

Abstract
Loss-of-function mutations and deletions in Wilms tumor 1 (WT1) gene are present in approximately 10% of T-cell acute lymphoblastic leukemia. Clinically, WT1 mutations are enriched in relapsed series and are associated to inferior relapse-free survival in thymic T-cell acute lymphoblastic leukemia cases. Here we demonstrate that WT1 plays a critical role in the response to DNA damage in T-cell leukemia. WT1 loss conferred resistance to DNA damaging agents and attenuated the transcriptional activation of important apoptotic regulators downstream of TP53 in TP53-competent MOLT4 T-leukemia cells but not in TP53-mutant T-cell acute lymphoblastic leukemia cell lines. Notably, WT1 loss positively affected the expression of the X-linked inhibitor of apoptosis protein, XIAP, and genetic or chemical inhibition with embelin (a XIAP inhibitor) significantly restored sensitivity to γ-radiation in both T-cell acute lymphoblastic leukemia cell lines and patient-derived xenografts. These results reveal an important role for the WT1 tumor suppressor gene in the response to DNA damage, and support the view that anti-XIAP targeted therapies could have a role in the treatment of WT1-mutant T-cell leukemia.
Introduction
The Wilms tumor 1 (WT1) tumor suppressor gene plays an important role in embryonic development and tumorigenesis.1 The WT1 protein contains a N-terminal transactivation domain and a C-terminus with four zinc-fingers that can act as an activator or a repressor depending on the cell context.2,3 The complexity of the protein is augmented by the generation of alternative isoforms that translates into a different capacity of DNA-binding. In mammals, exon 5 and nine nucleotides at the end of exon 9 are subjected to alternative splicing, generating four different predominant splice isoforms. Alternative splicing of exon 5 gives rise to proteins that differ in the presence or absence of a 17-amino-acid insertion (Ex5+ and Ex5−). A different splicing, which cuts three amino acids [Lysine, Serine, Threonine, (KTS)] at 3’ end of exon 9, generates proteins which vary for the KTS insert (KTS+ and KTS−). These two isoforms (KTS+ and KTS−) are conserved in all vertebrates and imbalanced expression of these variants are associated to developmental abnormalities. Importantly, WT1 isoforms lacking the KTS insertion bind to DNA more efficiently, and significantly affect transcription (reviewed by Hastie4). Notably, WT1 has been demonstrated to interact with a variety of proteins, such as TP53, STAT3 and TET2, that play crucial roles in transformation.5–8
Mutations and deletions in the WT1 gene were first described in inherited and sporadic Wilms tumors, a pediatric malignancy resulting from the transformation of pluripotent embryonic renal precursor cells.9,10 Subsequently, WT1 gene mutations were also found in acute myeloid and bi-phenotypic leukemia subtypes.11 More recently, WT1 mutations and/or deletions were also reported in approximately 10% of both pediatric and adult T-cell acute lymphoblastic leukemia (T-ALL).12 Leukemia-associated WT1 mutations typically consist of heterozygous frameshift-generating deletions and insertions in exon 7 leading to premature stop codons which may ultimately result in truncated proteins lacking the C-terminal DNA-binding domain or in loss-of-function due to nonsense-mediated RNA decay.13 WT1 mutations are particularly prevalent in patients with relapsed T-ALL,14 and have been associated with inferior relapse-free survival in cases with standard risk thymic T-ALL.15 Here we describe a previously unrecognized direct mechanistic role of WT1 loss in the attenuation of DNA damage-induced apoptosis in T-ALL.
Methods
Cell lines and patient-derived xenografts
MOLT4, PF382 and CCRF-HSB2 T-ALL cells and U2OS cells were obtained from the American Type Culture Collection (ATCC). The P12-Ichikawa T-ALL cells were from the German Collection of Microorganisms and Cell Cultures (Leibniz Institute DSMZ). T-ALL cell lines were cultured in vitro with complete RPMI medium supplemented with 10% FCS (Gibco). T-ALL patient-derived xenografts (T-ALL PDX) had been previously established from pediatric T-ALL samples in non-obese/severe combined immunodeficiency mice (NOD/SCID).16,17 T-ALL PDX were expanded in vivo via intravenous (i.v.) injection into NOD-scidIL2Rnull immunodeficient mice (NSG mice, Jackson Laboratory). For prolonged in vitro culture, T-ALL xenografts were maintained in complete RPMI medium supplemented with 20% FCS, cytokines (10 ng/mL IL-7, 20 ng/mL FLT3-L, and 50 ng/mL SCF, all from Peprotech) and 20 nM insulin (Sigma Aldrich). Procedures involving animals and their care conformed with institutional guidelines and were authorized by the animal ethical committee (Italian Ministry of Health).
Statistical analysis
Results were expressed as mean value±Standard Deviation (SD). Unpaired Student t-test was used to analyze data. P<0.05 was considered statistically significant.
Results
WT1 alterations confer resistance to DNA damage in T-ALL cells
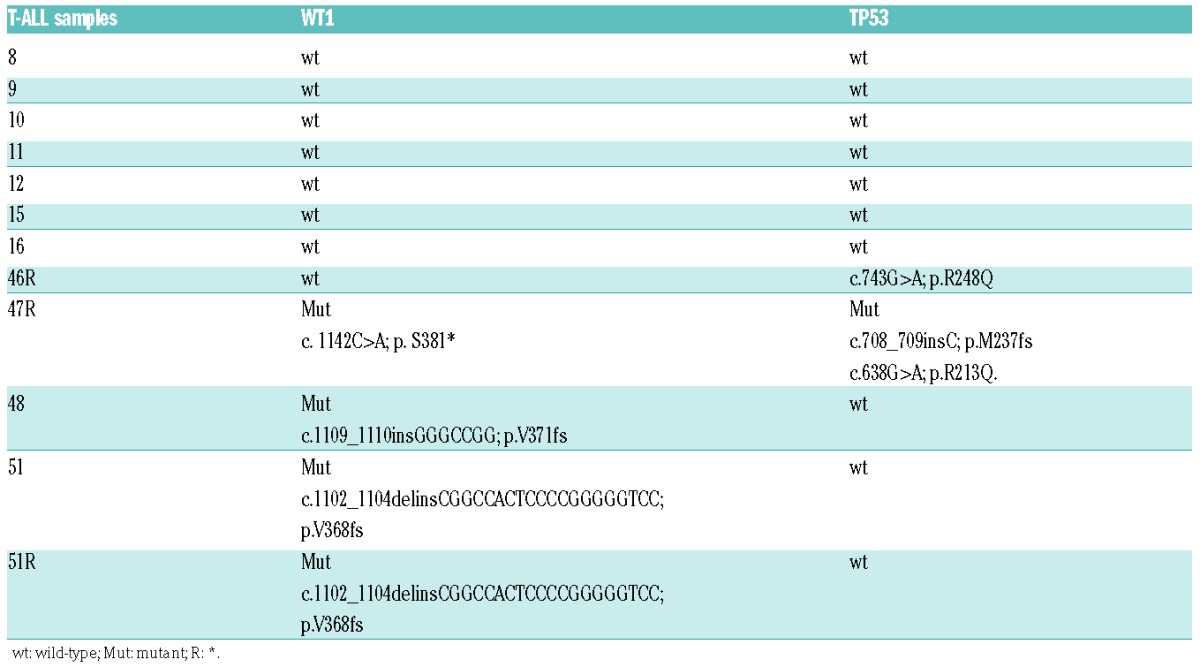
Given the association of WT1 mutations and loss with relapsed T-ALL, we hypothesized that WT1 inactivation could result in impaired response to DNA damaging agents in this disease. To test this, we investigated the effects of γ-radiation in a panel of T-ALL patient-derived xenografts (T-ALL PDX) including both WT1 wild-type [sample ns. 8, 9, 10, 11, 12, 15, previously obtained from T-ALL cells at diagnosis; sample 46R, previously obtained from T-ALL cells at relapse (R)] and WT1-mutant (sample ns. 48 and 51, and sample ns. 47R and 51R). Consistent with previous reports, all WT1 mutations in these samples consisted of truncating nonsense or frameshift alterations in exon 7 (Table 1). Of note, only 2 of these specimens (samples 46R, WT1 wild-type and 47R, WT1-mutant) presented inactivating mutations in TP53 (Table 1). Additional data, such as NOTCH1, FBXW7 mutations and PTEN expression, that are frequently altered in T-ALL samples, showed that alterations were homogenously distributed amongst the WT1 wild-type and WT1-mutant specimens (Online Supplementary Table S1).
Table 1.
WT1 and TP53 genetic status in T-cell acute lymphoblastic leukemia PDX. HGVS-nomenclature was used for the description of sequence variants.18
Cell viability assays in response to different doses of γ–radiation (0.5, 1, 2, 4 and 6 Gray) divided these T-ALL PDX into sensitive (median lethal dose = LD50<1.5 Gray) and resistant (LD50>1.5 Gray) (Figure 1A). Importantly, the T-ALL PDX resistant to DNA damage included all the WT1-mutant TP53 wild-type xenografts together with TP53-mutant samples 46R and 47R. Sample ns. 9 and 16 also clustered in the resistant group which, in contrast, did not present deletions in TP53 and WT1 loci, as assessed by Array-based Comparative Genomic Hybridization analysis (data not shown). In line with cell viability assays, apoptosis analysis at 24 hour (h) following 1 Gray of γ-radiation showed increased apoptosis in WT1 wild-type TP53 wild-type samples compared with WT1-mutant TP53 wild-type tumors (P<0.05) (Figure 1B and C). Overall, these results showed an association between WT1 mutations and resistance to DNA damage, thus suggesting a putative role of WT1 in DNA damage response.
Figure 1.

WT1 mutations are associated with increased resistance to γ-radiation-induced apoptosis in T-ALL PDX. (A) Cell viability analysis of WT1 wild-type TP53 wild-type (wt-WT1/wt-TP53), WT1 wild-type TP53-mutant (wt-WT1/mut-TP53), WT1-mutant TP53 wild-type (mut-WT1/wt-TP53) and WT1-mutant TP53-mutant (mut-WT1/mut-TP53) T-ALL PDX samples after 24 hours (h) from 0.5, 1, 2, 4 and 6 Gray of γ-radiation; analysis is shown as fold change (untreated cells fixed at 1). (B) Group column scatter analysis showing the specific apoptosis in wt-WT1/wt-TP53 and mut-WT1/wt-TP53 T-ALL PDX after 24 h from 1 Gray of γ-radiation (*P<0.05). (C) Representative FACS analysis of apoptosis, using Annexin-V/Sytox Red staining of wt-WT1/wt-TP53 (n=2; sample ns. 8 and 12) and mut-WT1/wt-TP53 (n=2; sample ns. 48 and 51) T-ALL PDX; apoptosis analysis was performed at 24 h from 1 Gray of γ-radiation.
In order to further investigate a potential mechanistic role of WT1 in DNA damage-induced apoptosis, we performed WT1-knockdown in MOLT4 T-ALL cells (Figure 2A, left panel). We used MOLT4 cells that, among several T-ALL cell lines tested, were reported to be wild-type for TP53 locus, even though results were contradictory.19 Sequencing analysis of our MOLT4 cells showed a heterozygous nonsense mutation in TP53 locus (R306*) that was not detectable at the cDNA level, as previously described.20 Analysis of cell viability in WT1-knockdown and shRNA scramble control cells treated with increased doses of γ-radiation showed significantly increased survival at 24 h in WT1-knockdown cells (Figure 2A, left panel). Similarly, analysis of γ-radiation-induced apoptosis showed significant protection from DNA damage-induced programmed cell death in WT1-knockdown T-ALL cells (Figure 2A, right panel). Moreover, analysis of cell cycle and DNA synthesis demonstrated a decreased number of cells in S phase in response to γ-radiation in Control but not in WT1-shRNA expressing cells (Figure 2B). These results were further validated using the CRIPSR-Cas9 strategy that allows us to directly target WT1 locus leading to complete WT1-knockout (Online Supplementary Figure S1A and B, left panels). Overall, these results implicate WT1 loss with increased resistance to DNA damage in T-ALL.
Figure 2.

WT1 loss protects from DNA damage in MOLT4 T-ALL cells. (A) Western blot analysis of WT1-knockdown in MOLT4 cells (upper panel); Western blot quantification of WT1 protein is reported on top of the gel. Values are normalized with respect to the loading control and relative intensity signal calculated with respect to Control (Ctrl) cells (fixed at 1). Cell viability assay and apoptosis analysis in MOLT4 cells infected either with sh-Scramble (Ctrl) or shRNA-WT1 (WT1-KD) after 24 hours (h)-treatment with increasing doses of γ-radiation (left and right panels, respectively). Quantitative data are shown as mean±Standard Deviation (SD); assays were performed in triplicates and reproduced at least three times. *P<0.05; **P<0.005; ***P<0.001. (B) FACS analysis of active proliferation (S-phase, EDU positive) in Ctrl and WT1-KD MOLT4 cells through direct measurement of DNA synthesis. The Click-iT® EdU Flow Cytometry Assay kit was used in combination with cell cycle analysis performed with Sytox Red staining. Analysis was performed after 12 h from 2 Gray of γ-radiation. Analysis is shown as fold change (untreated cells fixed at 1). Quantitative data are shown as mean±SD; assays were performed in triplicates. Two independent experiments were performed. (Right) A representative experiment is shown. Square regions identify cells in active proliferation.
WT1 resistance to DNA damage results in increased expression of pro-survival factors
To explore potential mechanisms implicated in the attenuation of DNA-damage-induced apoptosis following WT1 loss, we analyzed the expression of apoptosis-related proteins in Control and WT1-knockdown MOLT4 cells following 6 Gray of γ-radiation. These analyses revealed increased cleaved-Caspase3 in WT1 control cells compared with WT1-knockdown samples (Figure 3A and Online Supplementary Figure S2A). However, early stages of DNA-damage response upstream of TP53 seem to be intact in the absence of WT1, as TP53 phosphorylation was detected in both WT1 Control and WT1-knockdown T-ALL lymphoblasts. In addition, we noted increased levels of pro-survival factors survivin and XIAP as well as upregulation of HMOX2 (HO-2), an important factor in protection against oxidative stress, in response to γ-radiation in WT1-knockdown cells, as demonstrated by peptide array quantification (Online Supplementary Figure S2A). Western blot analysis using different validated antibodies and independent lysates, documented the strong downregulation of XIAP in MOLT4 cells treated with γ-radiation and the effective and sustained abrogation of this effect upon WT1-knockdown (Figure 3A, right panel, and Online Supplementary Figure S2B). This result was further confirmed in WT1-knockout MOLT4 cells (Online Supplementary Figure S2C). Analysis of the DNA-damage response in response to etoposide, a topoisomerase II inhibitor, showed similar results. In fact, WT1-knockdown cells showed increased cell viability and decreased apoptosis compared with Controls when treated with increasing doses of etoposide (Figure 3B, left and middle panels). As in the case of γ-radiation, WT1 inactivation resulted in decreased cleaved-Caspase3 and impaired XIAP downregulation following etoposide treatment (Figure 3B, right panel). In line with these results, increased survival of WT1 deficient MOLT4 T-ALL cells was also observed after cytarabine (ARA-C), vincristine and methotrexate treatment (Figure 3C).
Figure 3.

WT1-knockdown affects the expression of apotosis-related proteins following γ-radiation and chemotherapy. (A) Whole cell extracts from Control (Ctrl) and WT1-knockdown (WT1-KD) MOLT4 cells after 24 hours (h) from 6 Gray of γ-radiation were hybridized to human apoptosis arrays. Apoptotic proteins that were consistently modulated are labeled in red (left panel). Immunoblot analysis of cleaved-Caspase3 and XIAP in Ctrl or WT1-KD MOLT4 cells; analysis was performed after 24 h from 6 Gray of γ-radiation. β–Actin is shown as loading control (right panel). Western blot quantification of XIAP protein is reported on top of the gel. Values are normalized with respect to the loading Ctrl and the numbers represent the relative intensity signal calculated respect to untreated Ctrl cells (fixed at 1). (B) Cell viability assays in Ctrl or WT1-KD MOLT4 cells after 24 h treatment with increasing doses of etoposide (left). Analysis is shown as fold change (untreated cells fixed at 1). Quantitative data are shown as mean±Standard Deviation (SD); assays were performed in triplicates and reproduced at least three times. Apoptosis analysis using AnnexinV/PI staining in Ctrl or WT1-KD MOLT4 cells after 24 h treatment with increasing doses of etoposide (middle panel). Error bars represent SD for triplicate experiments. *P<0.05; **P<0.005; ***P<0.001. Immunoblot analysis of PARP, cleaved-Caspase 3 and XIAP in Ctrl or WT1-KD MOLT4 cells; analysis was performed after 24 h treatment with 0.5 μM etoposide (right panel). β–Actin is shown as loading Ctrl. (C) Cell viability assay in Ctrl or WT1-KD MOLT4 cells after 24 h treatment with increasing doses of cytarabine (ARA-C) (0.05, 0.25, 0.5 and 1 μM), vincristine (0.01, 1, 10 and 100 ng/mL), or methotrexate (0.01, 0.1, 0.3 and 3 μM). Analysis is shown as fold change (untreated cells fixed at 1). Quantitative data are shown as mean±SD; assays were performed in triplicates. *P<0.05; **P<0.005; ***P<0.001.
WT1 loss promotes cell survival by attenuating TP53 apoptotic response
The TP53 pathway is extremely efficient in detecting DNA lesions in cells. Once induced, TP53 regulates the expression of a wide range of genes, leading to the biological outcomes of repair, growth arrest or apoptosis.21 We thus analyzed a possible interaction between WT1 and TP53 pathways in Control or WT1-knockdown MOLT4 T-ALL cells treated with γ-radiation. Western blot analysis of phosphorylated TP53 (P-TP53, S15), acetylated TP53 (Ac-TP53, K382) and total TP53 demonstrated induction and activation of TP53 protein in both Control and WT1-knockdown MOLT4 cells (Figure 4A). Importantly, at early time points, TP53 activation resulted more strongly induced in Control with respect to WT1-knockdown MOLT4 cells. These results were confirmed also using WT1-knockout cells (Online Supplementary Figure S3A). This effect did not depend on impaired recognition of DNA damage due to WT1 loss. Indeed, phosphorylation of crucial proteins involved in the initial phases of DNA damage recognition and repair subsequent to DNA double strand breaks such as ATM, CHK2 and histone H2A variant X (P-H2AX, S139; also known as γH2AX), was comparable between Control and knockdown cells treated with γ-radiation (Figure 4B and C, and Online Supplementary Figure S3B).
Figure 4.

WT1 loss does not impair DNA damage recognition in MOLT4 cells. (A) Immunoblot of cleaved-Caspase3, P-TP53 (S15), Ac-TP53 (K382), and total TP53. β–Actin is shown as loading Control (Ctrl). (B) Immunoblot analysis of phosphorylated ataxia telangiectasia mutated (P-ATM; S1981), and P-CHK2 (T68) in Ctrl and WT1-KD MOLT4 cells following 1, 3 and 6 hours (h) from 6 Gray of γ-radiation. Tubulin is shown as loading Ctrl. Western blot quantification of P-TP53 (S15), Ac-TP53 (K382), TP53, P-ATM and P-CHK2 proteins is reported on top of the gel. Values are normalized with respect to the loading Ctrl. The numbers represent the relative intensity signal calculated with respect to untreated Ctrl cells (fixed at 1). For P-TP53, P-ATM and P-CHK2 proteins, that were not detected in untreated Ctrl cells, the relative intensity signal is calculated respected to 1 h (Ctrl cells fixed at 1). (C) Flow cytometry analysis of Histone H2AX phosphorylation (S139) in combination with PI incorporation and cell cycle analysis. Ctrl and WT1-KD cells were treated with 6 Gray of γ-radiation for 10 and 30 minutes. Three independent experiments were performed. One representative experiment is shown.
Following these results, we analyzed the expression of genes involved in DNA repair, apoptosis and cell cycle progression in control and WT1-knockdown MOLT4 cells under basal conditions and at 3, 6 and 12 h after γ-radiation. These analyses revealed that WT1-knockdown affected the expression of prominent genes involved in apoptosis, including BAX, BBC3, FAS and GADD45A (P<0.001) (Figure 5A). Similarly, upregulation of CDKN1A, a key TP53 target gene involved in cell cycle regulation, was also impaired in WT1-knockdown cells treated with γ-radiation compared with WT1 scramble shRNA Control (P<0.001). Among these, the BBC3 pro-apoptotic factor showed markedly impaired protein upregulation in WT1-knockdown irradiated cells (Figure 5B). We thus hypothesized that WT1 directly regulates BBC3 expression co-operating with TP53 in the DNA damage response. To test this possibility, we analyzed the effects of overexpression of the WT1 isoforms on the activity of a WT1-regulatory element located in intron 1–2 of the BBC3 gene in a luciferase reporter assay. In these experiments, expression of both WT1-(KTS-) isoforms induced an approximate 9-fold increase of BBC3 reporter activity (Online Supplementary Figure S4). Consistent with data in literature (reviewed by Hastie4), overexpression of both WT1-(KTS+) isoforms determined a weak transcriptional response (approx. 2-fold induction). Luciferase activity was seen to be abolished following transfection of mutant WT1 (E384*) isoforms, the expression of which was found to be barely detectable by Western blot analysis. Overall, these data suggest that, in response to DNA damage, WT1 loss de-regulates prominent TP53 targets affecting cell survival.
Figure 5.

WT1-knockdown impairs TP53 DNA damage response in MOLT4 cells. (A) Relative expression of specific genes involved in the DNA-damage response. Analysis is performed in both Control (Ctrl) and WT1-KD cells after different times [3, 6 and 12 hours (h)] from 6 Gray of γ-radiation. Expression is calculated relative to untreated sh-Scramble MOLT4 cells (Ctrl 0 h) fixed as 1. (B) Western blot analysis of CDKN1A, BBC3, BAX and XIAP in Ctrl and WT1-KD MOLT4 cells after 1, 3, 6 and 12 h from γ-radiation. β–Actin is shown as loading control. Western blot quantification of CDKN1A, BBC3, BAX and XIAP proteins is reported on top of the gel. Values are normalized respect to the loading control. The numbers represent the relative intensity signal calculated respect to untreated Ctrl cells (fixed at 1). For CDKN1A and BBC3 proteins, that were not detected in untreated Ctrl cells, the relative intensity signal is calculated respected to 3 h and 1 h, respectively (Ctrl cells fixed at 1).
Viability of TP53-mutant T-ALL cell lines is not significantly affected by WT1 loss when exposed to γ-radiation
In order to further validate the resistance to DNA damage conferred by WT1 loss in MOLT4 cells, we extended our analysis to additional T-ALL cell lines with known TP53 status (Online Supplementary Table S2). To this end, we infected CCRF-HSB2, which resulted wild-type for TP53 with a specific hairpin for WT1 (WT1-KD CCRF-HSB2). These cells showed decreased levels of WT1 as compared to control cells (Online Supplementary Figure S5A). Importantly, treatment of CCRF-HSB2 cells with increasing doses of γ-radiation and etoposide, showed that loss of WT1 conferred increased resistance to apoptosis, even if this was to a lesser extent than MOLT4 cells (P<0.005) (Online Supplementary Figure S5A and B). Notably, Western blot of XIAP protein showed higher levels upon WT1 loss at 1 μM etoposide (Online Supplementary Figure S5B, right panel). These experiments were also carried out on TP53-mutated T-ALL cell lines, PF382 and P12-Ichikawa, that presented deleterious mutations in the TP53 gene (Online Supplementary Table S2). Treatment of TP53-mutated P12-Ichikawa cells with increasing doses of γ-radiation revealed that WT1-knockdown cells were not protected from apoptosis compared to control cells (Figure 6A, top left panel). Molecular analysis showed that cleaved-PARP and XIAP were not significantly differentially expressed between Control and WT1-knockdown cells upon DNA damage (Figure 6A, bottom left). Similar results were obtained in TP53-mutated PF382 cells (Figure 6A, right panel). As expected from a TP53-mutated cell line, PF382 cells displayed TP53 stabilization in the absence of DNA damage and failed to induce the transcription of crucial apoptotic genes involved in TP53 response, such as BAX, BBC3, FAS, and GADD45A, following γ-radiation treatment (Figure 6B and C). Consistently, loss of WT1 in PF382 cells did not alter this profile (Figure 6C). CDKN1A resulted similarly up-regulated following γ-radiation in Control and WT1-knockdown PF382 cells (Figure 6C). In conclusion, differently to MOLT4 and CCRF-HSB2 cells that displayed functional TP53 activation and WT1 loss increased cell viability following DNA damage, TP53-mutated T-ALL cell lines, PF382 and P12 Ichikawa, were not significantly affected by WT1 loss when exposed to γ-radiation, suggesting a possible cross-talk between WT1 and functional TP53.
Figure 6.

WT1-knockdown does not affect survival in TP53-mutated T-cell acute lymphoblastic leukemia (T-ALL) cells. (A) Western blot analysis of WT1-knockdown in P12-Ichikawa and PF382 cells (top panels); cell viability analysis of Control (Ctrl) and WT1-knockdown P12-Ichikawa and PF382 cells after 24 hours (h) treatment with increasing doses of γ-radiation (middle panels). Analysis is shown as fold change (untreated cells fixed at 1). Western blot quantification of WT1 protein is reported on top of the gel. Values are normalized respect to the loading. The numbers represent the relative intensity signal calculated with respect to Ctrl cells (fixed at 1). Immunoblot analysis of PARP and XIAP in Ctrl and WT1-knockdown P12-Ichikawa and PF382 cells (lower panels); analysis was performed after 24 h from 6 Gray of γ-radiation. β–Actin is shown as a loading control. Western blot quantification of XIAP protein is reported on top of the gel. Values are normalized respect to the loading control. The numbers represent the relative intensity signal calculated respect to untreated Ctrl cells (fixed at 1). (B) Cell viability analysis in Ctrl or WT1-KD PF382 cells after 1, 3, 6, 12 and 24 h after 6 Gray of γ-radiation (left panel). Analysis is shown as fold change. Immunoblot of PARP, P-TP53 (S15), Ac-TP53 (K382), and total TP53. β–Actin is shown as loading control (right panel). Western blot quantification of P-TP53 (S15), Ac-TP53 (K382), and TP53, P-ATM and P-CHK2 proteins is reported on top of the gel. Values are normalized respect to the loading control. The numbers represent the relative intensity signal calculated respect to untreated Ctrl cells (fixed at 1). (C) Relative expression of specific genes involved in the DNA damage response. Analysis was performed in both Ctrl and WT1-KD after different times (3, 6 and 12 h) from 6 Gray of γ-radiation. Expression is calculated relative to untreated Ctrl PF382 cells (Ctrl 0 h) fixed as 1.
Increased resistance of WT1-deficient T-ALL cells to DNA damage can be rescued by pre-treatment with XIAP inhibitors
Molecular characterization of DNA damage response in WT1 deficient MOLT4 cells showed an impaired TP53 response characterized by de-regulation of several proteins involved at different steps of the apoptotic cascade downstream of TP53 activation. To further validate these results, we molecularly characterized the effects of γ-radiation on WT1 wild-type TP53 wild-type T-ALL PDX samples (n=2; sample ns. 8 and 12) and on WT1-mutated TP53 wild-type samples (n=2; sample ns. 48 and 51). All the tumors showed TP53 protein stabilization upon DNA damage (Figure 7A). Importantly, WT1 wild-type samples showed higher levels of cleaved-Caspase3 compared to WT1-mutant tumors. Following γ-radiation, BAX, survivin and HO-2 proteins were not significantly regulated either in WT1 wild-type nor in WT1-mutated T-ALL xenograft samples, while BBC3 was not differentially regulated between WT1 wild-type and WT1-mutated T-ALL, with WT1-mutated T-ALL PDX sample n. 51 undergoing a strong induction of BBC3 (Figure 7A and Supplementary Figure S6). On the other hand, XIAP protein resulted grossly down-regulated following γ-radiation in WT1 wild-type but not in WT1-mutant T-ALL PDX, in line with results obtained in MOLT4 and CCRF-HSB2. XIAP is one of the best characterized members of the inhibitor of apoptosis (IAP) family proteins and is considered to be a key regulator of apoptosis. We thus investigated whether inhibition of XIAP could contribute to rescue the effects of WT1 loss in T-ALL cells upon DNA damage. T-ALL PDX samples were treated with increasing doses of embelin, a natural compound that binds to XIAP and promotes apoptosis.22 Amongst 5 T-ALL PDX samples analyzed, 2 resulted resistant at 30 μM embelin (n. 12 and n. 15) whereas TALL PDX sample ns. 8, 48 and 51 showed decreased viability and increased apoptosis starting from 15 μM of embelin (Online Supplementary Figure S7). We thus pretreated WT1-mutated PDX sample n. 48 with 15 μM of embelin, followed by treatment with γ-radiation and etoposide at 0.5 Gray and 0.1 μM etoposide, respectively. Apoptosis analysis showed a significant rescue of resistance to both γ-radiation and etoposide treatments (P<0.001) (Figure 7B). Importantly, a complete rescue was achieved by γ-radiation and etoposide treatments in MOLT4 cells at 10 μM embelin. Indeed, apoptosis analysis revealed that pre-treatment with 7.5 μM and especially 10 μM embelin before DNA damaging agents (γ-radiation and etoposide), resulted in a significant increase in apoptosis with respect to that induced by γ-radiation or etoposide alone in WT1-knockdown MOLT4 cells (P<0.001) (Figure 7C). Notably, XIAP-knockdown in MOLT4 cells deficient for WT1 (WT1-KD) efficiently rescued resistance to apoptosis in the presence of increasing doses of etoposide (P<0.001 at 0.25 μM etoposide) (Online Supplementary Figure S8). Overall, these results identified XIAP as a critical player in regulating DNA damage resistance induced by WT1 loss, opening new therapeutic opportunities in WT1-mutant patients.
Figure 7.

XIAP is a critical player in WT1 resistance to DNA damage in T-cell acute lymphoblastic leukemia (T-ALL) cells. (A) Western blot analysis of TP53, cleaved-Caspase3, BBC3, BAX, survivin, HO-2 and XIAP in wt-WT1 (n=2; sample ns. 8 and 12) and mut-WT1 (n=2; sample ns. 48 and 51) TALL PDX following 3 hours (h) from 6 Gray of γ-radiation. Histogram plots representing Western blot quantification of TP53, cleaved-Caspase3, BBC3, BAX and XIAP proteins is reported on the right. Values are normalized respect to the loading control (Ctrl). The plots show the relative intensity signal calculated with respect to untreated Control cells (fixed at 1). (B) Apoptosis assay in WT1-mutated PDX sample n. 48 pre-treated for 24 h with 15 μM embelin and subjected to γ-radiation or etoposide treatment (0.5 Gray and 0.1 μM, respectively) in the presence or absence of embelin. (C) Apoptosis assay in MOLT4 cells pre-treated for 24 h with 7.5 μM or 10 μM embelin and subjected to γ-radiation or etoposide treatment (2 Gray and 0.2 μM, respectively). Analysis is shown at 48 h following γ-radiation and presented as percentage of apoptotic cells. *P<0.05; **P<0.005; ***P<0.001.
Discussion
The introduction of intensive combination chemotherapy protocols in T-ALL treatment has led to remarkable improvements in survival.23 Unfortunately, primary resistance to chemotherapy or acquired resistance are strongly associated with poor outcome and the molecular mechanisms that allow leukemia cells to resist chemotherapy are not yet completely understood. A recent study has demonstrated that refractory T-ALL clones can develop mutations in drug-resistance genes under chemotherapy pressure.24 Moreover, resistant clones may originate from minor sub-clones present in patients at diagnosis through a dramatic selection during chemotherapy.25,26 A better understanding of the genetic lesions that may contribute to chemo-resistance is imperative to improve treatment protocols and for the identification of more effective anti-leukemic drugs. In particular, it is necessary to better characterize genetic alterations associated to genes with poorly characterized function.
The WT1 gene is mutated in approximately 10% of TALL and AML cases, and these alterations are mainly heterozygous frameshift mutations that cluster in exon 7 and are predicted to lead to a truncated protein which lacks the zinc finger domain and the property to bind DNA.11,12,27 Monoallelic or subclonal deletions have also been observed in some cases in association with mutations.12,28,29 Even if WT1 alterations clearly suggest a role as a tumor suppressor mediated at least in part by de-regulation of its transcriptional activity, its role in T-ALL is still poorly understood. Most studies have focused their attention on acute myeloid leukemia (AML). Recently, WT1 inactivating mutations have been found to inversely correlate with TET2/IDH1/IDH2 mutations; indeed, WT1-mutant AML patients have reduced 5-hydrossy-methyl-cytosine levels, similar to TET2/IDH1/IDH2 mutant AML, suggesting a critical role for WT1 as an epigenetic regulator.7 Moreover, always in the context of AML, WT1 mutations were found to be associated with an increased risk of relapse and an inferior outcome, even if contradictory results have been reported,30–36 probably due to the difference between the AML subgroups analyzed. Interestingly, a case report of AML demonstrated that a minor sub-clone, characterized by a mutation in the WT1 gene, emerges as a dominant clone after repetitive chemotherapy, including stem cell transplant.37 In T-ALL, studies on the role of WT1 mutations are still limited. In pediatric and adult T-ALL, the presence of WT1 mutations in the entire cohort were not predictive of poor clinical outcome.12,27 Interestingly, one study observed that within the standard risk group of thymic T-ALL, the small group of WT1-mutant patients had inferior relapse-free survival compared to wild-type patients.15 Notably, a very recent study in a group of high-risk T-ALL composed of paired diagnosis and relapse samples reported that WT1 mutations resulted highly enriched with respect to unselected T-ALL cases.14 Moreover, WT1 was shown to be among the frequently altered genes in the Early T Progenitor ALL (ETP) subgroup.38 ETP ALL were originally identified as a high-risk subtype of leukemia that lacks several T-cell markers and is characterized by aberrant expression of myeloid and stem cell markers.39
Our study highlights an important role for WT1 as a critical regulator of the TP53-dependent apoptotic response following DNA damage, and indicates an association between WT1 loss-of-function and resistance both in TALL PDX and MOLT4 T-ALL cells. In fact, in the presence of DNA-damaging stimuli, such as γ-radiation and chemotherapy, loss of WT1 in MOLT4 cells conferred increased survival associated with an impaired induction of TP53 apoptotic response. Amongst the de-regulated genes, some of which have been previously described as WT1 targets, such as BAX and GADD45A, we identified BBC3 as a new important WT1 target. BBC3, which encodes for the pro-apoptotic factor Puma, is a potent antagonist of anti-apoptotic BCL2-related proteins contributing to the release and activation of the pro-apoptotic factors BAX and BAK. Although BBC3 was found to be a critical regulator of apoptosis in primary thymocytes in response to DNA damage and glucocorticoid treatment40 and a crucial factor in MOLT4 cells following WT1 loss, one mut-WT1 T-ALL xenograft in our series (n. 51) showed strong induction of BBC3 much like the wt-WT1 PDX samples, suggesting that other pro-apoptotic factors may be important in this sample.
An analysis of the WT1-knockout mouse first pointed out the involvement of WT1 in regulating apoptosis, which demonstrated that lack of WT1 expression determined severe abnormalities of renal development and that the metanephric blastema of WT1-null embryos was characterized by massive apoptosis compared to wild-type embryos.41 A number of subsequent studies have demonstrated that WT1 can directly or indirectly regulate several BCL-2 family members including pro-apoptotic factors, such as BAK and BAX, and the anti-apoptotic proteins BCL2 and BCL2A1 with different outcomes depending on the cell context (reviewed by Toska and Roberts2 and by Yang et al.3). Contradictory results regarding WT1 regulation may be due in part to the complex structure of the WT1 protein and its multiple isoforms. Another level of complexity is due to co-operativity between WT1 and its interacting proteins. WT1 was found to physically interact with TP53, and this interaction was demonstrated to modulate the ability of WT1 and TP53 to trans-activate their respective targets.5,42,43 In this scenario, it is plausible that the cellular context may substantially determine WT1 effects on specific targets. In our study, in fact, WT1 significantly directly or indirectly affects the transcription of important mediators of TP53-apoptotic response only in the presence of a functional TP53. MOLT4 cells carry a heterozygous nonsense mutation in TP53 locus (p.R306*) that was not detectable at the cDNA level as previously described by others;20 concerning this trait, unlike the majority of T-ALL cell lines, MOLT4 cells resemble primary T-ALL samples at diagnosis as they rarely display deleterious mutations in the TP53 gene.44 Indeed, treatment of MOLT4 cells with DNA-damaging stimuli determines TP53 stabilization and strong induction of TP53-specific target genes. This response is significantly down-regulated when MOLT4 cells are engineered to loose WT1, as in knockdown or knockout experiments. Our data suggest that WT1 loss can influence both stabilization and transcriptional activity of TP53. On the other hand, we found that TP53-mutated T-ALL cell lines, such as PF382 and P12-Ichikawa, were not significantly affected by WT1 loss in response to DNA damage, suggesting a possible link between TP53 and WT1, that remains a subject for future studies. The effects on TP53 response mediated by WT1 loss in MOLT4 cells also resulted in deregulation of downstream effectors such as XIAP. In fact, we found elevated levels of XIAP following γ-radiation and etoposide treatment in WT1-deficient MOLT4 cells. Importantly, pre-treatment of WT1-deficient MOLT4 cells with embelin, a natural compound that specifically inhibits XIAP22,45 and direct inactivation of XIAP using specific shRNAs, rescued the resistance of WT1-deficient MOLT4 towards γ-radiation and etoposide. Notably, in TALL PDX samples, as for MOLT4 cells, XIAP stability was maintained following DNA damage in WT1-mutant samples but not in WT1 wild-type xenografts, confirming a prominent role of XIAP downstream of WT1 resistance in T-ALL. In addition, also WT1-mutant T-ALL xenografts were seen to be sensitized to γ-radiation and etoposide when the treatments were performed in the presence of the XIAP inhibitor embelin.
XIAP acts by inhibiting caspase activation at an important point where intrinsic and extrinsic apoptotic pathways converge, making it an attractive therapeutic target. Pre-clinical studies concerning the use of embelin and small molecule XIAP inhibitors have demonstrated antitumor effects in xenograft and mouse models.46–49 Importantly, no specific toxicity was reported following XIAP inhibition in these studies.46 In conclusion, our study unveils an important role for the WT1 tumor suppressor gene in dampening TP53 DNA damage response and suggest that the combination of XIAP inhibitors with conventional chemotherapy or radiotherapy in T-ALL patients carrying WT1 mutations might be beneficial.
Supplementary Material
Acknowledgments
We are grateful to W. Pear (Perelman School of Medicine, University of Pennsylvania, Philadelphia) for providing MigR1 empty vector; E. Peta for performing some molecular analyses; S. Indraccolo for providing patient-derived xenografts (PDX); V. Agnusdei and M. Pinazza for maintaining PDX samples in immunodeficient mice; Cinzia Candiotto for TP53 mutational analysis in T-ALL cell lines; R. Grancara for computational assistance.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/103/2/266
Funding
This work was supported by the Italian Association for Cancer Research (AIRC) grant to VT (MFAG #13053), to AA (IG#14032), to PZ (IG #14256) and to GB (IG#19186); Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR) Ex 60% to E.P; Progetto di Ricerca di Ateneo (PRAT; #152403) to EP; Istituto Oncologico Veneto 5×1000 fund.
References
- 1.Miller-Hodges E, Hohenstein P. WT1 in disease: shifting the epithelial-mesenchymal balance. J Pathol. 2012;226(2):229–240. [DOI] [PubMed] [Google Scholar]
- 2.Toska E, Roberts SG. Mechanisms of transcriptional regulation by WT1 (Wilms’ tumour 1). Biochem J. 2014;461(1):15–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang L, Han Y, Suarez Saiz F, Minden MD. A tumor suppressor and oncogene: the WT1 story. Leukemia. 2007;21(5):868–876. [DOI] [PubMed] [Google Scholar]
- 4.Hastie ND. Life, sex, and WT1 isoforms–three amino acids can make all the difference. Cell. 2001;106(4):391–394. [DOI] [PubMed] [Google Scholar]
- 5.Maheswaran S, Park S, Bernard A, et al. Physical and functional interaction between WT1 and p53 proteins. Proc Natl Acad Sci USA. 1993;90(11):5100–5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rong Y, Cheng L, Ning H, et al. Wilms’ tumor 1 and signal transducers and activators of transcription 3 synergistically promote cell proliferation: a possible mechanism in sporadic Wilms’ tumor. Cancer Res. 2006;66(16):8049–8057. [DOI] [PubMed] [Google Scholar]
- 7.Rampal R, Alkalin A, Madzo J, et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 2014;9(5):1841–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Xiao M, Chen X, et al. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol Cell. 2015;57(4):662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Call KM, Glaser T, Ito CY, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell. 1990; 60(3):509–520. [DOI] [PubMed] [Google Scholar]
- 10.Gessler M, Poustka A, Cavenee W, Neve RL, Orkin SH, Bruns GA. Homozygous deletion in Wilms tumours of a zinc-finger gene identified by chromosome jumping. Nature. 1990;343(6260):774–778. [DOI] [PubMed] [Google Scholar]
- 11.King-Underwood L, Pritchard-Jones K. Wilms’ tumor (WT1) gene mutations occur mainly in acute myeloid leukemia and may confer drug resistance. Blood. 1998; 91(8):2961–2968. [PubMed] [Google Scholar]
- 12.Tosello V, Mansour MR, Barnes K, et al. WT1 mutations in T-ALL. Blood. 2009; 114(5):1038–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abbas S, Erpelinck-Verschueren CA, Goudswaard CS, Lowenberg B, Valk PJ. Mutant Wilms’ tumor 1 (WT1) mRNA with premature termination codons in acute myeloid leukemia (AML) is sensitive to nonsense-mediated RNA decay (NMD). Leukemia. 2010;24(3):660–663. [DOI] [PubMed] [Google Scholar]
- 14.Oshima K, Khiabanian H, da Silva-Almeida AC, et al. Mutational landscape, clonal evolution patterns, and role of RAS mutations in relapsed acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2016; 113(40): 11306–11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heesch S, Goekbuget N, Stroux A, et al. Prognostic implications of mutations and expression of the Wilms tumor 1 (WT1) gene in adult acute T-lymphoblastic leukemia. Haematologica. 2010;95(6):942–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agnusdei V, Minuzzo S, Frasson C, et al. Therapeutic antibody targeting of Notch1 in T-acute lymphoblastic leukemia xenografts. Leukemia. 2014;28(2):278–288. [DOI] [PubMed] [Google Scholar]
- 17.Pinazza M, Borga C, Agnusdei V, et al. An immediate transcriptional signature associated with response to the histone deacetylase inhibitor Givinostat in T acute lymphoblastic leukemia xenografts. Cell Death Dis. 2016;6:e2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.den Dunnen JT, Dalgleish R, Maglott DR, et al. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum Mutat. 2016;37(6):564–569. [DOI] [PubMed] [Google Scholar]
- 19.Leroy B, Girard L, Hollestelle A, Minna JD, Gazdar AF, Soussi T. Analysis of TP53 mutation status in human cancer cell lines: a reassessment. Hum Mutat. 2014; 35(6):756–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ikediobi ON, Davies H, Bignell G, et al. Mutation analysis of 24 known cancer genes in the NCI-60 cell line set. Mol Cancer Ther. 2006;5(11):2606–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008; 9(5):402–412. [DOI] [PubMed] [Google Scholar]
- 22.Nikolovska-Coleska Z, Xu L, Hu Z, et al. Discovery of embelin as a cell-permeable, small-molecular weight inhibitor of XIAP through structure-based computational screening of a traditional herbal medicine three-dimensional structure database. J Med Chem. 2004;47(10):2430–2440. [DOI] [PubMed] [Google Scholar]
- 23.Pui CH. Recent advances in acute lymphoblastic leukemia. Oncology. 2011;25(4):341, 346–347. [PubMed] [Google Scholar]
- 24.Tzoneva G, Perez-Garcia A, Carpenter Z, et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat Med. 2013;19(3):368–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mullighan CG, Phillips LA, Su X, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008;322(5906):1377–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clappier E, Gerby B, Sigaux F, et al. Clonal selection in xenografted human T cell acute lymphoblastic leukemia recapitulates gain of malignancy at relapse. J Exp Med. 2011; 208(4):653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Renneville A, Kaltenbach S, Clappier E, et al. Wilms tumor 1 (WT1) gene mutations in pediatric T-cell malignancies. Leukemia. 2010;24(2):476–480. [DOI] [PubMed] [Google Scholar]
- 28.Van Vlierberghe P, Homminga I, Zuurbier L, et al. Cooperative genetic defects in TLX3 rearranged pediatric T-ALL. Leukemia. 2008;22(4):762–770. [DOI] [PubMed] [Google Scholar]
- 29.Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481(7380):157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bachas C, Schuurhuis GJ, Reinhardt D, et al. Clinical relevance of molecular aberrations in paediatric acute myeloid leukaemia at first relapse. Br J Haematol. 2014;166(6):902–910. [DOI] [PubMed] [Google Scholar]
- 31.Becker H, Marcucci G, Maharry K, et al. Mutations of the Wilms tumor 1 gene (WT1) in older patients with primary cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. Blood. 2010;116(5):788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hollink IH, van den Heuvel-Eibrink MM, Zimmermann M, et al. Clinical relevance of Wilms tumor 1 gene mutations in childhood acute myeloid leukemia. Blood. 2009;113(23):5951–5960. [DOI] [PubMed] [Google Scholar]
- 33.Virappane P, Gale R, Hills R, et al. Mutation of the Wilms’ tumor 1 gene is a poor prognostic factor associated with chemotherapy resistance in normal karyotype acute myeloid leukemia: the United Kingdom Medical Research Council Adult Leukaemia Working Party. J Clin Oncol. 2008;26(33):5429–5435. [DOI] [PubMed] [Google Scholar]
- 34.Paschka P, Marcucci G, Ruppert AS, et al. Wilms’ tumor 1 gene mutations independently predict poor outcome in adults with cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J Clin Oncol. 2008;26(28):4595–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krauth MT, Alpermann T, Bacher U, et al. WT1 mutations are secondary events in AML, show varying frequencies and impact on prognosis between genetic subgroups. Leukemia. 2015;29(3):660–667. [DOI] [PubMed] [Google Scholar]
- 36.Ho PA, Zeng R, Alonzo TA, et al. Prevalence and prognostic implications of WT1 mutations in pediatric acute myeloid leukemia (AML): a report from the Children’s Oncology Group. Blood. 2010;116(5):702–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nyvold CG, Stentoft J, Braendstrup K, et al. Wilms’ tumor 1 mutation accumulated during therapy in acute myeloid leukemia: Biological and clinical implications. Leukemia. 2006;20(11):2051–2054. [DOI] [PubMed] [Google Scholar]
- 38.Liu Y, Easton J, Shao Y, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. 2017;49(8):1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coustan-Smith E, Mullighan CG, Onciu M, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009; 10(2):147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jeffers JR, Parganas E, Lee Y, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4(4):321–328. [DOI] [PubMed] [Google Scholar]
- 41.Kreidberg JA, Sariola H, Loring JM, et al. WT-1 is required for early kidney development. Cell. 1993;74(4):679–691. [DOI] [PubMed] [Google Scholar]
- 42.Idelman G, Glaser T, Roberts CT, Jr, Werner H. WT1-p53 interactions in insulin-like growth factor-I receptor gene regulation. J Biol Chem. 2003;278(5):3474–3482. [DOI] [PubMed] [Google Scholar]
- 43.Menke AL, Clarke AR, Leitch A, et al. Genetic interactions between the Wilms’ tumor 1 gene and the p53 gene. Cancer Res. 2002;62(22):6615–6620. [PubMed] [Google Scholar]
- 44.Hof J, Krentz S, van Schewick C, et al. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol. 2011;29(23):3185–3193. [DOI] [PubMed] [Google Scholar]
- 45.Chen J, Nikolovska-Coleska Z, Wang G, Qiu S, Wang S. Design, synthesis, and characterization of new embelin derivatives as potent inhibitors of X-linked inhibitor of apoptosis protein. Bioorg Med Chem Lett. 2006;16(22):5805–5808. [DOI] [PubMed] [Google Scholar]
- 46.Vogler M, Walczak H, Stadel D, et al. Small molecule XIAP inhibitors enhance TRAIL-induced apoptosis and antitumor activity in preclinical models of pancreatic carcinoma. Cancer Res. 2009;69(6):2425–2434. [DOI] [PubMed] [Google Scholar]
- 47.Dai Y, Desano J, Qu Y, et al. Natural IAP inhibitor Embelin enhances therapeutic efficacy of ionizing radiation in prostate cancer. Am J Cancer Res. 2011;1(2):128–143. [PMC free article] [PubMed] [Google Scholar]
- 48.Lunardi A, Ala U, Epping MT, et al. A co-clinical approach identifies mechanisms and potential therapies for androgen deprivation resistance in prostate cancer. Nat Genet. 2013;45(7):747–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang T, Lan J, Huang Q, et al. Embelin sensitizes acute myeloid leukemia cells to TRAIL through XIAP inhibition and NF-kappaB inactivation. Cell Biochem Biophys. 2015;71(1):291–297. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.