

Abstract
Exposure to hypoxia induces migration and proliferation of pulmonary arterial smooth muscle cells (PASMCs), leading to vascular remodeling and contributing to the development of hypoxic pulmonary hypertension. The mechanisms controlling PASMC growth and motility are incompletely understood, although aquaporin 1 (AQP1) plays an important role. In tumor, kidney, and stem cells, AQP1 has been shown to interact with β-catenin, a dual function protein that activates the transcription of crucial target genes (i.e., c-Myc and cyclin D1) related to cell migration and proliferation. Thus the goal of this study was to examine mechanisms by which AQP1 mediates PASMC migration and proliferation, with a focus on β-catenin. Using primary rat PASMCs from resistance level pulmonary arteries infected with adenoviral constructs containing green fluorescent protein (control; AdGFP), wild-type AQP1 (AdAQP1), or AQP1 with the COOH-terminal tail deleted (AdAQP1M), we demonstrated that increasing AQP1 expression upregulated β-catenin protein levels and the expression (mRNA and protein) of the known β-catenin targets c-Myc and cyclin D1. In contrast, infection with AdAQP1M had no effect on any of these variables. Using silencing approaches to reduce β-catenin levels prevented both hypoxia- and AQP1-induced migration and proliferation of PASMCs, as well as induction of c-Myc and cyclin D1 by AQP1. Thus our results indicate that elevated AQP1 levels upregulate β-catenin protein levels, via a mechanism requiring the AQP1 COOH-terminal tail, enhancing expression of β-catenin targets and promoting PASMC proliferation and migration.
Keywords: c-Myc, growth, hypoxia, Lin-7
exposure to chronic hypoxia (CH) can have adverse effects on the pulmonary vasculature. In addition to inducing sustained contraction, CH also causes vascular remodeling of the arteries arising, in part, from proliferation and migration of pulmonary arterial smooth muscle cells (PASMCs) (34). Together, the increased muscularity of the vasculature and enhanced production of contractile agents lead to development of pulmonary hypertension (10, 33).
The mechanisms underlying pulmonary vascular remodeling due to CH remain incompletely understood, although recent work from our laboratory suggests a role for aquaporin 1 (AQP1) (14). AQP1 belongs to a family of water-specific, small molecular weight membrane-channel proteins expressed in diverse tissues and cell types including endothelial, epithelial, tumor, and smooth muscle cells (5, 23, 30, 32). We previously showed that AQP1 expression is enhanced by hypoxia in PASMCs and that AQP1 was required for hypoxia-induced PASMC migration and proliferation (17). These results were consistent with reports in other cell types suggesting that AQP1 plays a role in cell migration and proliferation (4, 9, 12, 20, 25, 31).
AQP1 is composed of six bilayer spanning domains that form the channel portion of the protein and amino and carboxyl termini. While AQP1 is most often recognized as a channel that mediates water transport across biological membranes, we found that the mechanism by which AQP1 mediates cell growth and movement involves the COOH-terminal tail but appears to be independent of water transport. The exact mechanism underlying AQP1 COOH-terminal-mediated cellular function, however, remains unclear.
A recent study found an interaction between AQP1 and β-catenin via the scaffolding protein Lin-7 (22). Other studies have found a direct interaction between β-catenin and AQP1 (20, 21, 37). β-Catenin, a regulator of cell-cell adhesion found in cadherin complexes, also functions as an important transcription factor, regulating the expression of c-Myc, a multifunctional factor that both drives the synthetic functions necessary for rapid cell division and inhibits expression of genes with antiproliferative functions, and cyclin D1, a major regulator of cell cycle progression. In melanoma and endothelial cells, Lin-7 forms a stabilizing complex between AQP1 and β-catenin (22). With respect to the lung, in both pulmonary endothelial cells (2) and PASMCs (27), activation of the bone morphogenetic protein (BMP) signaling pathway increases β-catenin activation and mediates BMP-dependent growth. β-Catenin was also upregulated and contributed to proliferation in PASMCs from patients with pulmonary arterial hypertension (36). Whether Lin-7 and β-catenin are involved in AQP1-mediated changes in PASMC function has not been determined.
Since our previous studies demonstrated an essential role for the COOH-terminal tail of AQP1 in mediating PASMC migration and proliferation, in this study, we further explored the mechanism by which AQP1 regulates PASMC function. We hypothesized that upregulation of AQP1 by forced expression or hypoxia would increase β-catenin levels via interactions with Lin-7. We further hypothesized that loss of β-catenin would prevent AQP1- and hypoxia-mediated changes in PASMC migration and proliferation.
METHODS
All protocols were performed in accordance with standards set by and reviewed and approved by the Johns Hopkins University Animal Care and Use Committee.
Isolation and culture of PASMCs.
Male Wistar rats (250–300 g) were anesthetized with pentobarbital sodium (130 mg/kg ip). The chest was opened and the heart and lungs removed and transferred to a petri dish on ice where they were dissected in HEPES-buffered saline solution (HBSS) containing the following (in mM): 130 NaCl, 5 KCl, 1.2 MgCl2, 1.5 CaCl2, 10 HEPES, and 10 glucose, with pH adjusted to 7.2 using 5 M NaOH. Distal pulmonary arteries (dPAs; >4th generation) were dissected and cleaned of adventitia, after which the vessels were opened and gently rubbed with a cotton-topped swab to remove the endothelial layer. The dPAs were then placed in 4°C HBSS for at least 30 min and subsequently placed in reduced-calcium HBSS (20 μm CaCl2) at room temperature for at least 20 min. The vessels were digested in reduced-calcium HBSS containing: type I collagenase (1,750 U/ml), papain (9.5 U/ml), bovine serum albumin (2 mg/ml), and DTT (1 mM) at 37°C for 15–20 min. After digestion, dPAs were transferred to a small round bottom tube and triturated in Ca2+-free HBSS to obtain a single cell suspension. PASMCs were placed in culture dishes containing basal SmBM media (Lonza) supplemented with 0.3% FCS and 1% penicillin-streptomycin for 48 h to eliminate any contaminating endothelial cells, followed by 3–4 days of culture in SmGM complete media (Lonza) supplemented with 1% penicillin-streptomycin to expand cell numbers. PASMCs were returned to SmBM media with 0.3% FCS for 24–48 h before beginning experiments. In vitro exposure to hypoxia was achieved by placing culture dishes in an airtight chamber (Billups-Rothberg) gassed with 4% O2 and 5% CO2. The chamber and the nonhypoxic controls were placed in an incubator at 37°C.
AQP1 constructs and infection.
Adenoviral constructs containing a hemagglutinin (HA)-tagged wild-type AQP1 (AdAQP1), HA-tagged AQP1 with the last 37 amino acids deleted (COOH-terminal deletion; AdAQP1CT) and green fluorescent protein (GFP) were created as described previously (14). PASMCs were infected with 50 infection units/cell of virus before being used in protein expression measurements and functional assays. Cells infected with the same adenovirus containing GFP (50 infection units/cell) were used as controls.
siRNA transfection.
siRNA targeting β-catenin and nontargeting siRNA (siNT, control) were obtained as a “smart pool” from Dharmacon. PASMCs were incubated with 25–100 nM of siRNA for 6 h in serum- and antibiotic-free media. Serum was then added to media to bring the total concentration to 0.3% FCS, and cells were incubated for 24 h. After 24 h, cells were changed to fresh 0.3% FCS media for an additional 24–72 h, during which cells were infected with adenoviral constructs or exposed to hypoxia, followed by expression analysis or functional assays.
Conventional and real-time RT-PCR.
Total RNA was extracted from PASMCs using the RNeasy Plus Mini kit (Qiagen). Reverse transcription was performed using 500 ng of RNA and the iScript cDNA synthesis kit (Bio-Rad). Conventional PCR was used for screening of expressed mRNAs as previously described (17). For comparing expression levels between conditions, real-time PCR was performed using 1,000 ng of cDNA with QuantiTect SYBR Green PCR Master Mix (Qiagen) in an iCycler IQ real-time PCR detection system (Bio-Rad). The specific primer pairs for real-time PCR are listed in Table 1. PCR products were confirmed by 1) a single peak in the melt curve, 2) a single band of the correct size when products were run on an agarose gel, and 3) sequencing of the product. Relative concentrations of each gene were calculated using the Pfaffl method, and data are expressed as a power ratio of the gene of interest to the housekeeping gene efficiency curves run on the same plate as samples. For each experiment, the values were normalized to the value of control sample.
Table 1.
Primers for PCR
| Gene | Accession Number | Species | Primer Pair Sequence | Product Size, bp |
|---|---|---|---|---|
| AQP1 | NM_012778 | Rat | 118 | |
| Sense | 5′-CCGAGACTTAGGTGGCTCAG-3′ | |||
| Antisense | 5′-TTGATCCCACAGCCAGTGTA-3′ | |||
| Lin-7 | NM_053514 | Rat | 82 | |
| Sense | 5′-GTGGAAGGGGAACATCACGA-3′ | |||
| Antisense | 5′-GGGTGTATCTGACGACCAGC-3′ | |||
| β-Catenin | NM_053357 | Rat | 75 | |
| Sense | 5′-ACCATCGAGAGGGCTTGTTG-3′ | |||
| Antisense | 5′-CTGGCGACCCAAGCATTTTC-3′ | |||
| c-Myc | NM_012603 | Rat | 139 | |
| Sense | 5′-TGAAAAGAGCTCCTCGCGTT-3′ | |||
| Antisense | 5′-AAATAGGGCTGCACCGAGTC-3′ | |||
| Cyclin D1 | NM_171992 | Rat | 94 | |
| Sense | 5′-TCAAGTGTGACCCGGACTGC-3′ | |||
| Antisense | 5′-GGATCGATGTTCTGCTGGGC-3′ | |||
| Glut1 | NM_138827 | Rat | 292 | |
| Sense | 5′-GCCTGAGACCAGTTGAAAGCAC-3′ | |||
| Antisense | 5′-CTGCTTAGGTAAAGTTACAGGAG-3′ | |||
| Cyclophilin B | NM_022536 | Rat | 93 | |
| Sense | 5′-GGACGAGTGACCTTTGGACT-3′ | |||
| Antisense | 5′-TGACACGATGGAACTTGCTG-3′ |
Western blot analysis.
PASMCs were washed with PBS, and total protein was extracted in ice-cold T-PER buffer containing protease inhibitors (Roche Diagnostics). Proteins were quantified by use of the BCA protein assay (Pierce), and 8–20 μg total protein were resolved by 10% SDS-PAGE gels transferred onto polyvinylidene difluoride membranes, which were then blocked with 5% nonfat dry milk in Tris-buffered saline containing 0.2% Tween 20. Membranes were probed with primary antibodies: Lin-7 (No. ab174297; Abcam); AQP1 at 1:7,000 (No. AQP11-A; Alpha Diagnostic); β-catenin at 1:4,000 (No. 610153; BD Biosciences); c-Myc at 1:1000 (No. ab32072; Abcam); cyclin D1 at 1:1,000 (No. 2978; Cell Signaling Technology); glycogen synthase kinase-3β (GSK3β; No. 12456; Cell Signaling Technology); β-tubulin (No. T7816; Sigma); and hemagglutinin (HA; No. sc-7392; Santa Cruz Biotechnology). Bound antibodies were probed with horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG (Kirkegaard & Perry Laboratories) and detected by enhanced chemiluminescence. Membranes were then stripped and reprobed for β-tubulin (1:10,000; Sigma) as a housekeeping protein. Protein levels were quantified by densitometry using ImageJ. Representative full-length blots for all antibodies are provided in Fig. 1. Antibodies directed against AQP1 and c-Myc were previously validated by use of siRNA (17) and shRNA (38), respectively. The antibodies for GSK3β and cyclin D1 have been verified by Cell Signaling Technology using tissue from GSK3β −/− mice and siRNA, respectively (see company website for details). Lin-7, HA, and β-catenin produced single bands at the correct molecular weights that were absent in negative control tissues or were reduced by specific siRNA (see results).
Fig. 1.

Representative whole length gels used for anti-body validation. Each image provides a representative full-length gel probed with a different primary antibody. For antibodies where validation was previously performed, only lanes containing lysates from pulmonary arterial smooth muscle cells (PASMCs) are shown. For antibodies where previous validation was not performed, lanes showing specificity of the identified band of interest are provided using a control tissue that does not express the protein. AQP1, aquaporin 1; HA, hemagglutinin.
Cell migration assay.
One-hundred thousand cells were added to the top of a polycarbonate Transwell insert with 8-μm pores in 4 ml of basal media inserted in six-well plates. The cells were exposed under control or hypoxic conditions for 24 h. After exposure, the cells were fixed in 95% ethanol for 10 min, stained with Brilliant Blue (Pierce) for 5 min, and visualized via a microscope-mounted camera with Q-capture software. For each filter, five fields were randomly chosen and imaged to obtain a total cell count. Unmigrated cells were then scraped off from the top of the filter and the bottom layer imaged as migrated cells. The migration rate was calculated as the percentage of cells remaining on the filter normalized to the total amount of cells.
Cell proliferation assay.
Five thousand cells were seeded in each well of a 96-well plate in basal SmBM. After incubation for 24–72 h under control or hypoxic conditions, 5-bromo-2-deoxyuridine (Amersham Biosciences) was added to the cells for 24 h. The cells were fixed after removing the culture media. Proliferation was estimated from detection of peroxidase-labeled anti-5-bromo-2-deoxyuridine incorporated into newly synthesized cells after the color had developed and was read at 450 nm in a microtiter plate spectrophotometer. All samples were repeated in triplicate and averaged. Proliferation rate is shown as absorbance values of treatments normalized to that of the control cells on the same plate.
Statistical analysis.
Data are expressed as means ± SE, where n refers to the sample size. Since all experimental runs were performed on cells from different animals, n also refers to the number of animals. All experimental groups contain a minimum of five biological replicates. Statistical comparisons were performed using one-way ANOVA with a Holm-Sidak post hoc test. Differences were considered to be significant when P < 0.05.
RESULTS
Effect of AQP1 on β-Catenin Expression in PASMCs.
We first tested whether forced expression of AQP1 was sufficient to enhanced β-catenin levels in PASMCs. To increase AQP1 expression in PASMCs, cells were infected with AdGFP (control) or AdAQP1 (Fig. 2A, top). Using immunoblot analysis, we found that PASMCs infected with AdAQP1 expressed increased β-catenin protein levels, which were 67% higher compared with cells infected with AdGFP (Fig. 2A, bottom). Interestingly, upregulation of β-catenin was restricted to protein levels, as no effect on β-catenin mRNA levels was evident in PASMCs with forced expression of AQP1 (Fig. 2B).
Fig. 2.
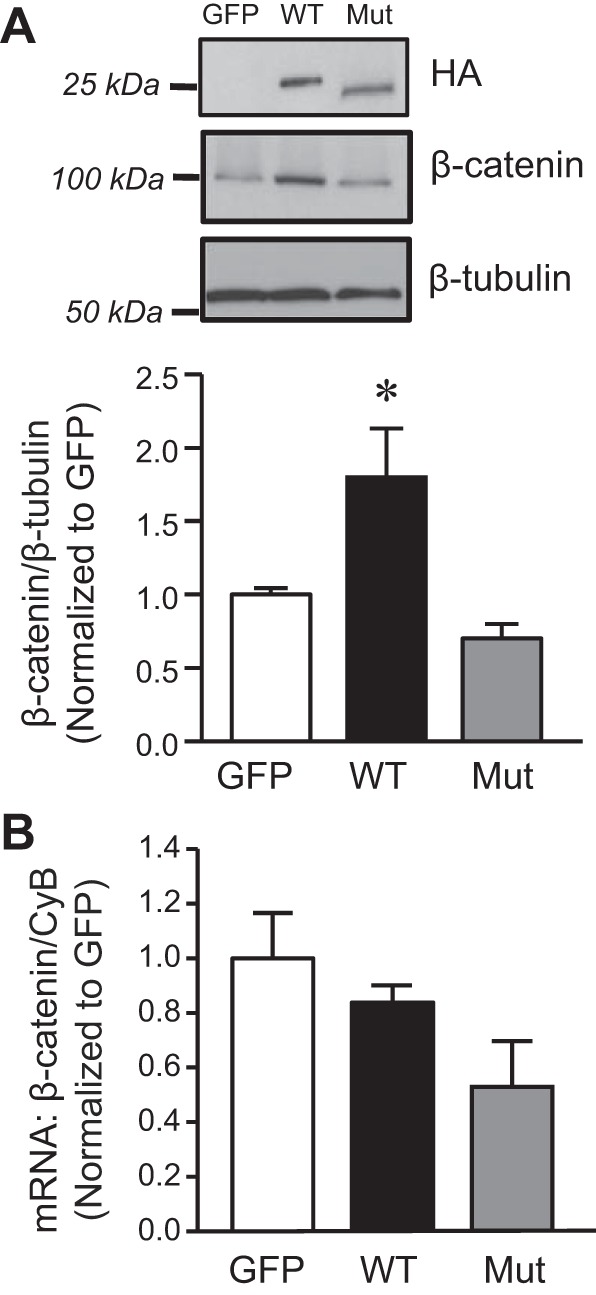
Effect of increased AQP1 expression on β-catenin levels in PASMCs. A, top: representative immunoblots showing the effect of infection with viruses containing constructs for green fluorescent protein (GFP; control), wild-type AQP1 (WT), or AQP1 with the COOH-terminal tail deleted (Mut) probed for the HA-tag (top) or β-catenin (middle) Infection with wild type (WT) and Mut resulted in similar levels of expressed AQP1. The mutant protein is visualized as a smaller MW band due to the loss of 37 amino acids (COOH-terminal tail). β-catenin protein levels were increased only in cells infected with WT AQP1. A, bottom: bar graph represents means ± SE values for the ratio of β-catenin protein to β-tubulin (normalized to GFP). B: bar graphs showing mean ± SE values for β-catenin mRNA normalized to cyclophilin B (CpB). Levels are expressed as fold increase from values measured in PASMCs infected with GFP. *Significant difference from GFP. n = 5 experiments per group.
To further explore potential mechanisms by which AQP1 might modulate β-catenin levels, we next investigated whether the COOH-terminal tail of AQP1 was involved by comparing β-catenin protein levels using protein isolated from PASMCs infected with AdAQP1M (Fig. 2B). Similar to our previous results (14), PASMCs infected with AdAQP1M exhibited similar levels of expressed AQP1 as cells infected with AdAQP1 (Fig. 2A) as measured using an antibody against the HA tag. However, β-catenin protein levels were much lower with AdAQP1M compared with cells infected with AdAQP1 and did not differ from those observed under control conditions (AdGFP infected cells). Compared with control conditions, β-catenin mRNA levels were reduced in cells infected with AdAQP1, although the level did not reach statistical significance (Fig. 2B). These data suggest that the COOH-terminal tail of AQP1 was required for stabilization of β-catenin protein.
Effect of AQP1 on the expression of β-catenin target genes in PASMCs.
After demonstrating that AQP1 upregulated β-catenin protein levels, we next confirmed that increasing AQP1 levels resulted in β-catenin activation as indicated by induction of β-catenin target genes. As shown in Fig. 3, cyclin D1 and c-Myc protein levels were quite low under control conditions. Cyclin D1 appeared as two bands on immunoblots, with a strong lower band and faint upper band. Since the antibody recognizes total protein, we hypothesize that the upper band is phosphorylated cyclin D1, which promotes protein expulsion from the nucleus and degradation (3). The mRNA (Fig. 3A) and protein (Fig. 3B) expression of both targets increased in PASMCs infected with AdAQP1 compared with cells infected with AdGFP. In contrast, no change in β-catenin target expression (mRNA or protein) was observed in PASMCs infected with AdAQP1M.
Fig. 3.
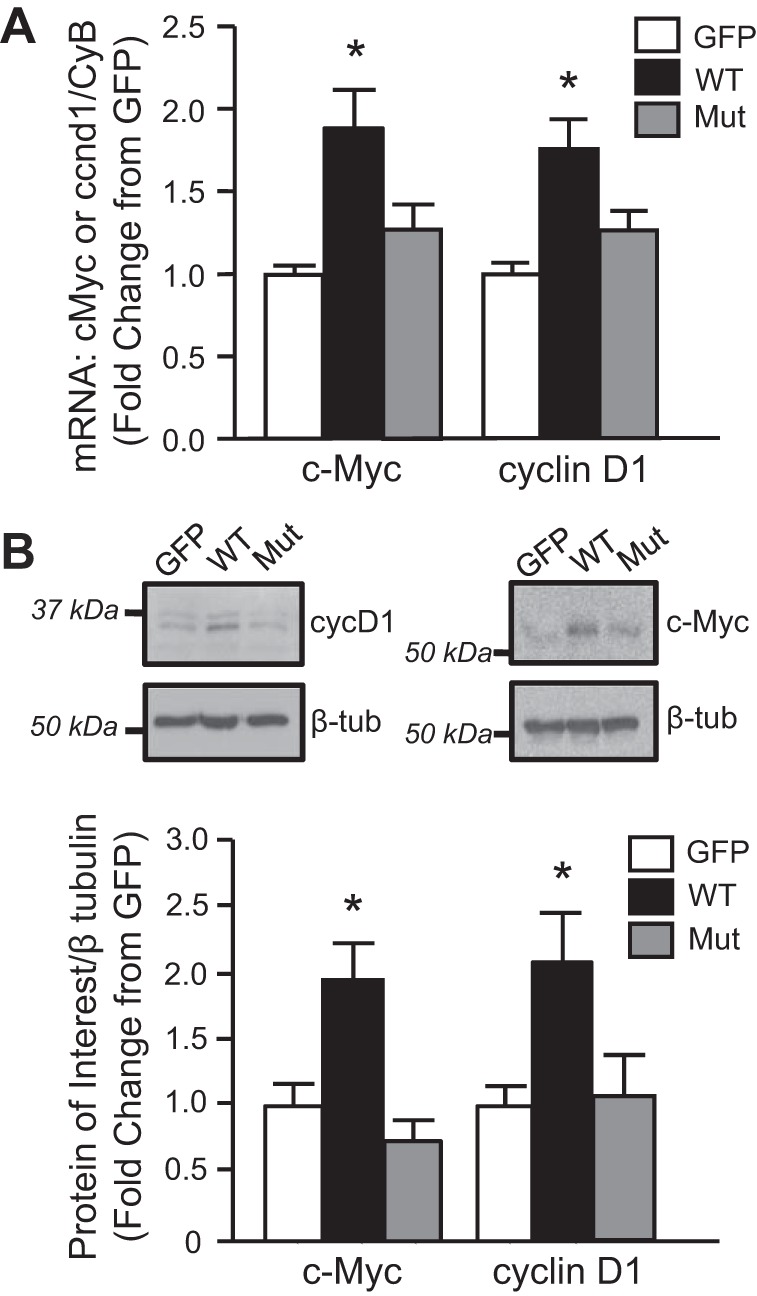
AQP1 increases the expression of β-catenin targets. A: bar graphs showing mean ± SE values for mRNA levels of the β-catenin targets, cyclin D1 and c-Myc, in PASMCs infected with viral constructs containing GFP (GFP; control), wild-type AQP1 (WT), or AQP1 with the COOH-terminal tail deleted (Mut); n = 11 for each group. B: representative immunoblot images show c-Myc and cyclin D1 protein levels. Bar graphs show means ± SE values for c-Myc and cyclin D1 protein normalized to β-tubulin. Data are expressed as fold change from GFP. *Significant difference from GFP value. n = 5–6 for each group.
Role of β-catenin in AQP1-mediated migration and proliferation of PASMCs.
To investigate whether the β-catenin cascade was involved in AQP1-mediated proliferation and migration, we performed loss-of-function experiments in which β-catenin was depleted. In PASMCs transfected with siRNA targeting β-catenin (siβ-catenin), we were able to achieve an efficient knockdown with an average of 84% reduction in β-catenin protein compared with cells transfected with a control non-targeting siRNA (siNT) (Fig. 4A). We then measured migration and proliferation in PASMCs treated with either siNT or siβ-catenin following infection with control construct (AdGFP) or AdAQP1. Migration was measured using a Transwell assay. Loss of β-catenin had no effect on total cell adherence to the Transwell filters (Fig. 4, B and C) or on basal migration (Fig. 4D) and proliferation (Fig. 4E) rates. In PASMCs transfected with siNT, infection with AdAQP1 significantly increased migration compared with cells infected with AdGFP (Fig. 4D), confirming our previous results (14). Depletion of β-catenin prevented the increase in migration induced by AdAQP1. With respect to proliferation, similar results were observed. Loss of β-catenin had no effect on basal proliferation rates but prevented the increase in proliferation observed in response to forced expression of AQP1 (Fig. 4E). To further explore the link between AQP1 and β-catenin, we examined the effect of AQP1 forced expression on β-catenin target genes when β-catenin was depleted (Fig. 4F). Loss of β-catenin had no effect on basal levels of c-Myc or cyclin D1, but prevented the AQP1-induced increased in the mRNA levels of both targets (Fig. 4F).
Fig. 4.
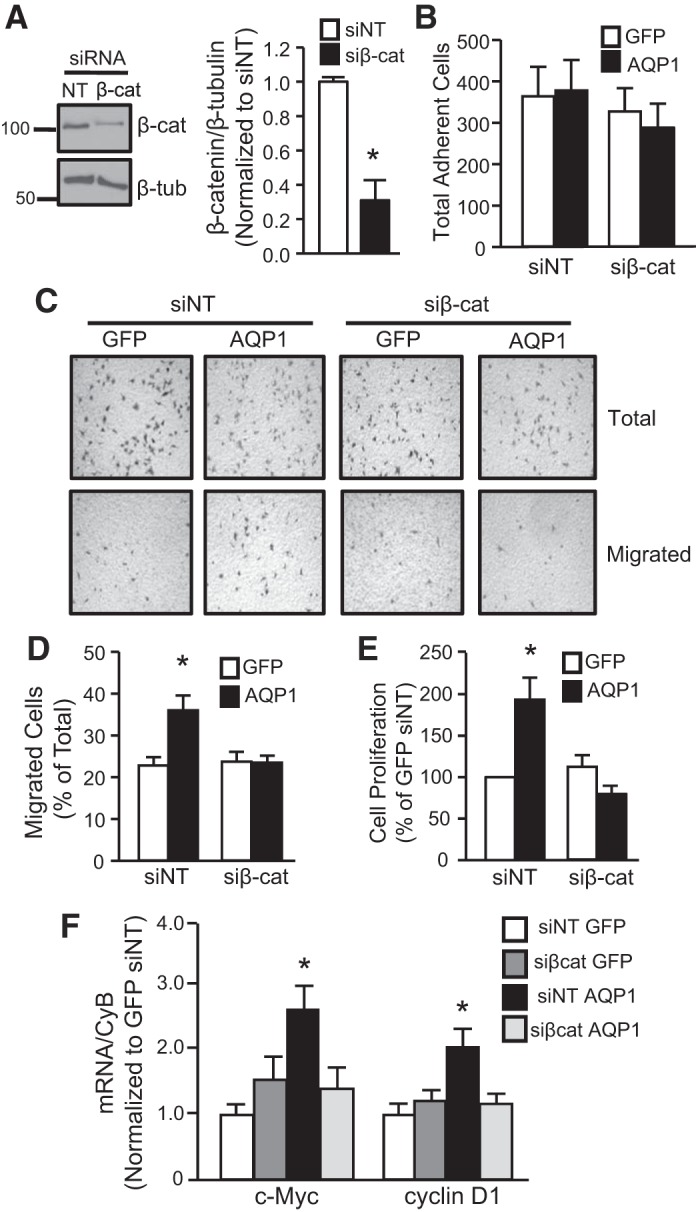
Role of β-catenin in AQP1-mediated PASMC proliferation and migration. A: representative immunoblot shows β-catenin and β-tubulin (housekeeping) protein levels in PASMCs transfected with siRNA targeting β-catenin (sißcat) or nontargeting siRNA (siNT). Bar graph shows fold change from siNT (means ± SE) values for β-catenin protein (normalized to β-tubulin). *Significant difference from siNT; n = 5 experiments. B: bar graph showing total cells adhering to the Transwell membrane in migration experiments (n = 5 each). C: representative images show cells on the Transwell membrane before (Total) and after (Migrated) removal of unmigrated cells. Cells were first transfected with control siRNA (siNT) or sißcat before infection with viral constructs containing GFP (control; GFP) or wild-type AQP1 (AQP1). D and E: bar graphs showing (D) percent migrated cells and proliferation (E; normalized to siNT GFP). *Significant difference from siNT GFP cells; n = 5 for each experimental group. F: bar graph showing effect on AQP1 on mRNA levels for β–catenin target genes (normalized to CyB) in cells transfected with siNT or sißcat. Data are expressed as fold change from siNT GFP; n = 7 for each group. *Significant difference from siNT GFP.
To determine the extent to which AQP1 was able to elicit β-catenin target expression, we also treated cells with CHIR-99021 (3 μM; 24 h; Selleckchem) an inhibitor of the serine-threonine kinase glycogen synthase kinase-3 (GSK3). The β-isoform GSK3β binds to and targets β-catenin for proteosomal degradation. When GSK3β was inhibited, β-catenin protein levels increased (Fig. 5A). Similarly, in these cells, protein levels of the β-catenin targets cyclin D1 and c-Myc were also increased (Fig. 5B). In all cases, the magnitude of increase was similar to what was observed in response to forced expression of AQP1. We also tested the effect of inhibiting GSK3β on cell function. Inhibiting GSK3β had no significant effect on cell adherence (Fig. 5C) but increased both migration (Fig. 5D) and proliferation (Fig. 5E) to a similar extent as AdAQP1.
Fig. 5.
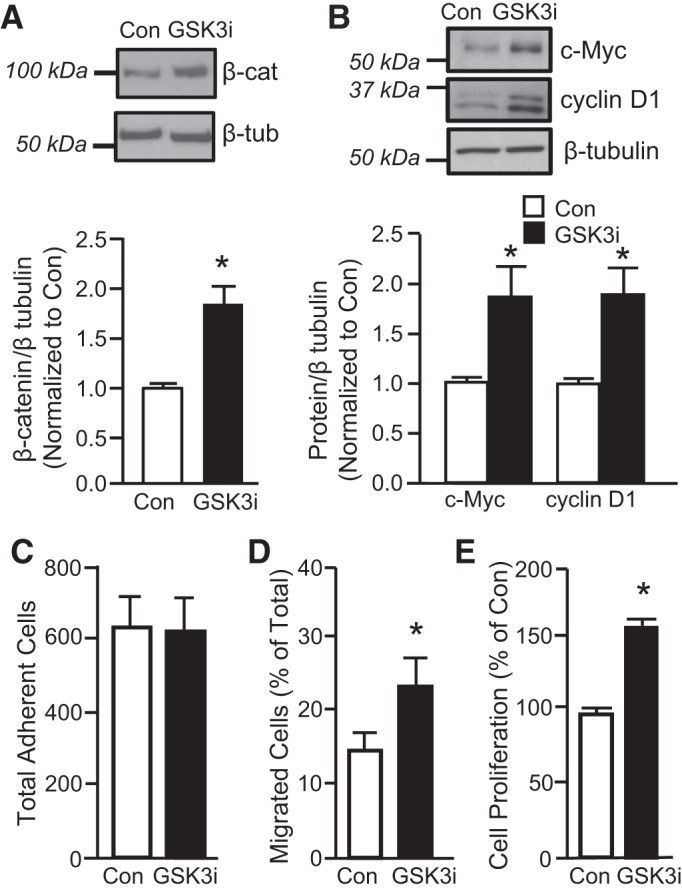
Effect of glycogen synthase kinase-3β (GSK3β) inhibition on β-catenin levels. A: representative immunoblots show β-catenin and β-tubulin (housekeeping) protein levels in PASMCs under control conditions (Con) or after treatment with the GSK3 inhibitor CHIR-99021 (GSK3i; 3 μM; 24 h). Bar graph shows means ± SE for the ratio of β-catenin protein to β-tubulin, expressed as fold change from Con; n = 5 per group. B: representative immunoblots showing c-Myc, cyclin D1 and β-tubulin protein levels under control conditions or after treatment with GSK3i. Bar graph shows means ± SE for the ratio of c-Myc or cyclin D1 protein to β-tubulin. Data are expressed as fold change values from Con. *Significant difference from Con value; n = 6 per group. C–E: effect of GSK3i on cell function. Inhibiting GSK3β had no effect on cell adherence (C) but enhanced migration (D; n = 6 per group) and proliferation (E; n = 5 per group). *Significant difference from Con.
Hypoxia-induced PASMC proliferation and migration requires β-catenin.
In previous experiments, we showed that hypoxia-induced migration and proliferation were dependent on AQP1. In the current study, we used forced expression of AQP1 to dissect the mechanisms by which induction of AQP1 might lead to changes in cell function; however, we also wanted to determine the role of β-catenin in mediating hypoxia-induced changes in PASMCs. We first confirmed that hypoxia (4% O2;5% CO2 for 24 or 48 h) increased AQP1 protein expression (Fig. 6A). Hypoxia also increased β-catenin protein levels in cells exposed to 4% O2 for 24 or 48 h; however, β-catenin mRNA levels were not changed by hypoxia (Fig. 6B), similar to results observed in cells where AQP1 expression was increased by forced expression. To confirm that the cells were indeed responsive to hypoxia, we measured the mRNA levels of GLUT1, a known hypoxia-responsive target, which increased significantly at both time points.
Fig. 6.
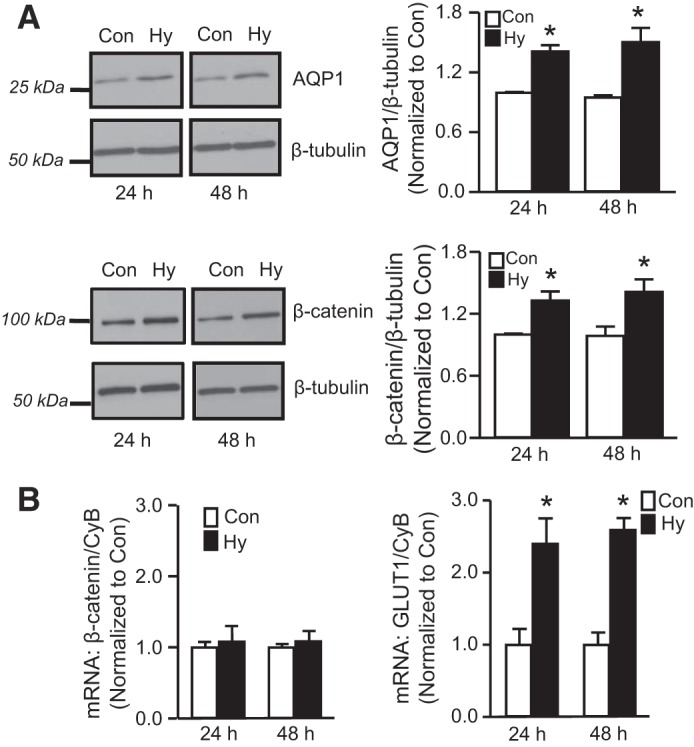
Effect of hypoxia on β-catenin expression. A: representative immunoblots demonstrating effect of hypoxia (Hy: 4% O2; 24 or 48 h) on protein levels of AQP1, β-catenin and β-tubulin in PASMCs. Bar graphs show means ± SE values for protein levels, expressed as fold change from control (Con). n = 6 per group. B: bar graphs show mean ± SE values for AQP1 and GLUT1 mRNA (normalized to cyclophilin B; CpB) in PASMCs maintained under control (Con) or hypoxic (Hy) conditions for 24 or 48 h. *Significant difference from Con value at the same time point. n = 5 experiments for each group.
We next depleted β-catenin before measuring migration and proliferation in PASMCs exposed to hypoxia. (Fig. 7, A–C). PASMCs were transfected with siNT or siβ-catenin, followed by exposure to 4% O2 for 24 h (migration) or 72 h (proliferation). In migration experiments, no significant difference in adherence was noted among the groups (Fig. 7A). In PASMCs transfected with siNT, hypoxia increased both migration (Fig. 7B) and proliferation (Fig. 7C). Depletion of β-catenin had no significant effect on PASMC migration or proliferation under control conditions but completely prevented a change in migration and proliferation with hypoxic exposure, confirming that β-catenin was necessary for hypoxia-induced proliferation and migration in PASMCs.
Fig. 7.
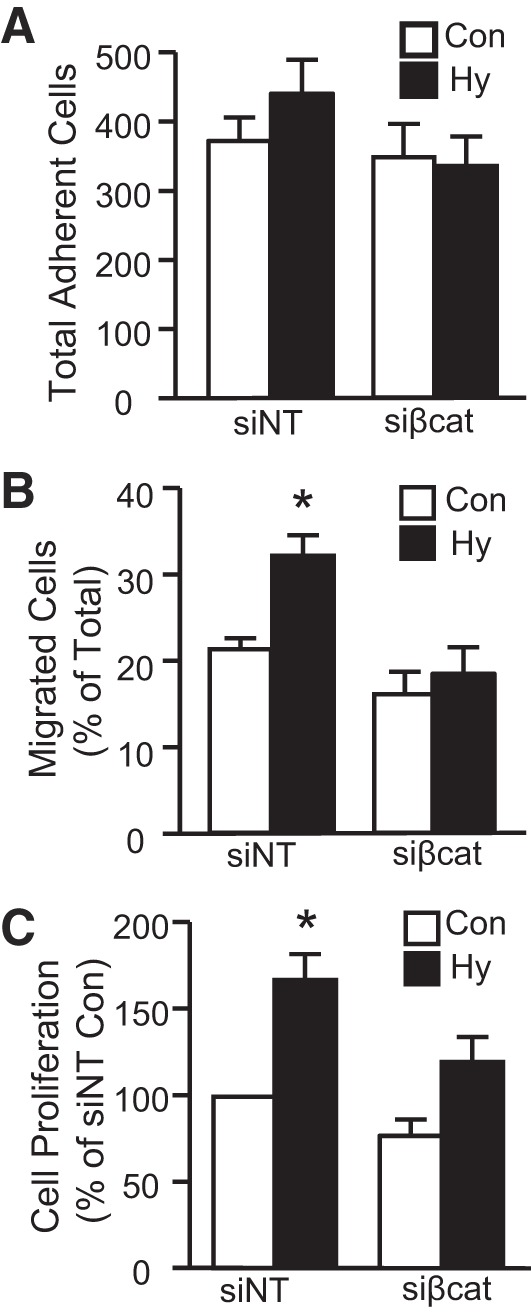
Role of β-catenin in hypoxia-induced PASMC migration and proliferation. Bar graphs show means ± SE values for total cell adherence (A), migration (B), and proliferation (C) of PASMCs treated with control (siNT) or β-catenin-targeting (sißcat) siRNA and then subjected to control (Con) or hypoxic (Hy; 4% O2 for 24 h) conditions. *Significant difference from Con siNT. n = 6–8 for each group.
Role of Lin-7 in AQP1-mediated regulation of β-catenin in PASMCs.
Since elevated AQP1 levels resulted in enhanced accumulation of β-catenin, we next wanted to determine the mechanism by which the increase in β-catenin occurred. In melanoma cells, AQP1 increased β-catenin levels via interactions with the scaffolding protein Lin-7 (22). To begin to determine whether Lin-7 also played a role in AQP1-mediated increases in β-catenin protein levels in PASMCs, we next evaluated Lin-7 mRNA and protein expression in PASMCs. As shown in Fig. 8A, agarose gel electrophoresis of PCR products showed that Lin-7 mRNA was expressed in brain and liver (positive controls), as well as PASMC samples. In contrast, immunoblot analysis revealed a lack of protein expression of Lin-7 in PASMCs, despite positive results in brain and liver (Fig. 8, B and C). Importantly, robust expression of Lin-7 protein could be observed using small amounts of total protein (6.25 µg) from brain lysates, yet increasing the amount of total protein loaded from PASMC lysates almost 10-fold (50 μg) failed to yield any appreciable bands for Lin-7 (Fig. 8B). Similar results were observed with isolates from different animals (Fig. 8C). To test whether Lin-7 expression might increase with stimulation, we also measured Lin-7 protein expression when AQP1 protein levels were increased by hypoxia or infection with AdAQP1. In all cases, Lin-7 protein was still absent on immunoblot (Fig. 8D), suggesting that Lin-7 protein is likely not expressed in PASMCs.
Fig. 8.

Lin-7 expression in rat pulmonary arterial smooth muscle cells (PASMCs). A: image showing PCR products for Lin-7A mRNA in PASMCs and control tissues, brain (Br) and liver (Liv). Each lane represents a sample from a different animal. B: representative immunoblot showing Lin-7 protein expression when increasing concentrations of total PASMC protein was loaded (6.25–50 µg) compared with brain (Br) lysate. C: representative immunoblot showing Lin-7 protein expression in PASMCs, Br and Liv and the corresponding β-tubulin (β-tub; housekeeping) for each sample. Each lane represents a sample from a different animal. For PASMCs, 50 µg of protein was loaded in each lane, compared with 6–10 µg for Br and Liv. D: immunoblot showing Lin-7 and β-tubulin protein expression in PASMCs under control conditions (C), exposed to hypoxia (Hy; 4% O2 for 24 h), or infected with viral constructs containing GFP (GFP), wild-type AQP1 (WT), or AQP1 lacking the COOH-terminal tail (Mut). Brain (Br) was used as a positive control. For PASMCs, 50 µg of total protein were loaded per lane, compared with 10 µg total protein for Br.
Since scaffolding with Lin-7 did not appear to be relevant for AQP1-induced β-catenin expression in our cells, we next explored whether AQP1 might directly bind β-catenin. Using co-immunoprecipitation, we determined whether AQP1 and β-catenin form a complex in PASMCs. In both control cells and those infected with AdAQP1, we could not reliably detect a specific band corresponding to AQP1 following immunoprecipitation for β-catenin nor could we reliably detect a band for β-catenin when lysates were immunoprecipitated for AQP1 (data not shown).
Given that GSK3β mediates degradation of β-catenin, we also determined whether increasing AQP1 levels might elevate β-catenin protein levels by reducing GSK3β expression. However, we found that neither AdAQP1 (Fig. 9A) nor hypoxia (Fig. 9B) altered GSK3β protein levels in PASMCs.
Fig. 9.
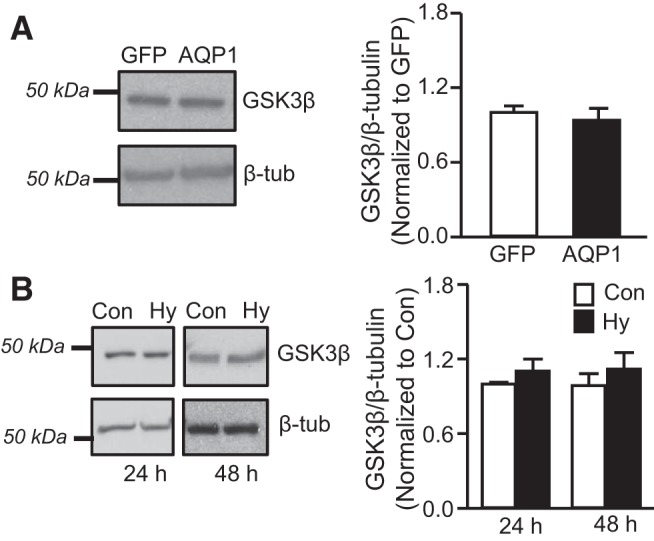
Effect of AQP1 and hypoxia on GSK3β expression. A: representative immunoblot showing GSK3β protein levels in PASMCs infected with AdGFP or AdAQP1. Bar graph shows mean ± SE values for the ratio of GSK3β to β-tubulin levels, expressed as fold change from GFP. n = 5 per group. B: Bar graphs show means ± SE values for the ratio of GSK3β to β-tubulin protein in PASMCs maintained under control (Con) or hypoxic (Hy; 4% O2) conditions for 24 or 48 h. Data are expressed as fold change from Con; n = 5 per group.
DISCUSSION
In this study, we demonstrated that AQP1, via the COOH-terminal tail, upregulates β-catenin protein levels and the expression of β-catenin target genes in PASMCs. Our study also revealed that β-catenin plays a critical role in both hypoxia- and AQP1-induced migration and proliferation of PASMCs.
Accumulating evidence supports that AQP1 is required for stimulating cell migration and participates in processes involving increased motility, including angiogenesis, tumor invasion, wound healing, and immune cell chemotaxis (9, 11, 25). One mechanism by which AQP1 is proposed to control cell migration is by actively moving to the leading edge of the cell and facilitating water transport through the cell plasma membrane to cause swelling and forward movement, or shrinkage and retraction (35, 39). However, our previous gain-of-function experiments performed in PASMCs demonstrated a clear role for the COOH-terminal tail of AQP1 in mediating both cell migration and proliferation (14). Since the water transport properties of AQP1 were not altered with deletion of the COOH-terminal tail (14), the effects of AQP1 on proliferation and migration appear independent of its role as a water channel.
A recent study in human melanoma and endothelial cells suggests that AQP1 interacts with a critical scaffold complex consisting of Lin-7 and β-catenin, and that this complex is important for cytoskeletal formation, adhesion and motility (22). β-Catenin is a dual function protein participating both in cell adhesion, as part of adherens junctions and focal adhesions, and in control of various prosurvival genes as a transcription factor (13). In PASMCs, β-catenin has been shown to be increased under hypoxic conditions or in response to growth factors and to modulate proliferative responses (24, 27, 36, 38). Moreover, activation and/or overexpression of β-catenin has been associated with development of pulmonary hypertension (16). As part of the Wnt signaling pathway, activated β-catenin translocates to the nucleus, increasing the expression of various transcriptional target activators, such as cyclin D1 and c-Myc, which are crucial in controlling cell proliferation and differentiation (1). Under basal conditions, β-catenin protein levels were low in our PASMCs and we could find little data to support activation at baseline. Indeed, there was limited expression of cyclin D1 or c-Myc in unstimulated cells, and depletion of β-catenin had no effect on migration or proliferation. However, conditions that increased AQP1 levels, including hypoxia and forced expression of AQP1, increased β-catenin protein levels, as well as the canonical β-catenin target genes cyclin D1 and c-Myc. These latter findings indicate that increasing AQP1 levels not only stabilizes the β-catenin protein but also appears to lead to activation as a transcription factor. The magnitude of increase in protein levels of both β-catenin and β-catenin targets was similar to that observed when β-catenin was maximally induced by inhibiting GSK3β, suggesting that the level of activation reached in response to increasing AQP1 was physiologically significant.
Although our data are consistent with activation of β-catenin as a transcription factor, we were unable to find evidence of β-catenin nuclear translocation in response to forced expression of AQP1, via either immunofluorescence or nuclear/cytosolic preparations, at the time points tested in this study (data not shown). Further experiments utilizing alternative methods and various time points, especially early time points, will be required to confirm that AQP1 induction leads to β-catenin transcriptional function.
That AQP1 might facilitate cell migration through β-catenin has been suggested in endothelial and cancer cells (21, 22). We confirmed a role for β-catenin in mediating both migration and proliferation in PASMCs when AQP1 levels were increased. Indeed, the effects of both forced expression of AQP1 and hypoxia were completely prevented when β-catenin was depleted, indicating that β-catenin was essential for the effects of AQP1. Our results are consistent with a previous report in human PASMCs, demonstrating that β-catenin was necessary for hypoxia-induced changes in PASMC total cell counts and metabolism (38). Importantly, the reductions in migration and proliferation observed when β-catenin levels were lowered were unlikely to be due to decreased cell adhesion, as similar numbers of attached cells were observed on membranes whether cells were treated with control siRNA or when β-catenin was depleted.
The mechanism by which AQP1 increased β-catenin levels is still unclear. Neither forced expression of AQP1 nor hypoxia increased β-catenin mRNA levels. That short-term in vitro exposure to hypoxia had no effect on β-catenin mRNA was not due to insufficient hypoxic exposure since mRNA levels of GLUT1, a canonical O2-sensitive target that is transcriptionally regulated by hypoxia (29), were significantly increased in the same cells. These data instead suggest that the effect of AQP1 must occur via posttranscriptional modifications. Interestingly, the effect of increasing AQP1 on β-catenin protein expression and β-catenin targets was lost when the COOH-terminal tail of AQP1 was deleted, perhaps explaining why the AQP1 COOH-terminal tail is necessary for promoting PASMC migration and proliferation.
In melanoma cells, AQP1 was found to stabilize β-catenin protein levels by forming a complex with the scaffolding protein Lin-7 preventing degradation. In mammals, the Lin-7 protein contains a PDZ domain that plays a role in organizing protein-protein interactions, including those that recruit cell adhesion molecules and regulate receptors and signaling proteins (26, 28). Depending on cell type, Lin-7 exists as three different isoforms in Homo sapiens (Lin-7A, Lin-7B, and Lin-7C), with Lin-7A the main isoform expressed in rat (22). Since the COOH-terminal tail of AQP1 contains potential regulatory and/or protein-protein interaction sites, including a putative motif for interaction with PDZ-binding proteins (7, 15), we speculated that an interaction between the COOH-terminal tail and Lin-7 might be the mechanism by which AQP1 increases β-catenin stability and activation. We found that PASMCs express mRNA for Lin-7A; however, to our surprise, immunoblot analysis failed to detect Lin-7 protein in PASMCs even when total protein loaded in the gels was increased 10-fold compared with tissues that express Lin-7. We consider it unlikely that our antibody or experimental conditions were faulty, as protein was readily detected in brain and liver samples, and the antibody cross-reacts with all isoforms of Lin-7. We initially speculated that perhaps Lin-7 expression was below the level of detection at baseline but could be increased under conditions where AQP1 levels were enhanced. However, this did not appear to be the case, as we also failed to detect Lin-7 protein in cells when AQP1 protein was increased by exposure to hypoxia or with forced expression using our adenoviral constructs. Our results are consistent with a dearth of published literature demonstrating Lin-7 expression in vascular smooth muscle. Thus, while Lin-7 plays a critical role in AQP1-mediated stabilization of β-catenin protein in melanoma cells (8), the fact that Lin-7 protein levels were either not expressed or were below the level of detection in PASMCs suggests that this may be a cell-type specific mechanism, although we cannot definitively rule out a role for Lin-7 in our cells at this time.
Another possibility to explain the role of the COOH terminus would be a direct interaction between AQP1 and β-catenin. In melanoma cells, binding of AQP1 and β-catenin was not observed (22). This is in contrast to results in human induced pluripotent stem cells (20), rat mesenchymal stem cells (21), and Madin-Darby canine kidney cells (37), where immunoprecipitation experiments indicated that AQP1 and β-catenin bind directly. However, in the latter study, binding of AQP1 to β-catenin was found to reduce total β-catenin protein levels. The reason for these differences are unclear but may reflect variability in cell type, culture conditions, or species. In preliminary experiments using coimmunoprecipitation, we were unable to confirm binding of AQP1 and β-catenin. Since experimental conditions can markedly alter the ability to detect protein-protein interactions, we cannot for certain rule out direct binding of these proteins as a mechanism of AQP1-induced β-catenin accumulation in our cells. Future experiments will be required to confirm or refute this possibility.
Thus the exact mechanism by which AQP1 increased β-catenin protein expression in PASMCs remains to be elucidated. β-Catenin is phosphorylated by GSK3β and requires initial prior phosphorylation by casein kinase 1 (19). Interaction with GSK3β targets β-catenin to ubiquitin E3 β-TrCP (18), resulting in ubiquitination and degradation. Additional posttranslational modifications, such as acetylation on Lys-19 and Lys-49, can also inhibit β-catenin degradation and enhance nuclear translocation, improving protein stability (6). In bone marrow mesenchymal stem cells, β-catenin interacts in a complex with AQP1 and FAK (21), possibly implying that this physical interaction could promote β-catenin stability and that there may be structural or adaptor proteins that interact with and increase β-catenin protein levels. Whether any of these mechanisms are involved in the AQP1-mediated increased in β-catenin protein stability in PASMCs will be the focus of future studies.
In summary, using gain- and loss-of-function approaches, we demonstrated that AQP1 mediates PASMC migration and proliferation through a mechanism involving upregulation of β-catenin protein that does not appear to require the scaffolding protein Lin-7. The precise relationship between AQP1 and β-catenin still requires further study; however, the AQP1-induced upregulation of β-catenin protein levels was posttranscriptional and required the AQP1 COOH-terminal tail. Together, our findings suggest the possibility that AQP1 may contribute to pulmonary vascular remodeling during development of hypoxic pulmonary hypertension through modulation of β-catenin signaling, although future in vivo experiments will be needed to confirm this possibility.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-073859 and HL-126514 and American Heart Association Mid-Atlantic Affiliate Grant-in-Aid AHA0755479. X. Yun was supported by funds from the Overseas Study Project of Guangzhou Elite Project.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
X.Y., N.L., and L.A.S. conceived and designed experiments; X.Y., H.J., and N.L. performed experiments; X.Y. and L.A.S. analyzed data; X.Y., J.W., and L.A.S. interpreted results of experiments; X.Y. and L.A.S. prepared figures; X.Y. drafted manuscript; X.Y., H.J., and L.A.S. edited and revised manuscript; X.Y., H.J., N.L., J.W., and L.A.S. approved final version of manuscript.
REFERENCES
- 1.Behrens J. Control of β-catenin signaling in tumor development. Ann N Y Acad Sci 910: 21–33, 2000. doi: 10.1111/j.1749-6632.2000.tb06698.x. [DOI] [PubMed] [Google Scholar]
- 2.de Jesus Perez VA, Alastalo TP, Wu JC, Axelrod JD, Cooke JP, Amieva M, Rabinovitch M. Bone morphogenetic protein 2 induces pulmonary angiogenesis via Wnt-β-catenin and Wnt-RhoA-Rac1 pathways. J Cell Biol 184: 83–99, 2009. doi: 10.1083/jcb.200806049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev 11: 957–972, 1997. doi: 10.1101/gad.11.8.957. [DOI] [PubMed] [Google Scholar]
- 4.Galán-Cobo A, Ramírez-Lorca R, Toledo-Aral JJ, Echevarría M. Aquaporin-1 plays important role in proliferation by affecting cell cycle progression. J Cell Physiol 231: 243–256, 2016. doi: 10.1002/jcp.25078. [DOI] [PubMed] [Google Scholar]
- 5.Galán-Cobo A, Sánchez-Silva R, Serna A, Abreu-Rodríguez I, Muñoz-Cabello AM, Echevarría M. Cellular overexpression of aquaporins slows down the natural HIF-2α degradation during prolonged hypoxia. Gene 522: 18–26, 2013. doi: 10.1016/j.gene.2013.03.075. [DOI] [PubMed] [Google Scholar]
- 6.Ge X, Jin Q, Zhang F, Yan T, Zhai Q. PCAF acetylates β-catenin and improves its stability. Mol Biol Cell 20: 419–427, 2009. doi: 10.1091/mbc.E08-08-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gonen T, Walz T. The structure of aquaporins. Q Rev Biophys 39: 361–396, 2006. doi: 10.1017/S0033583506004458. [DOI] [PubMed] [Google Scholar]
- 8.Harris BZ, Venkatasubrahmanyam S, Lim WA. Coordinated folding and association of the LIN-2, -7 (L27) domain. An obligate heterodimerization involved in assembly of signaling and cell polarity complexes. J Biol Chem 277: 34902–34908, 2002. doi: 10.1074/jbc.M205856200. [DOI] [PubMed] [Google Scholar]
- 9.Hayashi S, Takahashi N, Kurata N, Yamaguchi A, Matsui H, Kato S, Takeuchi K. Involvement of aquaporin-1 in gastric epithelial cell migration during wound repair. Biochem Biophys Res Commun 386: 483–487, 2009. doi: 10.1016/j.bbrc.2009.06.067. [DOI] [PubMed] [Google Scholar]
- 10.Hoeper MM, Humbert M, Souza R, Idrees M, Kawut SM, Sliwa-Hahnle K, Jing ZC, Gibbs JS. A global view of pulmonary hypertension. Lancet Respir Med 4: 306–322, 2016. doi: 10.1016/S2213-2600(15)00543-3. [DOI] [PubMed] [Google Scholar]
- 11.Hoque MO, Soria JC, Woo J, Lee T, Lee J, Jang SJ, Upadhyay S, Trink B, Monitto C, Desmaze C, Mao L, Sidransky D, Moon C. Aquaporin 1 is overexpressed in lung cancer and stimulates NIH-3T3 cell proliferation and anchorage-independent growth. Am J Pathol 168: 1345–1353, 2006. doi: 10.2353/ajpath.2006.050596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jagirdar RM, Apostolidou E, Molyvdas PA, Gourgoulianis KI, Hatzoglou C, Zarogiannis SG. Influence of AQP1 on cell adhesion, migration, and tumor sphere formation in malignant pleural mesothelioma is substratum- and histological-type dependent. Am J Physiol Lung Cell Mol Physiol 310: L489–L495, 2016. doi: 10.1152/ajplung.00410.2015. [DOI] [PubMed] [Google Scholar]
- 13.Königshoff M, Eickelberg O. WNT signaling in lung disease: a failure or a regeneration signal? Am J Respir Cell Mol Biol 42: 21–31, 2010. doi: 10.1165/rcmb.2008-0485TR. [DOI] [PubMed] [Google Scholar]
- 14.Lai N, Lade J, Leggett K, Yun X, Baksh S, Chau E, Crow MT, Sidhaye V, Wang J, Shimoda LA. The aquaporin 1 C-terminal tail is required for migration and growth of pulmonary arterial myocytes. Am J Respir Cell Mol Biol 50: 1010–1020, 2014. doi: 10.1165/rcmb.2013-0374OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laski ME. Structure-function relationships in aquaporins. Semin Nephrol 26: 189–199, 2006. doi: 10.1016/j.semnephrol.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 16.Laumanns IP, Fink L, Wilhelm J, Wolff JC, Mitnacht-Kraus R, Graef-Hoechst S, Stein MM, Bohle RM, Klepetko W, Hoda MA, Schermuly RT, Grimminger F, Seeger W, Voswinckel R. The noncanonical WNT pathway is operative in idiopathic pulmonary arterial hypertension. Am J Respir Cell Mol Biol 40: 683–691, 2009. doi: 10.1165/rcmb.2008-0153OC. [DOI] [PubMed] [Google Scholar]
- 17.Leggett K, Maylor J, Undem C, Lai N, Lu W, Schweitzer K, King LS, Myers AC, Sylvester JT, Sidhaye V, Shimoda LA. Hypoxia-induced migration in pulmonary arterial smooth muscle cells requires calcium-dependent upregulation of aquaporin 1. Am J Physiol Lung Cell Mol Physiol 303: L343–L353, 2012. doi: 10.1152/ajplung.00130.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu C, Kato Y, Zhang Z, Do VM, Yankner BA, He X. β-Trcp couples β-catenin phosphorylation-degradation and regulates Xenopus axis formation. Proc Natl Acad Sci USA 96: 6273–6278, 1999. doi: 10.1073/pnas.96.11.6273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. Control of β-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108: 837–847, 2002. doi: 10.1016/S0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- 20.Madonna R, Geng YJ, Shelat H, Ferdinandy P, De Caterina R. High glucose-induced hyperosmolarity impacts proliferation, cytoskeleton remodeling and migration of human induced pluripotent stem cells via aquaporin-1. Biochim Biophys Acta 1842: 2266–2275, 2014. doi: 10.1016/j.bbadis.2014.07.030. [DOI] [PubMed] [Google Scholar]
- 21.Meng F, Rui Y, Xu L, Wan C, Jiang X, Li G. Aqp1 enhances migration of bone marrow mesenchymal stem cells through regulation of FAK and β-catenin. Stem Cells Dev 23: 66–75, 2014. doi: 10.1089/scd.2013.0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monzani E, Bazzotti R, Perego C, La Porta CA. AQP1 is not only a water channel: it contributes to cell migration through Lin7/β-catenin. PLoS One 4: e6167, 2009. doi: 10.1371/journal.pone.0006167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mun GI, Jang SI, Boo YC. Laminar shear stress induces the expression of aquaporin 1 in endothelial cells involved in wound healing. Biochem Biophys Res Commun 430: 554–559, 2013. doi: 10.1016/j.bbrc.2012.11.114. [DOI] [PubMed] [Google Scholar]
- 24.Nie X, Qin G, Mao W, Wang W, Chang Y, Wei D, Zhou M, Wu B, Chen J. Axis inhibition protein 2 deficiency leads to hypoxic pulmonary hypertension through β-catenin signaling pathway. J Hypertens 34: 877–892, 2016. doi: 10.1097/HJH.0000000000000872. [DOI] [PubMed] [Google Scholar]
- 25.Papadopoulos MC, Saadoun S, Verkman AS. Aquaporins and cell migration. Pflugers Arch 456: 693–700, 2008. doi: 10.1007/s00424-007-0357-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perego C, Vanoni C, Massari S, Raimondi A, Pola S, Cattaneo MG, Francolini M, Vicentini LM, Pietrini G. Invasive behaviour of glioblastoma cell lines is associated with altered organisation of the cadherin-catenin adhesion system. J Cell Sci 115: 3331–3340, 2002. [DOI] [PubMed] [Google Scholar]
- 27.Perez VA, Ali Z, Alastalo TP, Ikeno F, Sawada H, Lai YJ, Kleisli T, Spiekerkoetter E, Qu X, Rubinos LH, Ashley E, Amieva M, Dedhar S, Rabinovitch M. BMP promotes motility and represses growth of smooth muscle cells by activation of tandem Wnt pathways. J Cell Biol 192: 171–188, 2011. doi: 10.1083/jcb.201008060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petrosky KY, Ou HD, Löhr F, Dötsch V, Lim WA. A general model for preferential hetero-oligomerization of LIN-2/7 domains: mechanism underlying directed assembly of supramolecular signaling complexes. J Biol Chem 280: 38528–38536, 2005. doi: 10.1074/jbc.M506536200. [DOI] [PubMed] [Google Scholar]
- 29.Pisarcik S, Maylor J, Lu W, Yun X, Undem C, Sylvester JT, Semenza GL, Shimoda LA. Activation of hypoxia-inducible factor-1 in pulmonary arterial smooth muscle cells by endothelin-1. Am J Physiol Lung Cell Mol Physiol 304: L549–L561, 2013. doi: 10.1152/ajplung.00081.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ribatti D, Ranieri G, Annese T, Nico B. Aquaporins in cancer. Biochim Biophys Acta 1550–1553: 1840, 2014. doi: 10.1016/j.bbagen.2013.09.025. [DOI] [PubMed] [Google Scholar]
- 31.Schwab A, Fabian A, Hanley PJ, Stock C. Role of ion channels and transporters in cell migration. Physiol Rev 92: 1865–1913, 2012. doi: 10.1152/physrev.00018.2011. [DOI] [PubMed] [Google Scholar]
- 32.Shankardas J, Patil RV, Vishwanatha JK. Effect of down-regulation of aquaporins in human corneal endothelial and epithelial cell lines. Mol Vis 16: 1538–1548, 2010. [PMC free article] [PubMed] [Google Scholar]
- 33.Shimoda LA, Laurie SS. Vascular remodeling in pulmonary hypertension. J Mol Med (Berl) 91: 297–309, 2013. doi: 10.1007/s00109-013-0998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res 99: 675–691, 2006. doi: 10.1161/01.RES.0000243584.45145.3f. [DOI] [PubMed] [Google Scholar]
- 35.Stroka KM, Jiang H, Chen SH, Tong Z, Wirtz D, Sun SX, Konstantopoulos K. Water permeation drives tumor cell migration in confined microenvironments. Cell 157: 611–623, 2014. doi: 10.1016/j.cell.2014.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takahashi J, Orcholski M, Yuan K, de Jesus Perez V. PDGF-dependent β-catenin activation is associated with abnormal pulmonary artery smooth muscle cell proliferation in pulmonary arterial hypertension. FEBS Lett 590: 101–109, 2016. doi: 10.1002/1873-3468.12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang W, Li F, Sun Y, Lei L, Zhou H, Lei T, Xia Y, Verkman AS, Yang B. Aquaporin-1 retards renal cyst development in polycystic kidney disease by inhibition of Wnt signaling. FASEB J 29: 1551–1563, 2015. doi: 10.1096/fj.14-260828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu XM, Wang L, Li JF, Liu J, Li J, Wang W, Wang J, Wang C. Wnt5a inhibits hypoxia-induced pulmonary arterial smooth muscle cell proliferation by downregulation of β-catenin. Am J Physiol Lung Cell Mol Physiol 304: L103–L111, 2013. doi: 10.1152/ajplung.00070.2012. [DOI] [PubMed] [Google Scholar]
- 39.Zhang H, Verkman AS. Aquaporin-1 water permeability as a novel determinant of axonal regeneration in dorsal root ganglion neurons. Exp Neurol 265: 152–159, 2015. doi: 10.1016/j.expneurol.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]