

PTEN-PI3K and IL-7R–mTORC1–Myc are two discrete signaling axes driving B cell development.
Abstract
Interleukin-7 (IL-7) drives early B lymphopoiesis, but the underlying molecular circuits remain poorly understood, especially how Stat5 (signal transducer and activator of transcription 5)–dependent and Stat5-independent pathways contribute to this process. Combining transcriptome and proteome analyses and mouse genetic models, we show that IL-7 promotes anabolic metabolism and biosynthetic programs in pro-B cells. IL-7–mediated activation of mTORC1 (mechanistic target of rapamycin complex 1) supported cell proliferation and metabolism in a Stat5-independent, Myc-dependent manner but was largely dispensable for cell survival or Rag1 and Rag2 gene expression. mTORC1 was also required for Myc-driven lymphomagenesis. PI3K (phosphatidylinositol 3-kinase) and mTORC1 had discrete effects on Stat5 signaling and independently controlled B cell development. PI3K was actively suppressed by PTEN (phosphatase and tensin homolog) in pro-B cells to ensure proper IL-7R expression, Stat5 activation, heavy chain rearrangement, and cell survival, suggesting the unexpected bifurcation of the classical PI3K-mTOR signaling. Together, our integrative analyses establish IL-7R–mTORC1–Myc and PTEN-mediated PI3K suppression as discrete signaling axes driving B cell development, with differential effects on IL-7R–Stat5 signaling.
INTRODUCTION
B lymphopoiesis is a highly ordered developmental process characterized by the sequential rearrangements of immunoglobulin heavy (IgH) and light chain (IgL) loci that accompany differentiation of lymphoid precursors to pro-B cells, pre-B cells, and immature B cells in the bone marrow (BM). The stepwise progression of B cell development is dependent upon a network of transcription factors and extrinsic signals (1), especially interleukin-7 (IL-7) (2). IL-7 is produced by BM stromal cells and signals through IL-7 receptor (IL-7R; composed of the IL-7Rα chain and a common γc chain) expressed on pro-B and pre-B cells to promote their proliferation, survival, and differentiation (3). Because of its potent trophic effect on lymphocytes, IL-7 therapy has been developed for clinical application. In particular, infusion of IL-7 induces a marked expansion of B cell precursors in human patients (4, 5), suggesting a strong trophic effect of IL-7 in adult human B cell precursors. Yet, the lack of full understanding of the IL-7–mediated signaling network hinders its rational utilization in clinics (6). Despite its importance in B lymphopoiesis, how IL-7 influences pro-B cell development, especially about the mechanisms linking IL-7R activation to transcriptional and translational events, has not been examined in a systematic manner. Furthermore, although IL-7 has been linked to lymphocyte metabolism (7–9), the detailed processes and molecular regulators are unclear.
Binding of IL-7 to IL-7R initiates phosphorylation of Janus kinase 1 (Jak1) and Jak3, which recruit and activate the transcription factor Stat5 (signal transducer and activator of transcription 5). IL-7R–mediated proliferation and survival of pro-B cells are critically dependent on the Jak1/3-Stat5 signaling axis (10–13). Ectopic expression of constitutively active Stat5 considerably, although incompletely, rescues B cell developmental defects in IL-7Rα–deficient mice (14), suggesting the involvement of other pathways downstream of IL-7R. Published studies indicate that phosphatidylinositol 3-kinase (PI3K) signaling mediates IL-7R signaling in pre-B cells (15), although for pro-B cell development, deletion of p110α and p110δ (16), pharmacological inhibition of PI3K, or deficiency of p85α does not exert a strong effect (17). Thus, the nature and function of Stat5-independent pathways in IL-7R signaling, and the extent to which PI3K contributes to early B cell development, remain elusive. Furthermore, PI3K has been traditionally associated with the activation of the mechanistic target of rapamycin (mTOR) (18, 19). Despite recent genetic studies of mTOR signaling in B cell development and function (20, 21), the upstream and downstream regulators of mTOR and the relationship with PI3K signaling in B lymphopoiesis are not well understood. Moreover, how mTOR, PI3K, and Stat5 signaling are integrated is largely unknown in lymphocyte development.
To understand the proteome landscape and signaling networks mediated by IL-7 signaling, we performed temporal proteomic profiling and network analysis of IL-7–stimulated pro-B cells using multiplexed tandem mass tag (TMT) and two-dimensional (2D) liquid chromatography–tandem mass spectrometry (LC/LC-MS/MS) (22, 23). Our proteomic profiling, together with transcriptome analysis, revealed enrichment of anabolic metabolism and activation of mTOR and Myc in IL-7–stimulated pro-B cells. Furthermore, using hCd2-iCre–mediated targeted mutagenesis (24), we genetically defined the roles and mechanisms of mTOR and its two complexes, mTORC1 and mTORC2, as well as Myc and PI3K signaling, in early B cell development. Our results showed that mTORC1, but not mTORC2, is critical for B lymphopoiesis, by linking IL-7R signaling to pro-B cell metabolism. These effects are separated from PI3K-Akt and are independent of Stat5 signaling and cell survival, but mTORC1 activity is required for IL-7–induced Myc protein translation. Further, mTORC1 and Myc form a feed-forward circuitry in lymphomagenesis, and Raptor deficiency completely blocks tumor development in a model of Myc-induced B cell lymphoma (25, 26). Unexpectedly, we found that active suppression of PI3K activity by PTEN (phosphatase and tensin homolog) is required for proper B cell development. Uncontrolled PI3K disrupts pro-B cell development, associated with impaired IL-7R–Stat5 signaling, reduced lineage transcription factor expression, diminished IgH chain rearrangement, and greatly elevated apoptosis. Together, our results uncover two signaling axes, namely, PTEN-mediated PI3K suppression and IL-7R–mTORC1–Myc, crucial for early B cell development with discrete mechanisms and effects on IL-7R–Stat5 signaling and immunoglobulin rearrangement, and point to the unexpected bifurcation of the conventional PI3K-mTOR signaling axis in IL-7–driven B lymphopoiesis.
RESULTS
IL-7 activates mTOR, Myc, and anabolic metabolism in pro-B cells
Cell metabolism is closely associated with lymphocyte activation and function, but its role in immune development remains poorly understood (27). To investigate metabolic regulation during B cell development, we examined mitochondria respiration and glycolysis in freshly isolated BM B cell subsets (the gating strategy is shown in fig. S1A) by measuring oxygen consumption rate (OCR) and extracellular acidification rate (ECAR), respectively. B cell precursors [Lin−B220+CD43+IgM−, including fraction A, B, C, and C′ cells (28)] had the highest OCR, relative to B220+CD25+ pre-B cells, immature B cells (B220+IgM+), and circulating mature B cells (B220hiIgM+; Fig. 1A). ECAR was modest in all cell subsets examined, but B cell precursors still contained the highest level (fig. S1B). We next examined additional metabolic parameters, including the expression of the nutrient receptor transferrin (CD71), which correlates with cell metabolic rate (29) (fig. S1C), proliferation via 5-ethynyl-2′-deoxyuridine (EdU) incorporation assay (fig. S1D), and glucose uptake, as indicated by staining with the glucose analog 2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxyglucose (2-NBDG; fig. S1E). We found that developing B cells gradually increased CD71 expression, proliferation, and glucose uptake during their developmental progression from fraction A [Lin−B220+CD43+IgM−BP-1−CD24−, according to the Hardy classification (30)] to fraction B (Lin−B220+CD43+IgM−BP-1−CD24+) and then fraction C and C′ (Lin−B220+CD43+IgM−BP-1+CD24+) stage (see fig. S1A for gating), whereas pre-B cells down-regulated all these parameters relative to fraction C/C′ cells (fig. S1, C to E). Accompanying the dynamic changes of metabolic activities, the expression of IL-7Rα was actively regulated during B cell development. Specifically, IL-7Rα expression was markedly increased on fraction B and fraction C/C′ cells compared to fraction A cells, followed by sequential down-regulation in later developmental stages (fig. S1F), suggesting coordinated regulation of IL-7R expression and metabolic programs during B cell development.
Fig. 1. Regulation of metabolic activity in developing B cells and IL-7–mediated proteome landscape in pro-B cells.
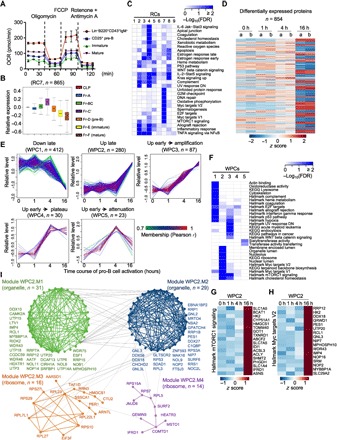
(A) OCR of Lin−B220+CD43+IgM− cells, CD25+ pre-B cells (Lin−B220+CD43−IgM−CD25+), immature B cells (B220+IgM+), and circulating mature B cells (B220hiIgM+) measured using a Seahorse XF24 analyzer. (B) RC7 from microarray data of common lymphoid progenitors (CLPs) and each developing B cell subset from ImmGen (gene expression relative to CLP). The central rectangle represents the first to third quartile, with notch in the plot corresponding to the median expression of all genes in the sample. End of the whiskers represents minimum and maximum level of expression of the genes in the cluster. (C) Functional annotations of RCs by Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Hallmark databases (FDR < 0.2). (D) Cluster analysis of DE proteins in temporal proteomic profiling of pro-B cells stimulated with 0, 1, 4, and 16 hours of IL-7. (E) Five WPCs during pro-B cell activation were defined by WGCNA. Each line indicates the relative abundance of each protein and is color-coded by the cluster membership determined by correlation analysis between individual protein and the consensus of the WPC (see the color bar). (F) Functional annotations of WPCs by GO, KEGG, and Hallmark databases (FDR < 0.2). (G and H) IL-7–mediated temporal expression of mTORC1 signaling (G) and Myc targets (H). (I) Four interconnected modules of organelles and ribosome components were derived from WPC2. The number and names of proteins and the representative functional term for each module are shown. See also fig. S1 and data S1 and S2.
Further bioinformatics analysis of transcriptome of developing B cell subsets in the Immunological Genome Project (ImmGen) (31) revealed 2499 genes that had significantly altered expression between any two subsets, that is, differentially expressed (DE) genes [false discovery rate (FDR) < 0.01, with at least 1.5 fold change (FC); data S1A]. We applied weighted gene correlation network analysis (WGCNA) (32) of these DE genes to identify RNA clusters (RCs) of highly correlated genes, which may share similar biological functions or mechanisms of regulation (33). We identified nine clusters that contained at least 10 DE genes (Fig. 1B and fig. S1G). The largest cluster, RC7 (865 genes), showed a gradual up-regulation from fraction A to fraction C′ cells and then down-regulation (Fig. 1B), a pattern similar to metabolic changes and IL-7Rα expression described above. Pathway analysis revealed that RC7 was enriched with mTORC1 signaling, Myc targets, metabolic pathways (including glycolysis, oxidative phosphorylation, and one-carbon metabolism), and cell cycle regulators (Fig. 1C, fig. S1H, and data S1B), further supporting the dynamic regulation of metabolic activities in early B cell development.
To comprehensively examine the direct roles and temporal effects of IL-7 on the proteome landscape and metabolic programs in pro-B cells, we applied multiplexed TMT and LC/LC-MS/MS approaches (22, 23, 34) to quantify the whole proteome of freshly isolated (0 hour) or IL-7–stimulated (1, 4, and 16 hours) pro-B cells (fig. S1I). We quantified 7733 unique proteins. The null comparisons of the experimental replicates showed random distribution (fig. S1J, left), whereas comparisons of the two time points (16 hours versus 0 hour) showed significant differences between freshly isolated and activated pro-B cells but consistency between replicates (fig. S1J, right). Clustering analysis of DE proteins (Fig. 1D) further verified the reproducibility of the results. We identified 854 DE proteins that significantly changed between any two time points using one-way analysis of variance (ANOVA; 10% FDR, z score of at least 2; Fig. 1D and data S2A). We then applied WGCNA for these DE proteins and identified five clusters of coexpressed proteins, named as whole protein clusters WPC1 to WPC5. WPC1, the largest cluster, showed late down-regulation at 16 hours of IL-7 stimulation, whereas WPC2, the second largest cluster, exhibited late up-regulation at 16 hours. In addition, WPC3 to WPC5 showed up-regulation at 4 hours but had distinct patterns of expression at 16 hours, namely, continued up-regulation (amplification), unchanged expression (plateau), or marked down-regulation (attenuation), respectively (Fig. 1E).
Pathway enrichment analysis revealed that WPC1 contained proteins involved in cytoskeletal regulation, including actin-binding proteins and multiple integrin molecules, suggesting a potential effect of IL-7 on cytoskeletal reorganization in pro-B cells (Fig. 1F and data S2B). Negative regulators of the cell cycle were also identified in WPC1, including CDKN2C and CDKN2D. BCL6 (B cell lymphoma 6), a transcriptional regulator suppressed by IL-7 signaling and induced by pre-BCR (B cell receptor) (35), was also identified in WPC1 (data S2B). WPC2 and WPC3 contained the majority of proteins up-regulated by IL-7. In addition to the modestly enriched Jak-Stat and IL-2–Stat5 pathways, mTORC1 signaling and cholesterol homeostasis pathways were more significantly enriched in WPC2 and WPC3, including some of the key enzymes in lipid biosynthesis such as HMGCS1, PMVK, MVD, SQLE, IDI1, ACSL3, and FASN (data S2B). Enzymes involved in amino acid metabolism, such as BCAT1, SLC7A5, and ASNS, were also identified in WPC2 and WPC3 (data S2B). Furthermore, Myc targets and organelle formation pathways were enriched in WPC2 (Fig. 1F and data S2B). The heat maps of mTORC1 signaling components (Fig. 1G and fig. S1K) and Myc targets (Fig. 1H) in WPC2 and WPC3 demonstrated a pronounced up-regulation of these pathways by IL-7 at the 16-hour time point, indicating a relatively late activation kinetics. WPC4 and WPC5 showed enrichment of the E2F target pathway and sialyltransferase activity (36), respectively, indicating dynamic effects of IL-7 on cell cycle progression and protein glycosylation in pro-B cells. Finally, despite close connection between PI3K and mTOR signaling pathways (19), the PI3K-Akt pathway was not identified in any of the WPCs (data S2B). Thus, in response to IL-7 stimulation, pro-B cells likely undergo changes in their metabolic features associated with the activation of mTORC1 and Myc pathways.
We further superimposed proteins within each WPC onto the protein-protein interaction (PPI) network to identify putative functional modules altered during temporal IL-7 stimulation on the basis of the assumption that coexpressed genes/proteins are more likely to share common molecular functions (33, 34). We identified 39 modules, with sizes ranging from 2 to 35 proteins (data S2C). The largest module (WPC1.M5) was composed of molecules involved in cell adhesion, migration, and cytoskeleton, including multiple integrin family members (data S2C). In WPC2, we found four interconnected modules (WPC2.M1 to WPC2.M4) with 86 combined components that were enriched for intracellular organelle or ribosome components (Fig. 1I), suggesting that IL-7 may coordinate up-regulation of organelle biogenesis. Collectively, our integrative network analyses predict that IL-7 likely regulates cell adhesion and migration and concomitantly activates mTORC1 and Myc signaling, biosynthetic metabolism, and organelle biogenesis in pro-B cells.
mTORC1, but not mTORC2, activity is critical for pro-B to pre-B cell transition
We next investigated the functional importance of mTOR signaling in B cell development. We crossed mice bearing conditional loxP-flanked Mtor (Mtorfl/fl) allele with hCd2-iCre (Cd2icre) transgenic mice, in which an optimized variant of Cre recombinase (iCre) is expressed under a human CD2 promoter and locus control region. This transgene drives iCre expression in lymphocyte lineages (24), leading to efficient recombination in all developing B cells including fraction A cells (37). Previous studies have shown that a hypomorphic Mtor allele with constitutive but partial loss of mTOR activity partly blocks pro-B to pre-B transition and reduces pre-B cells, but Cd19-Cre–driven [which initiates incomplete target gene deletion at the pro-B/pre-B cell stage (38)] deletion of Mtor does not affect B cell development in the BM (39, 40). In contrast, we observed an accumulation of B220+CD43+IgM− B cell precursors (Fig. 2A) in the absence of mTOR, which was attributed to increased fraction C/C′ cells with abnormally high expression of BP-1 (Fig. 2B). In addition, there were few CD25+ pre-B cells (Fig. 2C) and immature and circulating mature B cells in the BM (Fig. 2D) of Cd2icreMtorfl/fl mice. Thus, the mTOR kinase is essential for early B cell development.
Fig. 2. mTORC1, but not mTORC2, is required for pro-B cell development and pro-B to pre-B cell transition.
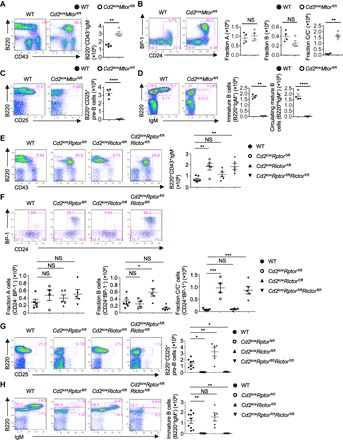
(A to D) Representative flow cytometry plots of BM cells from wild-type (WT) and Cd2icreMtorfl/fl mice. (A) Expression of B220 and CD43 on BM lymphocytes, with the percentages of B220+CD43+IgM− cells indicated (after further gating IgM+ cells out of B220+CD43+ cells; see fig. S1A for gating scheme). Right: Number of designated populations. (B) Expression of BP-1 and CD24 on B220+CD43+IgM− cells, with the percentages of fraction A (CD24−BP-1−), fraction B (CD24+BP-1−), and fraction C/C′ (CD24+BP-1+) cells indicated. Right: Number of indicated subset cells. (C) Expression of B220 and CD25 on BM lymphocytes, with the percentage of B220+CD25+ pre-B cells indicated. Right: Number of B220+CD25+ pre-B cells. (D) Expression of B220 and IgM on BM lymphocytes, with the percentages of pro-B/pre-B cells (B220+IgM−), immature B cells (B220+IgM+), and circulating mature B cells (B220hiIgM+) indicated. Right: Number of immature and mature B cells. (E to H) Representative flow cytometry plots of BM cells of WT, Cd2icreRptorfl/fl, Cd2icreRictorfl/fl, and Cd2icreRptorfl/flRictorfl/fl mice and the number of indicated populations. (E) Expression of B220 and CD43 on BM lymphocytes, with the percentage of B220+CD43+IgM− cells indicated. (F) Hardy classification of pro-B cell subsets in the B220+CD43+IgM− gate [from (E)], with the percentages of fraction A (CD24−BP-1−), fraction B (CD24+BP-1−), and fraction C/C′ (CD24+BP-1+) cells indicated. (G) Expression of B220 and CD25 on BM lymphocytes, with the percentage of B220+CD25+ pre-B cells indicated. (H) Expression of B220 and IgM on BM lymphocytes, with the percentages of pro-B/pre-B cells (B220+IgM−), immature B cells (B220+IgM+; statistics are on the right), and circulating mature B cells (B220hiIgM+) indicated. NS, not significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (one-way ANOVA with Tukey’s test). Results represent at least four independent experiments. Data are means ± SEM. Numbers indicate percentage of cells in gates. See also fig. S2.
mTOR signaling consists of mTORC1 and mTORC2 complexes. To investigate the extent to which mTORC1 and mTORC2 contribute to early B cell development, we generated Cd2icreRptorfl/fl and Cd2icreRictorfl/fl mice, in which Raptor and Rictor were deleted in developing B cells. Flow cytometry analysis of BM cells revealed that Cd2icreRptorfl/fl mice had a significant reduction of total B220+ cells (fig. S2A) and an increased number of B220+CD43+IgM− B cell precursors (Fig. 2E). Similar to Cd2icreMtorfl/fl mice, Cd2icreRptorfl/fl mice had normal numbers of fraction A and fraction B cells but significantly increased numbers of fraction C/C′ cells with unusually high expression of BP-1 (Fig. 2F). Strikingly, B220+CD25+ pre-B cells (Fig. 2G), immature B cells (Fig. 2H), and circulating mature B cells (Fig. 2H and fig. S2B) were all severely diminished in the absence of Raptor. Consequently, Cd2icreRptorfl/fl mice were devoid of peripheral B cells (fig. S2C), including follicular (CD21intCD23hi; fig. S2D) and marginal zone (CD21hiCD23low) B cells in the spleen (fig. S2E). Therefore, Raptor and mTOR kinase are required for fraction C/C′ to pre-B cell transition, and their deficiencies lead to disrupted pro-B cell development, a greatly diminished pre-B cell compartment, and development of profound lymphopenia of peripheral B cells.
In contrast, BM of Cd2icreRictorfl/fl mice contained a small (P > 0.05) increase of B220+CD43+IgM− B cell precursors (Fig. 2E) and a modest elevation of B220+CD25+ pre-B cells (Fig. 2G). These observations are consistent with a recent report using Mx1-Cre–mediated systemic deletion of Rictor (41). Within the pro-B cell compartment, Rictor deficiency did not substantially alter the distribution of pro-B cell subsets (Fig. 2F). Despite increased pre-B cells, Cd2icreRictorfl/fl mice had a normal immature B cell compartment (Fig. 2H), indicating that the modest perturbation during earlier B cell development did not affect immature B cell generation. However, Cd2icreRictorfl/fl mice had significantly reduced circulating mature B cells in the BM (fig. S2B) and diminished numbers of splenic B cells, including follicular and marginal zone B cells (fig. S2, C to E). These data indicate that mTORC2 activity is not essential for early B cell development but contributes to peripheral B cell maturation, consistent with a recent report using pan-hematopoietic and acute deletion systems (21). Combined deletion of both Raptor and Rictor led to developmental phenotypes that were largely similar to deletion of Raptor alone (Fig. 2, E to H, and fig. S2, A to E) or mTOR (Fig. 2, A to D). Furthermore, to examine whether B cell developmental defects due to Raptor deficiency were cell-intrinsic, we generated mixed BM chimeras that contained 50% of WT congenic cells and found that mTORC1-dependent B cell development was cell-intrinsic (fig. S2, F and G). Collectively, mTORC1 activity plays a predominant, intrinsic role in pro-B cell development and in the pro-B to pre-B transition. In contrast, mTORC2 has a relatively small suppressive effect on early B cell generation but contributes to peripheral B cell maturation.
mTORC1 mediates IL-7R signaling in a Stat5-independent but Myc-dependent manner
We next explored the mechanisms underlying mTORC1-mediated early B cell development. Raptor-deficient fraction C/C′ cells, but not fraction A or fraction B cells, had reduced CD71 expression (Fig. 3A) and EdU incorporation (Fig. 3B). However, Raptor deficiency did not affect cell apoptosis, as indicated by active caspase-3 staining (Fig. 3C). Similar phenotypes were observed in Cd2icreMtorfl/fl mice (fig. S3, A and B), indicating that mTORC1 impinges upon a specific set of physiological activities.
Fig. 3. Raptor-mTORC1 is essential to mediate IL-7 signaling and Myc induction but not Stat5 activation.
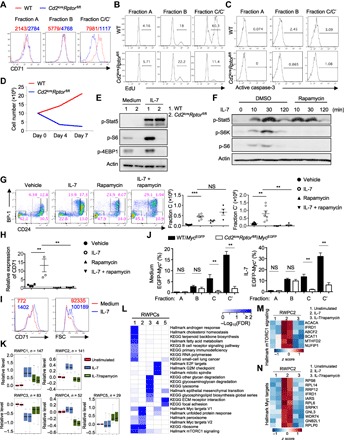
(A) Expression of CD71 on fraction A, B, and C/C′ cells from WT and Cd2icreRptorfl/fl mice. Numbers indicate the MFI of CD71 from a representative experiment. (B and C) EdU incorporation (at 1 hour after intravenous injection of EdU) (B) and active caspase-3 staining (C) in fraction A, B, and C/C′ cells from WT and Cd2icreRptorfl/fl mice, with the percentages of EdU+ and active caspase-3+ cells indicated from a representative experiment plotted in the graphs. (D) Cellularity of BM cells after IL-7 stimulation in vitro. Total BM cells were cultured in the presence of IL-7 (5 ng/ml) for 1 week, and cell numbers were counted on days 0, 4, and 7. (E) Immunoblot analysis of p-Stat5, p-S6, and p-4EBP1 in freshly sorted pro-B cells with or without IL-7 stimulation. (F) Immunoblot analysis of p-Stat5, p-S6K, and p-S6 in the in vitro cultured B cells. Lin− BM cells were cultured with OP9 stromal cells with IL-7 for 5 days, and cells were washed and rested overnight before they were restimulated with IL-7 for the indicated time points in the presence or absence of rapamycin. DMSO, dimethyl sulfoxide. (G) Expression of BP-1 and CD24 on BM Lin−B220+CD43+IgM− cells from mice injected with vehicle control, recombinant IL-7 (250 μg/kg), rapamycin (15 mg/kg), or IL-7 plus rapamycin, with the percentages of fraction A, B, C, and C′ cells indicated. Right: Number of fraction C and C′ cells. (H) Relative CD71 expression on fraction C cells from mice treated in (G). The MFI of CD71 from mice treated with vehicle control was set as 1. (I) Human BM pro-B cells (CD3−CD19+CD10+CD34+CD127+) were sorted from healthy donors. Cells were stimulated with recombinant human IL-7 (10 ng/ml) for 24 hours. Expression of CD71 (left) and cell size [measured by forward light scatter (FSC); right] were analyzed by flow cytometry. Numbers in plots indicate the MFI. (J) Frequency of EGFP-Myc+ cells in fraction A, B, C, and C′ cells from WT/MycEGFP and Cd2icreRptorfl/fl/MycEGFP mice cultured for 4 hours in medium (left) or with IL-7 (right). (K) Five RWPCs during pro-B cell activation were defined by WGCNA. The central rectangle represents the first to third quartile, with notch in the plot corresponding to the median expression of all proteins in the sample. End of the whiskers represents minimum and maximum level of expression of the proteins in the cluster. (L) Functional annotations of RWPCs by Hallmark and KEGG databases (FDR < 0.1). (M and N) Rapamycin-dependent IL-7–induced temporal expression of proteins in mTORC1 signaling (M) and Myc targets (N). **P < 0.01, ***P < 0.001 [Mann-Whitney test (J) and one-way ANOVA with Tukey’s test (G and H)]. Results represent three (A to C, E, and F) or two (D, G, and I) independent experiments or are pooled from four (J) or two (G and H) independent experiments. Data are means ± SEM. Numbers indicate percentage of cells in gates. See also figs. S3 to S5 and data S3.
Because IL-7 is the major growth factor driving early B cell proliferation, we tested in vitro IL-7–induced proliferative response of BM cells from WT and Cd2icreRptorfl/fl mice. WT BM cells proliferated in response to IL-7 stimulation, but Raptor-deficient BM cells showed no population expansion (Fig. 3D). To circumvent the potential complication from the altered composition of developing B cells, we purified lineage-negative hematopoietic progenitor cells from WT and Cd2icreRptorfl/fl mice and, as controls, those from Rag-1−/− and Jak3−/− mice and cultured them with OP9 stromal cells in the presence of IL-7. The normal and defective B cell differentiation from Rag-1−/− and Jak3−/− progenitor cells, respectively, indicated that B cell differentiation is independent of pre-BCR but is dependent upon IL-7R signaling in this experimental system (fig. S3C). Raptor-deficient progenitor cells, similar to Jak3-deficient cells, failed to differentiate into B cells (fig. S3C), indicating that IL-7R signaling is severely impaired in Raptor-deficient progenitor cells.
The failure of Raptor-deficient BM cells to respond to IL-7 prompted us to examine the IL-7R signaling machinery. Whereas WT mice substantially up-regulated IL-7Rα expression in fraction B and C/C′ cells relative to fraction A cells, Raptor-deficient fraction B and C/C′ cells showed only modest up-regulation of IL-7Rα expression (fig. S3D). However, Rictor-deficient, Raptor/Rictor-deficient, and mTOR-deficient B cell precursor subsets maintained largely normal IL-7Rα expression (fig. S3, D and E). Except for the differential IL-7Rα expression, Cd2icreRptorfl/flRictorfl/fl and Cd2icreMtorfl/fl mice share almost identical B cell developmental defects as Cd2icreRptorfl/fl mice, as described above, thereby largely excluding the possibility that the reduced IL-7Rα expression in Cd2icreRptorfl/fl mice is functionally important for their B cell developmental defects.
We therefore examined IL-7–induced Stat5 activation and other signaling events in pro-B cells. Raptor-deficient pro-B cells had largely normal Stat5 activation in response to IL-7 stimulation (Fig. 3E). Also, IL-7–induced Stat5 nuclear translocation was not altered by mTORC1 inhibition (fig. S3F). Further downstream, the IL-7R–Stat5 signaling pathway drives the expression of a number of transcription factors that orchestrate B cell lineage differentiation; for example, TCF3 (also known as E2A), through activation of Ebf1 and Pax5, promotes B cell development (2, 10, 42–44). The expression of Tcf3, Ebf1, and Pax5 in Raptor-deficient pro-B cells was largely normal, indicative of comparable Stat5-dependent function (fig. S3G). In contrast, IL-7 induced potent mTORC1 activation, as indicated by phosphorylation of S6 and 4EBP1 (eukaryotic translation initiation factor 4E-binding protein 1), which was completely abolished in Raptor-deficient cells (Fig. 3E). Similarly, Raptor/Rictor double deficiency and mTOR deficiency did not affect IL-7–induced Stat5 phosphorylation but abolished mTORC1 activation (fig. S3, H and I). We further tested the role of mTORC1 in IL-7 signaling via pharmacological approaches. IL-7 treatment of in vitro cultured B cells induced rapid S6 kinase (S6K) and S6 phosphorylation, which was blocked by rapamycin. However, rapamycin treatment did not affect IL-7–mediated Stat5 activation (Fig. 3F). Together, Raptor acts downstream of IL-7R to mediate mTORC1 activation but not Stat5 signaling, suggesting a unique program orchestrated by Raptor-mTORC1 in B cell development.
To directly examine the role of IL-7–mTORC1 signaling in early B cell development in vivo, we injected recombinant IL-7 into mice treated with or without rapamycin, followed by analysis of pro-B cell subsets after 2 days of IL-7 treatment. Exogenous IL-7 expanded both fraction C and C′ populations and enhanced CD71 expression (Fig. 3, G and H). Strikingly, treatment of rapamycin nearly completely blocked the expansion of fraction C′ cells and CD71 up-regulation induced by IL-7 (Fig. 3, G and H). Thus, transient inhibition of mTORC1 blocks IL-7–mediated pro-B cell developmental progression.
Recent studies establish an important role of IL-7 in B cell development in adult humans (45, 46). To test whether IL-7 can activate human pro-B cells, we sorted BM pro-B cells from healthy donors and stimulated them with IL-7. IL-7 stimulation induced CD71 up-regulation and cell growth, indicating that IL-7 may also enhance cell metabolism in human pro-B cells (Fig. 3I).
The proto-oncogene Myc is essential for B cell development (26, 47, 48). Given the concomitant activation of mTORC1 and Myc in response to IL-7 stimulation (Fig. 1, G and H), we investigated the effect of Raptor deficiency on Myc expression in developing B cell subsets. Myc mRNA was low and comparable in fraction A cells from WT and Cd2icreRptorfl/fl mice but was elevated in fraction B and C/C′ cells in Raptor-deficient mice compared with WT counterparts (fig. S4A). To accurately measure Myc protein expression on a single-cell level, we crossed WT and Cd2icreRptorfl/fl mice with MycEGFP mice, which express an N-terminal fusion enhanced green fluorescent protein (EGFP)–Myc protein that faithfully marks endogenous Myc expression (49). In WT cells, EGFP-Myc expression progressively increased from fraction A to fraction C′ cells, with the highest expression in the more mature fraction C′ cells, consistent with a previous study (Fig. 3J and fig. S4B) (48). The increased EGFP-Myc expression was markedly reduced in Raptor-deficient fraction C and C′ cells (Fig. 3J, left panel, and fig. S4B). IL-7 treatment further induced EGFP-Myc expression in WT fraction B and C/C′ cells but not in fraction A cells (Fig. 3J, right panel, and fig. S4B), in line with the expression of IL-7R in these cell subsets described above. IL-7–induced EGFP-Myc expression was greatly diminished in the absence of Raptor in fraction C and C′ cells (Fig. 3J and fig. S4B). Further, whereas the percentage of EGFP-Myc+ cells in fraction B cells from Cd2icreRptorfl/fl mice was not significantly reduced compared to that of WT cells, the mean fluorescence intensity (MFI) of EGFP-Myc in Raptor-deficient cells was significantly lower than that in WT cells (fig. S4C). Thus, although IL-7 is known to promote Myc transcription in developing B cells (50), our results identify a novel mTORC1-dependent regulation of Myc protein expression downstream of IL-7 signaling. The functional significance of this interaction was further verified by the drastically impaired B cell development in hCd2-iCre–mediated deletion of Myc in developing B cells (fig. S4, D to F). Myc deficiency blocked B cell development at an earlier stage than Raptor deficiency (fig. S4E), which may reflect, in part, the difference between complete and partial reduction of Myc expression in these models (Fig. 3J and fig. S4, B and C). These results collectively demonstrate that mTORC1 activity is required for IL-7–induced Myc expression but not for Stat5 activation in pro-B cells.
mTORC1 promotes B cell development through transcriptional and translational programs
We next determined mTORC1-dependent transcriptional programs. To circumvent potential secondary effects introduced by chronic deficiency of Raptor, we crossed Rptorfl/fl mice with Rosa26-Cre-ERT2 (a Cre-ER fusion gene was recombined into the ubiquitously expressed Rosa26 locus) mice to generate CreERRptorfl/fl mice, in which Rptor was acutely deleted after injection of tamoxifen. Using the acute deletion system, we performed functional genomics to identify IL-7–mediated molecular pathways that are controlled by mTORC1. Pro-B cells from tamoxifen-treated CreERRptorfl/fl and control (CreERRptorfl/+) mice were cultured in the presence or absence of IL-7 for 24 hours, and RNA was subjected to global gene expression profiling. Principal components analysis showed that Raptor-deficient pro-B cells had distinct gene expression profiles compared to control pro-B cells and that IL-7 had a more pronounced effect on the gene expression of control pro-B cells than that of Raptor-deficient cells (fig. S5A). Specifically, IL-7 treatment of control pro-B cells resulted in a total of 1315 up-regulated and 713 down-regulated probes (by greater than 0.5 log2 FC). By comparison, IL-7 treatment of Raptor-deficient cells resulted in only 452 increased and 213 decreased probes (fig. S5B). Gene set enrichment analysis (GSEA) identified cell cycle–related genes as one of the most down-regulated pathways in Raptor-deficient cells with or without IL-7 stimulation (fig. S5C), consistent with the highly reduced EdU incorporation (Fig. 3B) in Raptor-deficient pro-B cells. Furthermore, although IL-7 modestly promoted the expression of cell cycle genes in Raptor-deficient pro-B cells, possibly through intact Stat5 signaling, the magnitude of activation was much smaller than that in control pro-B cells (fig. S5C). In addition to the cell cycle pathway, the cholesterol biosynthesis pathway was also significantly down-regulated in Raptor-deficient cells (fig. S5D).
Because mTORC1 also controls gene expression at the translational level, as in the case of regulating Myc expression (Fig. 3J and fig. S4, A to C), we profiled the mTORC1-dependent proteome in IL-7–stimulated pro-B cells. The proteome of freshly isolated (0 hour), IL-7–stimulated (16 hours), or IL-7/rapamycin–treated pro-B cells was analyzed using multiplexed TMT and LC/LC-MS/MS proteomic profiling. Variance analysis revealed consistency between replicates (fig. S5E). We identified that 455 DE proteins (data S3A) significantly changed using one-way ANOVA (10% FDR, z score of at least 2 between IL-7 and IL-7/rapamycin treatment groups). We applied WGCNA for these proteins and identified five clusters of coexpressed proteins, named as rapamycin-dependent whole protein clusters RWPC1 to RWPC5 (Fig. 3K and data S3B). RWPC1 and RWPC2, the two largest clusters, showed up-regulation upon IL-7 treatment but partial and more complete suppression with rapamycin, respectively. RWPC4 also showed rapamycin-mediated suppression despite the lack of strong effects induced by IL-7, whereas RWPC3 and RWPC5 revealed rapamycin-mediated activation. Pathway enrichment analysis revealed that RWPC1 and RWPC2 were enriched with Myc and mTORC1 signaling targets, as well as different organelle components and multiple lipid biosynthesis pathways (Fig. 3L). The inhibitory effects of rapamycin on IL-7–induced mTORC1 and Myc activation and lipid biosynthesis were illustrated in the heat maps (Fig. 3, M and N, and fig. S5, F and G). These data demonstrate that IL-7–mediated multiple metabolic programs may depend upon mTORC1 signaling and provide additional validations of the conclusions from Fig. 1 as well as our extensive biochemical and genetic analyses.
Deficiency of mTORC1 protects mice from Myc-induced B cell lymphoma
Myc is one of the most studied proto-oncogenes, whereas aberrant up-regulation of mTORC1 activity is also observed in multiple types of tumors including B cell malignancies (51). To investigate how mTORC1 regulates B cell tumorigenesis and further explore the interplay between mTORC1 and Myc, we bred Cd2icreRptorfl/fl with Eμ-Myc transgenic mice, which develop B cell lymphoma induced by overexpression of Myc via the IgH enhancer (Eμ) (25). Strikingly, Raptor deficiency completely suppressed Myc-induced lymphomagenesis and early death (Fig. 4A). Lymphomas arising from Eμ-Myc mice are at various stages of B cell development (52). In the BM, B220+ cells from most pre-tumor Eμ-Myc mice showed a continuous expression of CD43, with an increased frequency of B220+CD43+IgM− B cell precursors, whereas B cell development in Cd2icreRptorfl/flEμ-Myc mice was largely arrested at this precursor stage (Fig. 4B and fig. S6A). Within the B cell precursor compartment, cells from Cd2icreRptorfl/flEμ-Myc mice showed a phenotype similar to that of Cd2icreRptorfl/fl mice, with high expression of BP-1 and an expanded fraction C/C′ cell population, whereas B cells from Eμ-Myc mice were either BP-1+CD24+ or BP-1−CD24+ (fig. S6, B and C). Eμ-Myc mice had reduced immature and circulating mature B cells, which was further exacerbated in Cd2icreRptorfl/flEμ-Myc mice (Fig. 4C and fig. S6D). Myc overexpression drove increased DNA synthesis (measured by EdU incorporation; fig. S6E), anabolic metabolism (indicated by enhanced CD71 and CD98 expression), and cell growth (measured by FSC profile; Fig. 4D and fig. S6F), which were all largely rectified by Raptor deficiency. Consequently, Cd2icreRptorfl/flEμ-Myc mice, like Cd2icreRptorfl/fl mice, had highly reduced B cells in circulation (Fig. 4E). Thus, these data demonstrate that mTORC1 signaling in early B cell progenitors is critical for Myc-induced B cell lymphoma.
Fig. 4. Loss of Raptor prevents Myc-induced B cell malignancy.
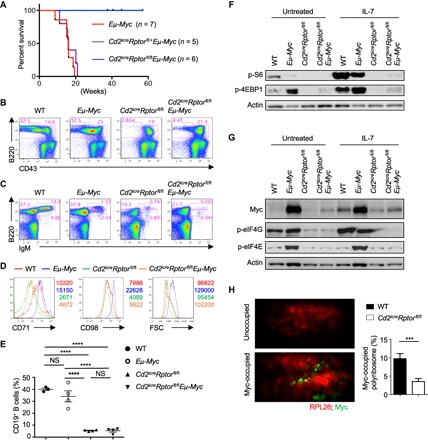
(A) Survival curve of Eμ-Myc, Cd2icreRptorfl/+Eμ-Myc, and Cd2icreRptorfl/flEμ-Myc mice (P = 0.0005, log-rank test). (B and C) Representative flow cytometry plots of BM cells of WT, Eμ-Myc, Cd2icreRptorfl/fl, and Cd2icreRptorfl/flEμ-Myc mice. (B) Expression of B220 and CD43 on BM lymphocytes, with the percentages of B220+CD43− and B220+CD43+ cells indicated. (C) Expression of B220 and IgM on BM lymphocytes, with the percentages of pro-B/pre-B cells (B220+IgM−), immature B cells (B220+IgM+), and circulating mature B cells (B220hiIgM+) indicated. (D) Analysis of CD71 and CD98 expression and cell size (via FSC) of pro-B cells. Numbers indicate the MFI. (E) Frequency of CD19+ B cells in the blood of WT, Eμ-Myc, Cd2icreRptorfl/fl, and Cd2icreRptorfl/flEμ-Myc mice. (F) Immunoblot analysis of p-S6 and p-4EBP1 in pro-B cells from WT, Eμ-Myc, Cd2icreRptorfl/fl, and Cd2icreRptorfl/flEμ-Myc mice. (G) Immunoblot analysis of Myc expression and phosphorylation of eIF4G and eIF4E in pro-B cells from WT, Eμ-Myc, Cd2icreRptorfl/fl, and Cd2icreRptorfl/flEμ-Myc mice. (H) 3D rendering of STORM imaging of polyribosomes (red) and Myc (green) in pro-B cells treated with IL-7. Left: Representative images of unoccupied (top) and Myc-occupied polyribosome (bottom). Right: The percentage of Myc-occupied polyribosomes in each imaged cell was enumerated. Data were pooled from three independent experiments, with a total of 35 cells from WT and 45 cells from Cd2icreRptorfl/fl mice examined. ***P < 0.001, ****P < 0.0001 [one-way ANOVA with Tukey’s test (E) and Mann-Whitney test (H)]. Results represent four (B and C), three (D, F, and H), or two (E and G) independent experiments. Data are means ± SEM. Numbers indicate percentage of cells in gates. See also fig. S6.
Mechanistically, Myc overexpression led to increased phosphorylation of 4EBP1 but reduced phosphorylation of S6 in pro-B cells (Fig. 4F). Because IL-7–mediated Myc up-regulation was dependent on mTORC1 (Fig. 3G), we tested whether Myc overexpression in Eμ-Myc mice was also dependent on mTORC1. Myc protein expression in Cd2icreRptorfl/flEμ-Myc pro-B cells was almost as low as that in WT cells, in contrast to the massive Myc expression found in Eμ-Myc cells (Fig. 4G). Various mTOR inhibitors also reduced Myc expression in Burkitt’s lymphoma cells (fig. S6G). In contrast, Myc mRNA level was similar between Eμ-Myc and Cd2icreRptorfl/flEμ-Myc pro-B cells (fig. S6H), suggesting that mTORC1 regulates Myc protein expression in Eμ-Myc mice. Furthermore, Myc protein stability was not affected by mTORC1 inhibition (fig. S6I). To directly examine Myc protein synthesis, we used stochastic optical reconstruction microscopy (STORM) to image the nascent Myc protein binding to polyribosomes in IL-7–stimulated pro-B cells (Fig. 4H and fig. S6J). We found a significant reduction of Myc protein binding to polyribosomes in Raptor-deficient pro-B cells (Fig. 4H), indicating that mTORC1 activity is required for efficient Myc protein synthesis.
Translation initiation is a major step for protein synthesis and an important target for cancer therapies (53). Eukaryotic translation initiation complex 4F (eIF4F) is a ternary complex composed of eIF4A, the cap-binding subunit eIF4E, and the scaffolding protein eIF4G. The binding of eIF4E to eIF4G is controlled by 4EBP1, whose phosphorylation and degradation are mediated by mTORC1 (53). How mTORC1 and Myc coordinate eIF4F activity is poorly defined (54). We found that Myc overexpression led to increased phosphorylation of eIF4E but did not significantly affect eIF4G phosphorylation (Fig. 4G). In contrast, the phosphorylation of both eIF4E and eIF4G was abolished in Cd2icreRptorfl/fl and Cd2icreRptorfl/flEμ-Myc pro-B cells (Fig. 4G) or markedly suppressed by mTOR inhibition (fig. S6G), suggesting that mTORC1 activity is required for eIF4G phosphorylation and Myc overexpression–induced eIF4E phosphorylation. Together, Myc overexpression promotes B cell transformation through a Myc-mTORC1 feed-forward circuitry, which can be interrupted through mTORC1 inhibition (fig. S6K).
Pro-B cell development requires active suppression of PI3K
PI3K-Akt is a well-established activator of mTOR in many cell types (19). However, deletion of p110α and p110δ (16), or p85α (17), does not affect pro-B cell development. Moreover, we observed elevated expression of PTEN, a crucial negative regulator of PI3K, in pro-B cells and pre-B cells than in other subsets (Fig. 5A), which correlated with increased IL-7Rα expression (fig. S1F). To test the functional importance of PI3K regulation, we deleted Pten using the hCd2-iCre system. Cd2icrePtenfl/fl mice contained a normal number of B220+CD43+IgM− cells (Fig. 5B), including normal numbers of fraction A and fraction B cells, but markedly reduced fraction C/C′ cells (Fig. 5C). Further, B220+CD25+ pre-B cell percentage and number were decreased substantially in the BM of Cd2icrePtenfl/fl mice (Fig. 5D). In addition, BM immature and circulating mature B cells (fig. S7A) were all profoundly lost in the absence of PTEN. These data show that deficiency of PTEN selectively blocks B cell development.
Fig. 5. Deletion of PTEN inhibits B cell development independent of mTORC1.
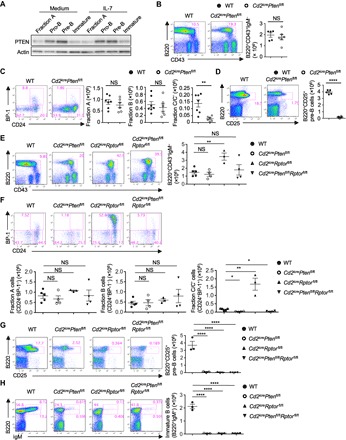
(A) Immunoblot analysis of PTEN in fraction A cells, pro-B cells, pre-B cells, and immature B cells with or without IL-7 stimulation for 4 hours. (B to D) Left: Representative flow cytometry plots of BM cells of WT and Cd2icrePtenfl/fl mice. Right: Absolute number of indicated populations. (B) Expression of B220 and CD43 on BM lymphocytes, with the percentage of B220+CD43+ cells indicated. Right: Number of B220+CD43+IgM− B cell precursors. (C) Expression of BP-1 and CD24 on B cell precursors, with the percentages of fraction A (CD24−BP-1−), fraction B (CD24+BP-1−), and fraction C/C′ (CD24+BP-1+) cells indicated. Right: Number of each subset. (D) Expression of B220 and CD25 on BM lymphocytes, with the percentage of B220+CD25+ pre-B cells indicated. Right: Number of B220+CD25+ pre-B cells. (E to H) Representative flow cytometry plots of BM cells of WT, Cd2icrePtenfl/fl, Cd2icreRptorfl/fl, and Cd2icrePtenfl/flRptorfl/fl mice. (E) Expression of B220 and CD43 on BM lymphocytes, with the percentage of B220+CD43+IgM− cells indicated. Right: Number of B220+CD43+IgM− cells. (F) Hardy classification of pro-B cell subsets in the B220+CD43+IgM− gate [from (B)], with the percentages of fraction A (CD24−BP-1−), fraction B (CD24+BP-1−), and fraction C/C′ (CD24+BP-1+) cells indicated. Bottom: Numbers of fraction A, B, and C/C′ cells. (G) Expression of B220 and CD25 on BM lymphocytes, with the percentage of B220+CD25+ pre-B cells indicated. Right: Number of B220+CD25+ pre-B cells. (H) Expression of B220 and IgM on BM lymphocytes, with the percentage of pro-B/pre-B cells (B220+IgM−), immature B cells (B220+IgM+), and mature B cells (B220hiIgM+) indicated. Right: Number of immature B cells. *P < 0.05, **P < 0.01, ****P < 0.0001 (one-way ANOVA with Tukey’s test). Results represent at least four independent experiments. Data are means ± SEM. Numbers indicate percentage of cells in gates. See also fig. S7.
Because PTEN has nuclear functions independent of PI3K-Akt (55, 56), we used hCd2-iCre to ectopically express a constitutively active form of the PI3K catalytic subunit p110α (encoded by Pik3ca) (57) in developing B cells (referred to as Pik3ca*). B cell development was blocked at the pro-B cell stage, with substantial loss of fraction C/C′ cells in Cd2icrePik3ca* mice (fig. S7B), and few pre-B cells emerged from Cd2icrePik3ca* mice (fig. S7C). Therefore, uncontrolled PI3K in pro-B cells blocks early B cell development, highlighting the importance of active suppression of PI3K in pro-B cells.
PI3K and mTORC1 independently control B cell development
The above observations were incongruent with the conventional model in which PI3K-Akt functions upstream to activate mTORC1 (19); for example, mTORC1 is required for mediating PTEN-deficient phenotypes in other contexts including hematopoietic stem cells (58). To conclusively test whether PI3K modulates B cell development through mTORC1, we crossed Cd2icrePtenfl/fl mice with Cd2icreRptorfl/fl mice to generate Cd2icrePtenfl/flRptorfl/fl mice. Deletion of PTEN and Raptor led to B cell developmental arrest at the pro-B cell stage associated with reduction of fraction C/C′ cells (Fig. 5, E and F) and profound loss of B220+CD25+ pre-B cells (Fig. 5G) and immature and mature B cells (Fig. 5H). Deletion of Raptor in Cd2icrePik3ca* mice led to a phenotype similar to that of Cd2icrePtenfl/flRptorfl/fl mice (fig. S7, D to F). Cd2icrePtenfl/flRptorfl/fl mice did not accumulate fraction C/C′ cells as observed in Cd2icreRptorfl/fl mice, possibly due to an earlier developmental block in the absence of PTEN (Fig. 5C). These results demonstrate that loss of mTORC1 does not restore B cell development in the absence of PTEN, and additionally, PI3K overactivation fails to rescue B cell developmental block induced by Raptor deficiency. We therefore conclude that mTORC1 and PTEN-PI3K pathways independently control early B cell development.
PTEN promotes pro-B cell survival and differentiation through Foxo1-dependent and Foxo1-independent mechanisms
Further mechanistic analyses of Cd2icrePtenfl/fl mice revealed a pronounced down-regulation of IL-7Rα expression, particularly in fraction B cells (Fig. 6A). It is well established that activation of the PI3K-Akt pathway suppresses Foxo1 activity, which is critical for IL-7Rα expression and B cell development (59). To test whether impaired Foxo1 activity could account for the B cell developmental defect in Cd2icrePtenfl/fl mice, we introduced a Rosa26-floxed stop mutant Foxo1 allele, which is refractory to Akt-induced inhibition and thus constitutively active (designated as Foxo1-CA) (60), into Cd2icrePtenfl/fl mice. Examination of IL-7Rα expression revealed that the reduced expression of IL-7Rα was largely restored in Cd2icrePtenfl/flFoxo1-CA mice (Fig. 6B). However, Cd2icrePtenfl/flFoxo1-CA mice had an even lower percentage of B220+CD43+IgM− precursor cells (Fig. 6C), with a small increase of fraction C/C′ cell proportion (Fig. 6D) and virtually no B220+CD25+ pre-B cells (Fig. 6E). Thus, Foxo1 likely contributes to IL-7Rα expression but does not account for B cell developmental block in Cd2icrePtenfl/fl mice.
Fig. 6. PTEN promotes early B cell development through Foxo1-dependent and Foxo1-independent mechanisms.
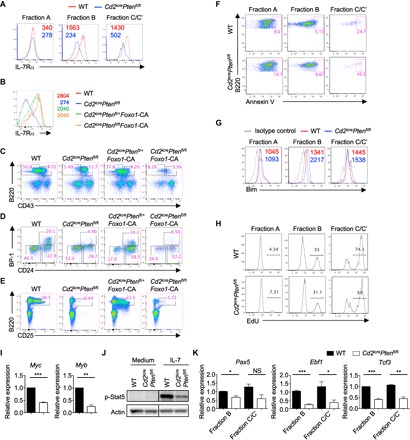
(A) Expression of IL-7Rα on fraction A, B, and C/C′ cells of WT and Cd2icrePtenfl/fl mice. Numbers indicate the MFI. (B to E) Representative flow cytometry plots of BM cells from WT, Cd2icrePtenfl/fl, Cd2icrePtenfl/+Foxo1-CA, and Cd2icrePtenfl/flFoxo1-CA mice. (B) Expression of IL-7Rα in pro-B cells. Numbers indicate the MFI. (C) Expression of B220 and CD43 on BM lymphocytes, with the percentage of B220+CD43+ and B220+CD43− cells indicated. (D) Hardy classification of pro-B cell subsets in the B220+CD43+IgM− gate (from C), with the percentages of fraction A (CD24−BP-1−), fraction B (CD24+BP-1−), and fraction C/C′ (CD24+BP-1+) cells indicated. (E) Expression of B220 and CD25 on BM lymphocytes, with the percentage of B220+CD25+ pre-B cells indicated. (F) Analysis of apoptotic cell death via annexin V staining on fraction A, B, and C/C′ cells. (G) Expression of Bim in fraction A, B, and C/C′ cells of WT and Cd2icrePtenfl/fl mice. Numbers indicate the MFI. (H) EdU incorporation in fraction A, B, and C/C′ cells of WT and Cd2icrePtenfl/fl mice. (I) Reverse transcription polymerase chain reaction (RT-PCR) analysis of Myc and Myb expression in pro-B cells. (J) Immunoblot analysis of p-Stat5 in pro-B cells of WT and Cd2icrePtenfl/fl mice. (K) RT-PCR analysis of Pax5, Ebf1, and Tcf3 expression in fraction B and C/C′ cells of WT and Cd2icrePtenfl/fl mice. *P < 0.05, **P < 0.01, ***P < 0.001 (Mann-Whitney test for cell frequencies and unpaired Student’s t test for cell numbers). Data represent four (A and F), three (J), or two (B to E, G to I, and K) independent experiments. Data are means ± SEM. Numbers indicate percentage of cells in gates. See also fig. S8.
To better understand how PTEN deficiency affects pro-B cell development, we compared gene expression profiles of pro-B cells from WT and Cd2icrePtenfl/fl mice. Ingenuity pathway analysis (IPA) showed that most of the top biological processes (based on P value) affected by PTEN deletion were down-regulated compared to WT cells, such as lymphocyte development and cellularity (fig. S8A). In addition, reduced cell viability was identified in PTEN-deficient pro-B cells, which was distinct from the prosurvival role of PI3K in mature B cells (57). We validated this finding by annexin V staining (Fig. 6F). PTEN-deficient B cell precursors also expressed more proapoptotic molecule Bim (Fig. 6G), whereas cell proliferation was largely normal (Fig. 6H). In addition, mRNA expression of two transcription factors critical for B cell development, Myc and Myb (61), was significantly reduced in PTEN-deficient pro-B cells (Fig. 6I). Moreover, immunoblot analysis showed that PTEN-deficient pro-B cells had impaired IL-7–induced Stat5 activation (Fig. 6J), consistent with the prosurvival effect of Stat5 signaling (12). As expected, PTEN-deficient pro-B cells had elevated Akt phosphorylation and mTORC1 activation (fig. S8B). The expression of Stat5-regulated genes Pax5, Ebf1, and Tcf3 was considerably reduced in PTEN-deficient early and fraction C/C′ cells (Fig. 6K). In addition, IL-7–induced population expansion in vitro was reduced in PTEN-deficient BM cells (fig. S8C). Therefore, PTEN function is required for the activation of Stat5, the expression of IL-7Rα and many Stat5-dependent transcription factors critical for B cell development, and the survival of pro-B cells. Deletion of PTEN selectively blocks B cell development at early to fraction C/C′ cell transition by affecting both Foxo1-dependent and Foxo1-independent pathways.
Raptor and PTEN deficiencies impair IgH rearrangement, but enforced expression of IgH and IgL does not rescue B cell developmental defects
B cell development is characterized by tightly controlled rearrangements of IgH and IgL chains, with intracellular expression of μ chain detected after successful rearrangement of the IgH locus in pro-B cells. IgH expression depends upon mTORC1 (20). Moreover, we found that deficiency of mTOR (Fig. 7A) or PTEN (Fig. 7B), greatly reduced IgH expression, indicating that heavy chain rearrangement could be impaired. Semiquantitative RT-PCR analysis showed that the expression of rearranged proximal VH7183-DJCμ and distal VHJ558-DJμ transcripts was substantially reduced in Raptor- and PTEN-deficient fraction B cells (Fig. 7, C and D). However, Raptor-deficient fraction C/C′ cells had a largely normal rearrangement, indicative of a temporary delay (Fig. 7C), whereas PTEN-deficient fraction C/C′ cells remained defective for IgH rearrangement (Fig. 7D). Thus, whereas both mTORC1 and PTEN are required for initiating intracellular heavy chain expression, only PTEN is essential for efficient IgH rearrangement in fraction C/C′ cells. Mechanistically, Ig rearrangement depends upon the induction of Rag1 and Rag2 expression. PTEN-deficient B cells, but not Raptor-deficient B cells, had highly reduced Rag1 and Rag2 expression (fig. S9, A and B), which may partly explain the differential defects in IgH rearrangement between these two genetic models.
Fig. 7. Loss of mTORC1 and PI3K overactivation exert differential effects on immunoglobulin gene rearrangement and expression.
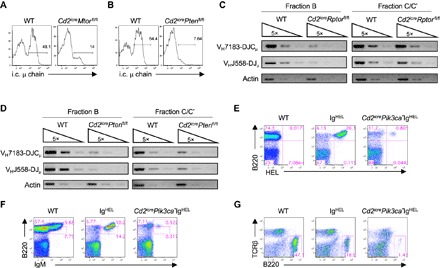
(A and B) Intracellular (i.c.) expression of Igμ chains in pro-B cells from Cd2icreMtorfl/fl (A) or Cd2icrePtenfl/fl mice (B). (C and D) Semiquantitative RT-PCR analysis of the expression of the rearranged VH7183-DJCμ and VHJ558-DJμ transcripts in fivefold serial dilutions of complementary DNA prepared from sorted fraction B and C/C′ cells from WT and Cd2icreRptorfl/fl (C) or Cd2icrePtenfl/fl mice (D). (E to G) Representative flow cytometry plots of BM cells (E and F) and splenocytes (G) of WT, IgHEL, and Cd2icrePik3ca*IgHEL mice. (E and F) Expression of B220 and HEL (E) and B220 and IgM (F) on BM lymphocytes. (G) Expression of T cell receptor β (TCRβ) and B220 on splenocytes. Results represent three (A and B) or two (C to G) independent experiments. Numbers indicate percentage of cells in quadrants or gates. See also fig. S9.
To test whether the impaired IgH and IgL expression accounts for B cell developmental defects in Raptor-deficient or Pik3ca*-expressing pro-B cells, we attempted to introduce rearranged IgH and IgL by crossing Cd2icreRptorfl/fl or Cd2icrePik3ca* mice with IgHEL mice, which carry a rearranged IgM/IgD transgene specific for the hen egg lysozyme (62). The IgHEL transgene was successfully introduced into Cd2icreRptorfl/fl mice (fig. S9C), but B cells failed to develop into circulating mature B cells (fig. S9D) in the BM or migrate to periphery (fig. S9E). In contrast, Cd2icrePik3ca*IgHEL mice expressed very little IgHEL transgene (Fig. 7E), and there were few mature B cells in the BM (Fig. 7F) or the spleen (Fig. 7G). The marked contrast between Cd2icreRptorfl/flIgHEL and Cd2icrePik3ca*IgHEL mice indicated that whereas loss of mTORC1 permits IgH transgene expression, suppression of PI3K is critically required for IgH expression. Furthermore, because the IgHEL transgene is able to rescue B cell development in Rag2-deficient mice (63), these results suggest that mechanisms other than defective Rag gene expression contribute to B cell developmental defects in Cd2icrePtenfl/fl mice. Finally, successful IgHEL expression cannot rescue B cell development in the absence of Raptor, indicating that mTORC1 is likely important for receptor-mediated downstream events.
DISCUSSION
Despite the well-recognized roles of IL-7R and Stat5 signaling in B cell development, the importance and mechanism of Stat5-independent pathways are poorly defined, and the global impact of IL-7 on pro-B cells is obscure. Further, how IL-7R–Stat5 signaling is regulated and the function of PI3K in pro-B cell development remain unclear. Here, we combined systems biology and mouse genetic models to investigate IL-7–dependent signaling programs in pro-B cells and dissect the functions of PI3K, mTORC1, and mTORC2 during early B cell development. Our comprehensive transcriptome and proteome analyses show that pro-B cell development is characterized by IL-7–mediated anabolic metabolism, organelle biogenesis, and mTORC1 and Myc activation. Deficiency of mTORC1 arrested B cell development mainly at the fraction C/C′ cell stage and disrupted pro-B cell subsets but without affecting Stat5 activation, B cell lineage transcription factor expression, or cell survival. In contrast, overactivation of PI3K due to PTEN deficiency blocked early B cell development at the fraction B stage, associated with impaired IL-7R–Stat5 signaling, reduced expression of B cell lineage transcription factors, and excessive apoptosis. Additional analyses of compound mutant mice in these two pathways allowed us to genetically dissociate mTORC1 and PI3K signaling, a conventional upstream signal of mTORC1. Notably, deletion of mTORC2 led to a modest increase of pro-B and pre-B cells but a normal immature B cell compartment. The B cell developmental defects in the absence of Raptor were largely phenocopied by loss of both mTORC1 and mTORC2, or mTOR itself, suggesting a predominant role of mTORC1, but not mTORC2, in early B cell development. Therefore, we have genetically defined two distinct signaling axes, mTORC1-Myc and PTEN-mediated PI3K suppression, crucial for early B cell development. Finally, mTORC1 promotes B cell development by activating Myc, and mTORC1 and Myc form a feed-forward circuitry to drive B cell lymphoma. Therefore, mTORC1 is a crucial regulator of both B cell development and malignancy and could serve as a legitimate therapeutic target.
The function of IL-7 in human B cell development appears to be age-dependent. Earlier studies on infant patients with severe combined immunodeficiency, including mutations in IL-7R, common γ chain, and Jak3, suggested that IL-7 signaling is not essential for human B cell development (64–67). Recent studies have revealed that IL-7 plays increasingly important roles for B cell production as ontogeny progresses, and it is essential for adult B lymphogenesis (45, 46, 68). Our data showed that the IL-7 and metabolic axis is operational in human pro-B cells, highlighting the relevance of our findings to human physiology.
Whereas the Jak1/3-Stat5 pathway is an established mechanism for IL-7–mediated pro-B cell development, the role of the PI3K-Akt pathway remains unclear. A recent study has shown that compound mutation of the PI3K catalytic subunits p110α and p110δ arrests B cell development at the pro-B to pre-B cell transition, but the pro-B cell compartment remains largely normal (16). However, p85α deficiency or pharmacological inhibition of PI3K does not affect pro-B cell proliferation (17). The PI3K-Akt pathway is the most well-known upstream activator for both mTORC1 and mTORC2 in many cellular contexts (19). Whereas p110α and p110δ double deficiency leads to increased VDJH rearrangement due to impaired down-regulation of Rag1 and Rag2 expression (16), Raptor-deficient B cells had reduced IgH rearrangement in fraction B cells, normal IgH rearrangement in fraction C/C′ cells, and normal Rag1 and Rag2 expression. These divergent phenotypes indicate that PI3K likely functions independently of mTORC1 during early B cell development. One possible upstream activator of mTORC1 could be 3-phosphoinositide-dependent protein kinase 1 (PDK1) because it activates mTORC1 in CD8+ T cells independent of PI3K (69), and PDK1 deficiency through Vav1-Cre–mediated deletion leads to some B cell phenotypes similar to mTORC1 deficiency (70).
Further underscoring the above point, although we anticipated an accelerated B cell development upon PI3K overactivation or PTEN deficiency, we were surprised to observe a profound block of B cell development at the fraction B stage, earlier than Raptor deficiency. It was reported earlier that PTEN is required for early B cell development, but the cellular and molecular mechanisms have been unclear (71). Mechanistically, PTEN deletion impaired IL-7R–Stat5 signaling and caused excessive apoptosis, distinct from the Stat5-independent activity of mTORC1. These observations are consistent with a prosurvival role of PTEN in pre-B acute lymphoblastic leukemia (ALL) (72) but are distinct from the prosurvival role of PI3K in mature B cells (57). The B cell developmental block caused by PTEN deletion is largely independent of Foxo1, a major target of PI3K-Akt and a critical factor for early B cell development (59). The expression of a constitutively active Foxo1 did not rescue B cell development but actually exacerbated the reduced B cell generation in PTEN-deficient mice. The underlying reason is unclear, but it may reflect the need of a precise temporal control of Foxo1 activity during early B cell development. These results suggest that B cell development requires two distinct and independent molecular signals: activation of mTORC1 to promote metabolic and proliferative activities, and proper control of PI3K activity via PTEN function to permit IL-7R-Stat5 signaling, cell survival, lineage transcription factor expression, and IgH rearrangement.
Whereas a previous study indicates that rapamycin can inhibit IL-7 signaling in ALL cells (73), how mTORC1 is regulated in normal B cell development has been unclear. Using both early and acute deletion systems, we provide the unequivocal evidence that mTORC1 activity is critically required for pro-B cell development downstream of IL-7R. Raptor-deficient B cell progenitors were unable to proliferate and differentiate in response to IL-7. Several lines of evidence suggest that Raptor-deficient pro-B cells have largely intact IL-7R–Stat5 signaling. First, IL-7–induced Stat5 phosphorylation was normal in the absence of Raptor. Second, freshly isolated Raptor-deficient pro-B cells did not exhibit increased cell death because one of the major functions of Stat5 is the maintenance of cell survival (12). Third, the expression of Stat5-dependent B cell lineage transcription factors Tcf3, Ebf1, and Pax5 was undisturbed in Raptor-deficient pro-B cells. Although IgH rearrangement was reduced in Raptor-deficient fraction B cells, it was recovered in fraction C/C′ cells. IgHEL transgene could be expressed in Raptor-deficient mice but not in PTEN-deficient mice, suggesting that mTORC1 is not absolutely required for IgH expression. Together, we conclude that the IL-7R–mTORC1 signaling pathway orchestrates a unique program essential for B cell development, distinct from Stat5 signaling, PTEN signaling, or IL-7R expression (fig. S9F).
IL-7 and pre-BCR coordinate early B cell development (3, 74). The loss of heavy chain expression precludes analysis of pre-BCR signaling in Raptor- or PTEN-deficient mice. However, the fact that IgHEL can be successfully expressed in Raptor-deficient mice, yet it fails to rescue B cell development, suggests that mTORC1 may also play a role in pre-BCR signaling. The potential function of mTOR in pre-BCR signaling warrants further investigation.
Our results reveal the intricate interplay between mTORC1 and Myc in both normal B cell development and lymphomagenesis. Similar to Cd2icreRptorfl/fl mice, Cd2icreMycfl/fl mice show developmental arrest at the pro-B cell stage (albeit with elevated severity), and these observations are recapitulated in Mb1-Cre–mediated deletion systems (20, 47). Further, overexpression of Myc leads to aggressive fatal B cell lymphoma (25) and can partially rescue B cell development in Jak3-deficient mice, indicating that Myc-mediated signaling events can partly compensate for defective IL-7–induced Jak3-Stat5 signaling (75). The mechanisms that control Myc expression in B cell physiology and malignancy have not been well studied. In cell lines, Stat5 has been shown to promote Myc mRNA transcription (76, 77). Here, we demonstrate that mTORC1 is required for IL-7–mediated Myc protein synthesis during normal B cell development and Myc overexpression during lymphomagenesis. Similar mTOR-dependent Myc translational regulation is also operative in myeloid cell lines (78). This raises an interesting model in which IL-7 promotes Myc expression through two independent pathways, namely, Stat5-dependent transcription and mTORC1-dependent translation (fig. S9F). Moreover, mTORC1 and Myc form a feed-forward circuitry in that Myc reinforces selective mTORC1 activity during lymphomagenesis. Specifically, Myc overexpression activates the 4EBP1, but not S6K (79) or S6, branch of mTORC1 and mTORC1-dependent eIF4E, but not eIF4G. Inhibition of mTORC1 interrupts this circuitry and blocks lymphoma development. Previous studies have shown that modulation of 4EBP1-mediated protein synthesis through ectopic expression of a degradation-resistant 4EBP1 mutant allele only has a partial inhibitory effect on tumor progression in Eμ-Myc tumor model (79). Instead, our data indicate that targeting mTORC1 at the pro-B stage completely blocks Myc-driven lymphoma formation. Aberrant activation of the mTORC1 pathway has been observed in many types of B cell lymphomas, and mTOR inhibitors are in early stages of clinical development (51). Our results provide mechanistic basis for targeting mTORC1 for B cell precursor malignancies.
MATERIALS AND METHODS
Animals
C57BL/6, CD45.1+, Rag1−/−, hCd2-iCre (Cd2icre), MycEGFP, Eμ-Myc, Mtorfl, and IgHEL mice were purchased from the Jackson Laboratory. ROSA26-Cre-ERT2, Rptorfl, Rictorfl, Ptenfl, Mycfl, and Pik3ca* mice have been previously described (57, 80–82). BM chimeras were generated by transferring 8 × 106 T cell–depleted BM cells into sublethally irradiated (5.5 Gy) Rag1−/− mice, followed by reconstitution for at least 2 months. For treatment of tamoxifen, mice were injected intraperitoneally with tamoxifen (1 mg per mouse) in corn oil daily for four consecutive days and analyzed 7 days after the last injection. IL-7 (250 μg/kg) was injected intraperitoneally daily for 2 days. Rapamune (15 mg/kg) was injected intraperitoneally daily for 3 days. All mice were kept in a specific pathogen–free condition in the Animal Resource Center at the St. Jude Children’s Research Hospital. All animal protocols were approved by the Institutional Animal Care and Use Committee of the St. Jude Children’s Research Hospital.
Human samples
Adult BM cells were purchased from Lonza Inc.
Flow cytometry
For analysis of surface markers, cells were stained in phosphate-buffered saline (PBS) containing 2% (w/v) fetal bovine serum (FBS) with indicated antibodies. The following antibodies were used: anti-B220 (RA3-6B2), anti-CD24 (M1/69), anti–BP-1 (6C3), anti-IgM (II/41), anti-IgD [11-26c (11–26)], anti–c-kit (ACK2), anti-CD21 (eBio8D9), anti-CD23 (B3B4), anti-CD93 (AA4.1), anti–IL-7Rα (A7R34), and anti-CD71 (R17217)—all were from eBioscience. Anti-CD25 (PC61) and Brilliant Violet 711–conjugated anti-B220 (RA3-6B2) were purchased from Sony Biotechnology. Anti-CD43 (S7) and anti-Igκ (187.1) were from BD Biosciences. Brilliant Violet 605–conjugated anti-B220 (RA3-6B2), anti-CD98 (RL388), anti-human CD19 (HIB19), anti-human CD10 (HI10a), anti-human CD34 (581), anti-human CD127 (A019D5), and anti-human CD71 (CY1G4) were from BioLegend. To exclude lineage-positive cells, cells were stained with a biotin mouse lineage panel (BD Biosciences), including anti-CD3e, anti–Ly-6G/Ly-6C, anti-CD11b, and anti–TER-119. Intracellular staining of Igμ was performed using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience). EdU (Life Technologies), active caspase-3 (BD Biosciences), and annexin V (BD Biosciences) staining were performed per the manufacturer’s instructions. Glucose uptake was measured by intravenous injection of 2-NBDG (Life Technologies) followed by flow cytometry analysis 30 min after injection. Flow cytometry data were acquired on an LSR II or LSRFortessa flow cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star).
Cell purification and culture
BM cells were extracted from the femur, tibia, and pelvis. Pro-B cell subsets were sorted on a Reflection or SY3200 cell sorter (i-Cyt). Sorted pro-B cells were cultured in Click’s medium (plus β-mercaptoethanol) supplemented with 10% (v/v) FBS and 1% penicillin-streptomycin. Cells were stimulated with IL-7 at 2 or 25 ng/ml and harvested at the indicated time points. For long-term IL-7 culture, total BM cells were cultured in Click’s medium with IL-7 (5 ng/ml). Cell numbers were determined using trypan blue exclusion. For the in vitro B cell differentiation assay, BM lineage-negative (B220−CD11b−Gr-1−CD4−CD8−TER119−) cells were purified by magnetic bead depletion. One hundred thousand cells were cultured with OP9 stromal cells in the presence of IL-7 (5 ng/ml). The numbers of B220+ cells were counted on day 9 by flow cytometry.
High-resolution fluorescence imaging
STORM was performed as previously described (83). Briefly, cells were cultured on Cell Tak–coated (5 μg/ml) LabTek II chambered coverslips in the presence of IL-7 for 2 hours before fixation for 10 min with 4% paraformaldehyde. Following rinsing with PBS, free reactive groups were quenched by incubation in sodium borohydride for 10 min. Cells were permeabilized for 3 min in PBS containing 0.1% Triton X-100, followed by blocking for 30 min in PBS buffer containing 2% bovine serum albumin (BSA) and 0.05% Tween 20. Samples were incubated with anti-Myc (250 ng/ml; Cell Signaling Technology, D84C12) and anti-RPL26 (25 ng/ml; Sigma, PLA0299), or anti-Stat5 (BioLegend, 9C8B50) in PBS containing BSA overnight at 4°C, followed by detection with Alexa Fluor 647–conjugated donkey anti-rabbit and Alexa Fluor 568–conjugated donkey anti-goat secondary antibodies (each at 500 ng/ml; Thermo Fisher Scientific) at room temperature for 1 hour. Samples were postfixed in 1% paraformaldehyde before image acquisition. Direct activator STORM (dSTORM) imaging was performed in tris-buffered saline buffer (pH 7.0) containing 10% glucose, 10 mM cysteamine, glucose oxidase, and catalase, as previously described (83). Images were acquired using a Nikon Ti-E inverted microscope equipped with a high-power Agilent laser launch, DU-897 high-speed electron-multiplying charge-coupled device camera, and 100× 1.45 numerical aperture (NA) oil objective. Image acquisition and analysis were facilitated using Nikon Elements software and algorithms for 3D molecule fitting and drift correction, as previously described (83, 84).
RNA and immunoblot analysis
RT-PCR analysis was performed as described (80) using primers and probe sets from Applied Biosystems or using the Power SYBR Green Master Mix from Life Technologies. Immunoblots were performed and quantified as previously described (80) using the following antibodies: anti–p-Stat5 (Tyr694), anti–p-S6K (108D2), anti–p-S6 (D57.2.2E), anti–p-4EBP1 (236B4), anti–p-Akt Ser473 (D9E), anti-Raptor (24C12), anti-Myc (D84C12), anti–p-eIF4E (Ser209), and anti–p-eIF4G (Ser1108) (all from Cell Signaling Technology) and actin (Sigma-Aldrich).
Metabolic assays
Total B cell precursors (Lin−B220+CD43+IgM−), B220+CD25+IgM− pre-B cells, and B220intIgM+ immature B cells were sorted by flow cytometry and used for Seahorse assays. Specifically, OCR and ECAR were measured in Seahorse XF Assay Media (unbuffered Dulbecco’s modified Eagle’s medium containing 25 mM glucose, 2 mM l-glutamine, and 1 mM sodium pyruvate) under basal condition and in response to 0.25 μM oligomycin, 1 μM fluorocarbonyl cyanide phenylhydrazone (FCCP), and 0.5 μM rotenone + 0.5 μM antimycin A with the XF-24 Extracellular Flux Analyzer (all from Seahorse Bioscience).
Whole proteome profiling by multiplex TMT-LC/LC-MS/MS
Pro-B cells for proteome profiling
Pro-B cells (Lin−B220+CD43+IgM−CD24+BP-1+/−) were sorted from BM cells of C57BL/6 mice by flow cytometry. Cells were stimulated with IL-7 (25 ng/ml) for 1, 4, and 16 hours (Fig. 1) or were pretreated with rapamycin (50 nM) for 30 min followed by IL-7 stimulation for 16 hours in the presence of rapamycin (Fig. 3, G and H).
Protein extraction, digestion, labeling, and pooling
Cells were washed twice with ice-cold PBS, and cell pellets from the 10 samples were lysed in fresh lysis buffer [50 mM Hepes (pH 8.5), 8 M urea, and 0.5% sodium deoxycholate]. The protein concentration of lysate was quantified by the BCA Protein Assay (Thermo Fisher Scientific). Proteins (10 μg) from each sample were loaded and run on a 10% SDS–polyacrylamide gel electrophoresis gel until all samples were inside of the gel. The gel was stained, and each sample was sliced and further chopped into 1-mm3 pieces. After gel detaining, the proteins were reduced with 5 mM dithiothreitol at 37°C for 30 min and alkylated with 10 mM iodoacetamide at room temperature in the dark for 30 min. Proteins were then digested in-gel with trypsin in 50 mM Hepes overnight at 37°C, and the protein-to-trypsin ratio (w/w) was 50:1. Peptides for each sample were extracted, dried, and labeled with 10-plex TMT reagents following the manufacturer’s instruction. Finally, the TMT-labeled samples were equally mixed.
Offline basic pH reversed-phase LC
The mixture of the 10 TMT-labeled samples was desalted, dried, and solubilized in 60 μl of buffer A (10 mM ammonium formate, pH 8) and separated on an XBridge C18 column (particle size, 3.5 μm; 4.6 mm × 25 cm; Waters) into 44 fractions with an 88-min gradient from 15 to 45% buffer B (95% acetonitrile, 10 mM ammonium formate, pH 8; flow rate, 0.4 ml/min). Each fraction was dried for whole proteome analysis.
Acidic pH reversed-phase LC-MS/MS
The analysis was performed on the basis of our optimized platform (85). For whole proteome analysis, the dried peptides were reconstituted in 5% formic acid and loaded on a reversed-phase column (75 μm × 30 cm, 1.9-μm C18 resin; Dr. Maisch GmbH) interfaced with a Q Exactive HF mass spectrometer (Thermo Fisher Scientific). Peptides were eluted by 12 to 36% buffer B gradient for 2.5 hours [buffer A: 0.2% formic acid and 3% DMSO; buffer B: buffer A plus 67% acetonitrile (flow rate of 0.25 μl/min)]. The column was heated at 65°C by a butterfly portfolio heater (Phoenix S&T) to reduce backpressure. Mass spectrometer was operated in data-dependent mode with a survey scan in Orbitrap [60,000 resolution, 1 × 106 automatic gain control (AGC) target, 50-ms maximal ion time] and 20 MS/MS high-resolution scans [60,000 resolution, 1 × 105 AGC target, 105-ms maximal ion time, high-energy collisional dissociation (HCD), 35 normalized collision energy, 1.0 mass/charge ratio (m/z) isolation window, and 20-s dynamic exclusion]. All raw data are available upon request and will be deposited in public databases upon manuscript acceptance.
Proteomic data analysis
The analysis was performed by our in-house JUMP search engine, which has been used in the data processing of numerous publications (23, 86–89). Briefly, acquired MS/MS raw files were converted into mzXML format and searched by the JUMP algorithm against a composite target/decoy database to estimate FDR. The target protein database was downloaded from the UniProt mouse database (52,490 protein entries), and the decoy protein database was generated by reversing all target protein sequences. Searches were performed with 10 ppm (parts per million) mass tolerance for both precursor ions and product ions, fully tryptic restriction, two maximal missed cleavages, and the assignment of a, b, and y ions. TMT tags on lysine residues and peptide N termini (+229.162932 Da) and carbamidomethylation of cysteine residues (+57.021 Da) were used for static modifications, and oxidation of methionine residues (+15.99492 Da) was used for dynamic modification. The assigned peptides were filtered by mass accuracy, minimal peptide length, matching scores, charge state, and trypticity to reduce protein FDR to below 1%.
TMT-based protein quantification
The analysis was performed by in-house JUMP software suite, as previously reported (34, 88). Briefly, TMT reporter ion intensities of each peptide spectrum match (PSM) were extracted, and the PSMs with very low intensity were removed. The raw intensities were then corrected on the basis of isotopic distribution of each labeling reagent and loading bias. The mean-centered intensities across samples were calculated, and protein relative intensities were derived by averaging related PSMs. Finally, protein absolute intensities were determined by multiplying the relative intensities by the grand mean of three most highly abundant PSMs.
Differential expression analysis of proteins
Differential expression events during B cell development were evaluated using R, as previously described (34). ANOVA was used for calculating the statistical significance followed by multiple testing correction using the Benjamini-Hochberg (BH) method (90, 91) on eight samples of pro-B cells. After applying threshold of corrected P value (<0.1), 2306 proteins were changed significantly during pro-B cell activation. We further applied an FC cutoff of 1.22, which was 2 SDs away from the mean of within replicate FC, that is, null comparison (in at least one pairwise comparison between time points). This resulted in 854 DE proteins.
WGCNA clustering analysis of proteomic data
Coexpression clustering analysis was performed using the WGCNA R package (32), as previously described (34). Briefly, using the eight samples of pro-B cells and the 854 DE proteins, the algorithm calculated a Pearson correlation matrix (with direction, that is, for building signed coexpression network), which was then converted to an adjacency matrix. Coexpression clusters were defined by the hybrid dynamic tree-cutting method (92) and merged if necessary. For each coexpression cluster, a consensus trend was calculated on the basis of the first principal component (also known as eigengene), and cluster membership was defined as Pearson correlation between individual protein and the consensus of the WPC. Proteins were assigned to the most correlated coexpression cluster (that is, WPC) with a cutoff of r of at least 0.7. Individual WPC was annotated using three pathway databases [GO, KEGG, and Hallmark downloaded from Molecular Signatures Database (MSigDB) (93)] by Fisher’s exact test (with BH FDR < 0.2).
PPI network analysis
Proteins in each WPC were superimposed onto the PPI database following the procedure that we previously described (34). The PPI database was constructed by merging the STRING PPI database (version 10) (94) with the affinity purification–MS–based BioPlex PPI database (using the latest version of June 2015) (95). Edges in the STRING database were filtered with the following requirements: (i) Edges with evidence of physical interactions (for example, through coimmunoprecipitation or yeast two-hybrid experiments) and (ii) edges of high confidence, as determined by the STRING score (with cutoff determined by requiring comparable degree distributions with BioPlex), were considered. The filtered STRING database was then combined with BioPlex to construct a composite PPI database. Modules were defined by a two-step procedure: (i) only retaining PPI edges of both nodes (that is, the two connected proteins) from the same WPC, resulting in a filtered PPI network defined by the DE proteins, and (ii) calculating a topological overlapping matrix (96) for this filtered PPI network and modularizing such a network into individual modules by the hybrid dynamic tree-cutting method (92). Individual module was annotated using three pathway databases (GO, KEGG, and Hallmark) by Fisher’s exact test and visualized by Cytoscape (97).
WGCNA clustering analysis of transcriptome data sets from ImmGen
Gene expression microarray data set corresponding to BM CLPs and B cell subsets, including fractions A, BC, C′, D, E, and F, were downloaded from Gene Expression Omnibus (GEO; GSE15907) using GEO2R. The expression matrix obtained was log2-transformed followed by differential expression analysis using one-way ANOVA. ANOVA P values were corrected for multiple testing using the BH method implemented in R. In addition, all pairwise comparisons were performed to calculate FCs for each gene. Finally, 2499 genes were called DE at BH-corrected P < 0.01 and absolute value of log2FC or |log2FC| > 1.5. Coexpression clustering analysis was performed on 2499 DE genes using WGCNA R package (32), which calculated a Pearson correlation matrix followed by calculation of an adjacency matrix by raising the correlation matrix to a power of 14 using scale-free topology criterion (98). Coexpression clusters were defined after the implementation of the dynamic tree-cutting method (92) with a minimum height of 0.15 to merge clusters. Each coexpression cluster was annotated using three pathway databases [GO, KEGG, and HALLMARK downloaded from MSigDB (93)] by Fisher’s exact test (BH FDR < 0.2).
Gene expression profiling and GSEA
B220+CD43+IgM− pro-B cells were sorted from WT and CreERRptorfl/fl mice 7 days after tamoxifen treatment. Cells were cultured in the presence or absence of IL-7 (25 ng/ml) for 24 hours. RNA samples were analyzed with the Mouse Gene 2.0 ST Signals array. Differentially expressed transcripts were identified by ANOVA (Partek Genomics Suite v6.5), and the BH method was used to estimate the FDR as previously described (80). GSEA was performed as previously described (80). Lists of DE genes at the 0.5 log2 (FC) cutoffs were used for IPA canonical pathway and upstream signaling analyses. Microarray data have been deposited into the GEO series database (GSE81844).
Statistical analysis
P values were calculated with Student’s t test, Mann-Whitney test, or ANOVA (GraphPad Prism), as specified in the figure legends. Tukey’s multiple comparisons test was performed following one-way ANOVA analysis.
Supplementary Material
Acknowledgments
We acknowledge M. Li for Foxo1-CA mice, the St. Jude Immunology FACS core facility for cell sorting, and N. Chapman and Y. Wang for scientific editing. Funding: This work was supported in part by NIH grants AI105887, AI101407, CA176624, and NS064599 and the American Asthma Foundation (to H.C.) and NIH grants PO1 HL44612 and R01 AI079087 (to D.W.) and R01 AG053987 (to J.P.). Author contributions: H.Z. designed and performed cellular, molecular, and biochemical experiments with all the mutant mice and wrote the manuscript; M.Y. designed and performed in vitro BM culture and Ig rearrangement assay; H.T. performed proteomic assay; Y.L., Y.D., and G.N. performed bioinformatics analysis; W.S. performed RT-PCR, flow cytometry, and immunoblot analysis; H.S. prepared samples for proteomic profiling and flow cytometry; C.G. performed Stat5 nuclear translocation, Myc, and polyribosome imaging studies; C.C. performed survival curve analysis; D.W. and J.P. designed experiments and provided direction; and H.C. designed experiments, wrote the manuscript, and provided overall direction. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/1/eaar5701/DC1
fig. S1. IL-7Rα mediates dynamic metabolic changes during B cell development.
fig. S2. mTORC1, but not mTORC2, is required for B cell development.
fig. S3. mTORC1 promotes B cell development independent of Stat5.
fig. S4. mTORC1 promotes Myc protein expression to support B cell development.
fig. S5. mTORC1-mediated transcriptional and translational programs in pro-B cells.
fig. S6. An mTORC1-Myc circuitry controls B cell malignancy.
fig. S7. Deficiency of PTEN or overactivation of PI3K blocks early B cell development.
fig. S8. Impaired differentiation and survival in PTEN-deficient pro-B cells.
fig. S9. Expression of Rag1 and Rag2 in Raptor- and PTEN-deficient B cells.
data S1. Bioinformatics analysis of gene expression in developing B cell subsets.
data S2. Whole proteome analysis of IL-7–stimulated pro-B cells.
data S3. Whole proteome analysis of IL-7– and rapamycin-treated pro-B cells.
REFERENCES AND NOTES
- 1.Rothenberg E. V., Transcriptional control of early T and B cell developmental choices. Annu. Rev. Immunol. 32, 283–321 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kikuchi K., Lai A. Y., Hsu C.-L., Kondo M., IL-7 receptor signaling is necessary for stage transition in adult B cell development through up-regulation of EBF. J. Exp. Med. 201, 1197–1203 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clark M. R., Mandal M., Ochiai K., Singh H., Orchestrating B cell lymphopoiesis through interplay of IL-7 receptor and pre-B cell receptor signalling. Nat. Rev. Immunol. 14, 69–80 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sereti I., Dunham R. M., Spritzler J., Aga E., Proschan M. A., Medvik K., Battaglia C. A., Landay A. L., Pahwa S., Fischl M. A., Asmuth D. M., Tenorio A. R., Altman J. D., Fox L., Moir S., Malaspina A., Morre M., Buffet R., Silvestri G., Lederman M. M. ACTG 5214 Study Team , IL-7 administration drives T cell–cycle entry and expansion in HIV-1 infection. Blood 113, 6304–6314 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sportès C., Babb R. R., Krumlauf M. C., Hakim F. T., Steinberg S. M., Chow C. K., Brown M. R., Fleisher T. A., Noel P., Maric I., Stetler-Stevenson M., Engel J., Buffet R., Morre M., Amato R. J., Pecora A., Mackall C. L., Gress R. E., Phase I study of recombinant human interleukin-7 administration in subjects with refractory malignancy. Clin. Cancer Res. 16, 727–735 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mackall C. L., Fry T. J., Gress R. E., Harnessing the biology of IL-7 for therapeutic application. Nat. Rev. Immunol. 11, 330–342 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacobs S. R., Michalek R. D., Rathmell J. C., IL-7 is essential for homeostatic control of T cell metabolism in vivo. J. Immunol. 184, 3461–3469 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rathmell J. C., Farkash E. A., Gao W., Thompson C. B., IL-7 enhances the survival and maintains the size of naive T cells. J. Immunol. 167, 6869–6876 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Wofford J. A., Wieman H. L., Jacobs S. R., Zhao Y., Rathmell J. C., IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood 111, 2101–2111 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yao Z., Cui Y., Watford W. T., Bream J. H., Yamaoka K., Hissong B. D., Li D., Durum S. K., Jiang Q., Bhandoola A., Hennighausen L., O’Shea J. J., Stat5a/b are essential for normal lymphoid development and differentiation. Proc. Natl. Acad. Sci. U.S.A. 103, 1000–1005 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoelbl A., Kovacic B., Kerenyi M. A., Simma O., Warsch W., Cui Y., Beug H., Hennighausen L., Moriggl R., Sexl V., Clarifying the role of Stat5 in lymphoid development and Abelson-induced transformation. Blood 107, 4898–4906 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malin S., McManus S., Cobaleda C., Novatchkova M., Delogu A., Bouillet P., Strasser A., Busslinger M., Role of STAT5 in controlling cell survival and immunoglobulin gene recombination during pro-B cell development. Nat. Immunol. 11, 171–179 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dai X., Chen Y., Di L., Podd A., Li G., Bunting K. D., Hennighausen L., Wen R., Wang D., Stat5 is essential for early B cell development but not for B cell maturation and function. J. Immunol. 179, 1068–1079 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Goetz C. A., Harmon I. R., O’Neil J. J., Burchill M. A., Farrar M. A., STAT5 activation underlies IL7 receptor-dependent B cell development. J. Immunol. 172, 4770–4778 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Ochiai K., Maienschein-Cline M., Mandal M., Triggs J. R., Bertolino E., Sciammas R., Dinner A. R., Clark M. R., Singh H., A self-reinforcing regulatory network triggered by limiting IL-7 activates pre-BCR signaling and differentiation. Nat. Immunol. 13, 300–307 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramadani F., Bolland D. J., Garcon F., Emery J. L., Vanhaesebroeck B., Corcoran A. E., Okkenhaug K., The PI3K isoforms p110α and p110δ are essential for pre–B cell receptor signaling and B cell development. Sci. Signal. 3, ra60 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Powers S. E., Mandal M., Matsuda S., Miletic A. V., Cato M. H., Tanaka A., Rickert R. C., Koyasu S., Clark M. R., Subnuclear cyclin D3 compartments and the coordinated regulation of proliferation and immunoglobulin variable gene repression. J. Exp. Med. 209, 2199–2213 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chi H., Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol. 12, 325–338 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saxton R. A., Sabatini D. M., mTOR signaling in growth, metabolism, and disease. Cell 169, 361–371 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Iwata T. N., Ramírez J. A., Tsang M., Park H., Margineantu D. H., Hockenbery D. M., Iritani B. M., Conditional disruption of raptor reveals an essential role for mTORC1 in B cell development, survival, and metabolism. J. Immunol. 197, 2250–2260 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee K., Heffington L., Jellusova J., Nam K. T., Raybuck A., Cho S. H., Thomas J. W., Rickert R. C., Boothby M., Requirement for Rictor in homeostasis and function of mature B lymphoid cells. Blood 122, 2369–2379 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niu M., Cho J.-H., Kodali K., Pagala V., High A. A., Wang H., Wu Z., Li Y., Bi W., Zhang H., Wang X., Zou W., Peng J., Extensive peptide fractionation and y1 ion-based interference detection method for enabling accurate quantification by isobaric labeling and mass spectrometry. Anal. Chem. 89, 2956–2963 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X., Li Y., Wu Z., Wang H., Tan H., Peng J., JUMP: A tag-based database search tool for peptide identification with high sensitivity and accuracy. Mol. Cell. Proteomics 13, 3663–3673 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Boer J., Williams A., Skavdis G., Harker N., Coles M., Tolaini M., Norton T., Williams K., Roderick K., Potocnik A. J., Kioussis D., Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur. J. Immunol. 33, 314–325 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Adams J. M., Harris A. W., Pinkert C. A., Corcoran L. M., Alexander W. S., Cory S., Palmiter R. D., Brinster R. L., The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 318, 533–538 (1985). [DOI] [PubMed] [Google Scholar]
- 26.Langdon W. Y., Harris A. W., Cory S., Adams J. M., The c-myc oncogene perturbs B lymphocyte development in Eμ-myc transgenic mice. Cell 47, 11–18 (1986). [DOI] [PubMed] [Google Scholar]
- 27.MacIver N. J., Michalek R. D., Rathmell J. C., Metabolic regulation of T lymphocytes. Annu. Rev. Immunol. 31, 259–283 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagasawa T., Microenvironmental niches in the bone marrow required for B-cell development. Nat. Rev. Immunol. 6, 107–116 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Zheng Y., Collins S. L., Lutz M. A., Allen A. N., Kole T. P., Zarek P. E., Powell J. D., A role for mammalian target of rapamycin in regulating T cell activation versus anergy. J. Immunol. 178, 2163–2170 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Hardy R. R., Carmack C. E., Shinton S. A., Kemp J. D., Hayakawa K., Resolution and characterization of pro-B and pre-pro-B cell stages in normal mouse bone marrow. J. Exp. Med. 173, 1213–1225 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heng T. S. P., Painter M. W. Immunological Genome Project Consortium , The Immunological Genome Project: Networks of gene expression in immune cells. Nat. Immunol. 9, 1091–1094 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Langfelder P., Horvath S., WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jansen R., Greenbaum D., Gerstein M., Relating whole-genome expression data with protein-protein interactions. Genome Res. 12, 37–46 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tan H., Yang K., Li Y., Shaw T. I., Wang Y., Blanco D. B., Wang X., Cho J.-H., Wang H., Rankin S., Guy C., Peng J., Chi H., Integrative proteomics and phosphoproteomics profiling reveals dynamic signaling networks and bioenergetics pathways underlying T cell activation. Immunity 46, 488–503 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duy C., Yu J. J., Nahar R., Swaminathan S., Kweon S.-M., Polo J. M., Valls E., Klemm L., Shojaee S., Cerchietti L., Schuh W., Jäck H.-M., Hurtz C., Ramezani-Rad P., Herzog S., Jumaa H., Koeffler H. P., de Alborán I. M., Melnick A. M., Ye B. H., Müschen M., BCL6 is critical for the development of a diverse primary B cell repertoire. J. Exp. Med. 207, 1209–1221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hennet T., Chui D., Paulson J. C., Marth J. D., Immune regulation by the ST6Gal sialyltransferase. Proc. Natl. Acad. Sci. U.S.A. 95, 4504–4509 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Siegemund S., Shepherd J., Xiao C., Sauer K., hCD2-iCre and Vav-iCre mediated gene recombination patterns in murine hematopoietic cells. PLOS ONE 10, e0124661 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hobeika E., Thiemann S., Storch B., Jumaa H., Nielsen P. J., Pelanda R., Reth M., Testing gene function early in the B cell lineage in mb1-cre mice. Proc. Natl. Acad. Sci. U.S.A. 103, 13789–13794 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang S., Pruitt M., Tran D., Du Bois W., Zhang K., Patel R., Hoover S., Simpson R. M., Simmons J., Gary J., Snapper C. M., Casellas R., Mock B. A., B cell–specific deficiencies in mTOR limit humoral immune responses. J. Immunol. 191, 1692–1703 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang S., Readinger J. A., DuBois W., Janka-Junttila M., Robinson R., Pruitt M., Bliskovsky V., Wu J. Z., Sakakibara K., Patel J., Parent C. A., Tessarollo L., Schwartzberg P. L., Mock B. A., Constitutive reductions in mTOR alter cell size, immune cell development, and antibody production. Blood 117, 1228–1238 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y., Hu T., Hua C., Gu J., Zhang L., Hao S., Liang H., Wang X., Wang W., Xu J., Liu H., Liu B., Cheng T., Yuan W., Rictor is required for early B cell development in bone marrow. PLOS ONE 9, e103970 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goetz C. A., Harmon I. R., O’Neil J. J., Burchill M. A., Johanns T. M., Farrar M. A., Restricted STAT5 activation dictates appropriate thymic B versus T cell lineage commitment. J. Immunol. 174, 7753–7763 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Hirokawa S., Sato H., Kato I., Kudo A., EBF-regulating Pax5 transcription is enhanced by STAT5 in the early stage of B cells. Eur. J. Immunol. 33, 1824–1829 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Mandel E. M., Grosschedl R., Transcription control of early B cell differentiation. Curr. Opin. Immunol. 22, 161–167 (2010). [DOI] [PubMed] [Google Scholar]
- 45.Milford T.-A. M., Su R. J., Francis O. L., Baez I., Martinez S. R., Coats J. S., Weldon A. J., Calderon M. N., Nwosu M. C., Botimer A. R., Suterwala B. T., Zhang X.-B., Morris C. L., Weldon D. J., Dovat S., Payne K. J., TSLP or IL-7 provide an IL-7Rα signal that is critical for human B lymphopoiesis. Eur. J. Immunol. 46, 2155–2161 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parrish Y. K., Baez I., Milford T.-A., Benitez A., Galloway N., Rogerio J. W., Sahakian E., Kagoda M., Huang G., Hao Q.-L., Sevilla Y., Barsky L. W., Zielinska E., Price M. A., Wall N. R., Dovat S., Payne K. J., IL-7 dependence in human B lymphopoiesis increases during progression of ontogeny from cord blood to bone marrow. J. Immunol. 182, 4255–4266 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vallespinós M., Fernández D., Rodríguez L., Alvaro-Blanco J., Baena E., Ortiz M., Dukovska D., Martínez D., Rojas A., Campanero M. R., Moreno de Alborán I., B lymphocyte commitment program is driven by the proto-oncogene c-myc. J. Immunol. 186, 6726–6736 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Sandoval G. J., Graham D. B., Bhattacharya D., Sleckman B. P., Xavier R. J., Swat W., Cutting edge: Cell-autonomous control of IL-7 response revealed in a novel stage of precursor B cells. J. Immunol. 190, 2485–2489 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang C.-Y., Bredemeyer A. L., Walker L. M., Bassing C. H., Sleckman B. P., Dynamic regulation of c-Myc proto-oncogene expression during lymphocyte development revealed by a GFP-c-Myc knock-in mouse. Eur. J. Immunol. 38, 342–349 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Morrow M. A., Lee G., Gillis S., Yancopoulos G. D., Alt F. W., Interleukin-7 induces N-myc and c-myc expression in normal precursor B lymphocytes. Genes Dev. 6, 61–70 (1992). [DOI] [PubMed] [Google Scholar]
- 51.Waibel M., Gregory G., Shortt J., Johnstone R. W., Rational combination therapies targeting survival signaling in aggressive B-cell leukemia/lymphoma. Curr. Opin. Hematol. 21, 297–308 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Wen R., Chen Y., Bai L., Fu G., Schuman J., Dai X., Zeng H., Yang C., Stephan R. P., Cleveland J. L., Wang D., Essential role of phospholipase Cγ2 in early B-cell development and Myc-mediated lymphomagenesis. Mol. Cell. Biol. 26, 9364–9376 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bhat M., Robichaud N., Hulea L., Sonenberg N., Pelletier J., Topisirovic I., Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 14, 261–278 (2015). [DOI] [PubMed] [Google Scholar]
- 54.So L., Lee J., Palafox M., Mallya S., Woxland C. G., Arguello M., Truitt M. L., Sonenberg N., Ruggero D., Fruman D. A., The 4E-BP–eIF4E axis promotes rapamycin-sensitive growth and proliferation in lymphocytes. Sci. Signal. 9, ra57 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song M. S., Carracedo A., Salmena L., Song S. J., Egia A., Malumbres M., Pandolfi P. P., Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell 144, 187–199 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shen W. H., Balajee A. S., Wang J., Wu H., Eng C., Pandolfi P. P., Yin Y., Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 128, 157–170 (2007). [DOI] [PubMed] [Google Scholar]
- 57.Srinivasan L., Sasaki Y., Calado D. P., Zhang B., Paik J. H., DePinho R. A., Kutok J. L., Kearney J. F., Otipoby K. L., Rajewsky K., PI3 kinase signals BCR-dependent mature B cell survival. Cell 139, 573–586 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kalaitzidis D., Sykes S. M., Wang Z., Punt N., Tang Y., Ragu C., Sinha A. U., Lane S. W., Souza A. L., Clish C. B., Anastasiou D., Gilliland D. G., Scadden D. T., Guertin D. A., Armstrong S. A., mTOR complex 1 plays critical roles in hematopoiesis and Pten-loss-evoked leukemogenesis. Cell Stem Cell 11, 429–439 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dengler H. S., Baracho G. V., Omori S. A., Bruckner S., Arden K. C., Castrillon D. H., DePinho R. A., Rickert R. C., Distinct functions for the transcription factor Foxo1 at various stages of B cell differentiation. Nat. Immunol. 9, 1388–1398 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ouyang W., Liao W., Luo C. T., Yin N., Huse M., Kim M. V., Peng M., Chan P., Ma Q., Mo Y., Meijer D., Zhao K., Rudensky A. Y., Atwal G., Zhang M. Q., Li M. O., Novel Foxo1-dependent transcriptional programs control Treg cell function. Nature 491, 554–559 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fahl S. P., Crittenden R. B., Allman D., Bender T. P., c-Myb is required for pro-B cell differentiation. J. Immunol. 183, 5582–5592 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mason D. Y., Jones M., Goodnow C. C., Development and follicular localization of tolerant B lymphocytes in lysozyme/anti-lysozyme IgM/IgD transgenic mice. Int. Immunol. 4, 163–175 (1992). [DOI] [PubMed] [Google Scholar]
- 63.Park H., Staehling K., Tsang M., Appleby M. W., Brunkow M. E., Margineantu D., Hockenbery D. M., Habib T., Liggitt H. D., Carlson G., Iritani B. M., Disruption of Fnip1 reveals a metabolic checkpoint controlling B lymphocyte development. Immunity 36, 769–781 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Macchi P., Villa A., Giliani S., Sacco M. G., Frattini A., Porta F., Ugazio A. G., Johnston J. A., Candotti F., O’Shea J. J., Vezzoni P., Notarangelo L. D., Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID). Nature 377, 65–68 (1995). [DOI] [PubMed] [Google Scholar]
- 65.Noguchi M., Yi H., Rosenblatt H. M., Filipovich A. H., Adelstein S., Modi W. S., McBride O. W., Leonard W. J., Interleukin-2 receptor γ chain mutation results in X-linked severe combined immunodeficiency in humans. Cell 73, 147–157 (1993). [DOI] [PubMed] [Google Scholar]
- 66.Puel A., Ziegler S. F., Buckley R. H., Leonard W. J., Defective IL7R expression in T−B+NK+ severe combined immunodeficiency. Nat. Genet. 20, 394–397 (1998). [DOI] [PubMed] [Google Scholar]
- 67.Russell S. M., Tayebi N., Nakajima H., Riedy M. C., Roberts J. L., Aman M. J., Migone T.-S., Noguchi M., Markert M. L., Buckley R. H., O’Shea J. J., Leonard W. J., Mutation of Jak3 in a patient with SCID: Essential role of Jak3 in lymphoid development. Science 270, 797–800 (1995). [DOI] [PubMed] [Google Scholar]
- 68.Bendall S. C., Davis K. L., Amir E.-a.D., Tadmor M. D., Simonds E. F., Chen T. J., Shenfeld D. K., Nolan G. P., Pe’er D., Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell 157, 714–725 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Finlay D. K., Rosenzweig E., Sinclair L. V., Feijoo-Carnero C., Hukelmann J. L., Rolf J., Panteleyev A. A., Okkenhaug K., Cantrell D. A., PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 209, 2441–2453 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Venigalla R. K. C., McGuire V. A., Clarke R., Patterson-Kane J. C., Najafov A., Toth R., McCarthy P. C., Simeons F., Stojanovski L., Arthur J. S. C., PDK1 regulates VDJ recombination, cell-cycle exit and survival during B-cell development. EMBO J. 32, 1008–1022 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Alkhatib A., Werner M., Hug E., Herzog S., Eschbach C., Faraidun H., Köhler F., Wossning T., Jumaa H., FoxO1 induces Ikaros splicing to promote immunoglobulin gene recombination. J. Exp. Med. 209, 395–406 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shojaee S., Chan L. N., Buchner M., Cazzaniga V., Cosgun K. N., Geng H., Qiu Y. H., von Minden M. D., Ernst T., Hochhaus A., Cazzaniga G., Melnick A., Kornblau S. M., Graeber T. G., Wu H., Jumaa H., Müschen M., PTEN opposes negative selection and enables oncogenic transformation of pre-B cells. Nat. Med. 22, 379–387 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brown V. I., Fang J., Alcorn K., Barr R., Kim J. M., Wasserman R., Grupp S. A., Rapamycin is active against B-precursor leukemia in vitro and in vivo, an effect that is modulated by IL-7-mediated signaling. Proc. Natl. Acad. Sci. U.S.A. 100, 15113–15118 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fleming H. E., Paige C. J., Pre-B cell receptor signaling mediates selective response to IL-7 at the pro-B to pre-B cell transition via an ERK/MAP kinase-dependent pathway. Immunity 15, 521–531 (2001). [DOI] [PubMed] [Google Scholar]
- 75.Dillon S. R., Schlissel M. S., Partial restoration of B cell development in Jak-3(−/−) mice achieved by co-expression of IgH and E(mu)-myc transgenes. Int. Immunol. 14, 893–904 (2002). [DOI] [PubMed] [Google Scholar]
- 76.Lord J. D., McIntosh B. C., Greenberg P. D., Nelson B. H., The IL-2 receptor promotes lymphocyte proliferation and induction of the c-myc, bcl-2, and bcl-x genes through the trans-activation domain of Stat5. J. Immunol. 164, 2533–2541 (2000). [DOI] [PubMed] [Google Scholar]
- 77.Pinz S., Unser S., Rascle A., Signal transducer and activator of transcription STAT5 is recruited to c-Myc super-enhancer. BMC Mol. Biol. 17, 10 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wall M., Poortinga G., Hannan K. M., Pearson R. B., Hannan R. D., McArthur G. A., Translational control of c-MYC by rapamycin promotes terminal myeloid differentiation. Blood 112, 2305–2317 (2008). [DOI] [PubMed] [Google Scholar]
- 79.Pourdehnad M., Truitt M. L., Siddiqi I. N., Ducker G. S., Shokat K. M., Ruggero D., Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc. Natl. Acad. Sci. U.S.A. 110, 11988–11993 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zeng H., Yang K., Cloer C., Neale G., Vogel P., Chi H., mTORC1 couples immune signals and metabolic programming to establish Treg-cell function. Nature 499, 485–490 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yang K., Shrestha S., Zeng H., Karmaus P. W. F., Neale G., Vogel P., Guertin D. A., Lamb R. F., Chi H., T cell exit from quiescence and differentiation into Th2 cells depend on raptor-mTORC1-mediated metabolic reprogramming. Immunity 39, 1043–1056 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang R., Dillon C. P., Shi L. Z., Milasta S., Carter R., Finkelstein D., McCormick L. L., Fitzgerald P., Chi H., Munger J., Green D. R., The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liedmann S., Hrincius E. R., Guy C., Anhlan D., Dierkes R., Carter R., Wu G., Staeheli P., Green D. R., Wolff T., McCullers J. A., Ludwig S., Ehrhardt C., Viral suppressors of the RIG-I-mediated interferon response are pre-packaged in influenza virions. Nat. Commun. 5, 5645 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Huang B., Wang W., Bates M., Zhuang X., Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 319, 810–813 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang H., Yang Y., Li Y., Bai B., Wang X., Tan H., Liu T., Beach T. G., Peng J., Wu Z., Systematic optimization of long gradient chromatography mass spectrometry for deep analysis of brain proteome. J. Proteome Res. 14, 829–838 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hanna J. A., Garcia M. R., Go J. C., Finkelstein D., Kodali K., Pagala V., Wang X., Peng J., Hatley M. E., PAX7 is a required target for microRNA-206-induced differentiation of fusion-negative rhabdomyosarcoma. Cell Death Dis. 7, e2256 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li Y., Wang X., Cho J.-H., Shaw T. I., Wu Z., Bai B., Wang H., Zhou S., Beach T. G., Wu G., Zhang J., Peng J., JUMPg: An integrative proteogenomics pipeline identifying unannotated proteins in human brain and cancer cells. J. Proteome Res. 15, 2309–2320 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mertz J., Tan H., Pagala V., Bai B., Chen P.-C., Li Y., Cho J. H., Shaw T., Wang X., Peng J., Sequential elution interactome analysis of the mind bomb 1 ubiquitin ligase reveals a novel role in dendritic spine outgrowth. Mol. Cell. Proteomics 14, 1898–1910 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pierce W. K., Grace C. R., Lee J., Nourse A., Marzahn M. R., Watson E. R., High A. A., Peng J., Schulman B. A., Mittag T., Multiple weak linear motifs enhance recruitment and processivity in SPOP-mediated substrate ubiquitination. J. Mol. Biol. 428, 1256–1271 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Benjamini Y., Hochberg Y., Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 57, 289–300 (1995). [Google Scholar]
- 91.Storey J. D., Tibshirani R., Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U.S.A. 100, 9440–9445 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Langfelder P., Zhang B., Horvath S., Defining clusters from a hierarchical cluster tree: The Dynamic Tree Cut package for R. Bioinformatics 24, 719–720 (2008). [DOI] [PubMed] [Google Scholar]
- 93.Liberzon A., Subramanian A., Pinchback R., Thorvaldsdóttir H., Tamayo P., Mesirov J. P., Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K. P., Kuhn M., Bork P., Jensen L. J., von Mering C., STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huttlin E. L., Ting L., Bruckner R. J., Gebreab F., Gygi M. P., Szpyt J., Tam S., Zarraga G., Colby G., Baltier K., Dong R., Guarani V., Vaites L. P., Ordureau A., Rad R., Erickson B. K., Wühr M., Chick J., Zhai B., Kolippakkam D., Mintseris J., Obar R. A., Harris T., Artavanis-Tsakonas S., Sowa M. E., De Camilli P., Paulo J. A., Harper J. W., Gygi S. P., The BioPlex network: A systematic exploration of the human interactome. Cell 162, 425–440 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ravasz E., Somera A. L., Mongru D. A., Oltvai Z. N., Barabasi A.-L., Hierarchical organization of modularity in metabolic networks. Science 297, 1551–1555 (2002). [DOI] [PubMed] [Google Scholar]
- 97.Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T., Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang B., Horvath S., A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 4, Article17 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/1/eaar5701/DC1
fig. S1. IL-7Rα mediates dynamic metabolic changes during B cell development.
fig. S2. mTORC1, but not mTORC2, is required for B cell development.
fig. S3. mTORC1 promotes B cell development independent of Stat5.
fig. S4. mTORC1 promotes Myc protein expression to support B cell development.
fig. S5. mTORC1-mediated transcriptional and translational programs in pro-B cells.
fig. S6. An mTORC1-Myc circuitry controls B cell malignancy.
fig. S7. Deficiency of PTEN or overactivation of PI3K blocks early B cell development.
fig. S8. Impaired differentiation and survival in PTEN-deficient pro-B cells.
fig. S9. Expression of Rag1 and Rag2 in Raptor- and PTEN-deficient B cells.
data S1. Bioinformatics analysis of gene expression in developing B cell subsets.
data S2. Whole proteome analysis of IL-7–stimulated pro-B cells.
data S3. Whole proteome analysis of IL-7– and rapamycin-treated pro-B cells.