

Cancer cells in the highly desmoplastic pancreatic ductal adenocarcinoma confront nutrient [i.e., amino acids (AA)] deprivation and hypoxia, but how pancreatic cancer (PaCa) cells adapt to these conditions is poorly understood. This study provides evidence that the maintenance of mitochondrial function, in particular, oxidative phosphorylation (OXPHOS), is a key mechanism that supports PaCa cell growth, both in normal conditions and under the environmental stresses. OXPHOS in PaCa cells critically depends on autophagy and AA supply. Furthermore, the oncogenic activation mutation in GTPase Kras upregulates OXPHOS through an autophagy-dependent mechanism.
Keywords: pancreatic cancer, autophagy, cell metabolism, mitochondria, cell growth
Abstract
Pancreatic ductal adenocarcinoma (PDAC) displays extensive and poorly vascularized desmoplastic stromal reaction, and therefore, pancreatic cancer (PaCa) cells are confronted with nutrient deprivation and hypoxia. Here, we investigate the roles of autophagy and metabolism in PaCa cell adaptation to environmental stresses, amino acid (AA) depletion, and hypoxia. It is known that in healthy cells, basal autophagy is at a low level, but it is greatly activated by environmental stresses. By contrast, we find that in PaCa cells, basal autophagic activity is relatively high, but AA depletion and hypoxia activate autophagy only weakly or not at all, due to their failure to inhibit mechanistic target of rapamycin. Basal, but not stress-induced, autophagy is necessary for PaCa cell proliferation, and AA supply is even more critical to maintain PaCa cell growth. To gain insight into the underlying mechanisms, we analyzed the effects of autophagy inhibition and AA depletion on PaCa cell metabolism. PaCa cells display mixed oxidative/glycolytic metabolism, with oxidative phosphorylation (OXPHOS) predominant. Both autophagy inhibition and AA depletion dramatically decreased OXPHOS; furthermore, pharmacologic inhibitors of OXPHOS suppressed PaCa cell proliferation. The data indicate that the maintenance of OXPHOS is a key mechanism through which autophagy and AA supply support PaCa cell growth. We find that the expression of oncogenic activation mutation in GTPase Kras markedly promotes basal autophagy and stimulates OXPHOS through an autophagy-dependent mechanism. The results suggest that approaches aimed to suppress OXPHOS, particularly through limiting AA supply, could be beneficial in treating PDAC.
NEW & NOTEWORTHY Cancer cells in the highly desmoplastic pancreatic ductal adenocarcinoma confront nutrient [i.e., amino acids (AA)] deprivation and hypoxia, but how pancreatic cancer (PaCa) cells adapt to these conditions is poorly understood. This study provides evidence that the maintenance of mitochondrial function, in particular, oxidative phosphorylation (OXPHOS), is a key mechanism that supports PaCa cell growth, both in normal conditions and under the environmental stresses. OXPHOS in PaCa cells critically depends on autophagy and AA supply. Furthermore, the oncogenic activation mutation in GTPase Kras upregulates OXPHOS through an autophagy-dependent mechanism.
pancreatic ductal adenocarcinomas (PDACs) are highly aggressive, invasive tumors with early metastatic potential, for which therapeutic options are limited. One of the main factors contributing to PDAC aggressiveness is its architecture. PDAC displays one of the most extensive and poorly vascularized desmoplastic stromal reactions of all carcinomas, thus limiting tumor vascularization (3, 13). Therefore, pancreatic cancer (PaCa) cells are confronted with hypoxia and nutrient deprivation, in particular, amino acid (AA) shortage (7, 30). This occurs without evidence of major cell death or the inhibition of cell growth. It is believed that in response to environmental stresses PaCa cells activate autophagy and undergo metabolism reprogramming to maintain a sufficient amount of ATP and macromolecules to survive and proliferate (7, 21, 44). However, there is little known about the role of autophagy and metabolism in the proliferation and survival of PaCa cells subjected to environmental stresses and underlying mechanisms.
Autophagy is a key catabolic mechanism that degrades unneeded or damaged cellular organelles (2, 14, 22, 36). In normal cells, autophagy operates at a low level and is greatly activated by starvation and hypoxia to produce building blocks for macromolecular synthesis and energy maintenance. The role of basal and stress-induced autophagy as a means of support for the proliferation of PaCa cells in conditions of oxygen and nutrient scarcity remains largely unexplored (2, 22). Despite a significant effort, the role of autophagy in pancreatic tumorigenesis is far from being well understood, and the data are controversial. Indeed, there have been many publications providing evidence for both pro- and anti-tumorigenic effects of inhibiting autophagy by genetic or pharmacologic means (16, 33, 40, 51, 52).
In most eukaryotic cells, autophagy is mediated by a multistep canonical pathway that is initiated by the stress-induced inhibition of mechanistic target of rapamycin (mTOR) kinase, resulting in the sequential recruitment of the Atg proteins mediating autophagy (6, 14, 32, 36). The process starts with the activation of unc-51-like autophagy-activating kinase 1 (ULK1; Atg1) kinase (4, 10, 21, 25). ULK1, in turn, phosphorylates Beclin1, which forms a complex with phosphoinositide-3-kinase class III (PIK3C3) that serves as a platform for autophagosome construction, followed by the recruitment of the Atg5–12 complex, which mediates membrane elongation and lipidation of light chain 3 (LC3; Atg8) necessary for the autophagosome seal (6, 32). Alternatively, the noncanonical pathway uses the existing cellular membrane to assemble the autophagosome and thus can bypass some early steps of the canonical pathway, for example, the one mediated by a complex of Beclin1 with PIK3C3 (6, 25). The pathways mediating autophagosome formation in PaCa cells remain largely unknown.
It has recently been shown that PaCa has fully reprogrammed pathways mediating glucose and glutamine metabolism (54). The data also suggest that mitochondria are involved in pancreatic tumorigenesis (28, 47). However, the role of autophagy in regulating PaCa cell mitochondrial metabolisms remains poorly investigated.
Activation mutation in GTPase Kras is an early and initiating event of PDAC development shown in over 90% of low-grade pancreatic intraepithelial neoplasia (PanIN) (8, 39, 53). Accumulating evidence indicates that Kras may regulate autophagic activity and metabolism of cancer cells (15, 27). However, little is known about the role of Kras in PaCa cell adaptation to metabolic stresses.
Here, we assess the role of autophagy and metabolism in maintaining growth of PaCa cells cultured in normal conditions and subjected to the environmental stresses: hypoxia and AA depletion.
MATERIALS AND METHODS
Antibodies.
Antibodies for LC3 (cat. 2775), Atg5 (cat. 2630), Beclin1 (D40C5; cat. 3495), ULK1 (R600; cat. 4773), phosphorylated (p)-ULK1 at Ser757 (cat. 6888), 4E-binding protein 1 (BP1; cat. 9644), p-4E-BP1 (cat. 2855), GAPDH (cat. 2118), ERK (cat. 9102), and p-ERK (cat. 4370) were from Cell Signaling Technology (Danvers, MA). Peroxidase-conjugated secondary antibodies were from Bio-Rad (Hercules, CA; cat. 170-6515). All antibodies were characterized before immunoblotting studies by serial dilutions to determine optimal conditions and negative controls to ensure specificity.
Other reagents.
Beclin small interfering (si)RNA was from Dharmacon (Lafayette, CO), DMEM/F12 medium and Earle’s balanced salt solutions were from Thermo Fisher Scientific (Waltham, MA). Premium-grade FBS was from VWR International (Radnor, PA); penicillin and streptomycin were from Omega Scientific (Tarzana, CA). E64D (cat. E8640) and pepstatin A (cat. P5318; E/P) were from Sigma-Aldrich (St. Louis, MO), cathepsin (Cat)B and CatL substrates were from Bachem (Torrance, CA), and CatD substrate was from Enzo Life Sciences (Farmingdale, NY). Protein determination reagent and nonfat dry milk were from Life Science Research, Bio-Rad Laboratories. SuperSignal West Pico and West Femto Chemiluminescent Substrate (cat. 34080) were from Thermo Fisher Scientific. Cell Death Detection ELISAPLUS kit was from Sigma-Aldrich. All other reagents were purchased from Sigma-Aldrich.
Cell culture.
Human pancreatic adenocarcinoma cell lines CAPAN-2, BxPC3, and PANC-1 were obtained from American Type Culture Collection (Manassas, VA). CAPAN-2 and BxPC3 were maintained in RPMI medium; PANC-1 cells were grown in DMEM/F12, supplemented with 4 mmol/l l-glutamine. Human pancreatic ductal epithelial (HPDE) cells and HPDE cells stably transfected with mutant KRASG12V were cultured, as previously described (29, 38). Cells used between passages 3 and 15 were maintained at 37°C in a humidified atmosphere containing 5% CO2 (basal, AA depletion) or subjected to hypoxia (1% O2, 5% CO2). For AA depletion, cells were cultured in Earle’s balanced salt solution (in the presence of 5.5 mM glucose). In all conditions, the medium was supplemented with 15% FBS, which was dialyzed to remove low molecular weight components, and with penicillin (100 U/ml) and streptomycin (100 μg/ml).
Inhibition of lysosomal protein degradation.
Two approaches are currently applied to inhibit lysosomal proteolysis (23, 24, 31). One is by inhibiting cathepsin activities using a combination of inhibitors of cysteine (E64D) and aspartic (pepstatin A) proteases. The second approach is by increasing lysosomal pH, leading to the inactivation of pH-dependent proteases. Cathepsin inhibition suppresses lysosomal proteolysis without affecting other organelles of the endocytic pathway or protein trafficking, because the lysosome is the predominant site of cathepsin activation in cells (5, 45). In contrast, as a weak base, chloroquine concentrates in all acidic organelles (including endosomes and Golgi vesicles), thus affecting its function to various extents (1). It also interferes with the pH-dependent sorting of lysosomal hydrolases (26). Based on these considerations, we chose cathepsin inhibitors vs. chloroquine to block lysosomal proteolysis.
Transient transfections.
Transient transfections of cells were performed with Beclin siRNA using the electroporation system Amaxa Nucleofactor (Lonza, Basel, Switzerland), according to the manufacturer’s protocol. The measurements were performed at 48 h post-transfection.
Transfection efficiencies are presented in Table 1.
Table 1.
Transfection efficiency
| Cell Line | Beclin1 siRNA | Atg5 siRNA |
|---|---|---|
| CAPAN-2 | 56 ± 3 | 59 ± 4 |
| BxPC3 | 70 ± 3 | 55 ± 7 |
| PANC-1 | 53 ± 1 | 58 ± 3 |
Values (in %) are means ± SE of 3 independent experiments. P < 0.05 vs. control siRNA.
Western blot analysis.
Immunoblot analysis was performed as we discussed (34). Briefly, cells were lysed, and proteins were separated by SDS-PAGE and transferred onto nitrocellulose membranes. Nonspecific binding was blocked, and the membranes were incubated with the primary antibody and then with the peroxidase-conjugated secondary antibody. Blots were developed using SuperSignal Chemiluminescent Substrate (Thermo Fisher Scientific). For detection and densitometric quantification of band intensities, we used FluorChem HD2 (ProteinSimple, San Jose, CA).
Cell metabolism.
The Seahorse XF24 analyzer (Agilent Technologies, Santa Clara, CA) simultaneously measures glycolysis and oxidative phosphorylation (OXPHOS) in the same cells. Glycolysis was determined through measurements of the extracellular acidification rate (ECAR) of the surrounding media, predominately from the excretion of lactic acid, and mitochondrial function by directly measuring the oxygen consumption rate (OCR) of cells. The decrease in OCR upon injection of the ATP synthase inhibitor oligomycin represents a portion of basal respiration that was being used to drive ATP production. Therefore, ATP-linked respiration was calculated as a difference between basal OCR and that in oligomycin-treated cells. The maximal OCR was obtained by adding the uncoupler carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), which stimulates the respiratory chain, to operate at maximum capacity. The combination of complex I inhibitor rotenone and complex III inhibitor antimycin A shuts down mitochondrial respiration. Therefore, for calculation of basal and maximal respiration, the values of OCR in the presence of rotenone + antimycin A were subtracted. OCR and ECAR were normalized per microgram of protein. Of note, we did not present data on the effect of hypoxia on the metabolic profile, as it was difficult to maintain cells under hypoxia during Seahorse measurements.
Immunofluorescence.
Cells were fixed for 15 min at −20°C in methanol/acetone (1:1), and the nonspecific binding was blocked with 5% goat or donkey serum (Abcam, Cambridge, MA) and 1% Aurion BSA (AURION Immuno Gold Reagents & Accessories, Wageningen, The Netherlands) in PBS containing 0.2% Triton. After incubation with the primary antibody and the fluorescent-labeled secondary antibody, cells were counterstained with 4′,6-diamidine-2′-phenylindole dihydrochloride and mounted with Antifade (Thermo Fisher Scientific), and immunofluorescence images were acquired with a Zeiss LSM 710 confocal microscope using a ×63 objective (Carl Zeiss Microscopy, Thornwood, NY).
Cell proliferation.
Cells were seeded in 96-well plates, treated for indicated times; 20 μl 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide solution was added to each well; and then the cells were incubated for 2 h at 37°C, and the optical density was measured spectrophotometrically at 540 nm.
Cathepsin activities.
Cathepsin activities were measured in cell homogenates in the presence of 2 mM EDTA and 2 mM DTT using fluorogenic substrates specific for CatB (Z-Arg-Arg-AMC; 50 μM), CatL (Z-Phe-Arg-AMC), and CatD [AMC-Gly-Lys-Pro-Ile-Leu-Phe-Phe-Arg-Leu-Lys(Dnp)-D-Arg-NH2; 50 µM]. CatB activity was measured in 50 mM phosphate buffer, pH 6.0, and CatL and CatD activities in 100 mM sodium acetate buffer, at pH 5.5 (CatL) and pH 4.0 (CatD). In measurements of CatL activity, the assay buffer also contained 50 μM CA-074me, an inhibitor of CatB.
Apoptosis.
Apoptosis was assessed by measuring internucleosomal DNA fragmentation with the Cell Death Detection ELISAPLUS kit.
Statistical analysis.
Data are presented as means ± SE. Statistical evaluation was performed using two-tailed Student’s t-test.
To assess the correlation between ATP-linked OCR and cell viability, the values of cell viability and ATP-linked respiration for cells cultured in normal conditions and subjected for 24 h to AA depletion, with and without autophagy inhibitors, were analyzed using GraphPad Prism 5 software (GraphPad Software, La Jolla, CA). For each cell line, the viability and ATP-linked OCR values in cells cultured in normal conditions were taken as 100%.
RESULTS
AA depletion and hypoxia activated autophagy in PaCa cells only weakly or not at all.
We analyzed autophagic responses to AA depletion and hypoxia in three human PDAC cell lines: well-differentiated CAPAN-2 cells, moderately differentiated BxPC3 cells, and poorly differentiated PANC-1 (9). Autophagy is associated with conversion from the cytosolic LC3-I (~18 kDa) to LC3-II (~16 kDa), which uniquely marks the autophagic membranes. We monitored autophagy by measuring the densitometric ratio of LC3-II/loading control. According to recent autophagy guidelines (24), this parameter provides more accurate assessment of the changes in autophagy compared with the LC3-I/LC3-II ratio, as the latter may depend on the autophagy-independent changes in LC3-I.
The number of autophagic vacuoles observed at any time is a function of the balance between the generation of autophagosomes and their degradation. Thus an increase in LC3-II could result from either increased autophagosome formation or decreased degradation. The established approach to discriminate between these two scenarios is a blockade of lysosomal degradation (23, 24, 31). In conditions in which lysosomal degradation is blocked, for example, by cathepsin inhibitors, the changes in LC3-II only reflect autophagy induction. To assess both autophagy induction (autophagosome formation) and the total level of autophagic vacuoles, we measured the changes in LC3-II in the presence and absence of lysosomal inhibitors.
The presence of basal autophagic activity in PaCa cells cultured in normal conditions was manifested by the pronounced LC3-II band on immunoblots (Fig. 1A). Lysosomal inhibitors increased the intensity of the LC3-II band, indicating constitutive autophagosome formation in cells cultured in normal conditions (Fig. 1, A and B). We confirmed that E/P, at the doses applied, blocked activities of major cathepsins in PaCa cells (Fig. 1C).
Fig. 1.

The extent of autophagy induction by environmental stresses varied among PaCa cell lines. CAPAN-2, BxPC3, and PANC-1 cells were cultured for 24 h (A–C, J, and K) or indicated times (D–I) in the absence or presence of lysosomal inhibitors 50 µM E64D + 10 µM pepstatin A (E/P). Cells were cultured in normal conditions (A–C) and in normal conditions (cont) in the medium devoid of amino acids (−AA) or under hypoxia (Hpx; D–K). A, B, and D–I: LC3-I and LC3-II levels were measured by immunoblot; GAPDH is the loading control. The densitometric intensity of the LC3-II band was normalized to that of GAPDH in the same sample, and the mean LC3-II/GAPDH ratio was normalized further to that in the control group. C: cathepsins (Cat)B, L, and D activities were measured with a fluorogenic assay. J: immunofluorescence analysis of LC3 (green) in CAPAN-2 cells; nuclei staining with 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI; blue). Original scale bars, 10 µm. K: effects of AA depletion and hypoxia on LC3 dots in PaCa cells, as shown in J. B–D, I, and K: values are means ± SE (at least 3 independent experiments). #P < 0.05 vs. no inhibitors; *P < 0.05 vs. control cells.
The autophagy activation by environmental stresses was only pronounced at 24 h (Fig. 1, D–I). The extent of autophagy activation varied between cell lines. Both AA depletion and hypoxia increased LC3-II (Fig. 1D) in CAPAN-2 cells. The effects were even more prominent in the presence of E/P, indicating that both AA depletion and hypoxia stimulated autophagy in these cells (Fig. 1E). In contrast, neither AA depletion nor hypoxia activated autophagy in BxPC3 cells (Fig. 1, F and G), and AA depletion but not hypoxia induced autophagy in PANC-1 cells (Fig. 1, H and I).
To corroborate the results of immunoblot analysis, we showed by immunostaining (Fig. 1, J and K) that changes in the number of LC3-positive puncta (i.e., autophagic vacuoles) correlate well with the changes in LC3-II intensity (Fig. 1D).
Expression of oncogenic Kras stimulated autophagy in nontransformed HPDE cells.
The differences in autophagic responses among PaCa cell lines may be caused by the differences in their mutational signatures: BxPC3 cells express wild-type, CAPAN-2 KrasG12V, and PANC-1 KrasG12D mutants (9). G12D and G12V are the two most common KRAS mutations observed in PaCa; expression of either of them in embryonic pancreatic cells results in PanIN development and invasive PDAC (19, 20).
To assess the role of oncogenic Kras in regulating autophagy, we compared autophagic responses of normal HPDE cells with HPDE cells that stably express KrasG12V (HPDE/Kras; Fig. 2).
Fig. 2.

Oncogenic Kras expression stimulated autophagy in nontransformed human ductal HPDE cells. A: immunoblots demonstrated a Kras-mediated increase in phosphorylated ERK (p-ERK) in HPDE/Kras vs. HPDE cells. *p-ERK1 and *p-ERK2 are p-ERK bands visualized with correspondingly longer and shorter exposure. The data are representative of 2 independent experiments that gave the same results. The densitometric intensity of the p-ERK band was normalized to that of total ERK in the same sample and further normalized to that in HPDE cells, as shown below the blot. B–J: HPDE and HPDE/Kras cell lines were cultured either in normal conditions (B and C) or subjected to stresses, as specified in Fig. 1, and cultured in the presence and absence of E64D + Pepstatin A (E/P) for 24 h (B, C, and H–J) or for indicated times (D–G). C and H–J: densitometric intensities of LC3-II bands were normalized to that of GAPDH in the same sample. The bands were normalized further to that in HPDE cells with no inhibitors (C), to control HPDE (H and I), or to control HPDE/Kras cells (J). Values are means ± SE (at least 3 independent experiments). *P < 0.05 vs. HPDE cells cultured without inhibitors; #P < 0.05 vs. HPDE/Kras cells cultured without inhibitors; ^P < 0.05 vs. HPDE cells cultured in the same conditions; $P < 0.05 vs. control of the same cell line (H–J).
Kras activity in HPDE/Kras cells was confirmed by increased phosphorylation of its substrate ERK (Fig. 2A). In HPDE cells cultured in normal conditions, the LC3-II band was barely detectable, but its intensity increased in the presence of E/P (Fig. 2B), indicating efficient autophagic flux. Expression of KrasG12V increased the LC3-II level in HPDE cells cultured in normal conditions. In the presence of E/P, the LC3-II level in HPDE/Kras was 2.5-fold greater than that in HPDE cells (Fig. 2, B and C), demonstrating that oncogenic Kras greatly stimulated autophagy in cells cultured in normal conditions.
In both HPDE and HPDE/Kras cells, AA depletion time dependently increased LC3-II, whereas the effect of hypoxia on LC3-II was minimal (Fig. 2, D–G). As in normal conditions, LC3-II levels remained greater in HPDE/Kras when both types of cells were subjected to AA depletion and hypoxia (Fig. 2, D and E).This was also observed in the presence of E/P (Fig. 2, F–H). However, the fold increase in LC3-II band intensities caused by AA depletion in E/P-treated HPDE/Kras was the same as in parent HPDE cells, indicating that Kras had no effect on autophagy activation by AA depletion (Fig. 2, I and J). Kras also did not promote autophagy induction by hypoxia (Fig. 2J). Thus expression of oncogenic Kras stimulated basal but not stress-induced autophagy.
Autophagy in PaCa cells was Beclin1/PIK3C3 independent.
Complex formation between Beclin1 and PIK3C3 is a necessary step in canonical autophagy (6, 14, 36). Beclin1 siRNA knockdown did not cause any significant reduction in basal autophagy and autophagic responses to AA depletion and hypoxia in PaCa cells (Fig. 3, A and B). Furthermore, the inhibition of PIK3C3 with 3-methyl adenine (6, 14, 36) did not reduce the LC3-II level in control and stressed cells (Fig. 3, C and D). Surprisingly, 3-methyladenine not only did not decrease but oppositely increased the LC3-II band intensity in PaCa cells (Fig. 3, C and D). The activation of autophagy by 3-methyladenine was reported in other cells as well and proved to be mediated by the inhibition of class I phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K), which is also a substrate of 3-methyladenine (12, 50). Unlike the class III PI3K, the class I PI3K inhibits autophagosome formation through stimulating the Akt–mTOR pathway (56). Thus the effects of 3-methyladenine on autophagy are dual: 3-methyladenine suppresses autophagy through inhibiting PIK3C3 but stimulates it by inhibiting class I PI3K. Because in the process of autophagy, class I PI3K acts well upstream of PIK3C3, the inhibitory effect of 3-methyladenine predominates if the Beclin1/PIK3C3 complex is involved in autophagosome formation. Thus the fact that in PaCa cells, as in some other cells (12, 50), 3-methyladenine stimulates autophagy is strong evidence that this pathway does not involve PIK3C3. As with other cells (50), we find that autophagy stimulation by 3-methyladenine was most pronounced in cells cultured in nutrient-rich conditions in which Akt and AA act in concert to stimulate mTOR (10). Collectively, the findings that Beclin siRNA did not inhibit (Fig. 3, A and B) and that 3-methyladenine either did not change or stimulated (Fig. 3, C and D) autophagy indicate that the Beclin1/PIK3C3 complex is not involved in autophagosome formation in PaCa cells. In contrast, Atg5 knockdown with siRNA reduced LC3-II in both control and stressed PaCa cells (Fig. 3E).
Fig. 3.

Autophagy operating in PaCa cells was Beclin1 and PIK3C3 independent. PaCa cells were transfected with control, Beclin1 siRNA (A and B), or Atg5 siRNA (E) or not transfected (C and D). Cells were cultured for 24 h in the presence or absence of 10 mM 3-methyladenine (3-MeAd) in normal conditions or subjected to stresses (as specified in Fig. 1). A, C, and E: Beclin1, Atg5, LC3-I, and LC3-II levels were measured by immunoblot. GAPDH is the loading control. B, D, and E: the densitometric intensity of the LC3-II band was normalized to that of GAPDH in the same sample, and the LC3-II/GAPDH ratio was normalized further to that in the control group. B and D: values are means ± SE (3 independent experiments). #P < 0.05 vs. control cells of the same cell line; *P < 0.05 vs. control cells cultured without 3-MeAd. E: values shown below the blots are representative of 2 or 3 independent experiments; the variations among the results of individual experiments were <15%. Densitometry shows that Atg5 knockdown decreased the LC3-II/GAPDH level in AA-depleted cells, correspondingly, by 40 ± 10% (n = 3, CAPAN-2), 30 ± 5% (n = 3, BxPC3), and 33 ± 3% (n = 2, PANC-1).
AA depletion and hypoxia did not inhibit mTOR in PaCa cells.
In nontransformed cells, environmental stresses activate autophagy through inhibiting mTOR kinase, followed by dephosphorylation (activation) of its substrate ULK1, which initiates autophagy (6, 32). We examined the role of mTOR in autophagy in PaCa cells using the pharmacologic mTOR inhibitor Torin1 (42). Activity of mTOR was assessed by measuring the changes in the phosphorylation of its substrate 4E-BP1. Torin1 markedly decreased p-4E-BP1 and mTOR-mediated p-ULK1 (at Ser757) and dramatically upregulated autophagy (Fig. 4), indicating its regulation by the mTOR-ULK1 circuit. However, AA depletion and hypoxia did not cause 4E-BP1 dephosphorylation (Fig. 4) and either did not activate autophagy or activated it to a much lesser extent than Torin1. The data indicate that the lack of mTOR inhibition may explain why AA depletion and hypoxia activated autophagy only weakly or not at all.
Fig. 4.

Environmental stresses failed to inhibit mTOR complex 1 activity in PaCa cells. PaCa cells were cultured for 24 h in normal conditions or subjected to stresses, as specified in Fig. 1, and cultured in the presence or absence of the mTOR inhibitor Torin1 (10 µM). A–C: phosphorylated (p)-4E-BP1, p-ULK1, total 4E-BP, LC3-I, and LC3-II were measured with immunoblot. D–F: intensity of p-4E-BP1, p-ULK1, and LC3-II bands was densitometrically quantified and normalized to that of GAPDH; all of the ratios were normalized further to that in the control group. Values are means ± SE (n = 3). #P < 0.05 vs. cells cultured in the same conditions but without Torin1; *P < 0.05 vs. control cells.
Although environmental stresses failed to inhibit mTOR, in some conditions, they reduced p-ULK1 (Fig. 4). Importantly, ULK1 dephosphorylation closely correlated with the extent of autophagy activation in stressed cells. For example, AA depletion and hypoxia (in BxPC3) and hypoxia (in PANC-1) did not dephosphorylate ULK1 and did not activate autophagy (Figs. 1, C, E, F, and H, and 4, B–F), whereas AA depletion and hypoxia in CAPAN-2 cells and AA depletion in PANC-1 cells reduced p-ULK1 and activated autophagy (Figs. 1, C, E, F, and H, and 4, A and C–F).
Autophagy inhibition and AA depletion greatly reduced OXPHOS in PaCa cells.
We characterized the metabolic profile of PaCa cells by simultaneously monitoring with the Seahorse XF24 OCR, resulting from OXPHOS and ECAR, an indirect indicator of glycolysis (Fig. 5A). To assess individual parameters of mitochondrial dysfunction, cells were injected at the indicated times with the ATP synthase inhibitor oligomycin, mitochondrial uncoupler FCCP, and a mix of rotenone (a complex I inhibitor) and antimycin A (a complex III inhibitor; Fig. 5A). Basal, ATP-linked, and maximal respiration was calculated, as described in materials and methods. The intensity of metabolic flux varied among PaCa cell lines (Fig. 5B). However, in all cells tested, OXPHOS plays a major role in metabolism, as evidenced by the high OCR/ECAR ratio (Fig. 5C), indicating a significant contribution of mitochondria.
Fig. 5.

Autophagy inhibition and AA depletion both decreased mitochondrial respiration. Oxygen consumption rate (OCR) reflecting mitochondrial OXPHOS and extracellular acidification rate (ECAR) reflecting glycolysis were measured using the Seahorse XF24 analyzer and normalized per microgram of protein. B and C: ECAR, basal OCR, and their ratios in PaCa cells cultured in normal conditions. A and D–G: ECAR, basal, ATP-linked, and maximal respiration in PaCa cells cultured in normal conditions (control), in the presence of E64D + pepstatin A (E/P), subjected to AA depletion (−AA), or a combination of AA depletion and inhibitors (−AA, E/P). Cells were injected at indicated times with ATP synthase inhibitor oligomycin (Oligo), FCCP, and rotenone and antimycin (RM). ATP-linked respiration was calculated as a difference between basal OCR and that in oligomycin-treated cells; the maximal OCR was obtained by cell treatment with FCCP. The values of nonmitochondrial respiration obtained in the presence of RM were subtracted. A: illustration of OCR recording in CAPAN-2 cells. D–G: values are means ± SE (n = 3–6). *P < 0.05 vs. cells subjected to autophagy inhibitors, AA depletion, or their combination; #P < 0.05 vs. cells cultured in normal conditions without inhibitors.
Neither autophagy inhibition nor AA depletion decreased the glycolysis rate in PaCa cells (Fig. 5D). In contrast, E/P and AA depletion greatly reduced both basal and ATP-linked respiration (Fig. 5, E and F), demonstrating a critical role of autophagy and AA supply in preserving mitochondrial functions of PaCa cells. Importantly, AA depletion similarly reduced respiration in cells treated with and without autophagy inhibitors, indicating that the lack of AA supply overwhelmed the ability of autophagy to maintain mitochondrial functions (Fig. 5, E and F). AA depletion and to a lesser extent, autophagy inhibition also reduced uncoupled respiration obtained in the presence of FCCP, which represents maximum activity of electron transport and substrate oxidation that is achievable by the cells under the assay conditions. The inhibition of maximal respiration indicates that mitochondrial capacity is diminished in stressed cells (Fig. 5G).
We next measured the effect of the expression of oncogenic Kras on the rate of OXPHOS in normal ductal HPDE cells (Fig. 6). AA withdrawal reduced OCR in both HPDE and HPDE/Kras cells (Fig. 6B), likely because in the absence of AAs, the amount of available respiratory substrates is not sufficient for mitochondrial functioning. The data suggest that to maintain ATP production, mitochondria of both HPDE and HPDE/Kras cells requires not only exogenous AAs but also endogenous AAs generated by autophagic recycling. Indeed, autophagy inhibition with E/P decreased ATP-linked OCR in both HPDE/Kras and parent HPDE cells (Fig. 6C).
Fig. 6.

Oncogenic Kras expression increased OXPHOS in nontransformed human ductal HPDE cells. HPDE and HPDE/Kras cells were cultured in normal conditions (A) or subjected for 24 h to AA depletion (B) or to treatment with lysosomal inhibitors E64D + pepstatin A (C). Basal, ATP-linked, and maximal respiration were measured using the Seahorse XF24 analyzer. Values are means ± SE (n = 3–6). *P < 0.05 vs. HPDE cells.
In nutrient-rich conditions, Kras expression increased basal, ATP-linked, and maximal respiration in HPDE cells (Fig. 6A), indicating that in these conditions, mitochondria of HPDE/Kras cells generates more ATP (ATP-linked respiration) and has a greater capacity to respond to increased energy demand (maximal respiration) than mitochondria of HPDE cells. Interestingly, E/P reduced maximal and basal respiration in HPDE/Kras but not in HPDE cells (Fig. 6C), indicating that autophagy is necessary for Kras to increase OXPHOS.
Autophagy promoted PaCa cell growth through maintaining OXPHOS.
Autophagy was critical in maintaining proliferation of PaCa cells cultured in normal conditions, as evidenced by markedly reduced proliferation of cells treated with E/P (Fig. 7A).
Fig. 7.

Autophagy inhibition and AA depletion both suppressed PaCa cell growth. A–C: PaCa cells were cultured for 48 h in the presence or absence of E64D + pepstatin A (E/P), 2 μM oligomycin (oligo), or FCCP in normal conditions or subjected to stresses, as in Fig. 1, and cell proliferation was measured by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay. B: correlation analysis between ATP-linked respiration and proliferation, measured with MTT, was performed using GraphPad Prism 5 software. For each cell line, the values of MTT and ATP-linked respiration in control cells were considered 100% (Pearson r = 0.8456). D: apoptosis was assessed by measuring DNA fragmentation in PaCa cells cultured for 48 h under specified conditions. A, C, and D: values are means ± SE (n = at least 4). *P < 0.05 vs. cells cultured in normal conditions without inhibitors; #P < 0.05 vs. cells subjected to the same stress conditions without inhibitors.
Autophagy was less efficient in sustaining proliferation of cells subjected to environmental stresses. Indeed, AA depletion reduced proliferation of cells cultured with and without autophagy inhibitors (Fig. 7A). Autophagy maintained proliferation of hypoxic BxPC3 and PANC-1 but not CAPAN-2 cells (Fig. 7A).
Importantly, stress-induced autophagy provided no additional advantage for PaCa cell growth. AA depletion inhibited proliferation of cells in which AA depletion did (CAPAN-2 and PANC-1) and did not (BxPC3) activate autophagy (Figs. 1 and 7A). The effect of hypoxia on cell viability was in opposition to the effects of hypoxia on autophagy: hypoxia activated autophagy but did not sustain proliferation in CAPAN-2 and sustained proliferation but did not activate autophagy in BxPC3 cells (Fig. 1). Thus autophagy, induced by AA depletion and hypoxia, was dispensable for the proliferation of hypoxic PaCa cells.
The effects of autophagy inhibition and AA depletion on cell growth strongly correlated with those on OXPHOS (Fig. 7B). To confirm the role of mitochondria in PaCa cell proliferation, we showed further that the inhibitor of mitochondrial ATP synthase oligomycin and mitochondrial uncoupler FCCP suppressed PaCa cell proliferation (Fig. 7C). Taken together, the results indicated that OXPHOS is critical for cell growth and that maintenance of OXPHOS is a main mechanism through which autophagy and AA supply supported PaCa cell growth.
Neither stresses nor autophagy had any effect on apoptosis in CAPAN-2 and BxPC3 cells (Fig. 7D). However, autophagy inhibition increased apoptosis more than fourfold in PANC-1 cells cultured in normal conditions and under hypoxia (Fig. 7D). Stimulation of cell death by autophagy inhibition was prevented by AA depletion (Fig. 7D).
DISCUSSION
In healthy tissues and cells, including normal pancreatic cells, basal autophagic activity is low, but autophagy is rapidly and greatly activated by starvation (6, 14, 36, 37). In contrast, in PaCa cells, basal autophagic activity was relatively high, but AA depletion and hypoxia activated autophagy only weakly or not at all. Autophagic activity in cancer cells is controlled by their mutational status (37). Expression of oncogenic Kras markedly increased autophagy in nontransformed HPDE cells cultured in normal conditions. In contrast, AA depletion stimulated autophagy in HPDE/Kras to the same extent as in HPDE cells, and hypoxia failed to activate autophagy in both cell types. Thus oncogenic Kras could mediate the strong basal autophagic activity and at the same time, be responsible for the lack of the autophagy stimulation by environmental stresses observed in PaCa cells. Of note, the PaCa cells that we used, as well as HPDE/Kras cells, have G12D (CAPAN-2) or G12V (PANC-1, HPDE/Kras) Kras mutations; these are the two most common KRAS mutations observed in PaCa; and expression of either of them in embryonic pancreatic cells results in PanIN development and invasive PDAC (19, 20).
It is well established that the principal mechanism through which environmental stresses activate autophagy in eukaryotic cells is by the inhibition of mTOR, resulting in ULK1 dephosphorylation (11, 32, 35, 49). The data indicate that the mTOR/p-ULK1 circuit was active and that mTOR greatly suppressed autophagy in PaCa cells. However, environmental stresses did not reduce mTOR activity, suggesting a weak or deficient regulation of mTOR by AAs and hypoxia. Insufficient mTOR inhibition, resulting in sustained p-ULK1, dampens the ability of environmental stresses to activate autophagy in PaCa cells. Defects in mTOR nutrient sensing were reported in various cancers (4, 17). For example, in cells with a mutated negative regulator of mTOR, GAP activity toward Rags, mTOR complex 1 was hyperactivated and resistant to AA starvation (4). Whether this is the case for PaCa remains to be determined.
ULK1 dephosphorylation could also be mediated through mTOR-independent mechanisms, such as activation of ULK1 phosphatase (48). A contribution of these mechanisms may explain that in CAPAN-2 cells, AA depletion and hypoxia dephosphorylated ULK1 without inhibiting mTOR. It cannot be excluded, however, that the observed decrease in p-ULK1 is caused by the transient mTOR inhibition, which we failed to detect.
Autophagy in PaCa cells was mediated through a noncanonical Beclin1/PIK3C3-independent and Atg5-mediated pathway. A similar noncanonical pathway operates in several other cell types (6, 18, 41, 43), yet it remains unknown whether the functions of canonical and noncanonical autophagy are the same.
Autophagy maintained growth of PaCa cells cultured in normal conditions but failed to support proliferation of CAPAN-2, BxPC3, and PANC-1 cells subjected to AA depletion and of CAPAN-2 cells subjected to hypoxia. Furthermore, stress-activated autophagy conferred no additional advantage to PaCa cell growth. For example, AA depletion similarly inhibited proliferation in cells in which AA depletion did and did not activate autophagy.
To gain insight into the mechanisms through which autophagy and AA supply maintained cell proliferation, we analyzed the effects of these treatments on a metabolic phenotype of PaCa cells. The characteristic feature of cancer cell metabolism is high levels of glucose uptake, even under adequate oxygen supply, a phenomenon termed the Warburg effect (46). Warburg suggested that cancer cells generate ATP through glycolysis instead of OXPHOS. Further studies, however, revealed a significant heterogeneity in cancer metabolism with many tumors, generating energy from both glycolytic and oxidative metabolism (46, 57). Furthermore, it is becoming increasingly clear that in rapidly proliferating cells, glucose intermediates are being shifted toward biosynthetic processes, rather than a source of energy supply (54).
PaCa cells displayed mixed metabolism of both glycolysis and OXPHOS (7, 55) with predominant mitochondrial contribution manifested by a high OCR/ECAR ratio. Both autophagy and AA supply are central to maintaining OXPHOS in PaCa cells. Indeed, AA withdrawal and autophagy inhibition caused a marked OCR reduction (Fig. 8), likely because both of these treatments decreased mitochondrial substrate availability. Expression of oncogenic Kras greatly increased OXPHOS in HPDE cells cultured in normal, nutrient-rich conditions but failed to do so in the absence of autophagy and AA supply. Thus the lack of AA supply and autophagic recycling overwhelmed the ability of PaCa cells to maintain mitochondrial energy production.
Fig. 8.
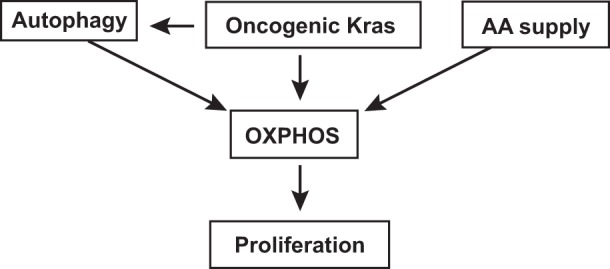
Schematic illustrating the effects of autophagy and OXPHOS on PaCa cell proliferation.
PaCa cell proliferation critically relied on OXPHOS (Fig. 8). There was a strong correlation between the rates of mitochondrial respiration and cell growth and the pharmacologic inhibitors of OXPHOS-suppressed PaCa cell growth. Moreover, autophagy inhibition and AA depletion, which caused a dramatic decrease in OCR, both suppressed PaCa cell growth. Thus the maintenance of OXPHOS is a key mechanism through which autophagy and AA supply facilitated cell growth (Fig. 8).
In summary, in healthy cells, basal autophagic activity is low, but it is greatly activated by environmental stresses. In contrast, in PaCa cells, basal autophagic activity was relatively high, but AA depletion and hypoxia failed to inhibit mTOR and either did not or only weakly activated autophagy. Basal autophagy was necessary, whereas stress-induced autophagy was dispensable for maintaining proliferation of PaCa cells. PaCa cells displayed a mixed glycolytic/oxidative metabolism, with a predominant mitochondrial contribution. Cell growth critically relied on OXPHOS, and both basal autophagy and AA supply were necessary to sustain it. In agreement with these results, we found that expression of oncogenic Kras upregulated autophagy and OXPHOS through an autophagy-dependent mechanism. The data indicate that the maintenance of OXPHOS is a key mechanism through which AA supply and autophagy promoted PaCa cell growth. Furthermore, the capacity of autophagy to preserve mitochondrial functions determines its effect on the proliferation in PaCa cells subjected to environmental stresses.
GRANTS
This work was supported by National Cancer Institute Grant P01CA163200-01A1 (Project 3 to A. S. Gukovskaya) and National Institute of Diabetes and Digestive and Kidney Diseases Grant P01DK098108 (to A. S. Gukovskaya).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.S.G. conceived and designed research; S.M., J.M.E., E.L., M.S., S.D.S., K.T., B.M.R., and O.A.M. performed experiments; S.M., M.S., O.A.M., P.J.G., and A.S.G analyzed data; J.M., M.M.L., and A.S.G. interpreted results of experiments; S.M. and A.S.G. prepared figures; A.S.G. drafted manuscript; S.M., M.S., J.M., M.M.L., and A.S.G. edited and revised manuscript; S.M., J.M.E., E.L., M.S., S.D.S., K.T., B.M.R., O.A.M., P.J.G., J.M., M.M.L., and A.S.G. approved final version of manuscript.
REFERENCES
- 1.Akpovwa H. Chloroquine could be used for the treatment of filoviral infections and other viral infections that emerge or emerged from viruses requiring an acidic pH for infectivity. Cell Biochem Funct 34: 191–196, 2016. doi: 10.1002/cbf.3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altman BJ, Rathmell JC. Metabolic stress in autophagy and cell death pathways. Cold Spring Harb Perspect Biol 4: a008763, 2012. doi: 10.1101/cshperspect.a008763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Apte M, Pirola RC, Wilson JS. Pancreatic stellate cell: physiologic role, role in fibrosis and cancer. Curr Opin Gastroenterol 31: 416–423, 2015. doi: 10.1097/MOG.0000000000000196. [DOI] [PubMed] [Google Scholar]
- 4.Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, Spear ED, Carter SL, Meyerson M, Sabatini DM. A tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 340: 1100–1106, 2013. doi: 10.1126/science.1232044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braulke T, Bonifacino JS. Sorting of lysosomal proteins. Biochim Biophys Acta 1793: 605–614, 2009. doi: 10.1016/j.bbamcr.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 6.Codogno P, Mehrpour M, Proikas-Cezanne T. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat Rev Mol Cell Biol 13: 7–12, 2011. 10.1038/nrm3249. [DOI] [PubMed] [Google Scholar]
- 7.Cohen R, Neuzillet C, Tijeras-Raballand A, Faivre S, de Gramont A, Raymond E. Targeting cancer cell metabolism in pancreatic adenocarcinoma. Oncotarget 6: 16832–16847, 2015. doi: 10.18632/oncotarget.4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collins MA, Pasca di Magliano M. Kras as a key oncogene and therapeutic target in pancreatic cancer. Front Physiol 4: 407, 2014. doi: 10.3389/fphys.2013.00407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deer EL, González-Hernández J, Coursen JD, Shea JE, Ngatia J, Scaife CL, Firpo MA, Mulvihill SJ. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 39: 425–435, 2010. doi: 10.1097/MPA.0b013e3181c15963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dibble CC, Cantley LC. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol 25: 545–555, 2015. doi: 10.1016/j.tcb.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dunlop EA, Tee AR. mTOR and autophagy: a dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol 36: 121–129, 2014. doi: 10.1016/j.semcdb.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 12.Dupont N, Nascimbeni AC, Morel E, Codogno P. Molecular mechanisms of noncanonical autophagy. Int Rev Cell Mol Biol 328: 1–23, 2017. doi: 10.1016/bs.ircmb.2016.08.001. [DOI] [PubMed] [Google Scholar]
- 13.Erkan M, Michalski CW, Rieder S, Reiser-Erkan C, Abiatari I, Kolb A, Giese NA, Esposito I, Friess H, Kleeff J. The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Clin Gastroenterol Hepatol 6: 1155–1161, 2008. http://www.cghjournal.org/article/S1542-3565(08)00498-9/fulltext. [DOI] [PubMed] [Google Scholar]
- 14.Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res 24: 24–41, 2014. doi: 10.1038/cr.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J, Gewirtz DA, Karantza V, Kimmelman A, Kumar S, Levine B, Maiuri MC, Martin SJ, Penninger J, Piacentini M, Rubinsztein DC, Simon HU, Simonsen A, Thorburn AM, Velasco G, Ryan KM, Kroemer G. Autophagy in malignant transformation and cancer progression. EMBO J 34: 856–880, 2015. doi: 10.15252/embj.201490784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gómez VE, Giovannetti E, Peters GJ. Unraveling the complexity of autophagy: potential therapeutic applications in pancreatic ductal adenocarcinoma. Semin Cancer Biol 35: 11–19, 2015. doi: 10.1016/j.semcancer.2015.09.011. [DOI] [PubMed] [Google Scholar]
- 17.Grabiner BC, Nardi V, Birsoy K, Possemato R, Shen K, Sinha S, Jordan A, Beck AH, Sabatini DM. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov 4: 554–563, 2014. doi: 10.1158/2159-8290.CD-13-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grishchuk Y, Ginet V, Truttmann AC, Clarke PG, Puyal J. Beclin 1-independent autophagy contributes to apoptosis in cortical neurons. Autophagy 7: 1115–1131, 2011. doi: 10.4161/auto.7.10.16608. [DOI] [PubMed] [Google Scholar]
- 19.Guerra C, Schuhmacher AJ, Cañamero M, Grippo PJ, Verdaguer L, Pérez-Gallego L, Dubus P, Sandgren EP, Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 11: 291–302, 2007. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 20.Hingorani SR, Petricoin EF III, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, Crawford HC, Putt ME, Jacks T, Wright CV, Hruban RH, Lowy AM, Tuveson DA. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4: 437–450, 2003. doi: 10.1016/S1535-6108(03)00309-X. [DOI] [PubMed] [Google Scholar]
- 21.Izuishi K, Kato K, Ogura T, Kinoshita T, Esumi H. Remarkable tolerance of tumor cells to nutrient deprivation: possible new biochemical target for cancer therapy. Cancer Res 60: 6201–6207, 2000. [PubMed] [Google Scholar]
- 22.Jiang X, Overholtzer M, Thompson CB. Autophagy in cellular metabolism and cancer. J Clin Invest 125: 47–54, 2015. doi: 10.1172/JCI73942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, Al-Zeer MA, Albert ML, Albin RL, Alegre-Abarrategui J, Aleo MF, Alirezaei M, Almasan A, Almonte-Becerril M, Amano A, Amaravadi R, Amarnath S, Amer AO, Andrieu-Abadie N, Anantharam V, Ann DK, Anoopkumar-Dukie S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8: 445–544, 2012. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, Adhihetty PJ, Adler SG, Agam G, Agarwal R, Aghi MK, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd ed.). Autophagy 12: 1–222, 2016. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ktistakis NT, Tooze SA. Digesting the expanding mechanisms of autophagy. Trends Cell Biol 26: 624–635, 2016. doi: 10.1016/j.tcb.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 26.Lerch MM, Saluja AK, Dawra R, Saluja M, Steer ML. The effect of chloroquine administration on two experimental models of acute pancreatitis. Gastroenterology 104: 1768–1779, 1993. doi: 10.1016/0016-5085(93)90658-Y. [DOI] [PubMed] [Google Scholar]
- 27.Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 330: 1340–1344, 2010. doi: 10.1126/science.1193494. [DOI] [PubMed] [Google Scholar]
- 28.Liou GY, Döppler H, DelGiorno KE, Zhang L, Leitges M, Crawford HC, Murphy MP, Storz P. Mutant KRas-induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Reports 14: 2325–2336, 2016. doi: 10.1016/j.celrep.2016.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu N, Furukawa T, Kobari M, Tsao MS. Comparative phenotypic studies of duct epithelial cell lines derived from normal human pancreas and pancreatic carcinoma. Am J Pathol 153: 263–269, 1998. doi: 10.1016/S0002-9440(10)65567-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mayers JR, Wu C, Clish CB, Kraft P, Torrence ME, Fiske BP, Yuan C, Bao Y, Townsend MK, Tworoger SS, Davidson SM, Papagiannakopoulos T, Yang A, Dayton TL, Ogino S, Stampfer MJ, Giovannucci EL, Qian ZR, Rubinson DA, Ma J, Sesso HD, Gaziano JM, Cochrane BB, Liu S, Wactawski-Wende J, Manson JE, Pollak MN, Kimmelman AC, Souza A, Pierce K, Wang TJ, Gerszten RE, Fuchs CS, Heiden MG, Wolpin BM. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat Med 20: 1193–1198, 2014. doi: 10.1038/nm.3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 140: 313–326, 2010. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, Gretzmeier C, Dengjel J, Piacentini M, Fimia GM, Cecconi F. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol 15: 406–416, 2013. doi: 10.1038/ncb2708. [DOI] [PubMed] [Google Scholar]
- 33.New M, Van Acker T, Long JS, Sakamaki JI, Ryan KM, Tooze SA. Molecular pathways controlling autophagy in pancreatic cancer. Front Oncol 7: 28, 2017. doi: 10.3389/fonc.2017.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nitsche C, Edderkaoui M, Moore RM, Eibl G, Kasahara N, Treger J, Grippo PJ, Mayerle J, Lerch MM, Gukovskaya AS. The phosphatase PHLPP1 regulates Akt2, promotes pancreatic cancer cell death, and inhibits tumor formation. Gastroenterology 142: 377–387, 2012. doi: 10.1053/j.gastro.2011.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Papinski D, Kraft C. Regulation of autophagy by signaling through the Atg1/ULK1 complex. J Mol Biol 428: 1725–1741, 2016. doi: 10.1016/j.jmb.2016.03.030. [DOI] [PubMed] [Google Scholar]
- 36.Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 20: 460–473, 2014. doi: 10.1089/ars.2013.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rabinowitz JD, White E. Autophagy and metabolism. Science 330: 1344–1348, 2010. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Radulovich N, Qian JY, Tsao MS. Human pancreatic duct epithelial cell model for KRAS transformation. Methods Enzymol 439: 1–13, 2008. doi: 10.1016/S0076-6879(07)00401-6. [DOI] [PubMed] [Google Scholar]
- 39.Rishi A, Goggins M, Wood LD, Hruban RH. Pathological and molecular evaluation of pancreatic neoplasms. Semin Oncol 42: 28–39, 2015. doi: 10.1053/j.seminoncol.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosenfeldt MT, O’Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, Adams PD, Anderson KI, Gottlieb E, Sansom OJ, Ryan KM. p53 status determines the role of autophagy in pancreatic tumour development. Nature 504: 296–300, 2013. doi: 10.1038/nature12865. [DOI] [PubMed] [Google Scholar]
- 41.Scarlatti F, Maffei R, Beau I, Codogno P, Ghidoni R. Role of non-canonical Beclin 1-independent autophagy in cell death induced by resveratrol in human breast cancer cells. Cell Death Differ 15: 1318–1329, 2008. doi: 10.1038/cdd.2008.51. [DOI] [PubMed] [Google Scholar]
- 42.Schenone S, Brullo C, Musumeci F, Radi M, Botta M. ATP-competitive inhibitors of mTOR: an update. Curr Med Chem 18: 2995–3014, 2011. doi: 10.2174/092986711796391651. [DOI] [PubMed] [Google Scholar]
- 43.Seo G, Kim SK, Byun YJ, Oh E, Jeong SW, Chae GT, Lee SB. Hydrogen peroxide induces Beclin 1-independent autophagic cell death by suppressing the mTOR pathway via promoting the ubiquitination and degradation of Rheb in GSH-depleted RAW 264.7 cells. Free Radic Res 45: 389–399, 2011. doi: 10.3109/10715762.2010.535530. [DOI] [PubMed] [Google Scholar]
- 44.Sousa CM, Kimmelman AC. The complex landscape of pancreatic cancer metabolism. Carcinogenesis 35: 1441–1450, 2014. doi: 10.1093/carcin/bgu097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Staudt C, Puissant E, Boonen M. Subcellular trafficking of mammalian lysosomal proteins: an extended view. Int J Mol Sci 18: 47, 2017. doi: 10.3390/ijms18010047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324: 1029–1033, 2009. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sánchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, Kost-Alimova M, Muller F, Colla S, Nezi L, Genovese G, Deem AK, Kapoor A, Yao W, Brunetto E, Kang Y, Yuan M, Asara JM, Wang YA, Heffernan TP, Kimmelman AC, Wang H, Fleming JB, Cantley LC, DePinho RA, Draetta GF. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514: 628–632, 2014. doi: 10.1038/nature13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wong PM, Feng Y, Wang J, Shi R, Jiang X. Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat Commun 6: 8048, 2015. doi: 10.1038/ncomms9048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wong PM, Puente C, Ganley IG, Jiang X. The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy 9: 124–137, 2013. doi: 10.4161/auto.23323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, Ong CN, Codogno P, Shen HM. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem 285: 10850–10861, 2010. doi: 10.1074/jbc.M109.080796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang S, Imamura Y, Jenkins RW, Cañadas I, Kitajima S, Aref A, Brannon A, Oki E, Castoreno A, Zhu Z, Thai T, Reibel J, Qian Z, Ogino S, Wong KK, Baba H, Kimmelman AC, Pasca Di Magliano M, Barbie DA. Autophagy inhibition dysregulates TBK1 signaling and promotes pancreatic inflammation. Cancer Immunol Res 4: 520–530, 2016. doi: 10.1158/2326-6066.CIR-15-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell’antonio G, Mautner J, Tonon G, Haigis M, Shirihai OS, Doglioni C, Bardeesy N, Kimmelman AC. Pancreatic cancers require autophagy for tumor growth. Genes Dev 25: 717–729, 2011. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ying H, Dey P, Yao W, Kimmelman AC, Draetta GF, Maitra A, DePinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev 30: 355–385, 2016. doi: 10.1101/gad.275776.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, Yan H, Wang W, Chen S, Viale A, Zheng H, Paik JH, Lim C, Guimaraes AR, Martin ES, Chang J, Hezel AF, Perry SR, Hu J, Gan B, Xiao Y, Asara JM, Weissleder R, Wang YA, Chin L, Cantley LC, DePinho RA. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149: 656–670, 2012. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu M, Zhou Q, Zhou Y, Fu Z, Tan L, Ye X, Zeng B, Gao W, Zhou J, Liu Y, Li Z, Lin Y, Lin Q, Chen R. Metabolic phenotypes in pancreatic cancer. PLoS One 10: e0115153, 2015. doi: 10.1371/journal.pone.0115153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu X, Long YC, Shen HM. Differential regulatory functions of three classes of phosphatidylinositol and phosphoinositide 3-kinases in autophagy. Autophagy 11: 1711–1728, 2015. doi: 10.1080/15548627.2015.1043076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zu XL, Guppy M. Cancer metabolism: facts, fantasy, and fiction. Biochem Biophys Res Commun 313: 459–465, 2004. doi: 10.1016/j.bbrc.2003.11.136. [DOI] [PubMed] [Google Scholar]