

Abstract
Pure red cell aplasia is an orphan disease, and as such lacks rationally established standard therapies. Most cases are idiopathic; a subset is antibody-mediated. There is overlap between idiopathic cases and those with T-cell large granular lymphocytic leukemia, hypogammaglobulinemia, and low-grade lymphomas. In each of the aforementioned, the pathogenetic mechanisms may involve autoreactive cytotoxic responses. We selected 62 uniformly diagnosed pure red cell aplasia patients and analyzed their pathophysiologic features and responsiveness to rationally applied first-line and salvage therapies in order to propose diagnostic and therapeutic algorithms that may be helpful in guiding the management of prospective patients, 52% of whom were idiopathic, while the others involved large granular lymphocytic leukemia, thymoma, and B-cell dyscrasia. T-cell-mediated responses ranged between a continuum from polyclonal to monoclonal (as seen in large granular lymphocytic leukemia). During a median observation period of 40 months, patients received a median of two different therapies to achieve remission. Frequently used therapy included calcineurin-inhibitors with a steroid taper yielding a first-line overall response rate of 76% (53/70). Oral cyclophosphamide showed activity, albeit lower than that produced by cyclosporine. Intravenous immunoglobulins were effective both in parvovirus patients and in hypogammaglobulinemia cases. In salvage settings, alemtuzumab is active, particularly in large granular lymphocytic leukemia-associated cases. Other potentially useful salvage options include rituximab, anti-thymocyte globulin and bortezomib. The workup of acquired pure red cell aplasia should include investigations of common pathological associations. Most effective therapies are directed against T-cell-mediated immunity, and therapeutic choices need to account for associated conditions that may help in choosing alternative salvage agents, such as intravenous immunoglobulin, alemtuzumab and bortezomib.
Introduction
Pure red cell aplasia (PRCA) can be inherited (Diamond Blackfan Anemia, [DBA]) or acquired (aPRCA). The latter is further subclassified as B19 parvovirus-associated (transient) aplastic crisis (TAC),1 drug-associated cases2 (e.g., allopurinol, azathioprine, diphenylhydantoin, rifampicin, valproic acid etc.), primary idiopathic aPRCA with various (immune) etiologies, and secondary aPRCA associated with other conditions, including B-cell dyscrasias (chronic lymphocytic leukemia [CLL]),3 Waldenström macroglobulinemia,4 monoclonal gammopathy of undetermined significance (MGUS)5 and multiple myeloma,6 T-cell lymphoproliferative disorders (large granular lymphocytic (LGL) leukemia)7,8 or solid organ malignancies (most commonly thymoma),9 and collagen vascular/autoimmune processes with aPRCA overlap.10–13 Myelodysplastic syndromes (MDS) can mimic PRCA morphologically.14 aPRCA may also be a forerunner of acquired aplastic anemia (AA).
The hallmark of PRCA is reticulocytopenic, malproductive anemia (absolute reticulocyte count < 10,000/ml [reticulocyte percentage, <1%]),15 which is diagnosed after the exclusion of obvious causes of anemia. The absence or profound depletion of erythroid precursors is sine qua non for PRCA. Secondary PRCA is often dominated by the underlying disease. B19-associated PRCA is due to the lytic activity of the virus on pronormoblasts,16 which is seen as large proerythroblasts with vacuolated cytoplasm and pseudopodia (“giant pronormoblasts”).
After the exclusion of viral etiologies, congenital diseases and drug reactions, idiopathic PRCA would be the most common cause, with the majority of cases thought to be mediated by autoreactive T-cells. This is likely via selective T- or natural killer (NK)-cell-mediated killing of erythroid colony (CFU-E) and burst (BFU-E) forming units, thereby inhibiting red cell precursor progression to mature erythrocytes.7,17–20 By analogy with acquired neutropenia, “idiopathic” PRCA is typically T-cell-mediated. In contrast, “autoimmune PRCA” mediated by antibodies is less common,10,13 and most antierythroid antibodies would typically result in immune hemolytic anemia. Anti-erythropoietin (EPO) antibody-mediated PRCA induced by recombinant EPO can be considered as a specific form of autoimmune-PRCA.21,22 Thymoma-associated PRCA may be either considered as secondary or primary with immune etiology. While some patients respond to T-cell-directed immunosuppression, other patients have underlying diseases that involve humoral immunity (e.g., relation to myasthenia gravis),9 and those are cases which are less responsive to such therapies. A similar association is suggested by the occurrence of PRCA in the context of Good’s syndrome (thymoma, combined variable immunodeficiency and PRCA).23
Although PRCA is rare, it is very diverse. This diversity seems to be related to overlapping pathologic and clinical associations for which systematic studies of these correlations have not been performed. Clearly, the rarity of the disease precludes the accumulation of clinical experience based on clinical trial evidence. Instead, current practices are mostly driven by retrospective analyses of case reports and series. Using data from a large number of PRCA patients seen in our center, we analyzed clinical presentations, pathophysiologic features, and clinical responses to different therapeutic options.
Methods
Patient populations
Based on uniformly defined criteria (Online Supplementary Table S1), 62 cases of PRCA, confirmed by bone marrow biopsy and treated at the Cleveland Clinic between the years 2000 and 2016, were included in the retrospective analysis (Online Supplementary Table S2 and Online Supplementary Figure S1). Informed consent was obtained per protocols approved by the Institutional Review Board in accordance with the Declaration of Helsinki. Clinical parameters of the patients, including demographics, blood counts, treatment specifics, and survival times, were obtained from medical records. The primary end-point was a hematological response in terms of an appropriate rise in reticulocytes/hemoglobin and becoming transfusion-independent, as determined by two separate measurements after the first eight weeks of initiating the treatment. This is the earliest time for assessment of response. By contrast, borrowed from more systematic studies in aplastic anemia and systemic immunosuppression, response was again assessed at the 3-month milestone, and in less common circumstances monitored for six months until defining it as a non-responder to a particular treatment. Normal blood counts were determined using hospital standards.
Response criteria
Complete response (CR) was defined by an appropriate rise in the reticulocyte count, commensurate to the level of hemoglobin rise, in addition to becoming transfusion-independent followed by subsequent normalization of hemoglobin levels after eight weeks of initiating the treatment (Online Supplementary Table S3). Partial response (PR) was defined when there was no appropriate rise in reticulocyte count and the patient was still anemic, but their transfusion requirement became less frequent than it was prior to initiating the drug. No response (NR) was defined when none of the above criteria were met at the end of eight weeks. Complete response rate (CRR) was calculated as the percentage of patients with CR over the total number of patients who received the drug. Overall response rate (ORR) was calculated as the percentage of patients with both CR and PR over the total number of patients who received the drug. We have distinguished primary and a co-associated PRCA cases. In those secondary cases with T-cell large granular lymphocytic (T-LGL) leukemia, the corresponding LGL leukemia treatment would not differ from that of “idiopathic” PRCA, and response in LGL would be determined according to the improvement of anemia. However, in cases associated with B-cell dyscrasia, B-cell modalities would include rituximab or bortezomib. In the latter cases, the response was monitored by monoclonal protein levels, but due to the low number of cases, a correlation between the level of decrease in M-protein and the overall response was not statistically established.
DNA Targeted Sequencing
Targeted sequencing was completed using Nextera Custom Enrichment library (Illumina). The custom targeted panel consisted of capture probes for genes known to be relevant in MDS, acute myeloid leukemia (AML) and other hematologic malignancies (both somatic and germline targets, N=169). The list of genes targeted can be found in Online Supplementary Table S4. The enriched targets were subjected to massive sequencing using Illumina MiSeq sequencer, with an average target depth of 260x.
The generation of BAM (.bam) files (alignment with Burrows-Wheeler Aligner [BWA] 0.7.9a) with its preprocessing and detection of somatic point mutations/insertions and deletions was carried out in accordance with the Genome Analysis Toolkit (GATK) best practices. Only variants with high-quality reads and a minimum depth of at least 20 and six positives were considered for further analysis. Sequencing results were annotated using ANNOVAR.24 Variants were considered somatic if they were not reported within the Exome Aggregation Consortium (ExAC) germline database (ExAC, Cambridge, MA, USA). Novel splicing, stop-gain and insertions-deletions variants were prioritized and studied further.
Statistical analysis
The R package forest plot was used to plot odds ratios (OR) computed using Fisher’s exact test and response rates, the 95% confidence intervals of which were computed using logistic regression, i.e., the R function generalized linear model (glm) was used with y ~ 1 as the model and the statistical distribution family set to binomial.
Results
Clinical features of PRCA
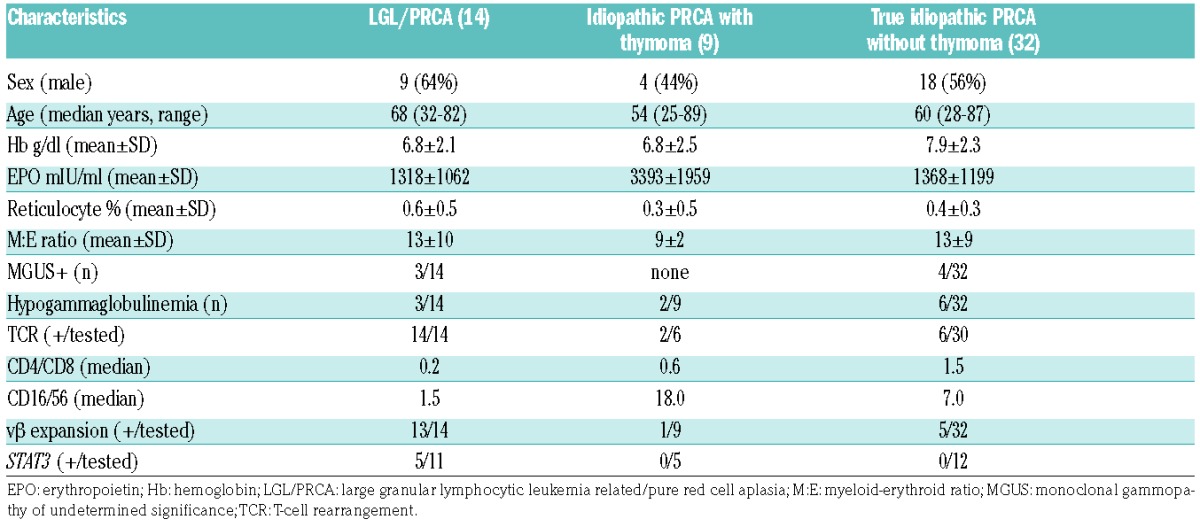
From January 2000 to December 2016, we saw and treated 62 patients with PRCA who fulfilled the diagnostic criteria of one of the clinical subentities. The clinical classification of PRCA patients (n=62) in our cohort is demonstrated in Online Supplementary Figure S1 and Online Supplementary Table S2. The pathogenesis of PRCA seen in our cohort of patients is illustrated in Figure 1. Excluding the DBA patients (Online Supplementary Table S5), the median age at presentation was 62 years (25–87 y) with a slight male predominance (n=34, 55%; P=0.6). Among the aPRCA patients (Online Supplementary Table S1), most were idiopathic (n=32, 52%) or followed by LGL leukemia (n=14, 22%) or thymoma (n=9, 15%); less common were B19-related PRCA patients (n=4, 6%; P=0.00004). Intriguingly, one patient initially treated for LGL-associated PRCA was later found to be positive for parvovirus on repeated DNA polymerase chain reaction (PCR) testing. The clinical characteristics of the acquired immune-mediated PRCA groups are listed in Table 1. The median follow-up of aPRCA patients in our cohort was 40 months (range: 1–133 months). The total number of aPRCA patients who were in remission during the last follow up was 46 (including partial and complete). The median number of different therapeutic approaches used to achieve remission was two (range: 1–8). Maintenance treatment with immunosuppressive agents was usually continued for a year or two, at the least, and gradually tapered off if the disease was stable. The OS and disease-free probability at 5/10 years in our cohort were 0.835 (95% confidence interval [CI] 0.695, 1)/0.674 (95% CI 0.472, 0.963) and 0.675 (95% CI 0.529, 0.861)/0.496 (95% CI 0.321, 0.764), respectively (Online Supplementary Table S2).
Figure 1.

Pathogenesis of pure red cell aplasia (PRCA) in our cohort. (A) Different causes of PRCA affect red cell production in different ways. Parvovirus causes direct cytolysis of erythroid precursor cells. Several drugs are implicated in causing PRCA, probably through a direct toxic effect. EPO antibodies can recognize antigens at the erythroid burst forming unit (BFU-E) level and cause PRCA. There is a wide range of antigen recognition by CTL exhibiting a polyclonal response, seen as negative TCR (T-cell gene rearrangement) by testing, which, however, still causes PRCA, probably by direct cytotoxicity. At the other end of the spectrum are LGLs that are monoclonal with STAT3 mutations and can recognize antigens at the erythroid precursor level, and thus cause PRCA. (B) Flow cytometry Vβ analysis of the peripheral blood of individual representative patients from the cohort shows clonal skewing. Illustration shows a spectrum of polyclonal to oligoclonal to monoclonal response of CTL in individual patients with PRCA.
Table 1.
Patient characteristics (including immunological/molecular characteristics) in different acquired immune-mediated PRCA groups.
Immunological characterization of acquired immune-mediated PRCA patients
The distributions of MGUS, hypogammaglobulinemia, presence of T-cell receptor (TCR) rearrangement, median CD4/CD8 ratio, median CD16/CD56 ratio, and Vβ expansion are listed in Table 1. There were eight cases in the idiopathic aPRCA group who had clonal TCR rearrangement but did not fulfill the criteria for LGL (Table 1 and Online Supplementary Table S6). The CD4/CD8 ratio was significantly lower among the LGL/PRCA group (0.2) compared to the idiopathic group with and without thymoma (0.9 and 1.5; P=0.0064) (Online Supplementary Figure S2). Some cases (n=6) of idiopathic aPRCA also demonstrated Vβ expansion. However, it appeared that there was a significant clinical overlap between LGL-associated and idiopathic cases of aPRCA.
Mutational status of acquired PRCA patients
The STAT3 mutation in the LGL/PRCA patients were positive in 5/11 patients tested, whereas 17 patients tested negative in the idiopathic group. Mutational analysis using a targeted myeloid next-generation sequencing (NGS) panel (Nextera, Illumina; see Methods) was performed in a selected subgroup of patients during diagnostic workup of their anemia. No somatic clonal mutations typical of MDS were found in 12 patients tested with idiopathic, otherwise typical aPRCA. Alterations found in genes affected in MDS were present in five typical idiopathic aPRCA patients and were considered to be of germline origin (Online Supplementary Table S7). None of these patients fulfilled the diagnostic criteria for MDS.
Immunosuppressive therapy
A rational treatment algorithm applied to our patients is illustrated in Figure 2. The choice of our first-line treatment differed based on the underlying cause of PRCA. The first-line CRR, overall CRR and ORR are illustrated in Figure 3 (see also Table 2 and Table 3).
Figure 2.

Treatment algorithm for immune-mediated PRCA in our patient cohort. Cyclosporine or cyclophosphamide with a steroid taper is the first-line choice of treatment in both idiopathic and LGL- related PRCA. Maintenance treatment with immunosuppressive therapy is usually needed, and varies based on the sustainability of the response obtained (see text for details). Methotrexate is used in a salvage setting in LGL/PRCA, but has no role in idiopathic PRCA. Alemtuzumab is one of the commonly used salvage options in refractory PRCA.
Figure 3.

Response probabilities across various treatment options in aPRCA. (A) Cyclosporine and tacrolimus are the most effective drugs for PRCA in our cohort. The details of IVIG response rates are elaborated in Table 3. Rituximab is used only in the salvage setting when the front-line options have failed, including commonly used salvage treatments such as alemtuzumab. ATG is used purely as a salvage option, but only shows mediocre, but acceptable, response rates. The other salvage treatment options include danazol, mycophenolate mofetil, bortezomib, erythropoietin, abatacept (Orencia), and tofacitinib (Xeljanz) etc., all of which show good response rates when used in the right setting (see text for details). (B) Response proportion for different treatment options: comparison between LGL vs. non-LGL PRCA groups. Cyclosporine and tacrolimus show better RR in the non-LGL group, whereas campath (alemtuzumab), though not statistically significant, works better in LGL-PRCA. Patients with parvovirus PRCA, and those presenting with low immunoglobulin levels in PRCA show excellent RR with IVIG (see also Table 3).
Table 2.
Treatment response rates in all aPRCA patients.

Table 3.
Treatment response rates in LGL and non-LGL aPRCA patients.
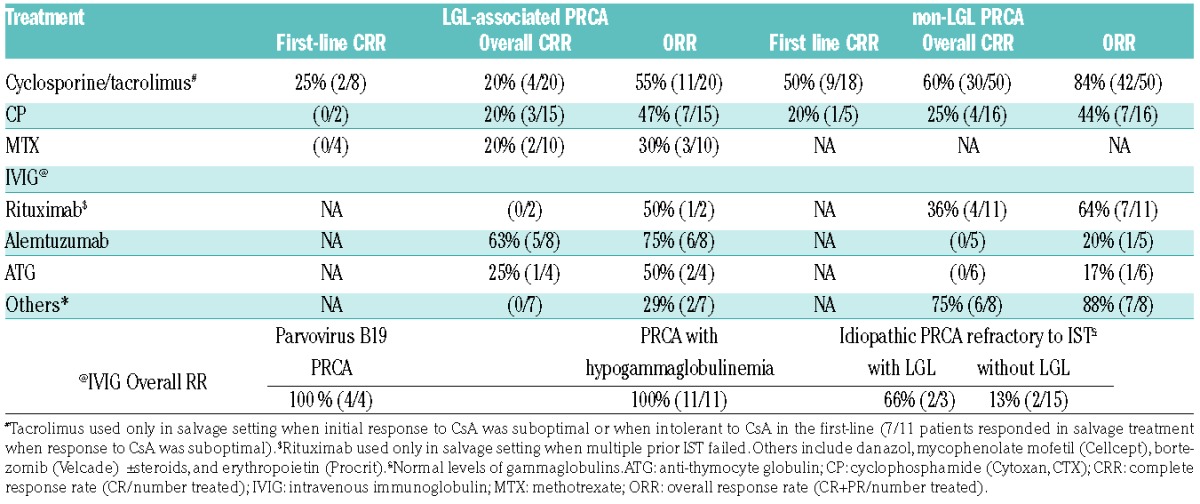
The most common first-line treatment used in idiopathic aPRCA was cyclosporine (CsA) combined with a steroid taper (prednisone [P]). This yielded an ORR of 76% (53/70) and first-line CRR and overall CRR of 40% (10/25) and 49% (34/70), respectively. Slight alterations in creatinine after initiation of CsA triggered periodic monitoring of kidney function. Further drug administration was either stopped or adjusted if creatinine trends worsened. ORR was better in non-LGL- vs. LGL-related PRCA (84% vs. 55%, P=0.01) (Figure 3B and Table 3). CsA was avoided as a first-line treatment, especially in patients with renal failure. Tacrolimus was used in salvage mode or in lieu of CsA when it was not tolerated by patients.
The second most common first-line choice of treatment was cyclophosphamide (CP, [Cytoxan, (CTX)]) with a steroid taper. ORR was 47% (14/30), first-line CRR 15% (1/7) and overall CRR 27% (8/30), i.e., lower than with CsA (P=0.6 and P=0.2, respectively). In subgroup analysis, there was no discernible difference in overall RR among the LGL and non-LGL group (Figure 3B and Table 3). Occasionally, methotrexate (MTX) was used as a first-line choice of treatment with a steroid taper, especially in the context of LGL-associated PRCA: the overall CRR remained low (20%, 2/10) along with the ORR (30%, 3/10) (Figure 3A and Table 2).
In our cohort, 13 refractory patients received Campath (alemtuzumab) therapy with an ORR of 54% (7/13) (Figure 3A and Table 2). Campath, used only as a salvage option on a compassionate basis, showed promising results in the LGL-associated PRCA group, with an ORR of 75% and overall CRR of 63% (Figure 3B and Table 3). Campath was given at a dose of 10 mg/week for 4–6 weeks following a test dose of 3 mg in the first week, and assessed for response with careful monitoring of the blood counts. Epstein-Barr virus (EBV) and cytomegalovirus (CMV) testing were carried out prior to starting campath so that appropriate antiviral prophylaxis could be given with the drug. Patients with good responses stayed on a maintenance dose of 10 mg subcutaneously every four to eight weeks based on their response; in most cases, clinical response was associated with the improvement of relative - or when present - absolute lymphocytosis and a decrease in the percentages of LGL in the blood. None of our patients had any adverse events following the use of campath at this dose. When anti-thymocyte globulin (ATG) was used in otherwise refractory aPRCA, ORR was low at 30% with only 1/10 patients showing a CR (Figure 3, Tables 2 and Table 3). Rituximab, used only in the salvage setting (mainly deployed in situations where the patients failed multiple T-cell-directed immunosuppressive therapy), showed similarly low RR (overall CRR 36% and ORR 64%; Table 2 and Table 3).
Special considerations
Intravenous immunoglobulin (IVIG), primarily used in parvovirus-related PRCA, showed a 100% ORR in four selected patients (Table 3), and 3/4 patients remained in complete remission during their last follow up (median follow-up of 23.4 months), except for one patient whose response only lasted for about three months, and who was still not in CR after several rounds of different immunosuppressive therapies (IST). IVIG was also found to be effective in patients with low immunoglobulins (including Good’s syndrome) with an excellent ORR (11/11, 100%), however, the response was short-lived, lasting less than three months for most, with the exception of three patients who remained in CR, requiring only monthly maintenance of IVIG. The response to IVIG was only mediocre when used in idiopathic aPRCA patients who were refractory to prior rounds of multiple IST (22%, 4/18; Table 3). In two patients with MGUS and PRCA, bortezomib (Velcade) was used and a CR and PR were achieved. Similarly, a response was seen in patients with PRCA and immunoplasmacytic lymphoma/von Waldenstrom’s macroglobulinemia. Out of nine patients with thymoma, one achieved a CR after thymectomy without additional treatment; 6/7 remained in CR after requiring maintenance IST. With respect to DBA patients in our cohort, 3/4 were responsive to prednisone and/or anabolic steroids until the end of last follow up.
Other salvage options used include anabolic steroids (danazol), mycophenolate mofetil (cellcept), abatacept (orencia), tofacitinib (xeljanz) and epoetin alfa (procrit). Orencia and cellcept were used primarily in LGL PRCA patients who failed to respond to campath, our first-line salvage option for them. In general, second-line salvage options, used in the right setting, showed a moderate ORR of 60%, but an overall RR of 88% in the non-LGL group (Figure 3, Table 2 and Table 3).
Discussion
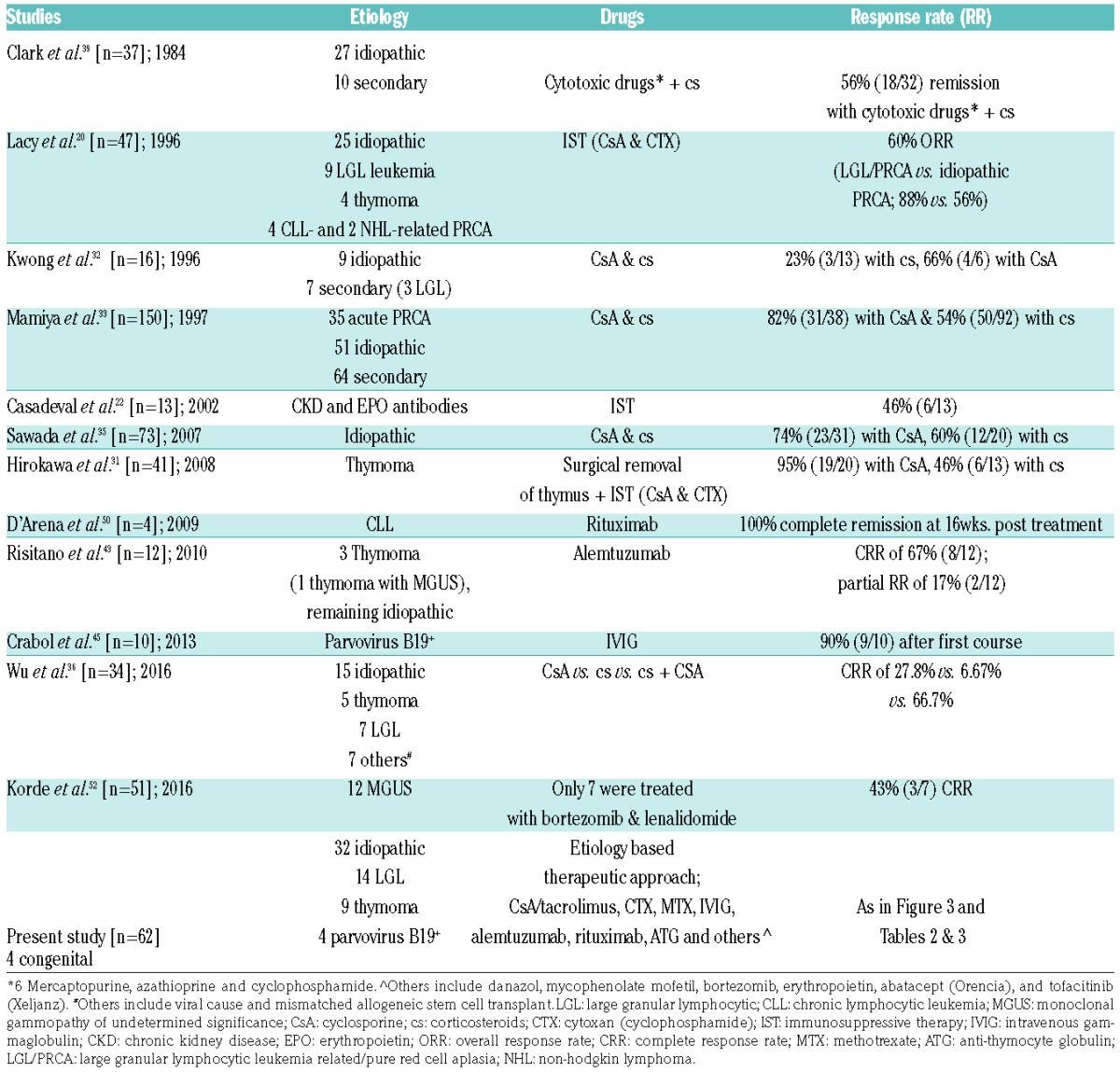
Progress in the management of rare diseases relies on the accumulation of empiric experiences, integrative analyses of several cases, and rarely, clinical trials. Guidelines for the management of PRCA are incomplete and, outside of a few referral centers, patients rely on the inquisitive minds of their hematologists. Given the rarity of this condition, we were inspired to further investigate the large number of cases of PRCA seen in our institution. A summary of our diagnostic approach and treatment results, and study outcomes previously published, are shown in Table 4. We also studied patient etiology, clinical phenotypes, immunological characteristics and genomic data. Our diagnostic approach was based on rational steps to exclude congenital disease (rare in adults), and malproductive anemia, which includes nutritional deficiencies, systemic diseases, and iatrogenic/drug-related diseases. Thereafter, infectious etiologies, in particular B19-mediated TAC, were distinguished in order to arrive at idiopathic- and pathophysiologically-related T-LGL cases. The diagnostic fractions obtained by this approach likely reflect those that would be seen in the community. Idiopathic aPRCA is present in a high percentage of cases and is likely T-cell-mediated. There are also reports of PRCA mediated by antibodies specific for erythroid progenitors.10,13 Mature red cells would have to lack such a tar get antigen as they would have been annihilated otherwise, as is the case in hemolytic anemia. While anti-EPO antibodies have been invoked in some cases treated with recombinant EPO, in our cohort not a single case suggested the presence of such antibodies. Nevertheless, rare responses to rituximab may imply the presence of erythroid antibodies in an occasional patient, but routine laboratory tests are not available to identify such patients. Lastly, the identification of clonal mutations, including subclonal hierarchy, with an assigned role in MDS may seem to suggest some are cases of “missed” MDS, or early developing MDS, but our results do not support this notion. Conversely, the presence of clonal events in older patients may simply be consistent with the diagnosis of clonal hematopoiesis of indeterminate potential (CHIP) or underscore, as in aplastic anemia, the contraction of normal progenitor and stem cell pools with a greater likelihood of detecting clonal events.25,26
Table 4.
Outcomes of PRCA reported from different studies.
The most common form of idiopathic aPRCA is mediated by a spectrum of T-cell responses, ranging from polyclonal to oligo and monoclonal, as seen in T-LGL where it constitutes a maximally skewed version of an originally polyclonal response. The presence of STAT3 mutations may support this notion,27 as STAT3 mutant clonal cells are usually a subfraction of clonally TCR-rearranged cytotoxic lymphocytes (CTL) as determined by quantitative analyses of TCR Vβ-chain sequencing (data not shown). Similarly, Vβ flow cytometry performed herein showed increased clonal skewing in some cases of aPRCA short of fully defined monoclonal expansion (Figure 1B). It is possible that a similar process is present in patients with B-cell dyscrasias. Abnormal B-cell or plasma cells can trigger a cross-reactive T-cell response28 that can evolve from polyclonal to clonal and be further fixed by STAT3 mutations. Such a scenario would explain the need to treat underlying B-cell/plasma cell lymphoproliferative processes, even if it is otherwise asymptomatic. Indeed, the presence of severe anemia may upstage the morphologically weighted severity of these conditions. Similar processes may also be operative in cases with thymoma, wherein a clearly IST-sensitive process is maintained by the presence of thymoma and interrupted by thymectomy. An alternative mechanism may be operative in hypogammaglobulinemia, wherein excessive, possibly cross-reactive responses are triggered by antigens that cannot be cleared due to the underlying immunodeficiency, as in cases of hypogammaglobulinemia in Good’s syndrome.29 Significantly, some cases of aPRCA have also been described in the context of NK-LGL,30 but while occasional cases did show nonspecific elevation of NK cells, we did not encounter such situations in our patients.
The proper distinction of pathogenetic mechanisms is essential for selecting the most effective therapeutic modality. Historically, CsA has by far been the most successfully used drug in the treatment of idiopathic aPRCA, with responses ranging from 66–95%.31–36 However, most of these studies did not examine a rational selection of treatments in various clinical contexts, nor did they compare efficacies to those of other available drugs. Herein, the systematic administration of CsA showed a similar response rate, however, a subgroup analysis revealed better responsiveness of non-LGL vs. T-LGL-associated PRCA. Tacrolimus showed activity as a salvage option in a few reports,37,38 and we were able to substantiate those findings in our study. While no head-to-head comparisons were conducted, previous reports suggested lower response rates to CTX vs. CsA, but prior studies utilized various combinations of CTX and involved relatively small numbers of patients.20,31,39 Nevertheless, given results comparable to CsA, CTX remains a front-line option in aPRCA, particularly in older patients and taking into consideration the oncogenic risks associated with chronic exposure to CTX. Despite the widespread application of MTX in T-LGL, its utility is mostly in the treatment of idiopathic or LGL-associated neutropenia, and results with MTX in aPRCA appear to be inferior to those of CsA or CTX.
Conceptually, the lack of response to IST may be due to insufficient intensity or alternate pathophysiology. Assuming that the latter forms are rare, salvage IST agents include campath and ATG. Campath has been effective in autoimmune disorders and in T-LGL,40–42 thus, we and others treat aPRCA with this drug,43,44 using low-dose subcutaneous regimens to limit infectious complications. Our experience with campath in aPRCA has shown that it yields the highest number of meaningful responses in patients, including those refractory to CTX or CsA and those who could not tolerate calcineurin inhibitors. Despite the small number of patients, our results indicate that the best response can be expected in T-LGL-associated PRCA. The concern of IST-related infectious complications with campath dictates antimicrobial-antiviral prophylaxis and careful monitoring. With these measures and low-dose subcutaneous treatment, the risks appear to be manageable. ATG has been reported to be used in a few patients with aPRCA. It was used in our series as a salvage option that had an acceptable success rate.
The identification of B19-related chronic PRCA or TAC has obvious therapeutic implications.45,46 Indeed, the results herein are similar to those of a published series:45 3/4 patients receiving IVIG for parvovirus PRCA responded with durable remissions. It is possible that patients with immunodeficiency, even in the absence of diagnostic signs of B19, may benefit from IVIG that could clear a hypothetically unrecognized viral agent. We recorded responses in hypogammaglobulinemic patients (including Goods syndrome), who received IVIG as preparation for IST. Responses to IVIG in auto-antibody mediated PRCA have been reported previously.47
Contrasting results have been reported with rituximab in aPRCA patients refractory to multiple IST.48,49 Ex juvantibus responsiveness to rituximab indicates a potentially B-cell mediated disease,50 or implies that PRCA may be a paraneoplastic syndrome of a coexisting B-cell dyscrasia.51 Consequently, one would not expect responses to rituximab in idiopathic or T-LGL-associated PRCA. Similarly, coexisting MGUS or plasma cell disorders have been successfully treated with bortezomib,52 and we were also able to make a similar observation in otherwise refractory aPRCA with co-associated monoclonal Immunoglobulin G (IgG) or Immunoglobulin M (IgM) gammopathy.
In summary, the succession of treatments was rationally decided upon in order to include the most likely target and least intense treatment, i.e., CsA or CTX, as a first-line choice, while salvage therapeutic options could include either B-cell-directed modality, anabolic steroids or higher intensity of immunosuppression. While the resulting salvage RR is likely due to the regimen applied, we cannot rule out the cumulative effects of such increased immunosuppression with sequentially used immunosuppressive agents.
Allogenic bone marrow transplant has been tried in the past for DBA patients who are refractory to steroids,53 however, the timing of transplant for acquired immune-mediated PRCA can still be debated as, to date, there are no randomized studies exploring the potential benefit of early bone marrow transplant. Arguably, it could be a potential option for medically refractory disease failing multiple different ISTs.54
In conclusion, the proper management of PRCA requires consideration of the clinical context, which can provide clues to the therapeutic modality, especially in the refractory setting.
Supplementary Material
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/103/2/221
Funding
This work was supported by the EPE1509JM - Edward P. Evans Foundation, R01HL123904 – NHLBI, R01HL118281 – NHLBI, R01HL128425 – NHLBI
References
- 1.Brown K, Young N. Parvovirus B19 infection and hematopoiesis. Blood Rev. 1995; 9(3):176–182. [DOI] [PubMed] [Google Scholar]
- 2.Thompson DF, Gales MA. Drug-induced pure red cell aplasia. Pharmacotherapy. 1996;16(6):1002–1008. [PubMed] [Google Scholar]
- 3.Stohlman F, Quesenberry P, Howard D, Miller M, Schur P. Erythroid aplasia: an autoimmune complication of chronic lymphocytic leukemia. Clin Res. 1971;19:566. [Google Scholar]
- 4.Masauzi N, Tanaka J, Watanabe M, et al. Primary Waldenström’s macroglobulinemia associated with pure red cell aplasia in which Ts/c lymphocytes inhibiting erythroid precursors were detected. [Rinsho ketsueki] Jpn J Clin Hematol. 1993; 34(3):355–361. [PubMed] [Google Scholar]
- 5.Kobayashi T, Hanada T, Sato Y, et al. A case of pure red cell aplasia with monoclonal gammopathy: immune-mediated inhibition of erythropoiesis. [Rinsho ketsueki] Jpn J Clin Hematol. 1987; 28(11): 2029–2033. [PubMed] [Google Scholar]
- 6.Orchard J, Myint H, Hamblin TJ. A patient with myeloma who still has pure red cell aplasia despite the most intensive immune modulation. Leuk Res. 1997;21(4):353–354. [DOI] [PubMed] [Google Scholar]
- 7.Abkowitz JL, Kadin ME, Powell JS, Adamson JW. Pure red cell aplasia: lymphocyte inhibition of erythropoiesis. Br J Haematol. 1986;63(1):59–67. [DOI] [PubMed] [Google Scholar]
- 8.Levitt LJ, Reyes GR, Moonka DK, et al. Human T cell leukemia virus-I-associated T-suppressor cell inhibition of erythropoiesis in a patient with pure red cell aplasia and chronic T gamma-lymphoproliferative disease. J Clin Invest. 1988;81(2):538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bailey RO, Dunn HG, Rubin AM, Ritaccio AL. Myasthenia gravis with thymoma and pure red blood cell aplasia. Am J Clin Pathol. 1988;89(5):687–693. [DOI] [PubMed] [Google Scholar]
- 10.Casadevall N, Dupuy E, Molho-Sabatier P, et al. Autoantibodies against erythropoietin in a patient with pure red-cell aplasia. N Engl J Med. 1996;334(10):630–633. [DOI] [PubMed] [Google Scholar]
- 11.Dessypris EN, Krantz SB, Roloff JS, Lukens JN. Mode of action of the IgG inhibitor of erythropoiesis in transient erythroblastopenia of children. Blood. 1982;59(1):114–123. [PubMed] [Google Scholar]
- 12.Duchmann R, Schwarting A, Poralla T, zum Büschenfelde K-HM, Hermann E. Thymoma and pure red cell aplasia in a patient with systemic lupus erythematosus. Scand J Rheumatol. 1995;24(4):251–254. [DOI] [PubMed] [Google Scholar]
- 13.Peschle C, Marmont AM, Marone G, et al. Pure red cell aplasia: studies on an IgG serum inhibitor neutralizing erythropoietin. Br J Haematol. 1975;30(4):411–417. [DOI] [PubMed] [Google Scholar]
- 14.Williamson P, Oscier D, Bell A, Hamblin T. Red cell aplasia in myelodysplastic syndrome. J Clin Pathol. 1991;44(5):431–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaznelson P. Zur entstehung der blutplättchen. Verh Dtsch Ges Inn Med. 1922;34:557–559. [Google Scholar]
- 16.Young N. Hematologic and hematopoietic consequences of B19 parvovirus infection. Semin Hematol. 1988;25(2):159–172. [PubMed] [Google Scholar]
- 17.Browman G, Freedman M, Blajchman M, McBride J. A complement independent erythropoietic inhibitor acting on the progenitor cell in refractory anemia. Am J Med. 1976;61(4):572–578. [DOI] [PubMed] [Google Scholar]
- 18.Charles RJ, Sabo KM, Kidd PG, Abkowitz JL. The pathophysiology of pure red cell aplasia: implications for therapy. Blood. 1996;87(11):4831–4838. [PubMed] [Google Scholar]
- 19.Hirayama Y, Nagai T, Ohta H, et al. A case of pure red cell aplasia accompanied with granular lymphocytic leukemia the tumor cells of which suppressed colony formation of BFU-E, and which was successfully treated by cyclophosphamide and cyclosporin A. [Rinsho ketsueki] Jpn J Clin Hematol. 1997;38(11):1206–1211. [PubMed] [Google Scholar]
- 20.Lacy MQ, Kurtin PJ, Tefferi A. Pure red cell aplasia: association with large granular lymphocyte leukemia and the prognostic value of cytogenetic abnormalities [see comments]. Blood. 1996;87(7):3000–3006. [PubMed] [Google Scholar]
- 21.Bergrem HDB, Eckardt KU, Kurtz A, Strisberg M. A case of antierythropoietin antibodies following recombinant human erythropoietin treatment. In: Erythropoietin: molecular physiology and clinical application. New York: Marcel Dekker; 1993;266(75). [Google Scholar]
- 22.Casadevall N, Nataf J, Viron B, et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002; 346(7):469–475. [DOI] [PubMed] [Google Scholar]
- 23.Murray W, Webb J. Thymoma associated with hypogammaglobulinaemia and pure red cell aplasia. Am J Med. 1966;41(6):974–980. [DOI] [PubMed] [Google Scholar]
- 24.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maciejewski J, Balasubramanian SK. Clinical implications of somatic mutations in aplastic anemia and myelodysplastic syndrome in genomic age (Article in press). Hematology Am Soc Hematol Educ Program. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshizato T, Dumitriu B, Hosokawa K, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med. 2015;373(1):35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koskela HL, Eldfors S, Ellonen P, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012; 366(20):1905–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bassan R, Pronesti M, Buzzetti M, et al. Autoimmunity and B cell dysfunction in chronic proliferative disorders of large granular lymphocytes/natural killer cells. Cancer. 1989;63(1):90–95. [DOI] [PubMed] [Google Scholar]
- 29.Good RA. Agammaglobulinaemia—a provocative experiment of nature. Bulletin of the University of Minnesota. 1954; 26:1–19. [Google Scholar]
- 30.Handgretinger R, Geiselhart A, Moris A, et al. Pure red-cell aplasia associated with clonal expansion of granular lymphocytes expressing killer-cell inhibitory receptors. N Engl J Med. 1999;340(4):278–284. [DOI] [PubMed] [Google Scholar]
- 31.Hirokawa M, Sawada K-i, Fujishima N, et al. Long-term response and outcome following immunosuppressive therapy in thymoma-associated pure red cell aplasia: a nationwide cohort study in Japan by the PRCA collaborative study group. Haematologica. 2008;93(1):27–33. [DOI] [PubMed] [Google Scholar]
- 32.Kwong Y, Wong K, Liang R, et al. Pure red cell aplasia: clinical features and treatment results in 16 cases. Ann Hematol. 1996; 72(3):137–140. [DOI] [PubMed] [Google Scholar]
- 33.Mamiya S, Itoh T, Miura AB. Acquired pure red cell aplasia in Japan. Eur J Haematol. 1997;59(4):199–205. [DOI] [PubMed] [Google Scholar]
- 34.Sawada K, Fujishima N, Hirokawa M. Acquired pure red cell aplasia: updated review of treatment. Br J Haematol. 2008; 142(4):505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sawada K-i, Hirokawa M, Fujishima N, et al. Long-term outcome of patients with acquired primary idiopathic pure red cell aplasia receiving cyclosporine A. A nationwide cohort study in Japan for the PRCA Collaborative Study Group. Haematologica. 2007;92(8):1021–1028. [DOI] [PubMed] [Google Scholar]
- 36.Wu X, Wang S, Shen W, et al. Adult patients with acquired pure red cell aplasia: treated by cyclosporine a and/or corticosteroids-single center experience. Blood. 2016;128(22):4818. [Google Scholar]
- 37.Hashimoto K, Harada M, Kamijo Y. Pure red cell aplasia induced by anti-erythropoietin antibodies, well-controlled with tacrolimus. Int J Hematol. 2016;104(4):502–505. [DOI] [PubMed] [Google Scholar]
- 38.Yoshida S, Konishi T, Nishizawa T, Yoshida Y. Effect of tacrolimus in a patient with pure red cell aplasia. Int J Lab Hematol. 2005;27(1):67–69. [DOI] [PubMed] [Google Scholar]
- 39.Clark DA, Dessypris EN, Krantz SB. Studies on pure red cell aplasia. XI. Results of immunosuppressive treatment of 37 patients. Blood. 1984;63(2):277–286. [PubMed] [Google Scholar]
- 40.Au W-Y, Lam CC, Chim C-S, Pang AW, Kwong Y-L. Alemtuzumab induced complete remission of therapy-resistant pure red cell aplasia. Leuk Res. 2005; 29(10):1213–1215. [DOI] [PubMed] [Google Scholar]
- 41.Rodon P, Breton P, Courouble G. Treatment of pure red cell aplasia and autoimmune haemolytic anaemia in chronic lymphocytic leukaemia with Campath 1H. Eur J Haematol. 2003;70(5):319–321. [DOI] [PubMed] [Google Scholar]
- 42.Schutzinger C, Gaiger A, Thalhammer R, et al. Remission of pure red cell aplasia in T-cell receptor [gamma][delta]-large granular lymphocyte leukemia after therapy with low-dose alemtuzumab. Leukemia. 2005; 19(11):2005–2008. [DOI] [PubMed] [Google Scholar]
- 43.Risitano AM, Selleri C, Serio B, et al. Alemtuzumab is safe and effective as immunosuppressive treatment for aplastic anaemia and single lineage marrow failure: a pilot study and a survey from the EBMT WPSAA. Br J Haematol. 2010;148(5):791–796. [DOI] [PubMed] [Google Scholar]
- 44.Thota S, Patel BJ, Sadaps M, et al. Therapeutic outcomes using subcutaneous low dose alemtuzumab for acquired bone marrow failure conditions. Br J Haematol. 2017. September 14 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crabol Y, Terrier B, Rozenberg F, et al. Intravenous immunoglobulin therapy for pure red cell aplasia related to human parvovirus B19 infection: a retrospective study of 10 patients and review of the literature. Clin Infect Dis. 2013;56(7):968–977. [DOI] [PubMed] [Google Scholar]
- 46.Kurtzman G, Frickhofen N, Kimball J, et al. Pure red-cell aplasia of 10 years’ duration due to persistent parvovirus B19 infection and its cure with immunoglobulin therapy. N Engl J Med. 1989;321(8):519–523. [DOI] [PubMed] [Google Scholar]
- 47.Linardaki GD, Boki KA, Fertakis A, Tzioufas AG. Pure red cell aplasia as presentation of systemic lupus erythematosus: antibodies to erythropoietin. Scand J Rheumatol. 1999;28(3):189–191. [DOI] [PubMed] [Google Scholar]
- 48.Auner HW, Wölfler A, Beham Schmid C, et al. Restoration of erythropoiesis by rituximab in an adult patient with primary acquired pure red cell aplasia refractory to conventional treatment. Br J Haematol. 2002;116(3):727–728. [DOI] [PubMed] [Google Scholar]
- 49.Dungarwalla M, Marsh J, Tooze J, et al. Lack of clinical efficacy of rituximab in the treatment of autoimmune neutropenia and pure red cell aplasia: implications for their pathophysiology. Ann Hematol. 2007; 86(3):191–197. [DOI] [PubMed] [Google Scholar]
- 50.D’Arena G, Vigliotti ML, Dell’Olio M, et al. Rituximab to treat chronic lymphoproliferative disorder associated pure red cell aplasia. Eur J Haematol. 2009;82(3):235–239. [DOI] [PubMed] [Google Scholar]
- 51.Alter R, Joshi SS, Verdirame JD, Weisenburger DD. Pure red cell aplasia associated with B cell lymphoma: demonstration of bone marrow colony inhibition by serum immunoglobulin. Leuk Res. 1990; 14(3):279–286. [DOI] [PubMed] [Google Scholar]
- 52.Korde N, Zhang Y, Loeliger K, et al. Monoclonal gammopathy associated pure red cell aplasia. Br J Haematol. 2016; 173(6):876–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roy V, Pérez WS, Eapen M, et al. Bone marrow transplantation for diamond-blackfan anemia. Biol Blood Marrow Transplant. 2005; z11(8):600–608. [DOI] [PubMed] [Google Scholar]
- 54.Tseng SB, Lin SF, Chang CS, et al. Successful treatment of acquired pure red cell aplasia (PRCA) by allogeneic peripheral blood stem cell transplantation. Am J Hematol. 2003;74(4):273–275. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.