significantly higher in patients with ARDS compared with healthy subjects (P < 0.0001; Table 2). Patients with ARDS were on average 13.0 yr older than healthy subjects (95% CI: 7.0, 19.0). There was no significant difference in the distribution of sex between the two groups. The percentage classified as white also was similar between the healthy group and patients with ARDS, 89.7% and 86.9%, respectively. The most common primary physiological injury in the patients with ARDS was pneumonia, in which 82 (78.8%) of the patients with ARDS had pneumonia at initial presentation (Table 2). The second most common primary physiological injury was sepsis observed for 10 (9.6%) of patients with ARDS in this cohort (Table 2).
Table 2.
Demographic summary of subjects by case status
| Variable | Overall | Healthy | ARDS | P Value |
|---|---|---|---|---|
| n | 134 | 30 | 104 | |
| Age | 50.66 ± 15.55 | 40.60 ± 12.40 | 53.56 ± 15.20 | <0.0001* |
| Sex, n (%) | 0.73† | |||
| Female | 64 (47.8) | 13 (43.3) | 51 (49) | |
| Male | 70 (52.2) | 17 (56.7) | 53 (51) | |
| Race/ethnicity‡, n (%) | — | |||
| White | 99 (87.6) | 26 (89.7) | 73 (86.9) | |
| American Indian | 2 (1.8) | 0 (0) | 2 (2.4) | |
| Asian | 3 (2.7) | 1 (3.4) | 2 (2.4) | |
| Black or African American | 7 (6.2) | 0 (0) | 7 (8.3) | |
| Hispanic | 2 (1.8) | 2 (6.9) | 0 (0) | |
| Primary physiological injury | ||||
| Pneumonia | 82 (78.8) | |||
| Aspiration | 6 (5.8) | |||
| Multiple transfusions | 2 (1.9) | |||
| Sepsis | 10 (9.6) | |||
| Unknown | 4 (3.8) | |||
P value corresponds to independent t-test.
P value corresponds to χ2 test.
Race not reported from 1 healthy subject and 20 patients with acute respiratory distress syndrome (ARDS).
MyD88L is higher among patients with ARDS compared with healthy control subjects.
To explore the relationship between isoforms of MyD88 and IRAK1 and both ARDS and inflammation, we measured production of MyD88L, MyD88S, IRAK1, and IRAK1c in addition to mRNA from two proinflammatory cytokines (IL-6 and TNF-α) in these PBMCs using qPCR. As expected and serving as a positive control, IL-6 mRNA levels were significantly increased in patients with ARDS compared with healthy subjects (Fig. 1). MyD88L mRNA levels were also found to be significantly higher in patients with ARDS compared with healthy controls (P = 0.024, Fig. 1). MyD88S mRNA levels were highly variable for both healthy subjects and patients with ARDS. There was no significant difference found between controls and patients with ARDS for MyD88S mRNA (P = 0.583, Fig. 1) or for the ratio of the two isoforms (not shown, P = 0.221). The substantial variability in MyD88S mRNA levels may have masked the change in splicing observed when MyD88L was monitored alone. The increase in the level of MyD88L mRNA and the relatively unchanged level of MyD88S mRNA in patients with ARDS suggest that MyD88 pre-mRNA splicing is altered in these patients; this is consistent with a direct assessment of the relationship between isoform levels (see below). In contrast to the altered level of MyD88L in patients with ARDS, there was no significant difference found between control subjects and patients with ARDS for IRAK1 (P = 0.139, Fig. 1), IRAK1c (P = 0.137, Fig. 1), or the ratio of the two isoforms (P = 0.557, not shown).
Fig. 1.

Increased expression of MyD88L in peripheral blood mononuclear cells (PBMCs) from patients with acute respiratory distress syndrome (ARDS). The boxplots depict the ln-transformed mRNA distribution of IL-6, TNF-α, MyD88S, MyD88L, IRAK1, and IRAK1c in PBMCs from patients with ARDS and healthy control subjects. Mean differences and P values (independent-sample t-test) are shown below for each variable.
MyD88 pre-mRNA splicing is altered in PBMCs from patients with ARDS.
To further investigate alternative splicing in patients with ARDS, we examined the relationships between each of the two isoforms of MyD88 and IRAK1 in PBMCs from patients with ARDS and healthy volunteers (Fig. 2, Table 3). Expression levels of IRAK1 were positively associated with IRAK1c mRNA levels in both patients with ARDS and healthy control subjects; a 10% increase in IRAK1c mRNA corresponded to an 11.0% increase in IRAK1 mRNA (95% CI: 10.2%, 11.8%; P < 0.0001). The relationship between expression of MyD88L and MyD88S differed between patients with ARDS and healthy subjects (P value for interaction = 0.032, Fig. 2, Table 3). Among patients with ARDS, a 10% increase in MyD88S expression was associated with an increase in expression of MyD88L by 2.4% (95% CI: 0.9%, 4.0%; P = 0.002). Among healthy subjects, MyD88L expression appears to decrease with increasing expression of MyD88S although this was not significant (P = 0.295; Fig. 2). These data are consistent with MyD88 pre-mRNA splicing but not IRAK1 pre-mRNA splicing, being altered in PBMCs from patients with ARDS.
Fig. 2.

MyD88 but not IRAK1 pre-mRNA splicing is altered in patients with acute respiratory distress syndrome (ARDS). Left: ln-transformed IRAK1 mRNA was compared with ln-transformed IRAK1c mRNA where the solid line displays predicted values from the linear regression model adjusted for mean age. Right: ln-transformed MyD88L mRNA levels were compared with ln-transformed MyD88S mRNA levels. Lines corresponding to predicted values from the final model for patients with ARDS (red) and healthy controls (blue) adjusted for mean age. Further statistical information relevant to this figure is presented in Table 3.
Table 3.
Changes in ln IRAK1 by ln IRAK1c and changes in ln MyD88L by ln MyD88S with corresponding 95% confidence intervals and P values
| IRAK1 Model | Change in ln IRAK1 (95% CI) | P Value | MyD88-L Model | Change in ln MyD88-L (95% CI) | P Value |
|---|---|---|---|---|---|
| IRAK1c Healthy* | 1.10 (1.02, 1.17) | <0.0001 | MyD88-S Healthy | −0.21 (−0.59, 0.18) | 0.294 |
| IRAK1c ARDS* | 1.10 (1.02, 1.17) | <0.0001 | MyD88-S ARDS | 0.25 (0.09, 0.42) | 0.002 |
| — | — | — | MyD88-S Difference | 0.46 (0.04, 0.88) | 0.032 |
| Age, yr | 0.00 (−0.01, 0.01) | 0.752 | Age, yr | 0.00 (−0.01, 0.01) | 0.929 |
Changes in IRAK1 by IRAK1c did not differ between healthy and acute respiratory distress syndrome (ARDS) subjects; the estimated change in ln IRAK1 for one unit change ln IRAK1c is the same regardless of affection status.
MyD88 pre-mRNA splicing is altered in PBMCs from patients with ILD undergoing an acute exacerbation.
To determine whether the alteration in MyD88 pre-mRNA splicing observed in PBMCs from patients with ARDS was unique to patients with ARDS, we measured MyD88L, MyD88S, IRAK1, and IRAK1c mRNA levels by qPCR in PBMCs from a second cohort of critically ill patients, patients with ILD undergoing an acute exacerbation (24). Like patients with ARDS, these patients also exhibit significant inflammation (24). MyD88L mRNA was significantly higher in PBMCs from patients with ILD compared with both patients with ARDS and healthy control subjects (ANOVA P < 0.001, Fig. 3). There was no significant difference found among patients with ILD, patients with ARDS, and control subjects for MyD88S mRNA (ANOVA P = 0.319, Fig. 3). This increase in MyD88L mRNA without a corresponding increase in MyD88S mRNA suggests that MyD88 pre-mRNA splicing is altered in these patients in a manner that is comparable to that observed in patients with ARDS.
Fig. 3.

Altered MyD88 pre-mRNA splicing and IRAK1 expression in peripheral blood mononuclear cells (PBMCs) from patients with interstitial lung disease (ILD). The boxplots depict the ln-transformed mRNA distribution of MyD88S, MyD88L, IRAK1, and IRAK1c in PBMCs from patients with ILD (I), patients with ARDS (A), and healthy control subjects (H). One-way ANOVA P values are shown below for each variable. The ILD cohort included 22 patients of 63.9 ± 14.5 (means ± SD) yr of age; 16 of the 22 (72.7%) patients with ILD were male. Data for healthy subjects and patients with ARDS are the same as in Fig. 1.
Although IRAK1 expression and splicing was relatively unchanged in patients with ARDS compared with healthy subjects (Fig. 1), both IRAK1c and IRAK1 mRNA levels were significantly higher in patients with ILD compared with patients with ARDS and healthy control subjects (ANOVA P < 0.001 for both, Fig. 3). This substantial increase in both IRAK1 isoforms is consistent with a significant increase in IRAK1 expression rather than a change in IRAK1 pre-mRNA splicing in the patients with ILD. Thus, although MyD88 splicing is altered in a similar fashion in both critically ill cohorts, the regulation of IRAK1 isoform expression differs between patients with ARDS and ILD.
IRAK1c mRNA levels in PBMCs from patients with ARDS are associated with survival.
To determine whether MyD88 or IRAK1 isoform mRNA expression levels in PBMCs could serve as a marker for clinical outcome in patients with ARDS, we examined MyD88L, MyD88S, IRAK1, and IRAK1c mRNA levels for an association with survival and APACHE III score. We did not detect a significant association between MyD88L or MyD88S with these clinical parameters in the patients with ARDS (Table 4). In contrast, higher IRAK1c levels were associated with survival (Fig. 4). Within patients with ARDS, the odds of being dead at or before 28 days decreased by 57% for a one-unit increase in log IRAK1c mRNA levels (95% CI: 16%, 82%; P = 0.032). Thus decreased IRAK1c isoform expression is associated with poor health indicators. IRAK1 mRNA levels were not associated with survival or APACHE III score (Table 4).
Table 4.
Odds ratio estimates of death at 28 days and parameter estimates of APACHE III Score by log transformed isoform expression with corresponding 95% CI and P values
| Characteristic | Dead vs. Alive OR (95% CI) |
P Value | APACHE III Score Est (95% CI) |
P Value |
|---|---|---|---|---|
| MyD88-S | 1.21 (0.71, 2.08) | 0.478 | 1.36 (−3.55, 6.28) | 0.583 |
| MyD88-L | 1.07 (0.61, 1.99) | 0.819 | 0.92 (−4.26, 6.1) | 0.724 |
| IRAK1c | 0.43 (0.18, 0.84) | 0.032 | −3.11 (−6.85, 0.63) | 0.102 |
| IRAK1 | 0.51 (0.21, 0.9) | 0.064 | −2.12 (−5.3, 1.06) | 0.189 |
Fig. 4.
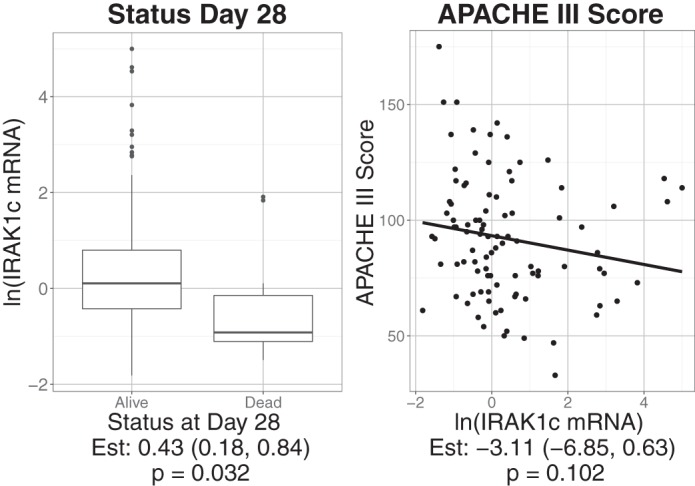
Decreased IRAK1c mRNA is associated with worse clinical outcomes in patients with acute respiratory distress syndrome (ARDS). Left: boxplot of ln-transformed IRAK1c mRNA levels compared with survival at day 28. Right: ln-transformed IRAK1c mRNA levels were compared with APACHE III scores with the solid line displaying the predicted values from single variable linear regression. Estimates reflect changes in clinical outcomes with corresponding 95% confidence intervals and P values reported below each graph.
Interaction between MyD88S and IL-6 in PBMCs from patients with ARDS.
Cell line studies have demonstrated that inflammatory stimuli induce MyD88S and IRAK1c production (11, 26, 54, 64), and, in turn, MyD88S and IRAK1c then limit resulting inflammation. We therefore examined the relationship between MyD88S and IRAK1c mRNA levels and IL-6 mRNA levels in PBMCs from patients with ARDS and healthy subjects. There was a significantly different relationship between IL-6 mRNA and MyD88S mRNA observed for patients with ARDS compared with that observed in the healthy subjects (P value for interaction = 0.009; Table 5, Fig. 5A). Among patients with ARDS, a 10% increase in MyD88S expression was associated with a 7.0% increase in expression of IL-6 (95% CI: 2.8%, 11.2%; P = 0.001). Among healthy subjects, there was an apparent decrease in IL-6 mRNA with an increase in expression of MyD88S (P = 0.145; Fig. 5A, Table 5). Taken together, the relationship between IL-6 and MyD88S is different for patients with ARDS compared with healthy controls.
Table 5.
Changes in IL-6 mRNA expression (log) by MyD88S mRNA expression (log) (case status and interaction adjusted for age) with corresponding 95% CIs and P values
| Variable | Change in ln IL-6 (95% CI) | P Value |
|---|---|---|
| Healthy | −0.73 (−1.72, 0.25) | 0.145 |
| ARDS | 0.71 (0.29, 1.12) | <0.001 |
| Difference | 1.44 (0.37, 2.51) | 0.009 |
| Age | −0.01 (−0.04, 0.02) | 0.433 |
ARDS, acute respiratory distress syndrome.
Fig. 5.

The interaction between MyD88S and IL-6 is opposite between peripheral blood mononuclear cells (PBMCs) of patients with acute respiratory distress syndrome (ARDS) and healthy subjects. The graphs depict ln-transformed IL-6 mRNA levels compared with either ln-transformed MyD88S mRNA levels (A), ln-transformed MyD88L mRNA levels (B), ln-transformed IRAK1c mRNA levels (C), and ln-transformed IRAK1 mRNA levels (D). Lines corresponding to predicted values from the final model (see materials and methods) for patients with ARDS (red) and healthy control subjects (blue) are adjusted for mean age. Further statistical information relevant to A is presented in Table 5.
The differential relationship among MyD88S and IL-6 expression between patients with ARDS and healthy control subjects was not observed for MyD88L (Fig. 5B). Instead, a significantly positive correlation was found for IL-6 expression and expression of MyD88L (P < 0.0001) for both patients with ARDS and healthy subjects. This suggests that the change in the relationship between MyD88S and IL-6 in PBMCs likely represents a change in MyD88 alternative pre-mRNA splicing. As further confirmation of the specificity of the interaction between MyD88S and IL-6 in patients with ARDS, we observed a significantly positive correlation between IL-6 expression and expression of both IRAK1 and IRAK1c for both patients with ARDS and healthy subjects (Fig. 5, C and D). Thus there are differences in the regulation of alternative pre-mRNA splicing of MyD88 and IRAK1 in patients with ARDS.
DISCUSSION
MyD88 and IRAK1 are key signaling proteins that activate NF-κB and inflammatory cytokine production (30, 65). The alternative MyD88 and IRAK1 pre-mRNA splicing induced by inflammatory stimuli results in a negative feedback loop that limits inflammation; this suggests that altered MyD88 or IRAK1 pre-mRNA splicing could affect the pathogenesis of diseases with an inflammatory component. Although the regulation of these alternative splicing events has been investigated in vitro, there has been limited study of MyD88 or IRAK1 splicing in vivo, particularly in a disease setting. There have been a few reports of the levels of individual MyD88 isoforms in PBMCs from septic patients, but no reports measured both isoforms simultaneously (1, 57, 58). We are unaware of any prior studies monitoring alternative splicing of IRAK1 in any disease. We now for the first time examine both MyD88 and IRAK1 isoforms in a large cohort of patients with ARDS.
MyD88L was increased in PBMCs from these patients without a corresponding increase in MyD88S levels, suggesting that MyD88 pre-mRNA splicing is being altered in patients with ARDS in the proinflammatory direction. This was confirmed by monitoring both splice forms simultaneously in individual patients. This effect was unique to MyD88, as IRAK1 pre-mRNA splicing was not altered in patients with ARDS compared with healthy subjects. Our observed increase in MyD88L in PBMCs from patients with ARDS is consistent with observations made in septic patients (57, 58). In another study, MyD88S levels were increased roughly 10-fold in PBMCs from septic patients (1), which is quite different from our results in ARDS, in which we did not observe a change in MyD88S levels. The difference in our study and this prior study could be due to differences in disease, cohort, or PBMC isolation protocol. Moreover, timing may also be a key factor. Although our samples were obtained shortly after intubation, it is unknown how long the patients were ill before hospital admission.
Our studies of a small cohort of patients with ILD undergoing an acute exacerbation suggests that the alterations in MyD88 pre-mRNA splicing observed in patients with ARDS may not be unique to ARDS but instead could be common to multiple critical diseases with a significant inflammatory component. In both patients with ARDS and ILD, we observed an increase in production of the proinflammatory form of MyD88 without a significant change in the anti-inflammatory form of MyD88. The regulation of IRAK1 isoform levels differed in the two critically ill cohorts. Although IRAK1 expression and pre-mRNA splicing was largely unchanged in patients with ARDS, we observed a very substantial increase in IRAK1 expression (of both isoforms) in patients with ILD, indicating that the regulation of IRAK1 may differ in different inflammatory diseases. The increase in MyD88L levels was larger in the patients with ILD than the patients with ARDS; this may reflect the chronic nature of ILD or may reflect the critical nature of the acute exacerbation.
We also found that increased IRAK1c levels were associated with improved patient outcome in the ARDS cohort. This may be due to the fact that IRAK1c limits detrimental inflammation. It is interesting that IRAK1 splicing did not differ in the patients with ARDS compared with the control subjects; this may indicate that preexisting differences in IRAK1 isoform levels influence disease outcome in patients with ARDS.
We also found that the relationship between MyD88S and IL-6 differed in PBMCs from patients with ARDS and healthy volunteers. Cell-based studies have indicated that inflammatory stimuli induce MyD88S production, which in turn inhibits inflammation, forming a negative feedback loop that ensures that inflammation is self-limiting (11, 26). In healthy control subjects, we observed that increased MyD88S levels correlated with decreased IL-6 levels. Thus MyD88S may help keep unwanted inflammation in check in healthy volunteers, maintaining balance given frequent environmental exposure to inflammatory stimuli in our pathogen-rich world. In contrast, in PBMCs from patients with ARDS, we observed a positive correlation between MyD88S levels and IL-6 levels. Thus, although the increased inflammation present in patients with ARDS is associated with increased MyD88S production, the data suggest that the inflammatory processes in such patients are so great that MyD88S cannot overcome this state.
The alternate splice forms of MyD88 and IRAK1 are both induced by TLR agonists in vitro (11, 26, 54, 64). Moreover, both MyD88 and IRAK1 function sequentially in common TLR signaling pathways (30, 65). Despite these commonalities, we observed differences in the regulation and potential effects of this alternate splicing in patients with ARDS. We observed that MyD88 splicing was altered in patients with ARDS compared with healthy control subjects, whereas IRAK1 splicing was not. This suggests that the regulation of these two alternative splicing events is more complex than the in vitro studies have so far indicated (11, 26, 54, 64); perhaps multiple mechanisms regulate alternative splicing in the TLR signaling pathway.
Although IRAK1c levels correlated with patient outcome, MyD88S levels did not. Upon LPS stimulation, the LPS receptor TLR4 recruits a large signaling complex that includes MyD88, another IRAK kinase IRAK4, IRAK1, and the ubiquitin ligase TRAF6 (51). This signaling complex contains numerous MyD88 and IRAK1 peptides (16, 33, 42). Like IRAK1, IRAK1c still interacts with other members of the signaling complex, including MyD88, IRAK1, and TRAF6 (54, 64). However, IRAK1c exhibits several functional differences from IRAK1: IRAK1c cannot be phosphorylated by IRAK4, and IRAK1c in turn cannot itself act as a kinase (54). Moreover, IRAK1c, unlike IRAK1, remains stable after LPS challenge, allowing it to form long-lived but nonfunctional signaling complexes (51). MyD88S likewise binds to the TLR signaling complex and can still interact with IRAK1; however, MyD88S cannot interact with IRAK4, leading to loss of phosphorylation and activation of IRAK1 (8, 26). Thus, although both MyD88S and IRAK1c act as dominant negative inhibitors of LPS-induced inflammation, there are subtle differences in how they inhibit signaling. This is exemplified by the observation that, although IRAK1c inhibits both the NF-κB and MAPK arms of LPS response (54), MyD88S has been reported to inhibit NF-κB but not MAPK signaling (27). These subtle differences could explain the different effects of these two inhibitory isoforms in patients with ARDS.
There are several limitations to our study. Previous studies have demonstrated that PBMCs are not the primary and/or sole source of plasma cytokines in septic patients (17, 43, 44). In the future, we intend to investigate alternative splicing in lung immune cells from patients with ARDS, which should have a more proximal effect on the disease. Additionally, we do not know which arm of the studies (treatment or placebo) these patients were in. However, we note that none of the treatments affected the primary end points of the studies, and almost all patient samples were obtained shortly after the diagnosis of ARDS and entrance into the study. Moreover, MyD88L mRNA levels were not altered between studies (P = 0.37, data not shown). We also have not analyzed critically ill control subjects at risk for ARDS but without ARDS and as such cannot determine whether the observed splicing changes are unique to ARDS or may reflect the effect of the underlying disease. An additional limitation is that these association studies do not prove causation; however, they do suggest models that can be tested in other cohorts and eventually in mice.
In conclusion, we find that the alternate splice form of IRAK1, IRAK1c, is predictive of patient outcome in patients with ARDS and that MyD88 pre-mRNA splicing is altered in the proinflammatory direction in patients with ARDS compared with healthy volunteers. These data indicate that further research into alternative pre-mRNA splicing of genes that control inflammation could lead to novel diagnostic and therapeutic approach for patients with ARDS and other inflammatory disorders.
GRANTS
This work was funded by NIH grants R01ES025161, R01HL090991, P01HL068743, the NHLBI ARDS Network, the Wendy Siegel Fund for Leukemia and Cancer Research, the Cystic Fibrosis Foundation (NICK14G0), and the Rebecca Runyon Bryan Chair for Cystic Fibrosis.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.Z.B., B.R.H., K.C.M., T.E.F., and S.A. analyzed data; R.Z.B., B.R.H., K.C.M., T.E.F., and S.A. interpreted results of experiments; R.Z.B. and S.A. prepared figures; R.Z.B., T.E.F., and S.A. drafted manuscript; R.Z.B., B.R.H., K.C.M., E.L.B., M.M., E.A., T.J.H., J.A.N., T.E.F., and S.A. edited and revised manuscript; R.Z.B., B.R.H., K.C.M., E.L.B., M.M., E.A., T.J.H., J.A.N., T.E.F., and S.A. approved final version of manuscript; B.R.H., K.C.M., E.L.B., M.M., E.A., T.J.H., J.A.N., and S.A. performed experiments; S.A. conceived and designed research.
ACKNOWLEDGMENTS
We sincerely thank all participating patients with ARDS and their families for contributions to ARDS research.
REFERENCES
- 1.Adib-Conquy M, Adrie C, Fitting C, Gattolliat O, Beyaert R, Cavaillon JM. Up-regulation of MyD88s and SIGIRR, molecules inhibiting Toll-like receptor signaling, in monocytes from septic patients. Crit Care Med 34: 2377–2385, 2006. doi: 10.1097/01.CCM.0000233875.93866.88. [DOI] [PubMed] [Google Scholar]
- 2.Arcaroli J, Silva E, Maloney JP, He Q, Svetkauskaite D, Murphy JR, Abraham E. Variant IRAK-1 haplotype is associated with increased nuclear factor-kappaB activation and worse outcomes in sepsis. Am J Respir Crit Care Med 173: 1335–1341, 2006. doi: 10.1164/rccm.200603-341OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bauer TT, Montón C, Torres A, Cabello H, Fillela X, Maldonado A, Nicolás JM, Zavala E. Comparison of systemic cytokine levels in patients with acute respiratory distress syndrome, severe pneumonia, and controls. Thorax 55: 46–52, 2000. doi: 10.1136/thorax.55.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, Lamy M, Legall JR, Morris A, Spragg R. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med 149: 818–824, 1994. doi: 10.1164/ajrccm.149.3.7509706. [DOI] [PubMed] [Google Scholar]
- 5.Bhatia M, Moochhala S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J Pathol 202: 145–156, 2004. doi: 10.1002/path.1491. [DOI] [PubMed] [Google Scholar]
- 6.Bouros D, Alexandrakis MG, Antoniou KM, Agouridakis P, Pneumatikos I, Anevlavis S, Pataka A, Patlakas G, Karkavitsas N, Kyriakou D. The clinical significance of serum and bronchoalveolar lavage inflammatory cytokines in patients at risk for acute respiratory distress syndrome. BMC Pulm Med 4: 6, 2004. doi: 10.1186/1471-2466-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burns K, Janssens S, Brissoni B, Olivos N, Beyaert R, Tschopp J. Inhibition of interleukin 1 receptor/Toll-like receptor signaling through the alternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4. J Exp Med 197: 263–268, 2003. doi: 10.1084/jem.20021790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burns K, Martinon F, Esslinger C, Pahl H, Schneider P, Bodmer JL, Di Marco F, French L, Tschopp J. MyD88, an adapter protein involved in interleukin-1 signaling. J Biol Chem 273: 12203–12209, 1998. doi: 10.1074/jbc.273.20.12203. [DOI] [PubMed] [Google Scholar]
- 9.Chiumello D, Coppola S, Froio S, Gotti M. What’s next after ARDS: long-term outcomes. Respir Care 61: 689–699, 2016. doi: 10.4187/respcare.04644. [DOI] [PubMed] [Google Scholar]
- 10.Chun CD, Liles WC, Frevert CW, Glenny RW, Altemeier WA. Mechanical ventilation modulates Toll-like receptor-3-induced lung inflammation via a MyD88-dependent, TLR4-independent pathway: a controlled animal study. BMC Pulm Med 10: 57, 2010. doi: 10.1186/1471-2466-10-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Arras L, Alper S. Limiting of the innate immune response by SF3A-dependent control of MyD88 alternative mRNA splicing. PLoS Genet 9: e1003855, 2013. doi: 10.1371/journal.pgen.1003855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Arras L, Laws R, Leach SM, Pontis K, Freedman JH, Schwartz DA, Alper S. Comparative genomics RNAi screen identifies Eftud2 as a novel regulator of innate immunity. Genetics 197: 485–496, 2014. doi: 10.1534/genetics.113.160499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fanelli V, Vlachou A, Ghannadian S, Simonetti U, Slutsky AS, Zhang H. Acute respiratory distress syndrome: new definition, current and future therapeutic options. J Thorac Dis 5: 326–334, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fujishima S. Pathophysiology and biomarkers of acute respiratory distress syndrome. J Intensive Care 2: 32, 2014. doi: 10.1186/2052-0492-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao L, Barnes KC. Recent advances in genetic predisposition to clinical acute lung injury. Am J Physiol Lung Cell Mol Physiol 296: L713–L725, 2009. doi: 10.1152/ajplung.90269.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gay NJ, Gangloff M, O’Neill LA. What the Myddosome structure tells us about the initiation of innate immunity. Trends Immunol 32: 104–109, 2011. doi: 10.1016/j.it.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 17.Gille-Johnson P, Smedman C, Gudmundsdotter L, Somell A, Nihlmark K, Paulie S, Andersson J, Gårdlund B. Circulating monocytes are not the major source of plasma cytokines in patients with sepsis. Shock 38: 577–583, 2012. doi: 10.1097/SHK.0b013e3182746e52. [DOI] [PubMed] [Google Scholar]
- 18.Gray P, Michelsen KS, Sirois CM, Lowe E, Shimada K, Crother TR, Chen S, Brikos C, Bulut Y, Latz E, Underhill D, Arditi M. Identification of a novel human MD-2 splice variant that negatively regulates Lipopolysaccharide-induced TLR4 signaling. J Immunol 184: 6359–6366, 2010. doi: 10.4049/jimmunol.0903543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graziewicz MA, Tarrant TK, Buckley B, Roberts J, Fulton L, Hansen H, Orum H, Kole R, Sazani P. An endogenous TNF-alpha antagonist induced by splice-switching oligonucleotides reduces inflammation in hepatitis and arthritis mouse models. Mol Ther 16: 1316–1322, 2008. doi: 10.1038/mt.2008.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hager DN. Recent advances in the management of the acute respiratory distress syndrome. Clin Chest Med 36: 481–496, 2015. doi: 10.1016/j.ccm.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 21.Hardy MP, O’Neill LA. The murine IRAK2 gene encodes four alternatively spliced isoforms, two of which are inhibitory. J Biol Chem 279: 27699–27708, 2004. doi: 10.1074/jbc.M403068200. [DOI] [PubMed] [Google Scholar]
- 22.Haslett C, Guthrie LA, Kopaniak MM, Johnston RB Jr, Henson PM. Modulation of multiple neutrophil functions by preparative methods or trace concentrations of bacterial lipopolysaccharide. Am J Pathol 119: 101–110, 1985. [PMC free article] [PubMed] [Google Scholar]
- 23.Herridge MS, Moss M, Hough CL, Hopkins RO, Rice TW, Bienvenu OJ, Azoulay E. Recovery and outcomes after the acute respiratory distress syndrome (ARDS) in patients and their family caregivers. Intensive Care Med 42: 725–738, 2016. doi: 10.1007/s00134-016-4321-8. [DOI] [PubMed] [Google Scholar]
- 24.Huie TJ, Olson AL, Cosgrove GP, Janssen WJ, Lara AR, Lynch DA, Groshong SD, Moss M, Schwarz MI, Brown KK, Frankel SK. A detailed evaluation of acute respiratory decline in patients with fibrotic lung disease: aetiology and outcomes. Respirology 15: 909–917, 2010. doi: 10.1111/j.1440-1843.2010.01774.x. [DOI] [PubMed] [Google Scholar]
- 25.Iwami KI, Matsuguchi T, Masuda A, Kikuchi T, Musikacharoen T, Yoshikai Y. Cutting edge: naturally occurring soluble form of mouse Toll-like receptor 4 inhibits lipopolysaccharide signaling. J Immunol 165: 6682–6686, 2000. doi: 10.4049/jimmunol.165.12.6682. [DOI] [PubMed] [Google Scholar]
- 26.Janssens S, Burns K, Tschopp J, Beyaert R. Regulation of interleukin-1- and lipopolysaccharide-induced NF-kappaB activation by alternative splicing of MyD88. Curr Biol 12: 467–471, 2002. doi: 10.1016/S0960-9822(02)00712-1. [DOI] [PubMed] [Google Scholar]
- 27.Janssens S, Burns K, Vercammen E, Tschopp J, Beyaert R. MyD88S, a splice variant of MyD88, differentially modulates NF-kappaB- and AP-1-dependent gene expression. FEBS Lett 548: 103–107, 2003. doi: 10.1016/S0014-5793(03)00747-6. [DOI] [PubMed] [Google Scholar]
- 28.Jaresova I, Rozkova D, Spisek R, Janda A, Brazova J, Sediva A. Kinetics of Toll-like receptor-4 splice variants expression in lipopolysaccharide-stimulated antigen presenting cells of healthy donors and patients with cystic fibrosis. Microbes Infect 9: 1359–1367, 2007. doi: 10.1016/j.micinf.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 29.Johnson ER, Matthay MA. Acute lung injury: epidemiology, pathogenesis, and treatment. J Aerosol Med Pulm Drug Deliv 23: 243–252, 2010. doi: 10.1089/jamp.2009.0775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11: 373–384, 2010. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 31.Kovach MA, Standiford TJ. Toll like receptors in diseases of the lung. Int Immunopharmacol 11: 1399–1406, 2011. doi: 10.1016/j.intimp.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leeman JR, Gilmore TD. Alternative splicing in the NF-kappaB signaling pathway. Gene 423: 97–107, 2008. doi: 10.1016/j.gene.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 465: 885–890, 2010. doi: 10.1038/nature09121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu G, Tsuruta Y, Gao Z, Park YJ, Abraham E. Variant IL-1 receptor-associated kinase-1 mediates increased NF-kappa B activity. J Immunol 179: 4125–4134, 2007. doi: 10.4049/jimmunol.179.6.4125. [DOI] [PubMed] [Google Scholar]
- 35.Lynch KW. Consequences of regulated pre-mRNA splicing in the immune system. Nat Rev Immunol 4: 931–940, 2004. doi: 10.1038/nri1497. [DOI] [PubMed] [Google Scholar]
- 36.Malcolm KC, Kret JE, Young RL, Poch KR, Caceres SM, Douglas IS, Coldren CD, Burnham EL, Moss M, Nick JA. Bacteria-specific neutrophil dysfunction associated with interferon-stimulated gene expression in the acute respiratory distress syndrome. PLoS One 6: e21958, 2011. doi: 10.1371/journal.pone.0021958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marshall RP, Webb S, Hill MR, Humphries SE, Laurent GJ. Genetic polymorphisms associated with susceptibility and outcome in ARDS. Chest 121, Suppl: 68S–69S, 2002. doi: 10.1378/chest.121.3_suppl.68S. [DOI] [PubMed] [Google Scholar]
- 38.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest 122: 2731–2740, 2012. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meduri GU, Headley S, Kohler G, Stentz F, Tolley E, Umberger R, Leeper K. Persistent elevation of inflammatory cytokines predicts a poor outcome in ARDS. Plasma IL-1 beta and IL-6 levels are consistent and efficient predictors of outcome over time. Chest 107: 1062–1073, 1995. doi: 10.1378/chest.107.4.1062. [DOI] [PubMed] [Google Scholar]
- 40.Meduri GU, Kohler G, Headley S, Tolley E, Stentz F, Postlethwaite A. Inflammatory cytokines in the BAL of patients with ARDS. Persistent elevation over time predicts poor outcome. Chest 108: 1303–1314, 1995. doi: 10.1378/chest.108.5.1303. [DOI] [PubMed] [Google Scholar]
- 41.Mendoza-Barberá E, Corral-Rodríguez MA, Soares-Schanoski A, Velarde M, Macieira S, Messerschmidt A, López-Collazo E, Fuentes-Prior P. Contribution of globular death domains and unstructured linkers to MyD88.IRAK-4 heterodimer formation: an explanation for the antagonistic activity of MyD88s. Biochem Biophys Res Commun 380: 183–187, 2009. doi: 10.1016/j.bbrc.2009.01.069. [DOI] [PubMed] [Google Scholar]
- 42.Motshwene PG, Moncrieffe MC, Grossmann JG, Kao C, Ayaluru M, Sandercock AM, Robinson CV, Latz E, Gay NJ. An oligomeric signaling platform formed by the Toll-like receptor signal transducers MyD88 and IRAK-4. J Biol Chem 284: 25404–25411, 2009. doi: 10.1074/jbc.M109.022392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Munoz C, Carlet J, Fitting C, Misset B, Blériot JP, Cavaillon JM. Dysregulation of in vitro cytokine production by monocytes during sepsis. J Clin Invest 88: 1747–1754, 1991. doi: 10.1172/JCI115493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Munoz C, Misset B, Fitting C, Blériot JP, Carlet J, Cavaillon JM. Dissociation between plasma and monocyte-associated cytokines during sepsis. Eur J Immunol 21: 2177–2184, 1991. doi: 10.1002/eji.1830210928. [DOI] [PubMed] [Google Scholar]
- 45.Truwit JD, Bernard GR, Steingrub J, Matthay MA, Liu KD, Albertson TE, Brower RG, Shanholtz C, Rock P, Douglas IS, deBoisblanc BP, Hough CL, Hite RD, Thompson BT; National Heart, Lung, and Blood Institute ARDS Clinical Trials Network . Rosuvastatin for sepsis-associated acute respiratory distress syndrome. N Engl J Med 370: 2191–2200, 2014. doi: 10.1056/NEJMoa1401520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network; Matthay MA, Brower RG, Carson S, Douglas IS, Eisner M, Hite D, Holets S, Kallet RH, Liu KD, MacIntyre N, Moss M, Schoenfeld D, Steingrub J, Thompson BT. Randomized, placebo-controlled clinical trial of an aerosolized beta(2)-agonist for treatment of acute lung injury. Am J Respir Crit Care Med 184: 561–568, 2011. doi: 10.1164/rccm.201012-2090OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rice TW, Wheeler AP, Thompson BT, Steingrub J, Hite RD, Moss M, Morris A, Dong N, Rock P; National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network . Initial trophic vs full enteral feeding in patients with acute lung injury: the EDEN randomized trial. JAMA 307: 795–803, 2012. doi: 10.1001/jama.2012.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nick JA, Caceres SM, Kret JE, Poch KR, Strand M, Faino AV, Nichols DP, Saavedra MT, Taylor-Cousar JL, Geraci MW, Burnham EL, Fessler MB, Suratt BT, Abraham E, Moss M, Malcolm KC. Extremes of interferon-stimulated gene expression associate with worse outcomes in the acute respiratory distress syndrome. PLoS One 11: e0162490, 2016. doi: 10.1371/journal.pone.0162490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O’Connor BP, Danhorn T, De Arras L, Flatley BR, Marcus RA, Farias-Hesson E, Leach SM, Alper S. Regulation of toll-like receptor signaling by the SF3a mRNA splicing complex. PLoS Genet 11: e1004932, 2015. doi: 10.1371/journal.pgen.1004932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ohta S, Bahrun U, Tanaka M, Kimoto M. Identification of a novel isoform of MD-2 that downregulates lipopolysaccharide signaling. Biochem Biophys Res Commun 323: 1103–1108, 2004. doi: 10.1016/j.bbrc.2004.08.203. [DOI] [PubMed] [Google Scholar]
- 51.Ostuni R, Zanoni I, Granucci F. Deciphering the complexity of Toll-like receptor signaling. Cell Mol Life Sci 67: 4109–4134, 2010. doi: 10.1007/s00018-010-0464-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parsons PE, Eisner MD, Thompson BT, Matthay MA, Ancukiewicz M, Bernard GR, Wheeler AP; NHLBI Acute Respiratory Distress Syndrome Clinical Trials Network . Lower tidal volume ventilation and plasma cytokine markers of inflammation in patients with acute lung injury. Crit Care Med 33: 1–6, 2005. doi: 10.1097/01.CCM.0000149854.61192.DC. [DOI] [PubMed] [Google Scholar]
- 53.Ragaller M, Richter T. Acute lung injury and acute respiratory distress syndrome. J Emerg Trauma Shock 3: 43–51, 2010. doi: 10.4103/0974-2700.58663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rao N, Nguyen S, Ngo K, Fung-Leung WP. A novel splice variant of interleukin-1 receptor (IL-1R)-associated kinase 1 plays a negative regulatory role in Toll/IL-1R-induced inflammatory signaling. Mol Cell Biol 25: 6521–6532, 2005. doi: 10.1128/MCB.25.15.6521-6532.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rice TW, Wheeler AP, Thompson BT, deBoisblanc BP, Steingrub J, Rock P; NIH NHLBI Acute Respiratory Distress Syndrome Network of Investigators . Enteral omega-3 fatty acid, gamma-linolenic acid, and antioxidant supplementation in acute lung injury. JAMA 306: 1574–1581, 2011. doi: 10.1001/jama.2011.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med 353: 1685–1693, 2005. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 57.Salomao R, Brunialti MK, Gomes NE, Mendes ME, Diaz RS, Komninakis S, Machado FR, da Silva ID, Rigato O. Toll-like receptor pathway signaling is differently regulated in neutrophils and peripheral mononuclear cells of patients with sepsis, severe sepsis, and septic shock. Crit Care Med 37: 132–139, 2009. doi: 10.1097/CCM.0b013e318192fbaf. [DOI] [PubMed] [Google Scholar]
- 58.Salomão R, Martins PS, Brunialti MK, Fernandes ML, Martos LS, Mendes ME, Gomes NE, Rigato O. TLR signaling pathway in patients with sepsis. Shock 30, Suppl 1: 73–77, 2008. doi: 10.1097/SHK.0b013e318181af2a. [DOI] [PubMed] [Google Scholar]
- 59.Sazani P, Kole R. Therapeutic potential of antisense oligonucleotides as modulators of alternative splicing. J Clin Invest 112: 481–486, 2003. doi: 10.1172/JCI200319547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schütte H, Lohmeyer J, Rosseau S, Ziegler S, Siebert C, Kielisch H, Pralle H, Grimminger F, Morr H, Seeger W. Bronchoalveolar and systemic cytokine profiles in patients with ARDS, severe pneumonia and cardiogenic pulmonary oedema. Eur Respir J 9: 1858–1867, 1996. doi: 10.1183/09031936.96.09091858. [DOI] [PubMed] [Google Scholar]
- 61.Singh RK, Cooper TA. Pre-mRNA splicing in disease and therapeutics. Trends Mol Med 18: 472–482, 2012. doi: 10.1016/j.molmed.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Song Z, Yao C, Yin J, Tong C, Zhu D, Sun Z, Jiang J, Shao M, Zhang Y, Deng Z, Tao Z, Sun S, Bai C. Genetic variation in the TNF receptor-associated factor 6 gene is associated with susceptibility to sepsis-induced acute lung injury. J Transl Med 10: 166, 2012. doi: 10.1186/1479-5876-10-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sperry JL, Zolin S, Zuckerbraun BS, Vodovotz Y, Namas R, Neal MD, Ferrell RE, Rosengart MR, Peitzman AB, Billiar TR. X chromosome-linked IRAK-1 polymorphism is a strong predictor of multiple organ failure and mortality postinjury. Ann Surg 260: 698–703, 2014. doi: 10.1097/SLA.0000000000000918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Su J, Richter K, Zhang C, Gu Q, Li L. Differential regulation of interleukin-1 receptor associated kinase 1 (IRAK1) splice variants. Mol Immunol 44: 900–905, 2007. doi: 10.1016/j.molimm.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 65.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 140: 805–820, 2010. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 66.Terpstra ML, Aman J, van Nieuw Amerongen GP, Groeneveld AB. Plasma biomarkers for acute respiratory distress syndrome: a systematic review and meta-analysis. Crit Care Med 42: 691–700, 2014. doi: 10.1097/01.ccm.0000435669.60811.24. [DOI] [PubMed] [Google Scholar]
- 67.Tolle LB, Standiford TJ. Danger-associated molecular patterns (DAMPs) in acute lung injury. J Pathol 229: 145–156, 2013. doi: 10.1002/path.4124. [DOI] [PubMed] [Google Scholar]
- 68.Toubiana J, Courtine E, Pène F, Viallon V, Asfar P, Daubin C, Rousseau C, Chenot C, Ouaaz F, Grimaldi D, Cariou A, Chiche JD, Mira JP. IRAK1 functional genetic variant affects severity of septic shock. Crit Care Med 38: 2287–2294, 2010. doi: 10.1097/CCM.0b013e3181f9f9c7. [DOI] [PubMed] [Google Scholar]
- 69.Touznik A, Lee JJ, Yokota T. New developments in exon skipping and splice modulation therapies for neuromuscular diseases. Expert Opin Biol Ther 14: 809–819, 2014. doi: 10.1517/14712598.2014.896335. [DOI] [PubMed] [Google Scholar]
- 70.van Ommen GJ, Aartsma-Rus A. Advances in therapeutic RNA-targeting. N Biotechnol 30: 299–301, 2013. doi: 10.1016/j.nbt.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 71.Veltrop M, Aartsma-Rus A. Antisense-mediated exon skipping: taking advantage of a trick from Mother Nature to treat rare genetic diseases. Exp Cell Res 325: 50–55, 2014. doi: 10.1016/j.yexcr.2014.01.026. [DOI] [PubMed] [Google Scholar]
- 72.Vickers TA, Zhang H, Graham MJ, Lemonidis KM, Zhao C, Dean NM. Modification of MyD88 mRNA splicing and inhibition of IL-1beta signaling in cell culture and in mice with a 2′-O-methoxyethyl-modified oligonucleotide. J Immunol 176: 3652–3661, 2006. doi: 10.4049/jimmunol.176.6.3652. [DOI] [PubMed] [Google Scholar]
- 73.Wang CY, Calfee CS, Paul DW, Janz DR, May AK, Zhuo H, Bernard GR, Matthay MA, Ware LB, Kangelaris KN. One-year mortality and predictors of death among hospital survivors of acute respiratory distress syndrome. Intensive Care Med 40: 388–396, 2014. doi: 10.1007/s00134-013-3186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ware LB. Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Semin Respir Crit Care Med 27: 337–349, 2006. doi: 10.1055/s-2006-948288. [DOI] [PubMed] [Google Scholar]
- 75.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 342: 1334–1349, 2000. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 76.Wells CA, Chalk AM, Forrest A, Taylor D, Waddell N, Schroder K, Himes SR, Faulkner G, Lo S, Kasukawa T, Kawaji H, Kai C, Kawai J, Katayama S, Carninci P, Hayashizaki Y, Hume DA, Grimmond SM. Alternate transcription of the Toll-like receptor signaling cascade. Genome Biol 7: R10, 2006. doi: 10.1186/gb-2006-7-2-r10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wheeler AP, Bernard GR. Acute lung injury and the acute respiratory distress syndrome: a clinical review. Lancet 369: 1553–1564, 2007. doi: 10.1016/S0140-6736(07)60604-7. [DOI] [PubMed] [Google Scholar]
- 78.Xiang M, Fan J. Pattern recognition receptor-dependent mechanisms of acute lung injury. Mol Med 16: 69–82, 2010. doi: 10.2119/molmed.2009.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yılmaz-Eliş AS, Aartsma-Rus A, ’t Hoen PA, Safdar H, Breukel C, van Vlijmen BJ, van Deutekom J, de Kimpe S, van Ommen GJ, Verbeek JS. Inhibition of IL-1 signaling by antisense oligonucleotide-mediated exon skipping of IL-1 receptor accessory protein (IL-1RAcP). Mol Ther Nucleic Acids 2: e66, 2013. doi: 10.1038/mtna.2012.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yilmaz-Elis S, Aartsma-Rus A, Vroon A, van Deutekom J, de Kimpe S, ’t Hoen PA, van Ommen GJ, Verbeek JS. Antisense oligonucleotide mediated exon skipping as a potential strategy for the treatment of a variety of inflammatory diseases such as rheumatoid arthritis. Ann Rheum Dis 71, Suppl 2: i75–i77, 2012. doi: 10.1136/annrheumdis-2011-200971. [DOI] [PubMed] [Google Scholar]