

Abstract
The development of a new decarboxylative cross-coupling protocol that affords terminal and substituted alkynes from various carboxylic acids is described using both nickel- and iron-based catalysts. The use of N-hydroxytetrachlorophthalimide (TCNHPI) esters is crucial to the success of the transformation, and the reaction is amenable to in situ carboxylic acid activation. Additionally, an inexpensive, commercially-available alkyne source is employed in this formal homologation process that serves as a surrogate for other well-established alkyne syntheses. The reaction can be conducted on a mole scale, and the conditions are mild, operationally simple, and broad in scope while providing succinct avenues to previously reported synthetic intermediates.
Keywords: nickel catalysis, iron catalysis, redox-active esters, alkynylation, homologation
Graphical abstract
All kinds of Alkynes: A convenient method for the decarboxylative alkynylation of redox active esters has been developed. The reaction is broad, functional group tolerant, and provides access to alkynes that are both terminal and substituted using either Ni or Fe catalysis. The method is used to swiftly synthesize a variety of alkynyl intermediates that were previously difficult to access using conventional alkynylation strategies.
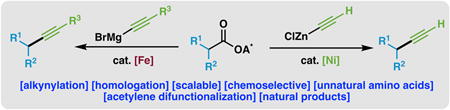
Alkynes are amongst the most versatile functionalities in organic chemistry.1 In addition to an array of utility in the fossil fuel industry,2a materials science,2b chemical biology,2c and drug discovery,2d alkynes have a privileged role in organic synthesis, serving as both nucleophiles and electrophiles. The relatively limited range of methods available for their construction is therefore puzzling. This has been particularly true of terminal alkynes, which often have to be accessed from aldehyde synthetic precursors.3 Homologation reactions of this variety have largely been limited to classic reactions including the Corey-Fuchs alkynylation,4 the Seyferth-Gilbert homologation, and their respective modifications (e.g. Bestmann-Ohira protocol).5 While these methods have been widely adopted, they have considerable limitations, including the use of strongly basic conditions and/or costly reagents that are not easily commutable to complex systems or to those where an economically prudent production process is needed. Additionally, it is commonplace that one or more concession steps are required to access the aldehyde substrates employed in these procedures.3 In this Communication, a simple protocol for alkynylation is disclosed wherein native carboxylic acids6 are swiftly transformed into both substituted and terminal alkynes in a practical, straightforward fashion.
Recently, research efforts in this laboratory have explored the utility of carboxylic acids and their redox-active ester (RAE) derivatives as electrophiles in a variety of cross-coupling protocols including arylation,7a,c,d,f alkylation,7b,e decarboxylation,7e borylation,7g and alkenylation.7h The success of these decarboxylative processes prompted the investigation of alkynyl nucleophiles as suitable cross-coupling partners towards forging sp–sp3 C–C linkages. Inspiration to achieve such a reaction was motivated by several useful alkynes that are extremely laborious to access. An illustrative example is depicted in Figure 1A, wherein an alkynyl amino acid derivative (3) was accessed from a parent amino acid (1) in 4 steps.8 This type of unnatural amino acid has proven to be of critical value to practitioners in chemical biology and drug discovery.2d In contrast, a mild decarboxylative alkynylation strategy could dramatically simplify the pathway to 3 because the native carboxylate of a glutamic acid (2) could be viewed as a progenitor of the alkyne unit without redox-manipulations. Whereas other decarboxylative alkynylation strategies require the use of engineered alkynyl coupling partners (e.g. sulfonyl, iodine (III) reagents),9a–k strong oxidants,9a or photo-induced electron transfer (PET) reagents,9g–k this design would provide convenient access to terminal alkynes with little to no need for subsequent manipulation or removal of protecting groups (Figure 1C).
Figure 1.

(A) Synthesis of an unnatural alkynyl amino acid derivative. B) Traditional synthetic routes from carboxylic acids to terminal alkynes and design of a new terminal alkyne cross-coupling synthetic strategy. (C) Optimization of reaction conditions. TCNHPI = N-hydroxytetrachloro-phthalimide, DIC = N,N′-diisopropylcarbodiimide.
Our investigation of the title transformation commenced with an evaluation of various metal salts, ligands, solvents, and activated esters. After considerable optimization (see Supporting Information), it was found that the piperidine derived TCNHPI ester 4 smoothly underwent the desired decarboxylative alkynylation forming 6 in 82% isolated yield10 (Figure 1C) utilizing an alkynyl zinc reagent (5) derived from ethynylmagnesium bromide and ZnCl2. The optimal catalyst was derived from a 1:1 ratio of NiCl2•6H2O and L1. Other ligands gave inferior yields, and changing the solvent from DMF to other polar solvents had a deleterious effect on reaction efficiency (entries 5-7). Additionally, control experiments without catalyst or ligand resulted in little product formation (Entry 9). In situ activation of the carboxylic acid with DIC and TCNHPI resulted in only a slightly reduced yield (73%), allowing for the transformation to be simplified into a one-pot process (Figure 1C, entry 1). Simple iron salts such as FeBr2•H2O (entry 8) with Grignard reagents also delivered workable yields, a fortuitous finding as this catalytic system provided superior yields for the cross-coupling of substituted alkynes (vide infra).
With the optimized conditions in hand for terminal alkyne synthesis, the scope of the alkynylation reaction was evaluated (Scheme 1). Under the standard conditions, ethynylzinc chloride (5) was smoothly coupled with a variety of secondary and primary RAE substrates (9). Simple cyclic (6, 12, 13, 20) and acyclic (14–15) alkynes were easily generated, and amino acid substrates with sensitive functionality were also easily converted to their requisite alkynes (16–17). Alkynes with α-heteroatom substitution were also products of homologation, albeit with lower yield (18–19). Furthermore, heterocyclic moieties were tolerated under the mild reaction conditions, with the 3-pyridyl product (15) formed in good yield following an in situ activation protocol. Complex steroidal derivatives (23–24) proved to be exceptional coupling products with notably sensitive acetate and ketone functionalities left unperturbed in the coupling. Perhaps most impressively, the tetrapeptide 25 was successfully synthesized with this methodology, demonstrating the potential application of this reaction to late-stage diversification of peptidyl side-chain carboxylic acids. The synthesis of alkynes with benzylic substitution (10%, 42), proline (0%, 43) and tertiary substitution (0%, 44) represent current limitations of this coupling.
Scheme 1.

Scope of the Ni and Fe-catalyzed decarboxylative alkynylation with TCNHPI redox-active esters. a) Reaction conditions with
 : RAE (1.0 equiv), NiCl2•6H2O (20 mol %), L1 (20 mol%), ethynylzinc chloride (2.5 equiv), THF/DMF, rt, 12 h. Reaction conditions with
: RAE (1.0 equiv), NiCl2•6H2O (20 mol %), L1 (20 mol%), ethynylzinc chloride (2.5 equiv), THF/DMF, rt, 12 h. Reaction conditions with
 : RAE (1.0 equiv), FeBr2•H2O (20 mol%), alkynyl Grignard (1.5 equiv), NMP/THF, 15 min, −15 °C. b) 4.0 mmol scale. c) NMR yield using internal standard. d) in situ reaction with TCNHPI (1.1 equiv) and DIC (1.1 equiv). e) 2.0 mmol scale. f) For details, see Supporting Information. g) in situ reaction with TCNHPI (1.1 equiv) and DCC (1.1 equiv).
: RAE (1.0 equiv), FeBr2•H2O (20 mol%), alkynyl Grignard (1.5 equiv), NMP/THF, 15 min, −15 °C. b) 4.0 mmol scale. c) NMR yield using internal standard. d) in situ reaction with TCNHPI (1.1 equiv) and DIC (1.1 equiv). e) 2.0 mmol scale. f) For details, see Supporting Information. g) in situ reaction with TCNHPI (1.1 equiv) and DCC (1.1 equiv).
While Ni catalysis proved successful with ethynylzinc chloride (5), expanding the transformation to include substituted alkynes proved more challenging. Additional optimization (see Supporting Information), revealed that using FeBr2•H2O with an NMP/THF solvent mixture and 1.5 equivalents of alkynyl Grignard proved fruitful for coupling a variety of substituted alkynes (26–34).11 As shown in Scheme 1, substituted alkynes bearing silyl (29), alkyl (27, 28, 30-33), and aryl (26, 34) groups were efficiently generated. Additionally, a bis-13C trimethylsilylacetylene nucleophile was successfully coupled (35), demonstrating that this reaction can potentially provide access to valuable isotopically labeled intermediates. The Fe-catalyzed alkynylation reaction was also successful in producing acyclic alkynes (36–37, 41) and more challenging coupling products including a tartrate derived alkyne (38, accessed via in situ activation), a spirocyclobutanone (39), and a glutamic acid derivative (40). It is noteworthy that, in the latter three examples, the carbonyl functionalities remained intact even in the presence of a slight excess of Grignard nucleophile, demonstrating the chemoselectivity of the cross-coupling reaction.11,12 Currently, the reactivity difference remains unclear when comparing the reactions catalyzed by either Ni or Fe.
Following the evaluation of the scope, both a mechanistic inquiry and simple product diversification were investigated. As with prior research on RAEs in cross-coupling, it was postulated that this transformation proceeded via a radical pathway.6d,13 In order to probe this hypothesis, the activation and cross-coupling of cyclopropane substrate 45 under Fe catalysis was examined, resulting in the isolation of the ring-opened product 46 in 56% yield (Scheme 2A). As a further indication of the decarboxylative nature of this reaction, 3,4,5,6-tetrachlorophthalimide was also isolated in 52% yield. If desired, TCNHPI can be regenerated from this byproduct using a known protocol.14 Further mechanistic studies are currently ongoing.
Scheme 2.

(A) Radical ring opening of a cyclopropyl methylene RAE (45). (B) Formal Arndt-Eistert transformation. (C) Functionalization of terminal alkyne 26. (D) Copper-assisted azide-alkyne cycloaddition on peptide 44.
Decarboxylative alkynylation opens up many interesting opportunities for synthesis. For instance, the simple exchange of a carboxylate with a TMS-substituted alkyne enables a formal Arndt-Eistert homologation after simple hydroboration/oxidation of 29 to carboxylic acid 47 (Scheme 2B).15 Next, the difunctionalization of acetylene could be achieved in a simple way. Thus, terminal alkyne 14 was succesfully employed in a hydrozirconation/RAE cross-coupling protocol to access difunctionalized alkene 48 in 58% yield (Scheme 2C).6h Alternatively, alkyne 14 could be directly cross-coupled under Fe catalysis to afford the disubstituted alkyne 49. This example showcases a new tactic for the construction of C–C bonds starting with commodity chemicals (simple acids and acetylene) and sustainable coinage metal catalysts. Lastly, a Click reaction with benzyl azide was carried out on peptidyl alkyne 25 to form triazole 50, illustrating a direct application of peptide alkynes relevant to peptide-labeling and bioconjugation.16
Most importantly, known synthesis pathways can be dramatically improved by employing the decarboxylative alkynylation technology. For example, alkyne 53 was a late-stage intermediate in a recent (2011) synthesis of (+)-sapinofuranone B (Scheme 3A).17 While this key intermediate was prepared in 11 steps (25% overall yield), it was envisioned that an iterative bis-functionalization of a tartaric acid derivative could provide faster access. First, in situ activation of mono-acid 51 and reaction with TIPS-protected ethynyl Grignard under Fe catalysis proceeded in 43% yield with complete diastereocontrol. This silylalkyne intermediate was saponified, and the resulting carboxylic acid was subjected to a decarboxylative Giese reaction and in situ TIPS removal delivering 53 in 47% yield (7:1 dr).18
Scheme 3.

(A) Brief synthesis of intermediate 53 en route to the synthesis of (+)-sapinofuranone B. (C) Scalable synthesis of alkynyl amino acid derivative and subsequent diversification (See SI for experimental details).
Secondly, decarboxylative alkynylation provided a mild and efficient route to various unnatural amino acids. The reaction of protected glutamic acid 2 was successful via isolation of the RAE (72% yield) and in situ activation (69% yield). This simple protocol provides for a significant improvement to the state of the art in preparing this valuable intermediate.7 Furthermore, this reaction was conducted on 1 mol scale and the product was isolated in 73% yield with no erosion of enantiopurity. The alkyne (3) was then saponified to afford amino acid 58 in quantitative yield, providing facile access to this expensive unnatural amino acid (ca. $1000/g).19
As a demonstration of the utility of alkynyl amino ester 3, it was subjected to various post-alkynylation functionalizations. For example, indole 54 and benzofuran 55 were synthesized in good yield via a Sonagashira reaction and Larock annulation, respectively, using Pd catalysis.20 Both symmetrical (56) and unsymmetrical diynes (57) were synthesized under Cu catalysis using a Glaser coupling protocol.20 In particular diyne 57 could be a useful unnatural amino acid for bioimaging using Raman spectroscopy (ATRI).22
In summary, a decarboxylative alkynylation protocol has been developed using both Ni and Fe catalysis. The reaction employs an economical catalyst system coupled with the use of readily available cross-coupling partners. The title transformation stands as an expedient alternative to age-old methods for the homologation of carbonyl compounds and benefits from employing ubiquitous carboxylic acid starting materials as electrophilic coupling partners. Substituted alkynyl Grignards can also be coupled under functional group tolerant conditions providing access to a variety of synthetically useful alkyne products. The utility of the products was demonstrated through various post-alkynylation transformations and synthetic applications. From a strategic standpoint, we anticipate that the use of alkynyl organometallics, commodity acids, and cheap Earth-abundant catalysts to make C–C bonds should find widespread practical use in various disciplines of chemical science.
Supplementary Material
Acknowledgments
Financial support for this work was provided by NIH (grant number GM-118176), Bristol-Myers Squibb, NIH (F32GM117816 postdoctoral fellowship to L. R. M), Department of Defense (NDSEG fellowship to J. T. E.), and the Arnold and Mabel Beckman Foundation (Postdoctoral Fellowship to J. M. S.) We are grateful to D.-H. Huang and L. Pasternack (The Scripps Research Institute) for assistance with nuclear magnetic resonance spectroscopy. We are also grateful to Dr. Arnold Rheingold, Milan Gembicky, and Curtis Moore for x-ray crystallographic analysis.
References
- 1.(a) Lam J, Breteler H, Arnason T, Hansen L, editors. Chemistry and Biology of Naturally-Occurring Acetylenes and Related Compounds. Elsevier; Amsterdam: 1988. [Google Scholar]; (b) Patai S, editor. Chemistry of Triple-Bonded Functional Groups. Wiley-VCH; New York: 1994. [Google Scholar]; (c) Stang PJ, Diederich F, editors. Modern Acetylene Chemistry. Wiley-VCH; Weinheim, Germany: 1995. [Google Scholar]
- 2.(a) Pässler P, et al. “Acetylene,” Ullman's Encyclopedia of Industrial Chemistry. Wiley-VCH; Weinheim, Germany: 2002. [Google Scholar]; (b) Liu Y, Lam JWY, Tang BZ. Natl Sci Rev. 2015;2:493. [Google Scholar]; (c) Jewett JC, Bertozzi C. Chem Soc Rev. 2010;39:1272. doi: 10.1039/b901970g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Thirumurugan P, Matasiuk D, Jozwiak K. Chem Rev. 2013;113:4905. doi: 10.1021/cr200409f. [DOI] [PubMed] [Google Scholar]
- 3.For a review, see: Habrant D, Rauhala V, Koskinen AMP. Chem Soc Rev. 2010;39:2007. doi: 10.1039/b915418c.
- 4.Corey EJ, Fuchs PL. Tetrahedron Lett. 1972;13:3769. [Google Scholar]
- 5.(a) Seyferth D, Marmor RS, Hilbert P. J Org Chem. 1971;36:1379. [Google Scholar]; (b) Gilbert JC, Weerasooriya U. J Org Chem. 1982;47:1837. [Google Scholar]; (c) Ohira S. Synth Commun. 1989;19:561. [Google Scholar]; (d) Müller S, Liepold B, Roth G, Bestmann HJ. Synlett. 1996:521. [Google Scholar]; (d) Roth GJ, Liepold B, Muller SG, Bestmann HJ. Synthesis. 2004:59. [Google Scholar]
- 6.For reviews on decarboxylative cross-coupling, see: Dzik WI, Lange PP, Gooßen LJ. Chem Sci. 2012;3:2671.Cornella J, Larrosa I. Synthesis. 2012;44:653.Rodríguez N, Gooßen LJ. Chem Soc Rev. 2011;40:5030. doi: 10.1039/c1cs15093f.Weaver JD, Recio A, Grenning AJ, Tunge JA. Chem Rev. 2011;111:1846. doi: 10.1021/cr1002744.Gooßen LJ, Rodríguez N, Gooßen K. Angew Chem, Int Ed. 2008;47:3100. doi: 10.1002/anie.200704782.
- 7.Cornella J, Edwards JT, Qin T, Kawamura S, Wang J, Pan CM, Gianatassio R, Schmidt M, Eastgate MD, Baran PS. J Am Chem Soc. 2016;138:2174. doi: 10.1021/jacs.6b00250.Qin T, Cornella J, Li C, Malins LR, Edwards JT, Kawamura S, Maxwell BD, Eastgate MD, Baran PS. Science. 2016;352:801. doi: 10.1126/science.aaf6123.Wang J, Qin T, Chen TG, Wimmer L, Edwards JT, Cornella J, Vokits B, Shaw SA, Baran PS. Angew Chem Int Ed. 2016;55:9676. doi: 10.1002/anie.201605463.Toriyama F, Cornella J, Wimmer L, Chen TG, Dixon DD, Creech G, Baran PS. J Am Chem Soc. 2016;138:11132. doi: 10.1021/jacs.6b07172.Qin T, Malins LR, Edwards JT, Merchant RR, Novak AJE, Zhong JZ, Mills RB, Yan M, Yuan C, Eastgate MD, Baran PS. Angew Chem Int Ed. 2017;56:260. doi: 10.1002/anie.201609662.Sandfort F, O'Neill MJ, Cornella J, Wimmer L, Baran PS. Angew Chem Int Ed. 2017;56:3319. doi: 10.1002/anie.201612314.Li C, Wang J, Barton LM, Yu S, Tian M, Peters DS, Kumar M, Yu AW, Johnson KA, Chatterjee AK, Yan M, Baran PS. Science. 2017 doi: 10.1126/science.aam7355.Edwards JT, Merchant RR, McClymont KS, Knouse KW, Qin T, Malins LR, Vokits B, Shaw SA, Bao DH, Wei FL, Zhou T, Eastgate MD, Baran PS. Nature. 2017;545:213. doi: 10.1038/nature22307.; (i) For other example of using RAE cross-coupling, see: Huihui KMM, Caputo JA, Melchor Z, Olivares AM, Spiewak AM, Johnson KA, DiBenedetto TA, Kim S, Ackerman LKG, Weix DJ. J Am Chem Soc. 2016;138:5016. doi: 10.1021/jacs.6b01533.Candish L, Teders M, Glorius F. J Am Chem Soc. 2017 doi: 10.1021/jacs.7b03127.Hu D, Wang L, Li P. Org Lett. 2017;19:2770. doi: 10.1021/acs.orglett.7b01181.; For a seminal use of RAEs of the phthalimide-type, see: Okada K, Okamoto K, Oda M. J Am Chem Soc. 1988;110:8736.
- 8.Murakami Y, Suzuki R, Yanuma H, He J, Ma S, Turino G, Lin YY, Usuki T. Org Biomol Chem. 2014;48:9887. doi: 10.1039/c4ob01438c. [DOI] [PubMed] [Google Scholar]
- 9.For examples of decarboxylative alkynylation of alkyl carboxylic acids, see: Bi HP, Zhao L, Liang YM, Li C. Angew Chem Int Ed. 2009;48:792. doi: 10.1002/anie.200805122.Zhang C, Seidel D. J Am Chem Soc. 2010;132:1792. doi: 10.1021/ja910719x.Bi HP, Teng Q, Guen M, Chen WW, Liang YM, Yao X, Li C. J Org Chem. 2010;75:783. doi: 10.1021/jo902319h.Liu X, Wang Z, Cheng X, Li C. J Am Chem Soc. 2012;134:14330. doi: 10.1021/ja306638s.Feng YS, Xu ZQ, Mao L, Zhang FF, Xu HJ. Org Lett. 2013;15:1472. doi: 10.1021/ol400197y.Zhou QQ, Guo W, Ding W, Wu X, Chen X, Lu LQ, Xiao WJ. Angew Chem Int Ed. 2015;54:11196. doi: 10.1002/anie.201504559.Yang J, Zhang J, Qi L, Hu C, Chen Y. Chem Comm. 2015;51:5275. doi: 10.1039/c4cc06344a.Le Vaillant F, Cournat T, Waser J. Angew Chem Int Ed. 2015;54:11200. doi: 10.1002/anie.201505111.Yang C, Yang JD, Li YH, Li X, Cheng JP. J Org Chem. 2016;81:12357. doi: 10.1021/acs.joc.6b02385.Schwarz J, König B. ChemPhotoChem. doi: 10.1002/cptc.201700034.Jiang M, Jin Y, Yang H, Fu H. Sci Rep. 2016;6:26161. doi: 10.1038/srep26161.; For a recent example with arenes, see: Okita T, Kumazawa K, Takise R, Muto K, Itami K, Yamaguchi J. Chem Lett. 2017;46:218.
- 10.For a recent example of Ni-catalyzed alkynylation, see: Vechorkin O, Godiniat A, Scopelliti R, Hu X. Angew Chem Int Ed. 2011;50:11777. doi: 10.1002/anie.201105964.
- 11.For recent examples of Fe-catalyzed alkynylation, see: Hatakeyama T, Okada Y, Yoshimoto Y, Nakamura M. Angew Chem Int Ed. 2011;50:10973. doi: 10.1002/anie.201104125.Cheung CW, Ren P, Hu X. Org Lett. 2014;16:2566. doi: 10.1021/ol501087m.
- 12.For other examples of alkynyl Grignard cross-coupling with alkyl halides, see: Yang LM, Huang LF, Luh TY. Org Lett. 2004;6:1461. doi: 10.1021/ol049686g.Ohmiya H, Yorimitsu H, Oshima K. Org Lett. 2006;8:3093. doi: 10.1021/ol0611144.Hammann JM, Hass D, Tullman CP, Karaghiosoff K, Knochel P. Org Lett. 2016;18:4778. doi: 10.1021/acs.orglett.6b02119.
- 13.(a) Kochi JK. Acc Chem Res. 1974;7:351. and references therein. [Google Scholar]; (b) Yan M, Lo JC, Edwards JT, Baran PS. J Am Chem Soc. 2016;138:12692. doi: 10.1021/jacs.6b08856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Einhorn C, Einhorn J, Marcadal-Abbadi C. Synth Commun. 2001;5:741. [Google Scholar]
- 15.Alemany C, Bach J, Garcia J, López M, Rodriguez AB. Tetrahedron. 2000;56:9305. [Google Scholar]
- 16.Himo F, Lovell T, Hilgraf R, Rostovtsev VV, Noodleman L, Sharpless KB, Fokin VV. J Am Chem Soc. 2005;127:210. doi: 10.1021/ja0471525. [DOI] [PubMed] [Google Scholar]
- 17.(a) Yadav JS, Reddy JSS, Mandal SS, Srihari P. Synlett. 2010:2636. [Google Scholar]; (b) Yadav JS, Reddy JSS, Mandal SS, Srihari P. Tetrahedron. 2011;67:4620. [Google Scholar]
- 18.As this reaction proceeds via radical intermediates, the selectivity reflects the inherent selectivity of the substrate. For similar results, see: (a) Reference 7e; Barton DHR, Gateau-Olesker A, Géro SD, Lacher B, Tachdjian C, Zard SZ. Tetrahedron. 1993;49:4589.
- 19.(a) Price = $1096/g on 05/16/2017 from ChemPep, Inc.; (b) Price = $3280/5g on 05/16/2017 from Affinity Research Chemicals Inc.
- 20.(a) van Esseveldt BCJ, Vervoort PWH, van Delft FL, Rutjes FPJT. J Org Chem. 2005;70:1791. doi: 10.1021/jo0484023. [DOI] [PubMed] [Google Scholar]; (b) van Esseveldt BCJ, van Delft FL, Smits JMM, de Gelder R, Schoemaker HE, Rutjes FPJT. Adv Synth Catal. 2004;346:823. [Google Scholar]
- 21.Balaraman K, Kesavan V. Synthesis. 2010:3461. [Google Scholar]
- 22.(a) Yamakoshi H, Dodo K, Okada M, Ando J, Palonpon A, Fujita K, Kawata S, Sodeoka M. J Am Chem Soc. 2011;133:6102. doi: 10.1021/ja108404p. [DOI] [PubMed] [Google Scholar]; (b) Yamakoshi H, Dodo K, Palonpon A, Ando J, Fujita K, Kawata S, Sodeoka M. J Am Chem Soc. 2012;134:20681. doi: 10.1021/ja308529n. [DOI] [PubMed] [Google Scholar]
- 23.Crystallographic data (excluding structure factors) for structures 6, 20, 23 and 29 reported in this paper have been deposited in the Cambridge Crystallographic Data Centre as supplementary publication nos. CCDC-1549218, CCDC-1549219, CCDC-1549220 and CCDC-1549221.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.