

Abstract
Dissection of pleiotropic effects of missense mutations, rarely investigated in inherited diseases, is fundamental to understanding genotype-phenotype relationships. Missense mutations might impair mRNA processing in addition to protein properties. As a model for hemophilia A, we investigated the highly prevalent F8 c.6046c>t/p.R2016W (exon 19) mutation. In expression studies exploiting lentiviral vectors, we demonstrated that the amino acid change impairs both Factor VIII (FVIII) secretion (antigen 11.0±0.4% of wild-type) and activity (6.0±2.9%). Investigations in patients’ ectopic F8 mRNA and with minigenes showed that the corresponding nucleotide change also decreases correct splicing to 70±5%, which is predicted to lower further FVIII activity (4.2±2%), consistently with patients’ levels (<1–5%). Masking the mutated exon 19 region by antisense U7snRNA supported the presence of a splicing regulatory element, potentially affected by several missense mutations causing hemophilia A. Among these, the c.6037g>a (p.G2013R) reduced exon inclusion to 41±3% and the c.6053a>g (p.E2018G) to 28±2%, similarly to a variant affecting the 5′ splice site (c.6113a>g, p.N2038S, 26±2%), which displayed normal protein features upon recombinant expression. The p.G2013R reduced both antigen (7.0±0.9%) and activity (8.4±0.8%), while the p.E2018G produced a dysfunctional molecule (antigen: 69.0±18.1%; activity: 19.4±2.3%). In conclusion, differentially altered mRNA and protein patterns produce a gradient of residual activity, and clarify genotype-phenotype relationships. Data detail pathogenic mechanisms that, only in combination, account for moderate/severe disease forms, which in turn determine the mutation profile. Taken together we provide a clear example of interplay between mRNA and protein mechanisms of disease that operate in shaping many other inherited disorders.
Introduction
It is widely accepted that mutations can have pleiotropic effects that define the overall pathological phenotype.1,2 However, understanding in detail the contribution of each of these effects is complex and compromises clarification of genotype-phenotype relationships. Missense mutations are the second most common cause of severe hemophilia A (HA)3 (http://www.eahad-db.org/; http://factorviii-db.org/) and the most frequent cause of the rare coagulation factor deficiencies,4,5 as well as of other human diseases, and are known to impair several properties of mutated proteins.6,7 However, coding sequences overlap with exonic pre-mRNA splicing regulatory elements and thus missense changes may also affect pre-mRNA processing.8,9
To quantitatively evaluate the interplay between the effects of missense mutations on mRNA and protein biosynthesis and their impact on HA coagulation phenotype, as a relevant model we initially investigated the F8 p.R2016W (c.6046c>t) missense mutation (HGVS numbering; reference sequences NP_000123.1 and NM_000132.3 for protein and cDNA, respectively).10 This mutation in exon 19 is one of the most highly reported amino acid substitutions (>60 patients), particularly in Italy.11,12 It is suggested to affect splicing13 and candidate to impair FVIII protein structure, but has never been characterized. Here, we evaluated the potential pleiotropic effects by extensive expression studies and dissected the effects on FVIII protein secretion and activity as well as on exon 19 definition. Intriguingly, several missense mutations clustered in the p.R2016W region impacted on each mechanism to a different extent, consistent with the variable degree of HA severity.
Methods
Lentiviral vectors and FVIII protein expression studies
Mutations were introduced by site-specific mutagenesis9 into the codon-optimized human FVIII cDNA lacking B-domain (coFVIII).14 The F8 exon 19 deleted cDNA fragment (FVIIIΔ19) was made by overlapping PCR15 and subsequent cloning through the MluI and SbfI restriction sites.
The coFVIII variants were cloned in the lentivirus plasmid pLNT backbone (Figure 1A) to produce lentiviruses (LV).14 CHO cells16 were transduced with LV-rFVIII at MOI 2. Cells and media were harvested 72 hours (h) post infection to evaluate lentiviral copy number, FVIII antigen (ELISA) and chromogenic activity levels, as described.14
Figure 1.

The splicing-defective F8 missense mutations differentially impair Factor VIII (FVIII) protein secretion and function. (A) Schematic representation (upper part) of the lentiviral vector backbone harboring the codon-optimized cDNA of human FVIII lacking the B-domain (coFVIII) and sequence of the affected region and of the investigated mutations (middle part). The alignment of FVIII sequence across species is reported (lower part) together with affected residues (red). (B) Secreted FVIII antigen (upper) and co-factor activity (lower) levels of rFVIII variants expressed as % of rFVIIIwt. Secreted protein levels were normalized on virus copy number per cell determined by qPCR.14 Results are reported as mean±Standard Deviation from three independent experiments. (C) Structure of the human FVIII (PDB: 2R7E). Domain overview (A1-A2-A3-C1-C2 domains, inset) and interface between the A1 (blue) and A3 (white) domains (ribbon). The clustered residues under investigation (HGVS numbering) are represented by ball and stick. The R2016 residue is shown in space-filling.
Creation of F8 minigenes and splicing assays
The genomic cassette consisting of F8 exon 19 (117 bp) and the surrounding intron 18 (343 nucleotides) and intron 19 (332 nucleotides) sequences was amplified from DNA of a normal subject and cloned in the pTB vector17 through NdeI restriction sites (Figure 2) within primers. Mutations were introduced by mutagenesis to create minigene variants. Expression vectors for the U1snRNA and U7snRNA variants were created as described.18,19
Figure 2.

Features of the F8 minigene and of the sequences under investigation. Schematic representation (bottom) of the F8 exon 19 region cloned into the pTB vector through the NdeI restriction sites (indicated). The sequences of the exon-intron boundary and of the engineered U1snRNA (U1 F8ex19) are reported together with 3′ss and 5′ss scores (numbers) (splice site prediction by neural network; www.fruitfly.org). (Top) The region masked by the engineered U7 F8exon19 is magnified. The investigated mutations are indicated as nucleotide and amino acid changes.
HepG2 or HEK293 cells were transfected9,20 with F8 minigenes alone or equimolar amounts of pU1snRNA/pU7snRNAs to evaluate exon 19 inclusion by RT-PCR with primers alpha 2,3 and Bra2.17
Evaluation of FVIII plasma levels and F8 splicing profile in HA patients
FVIII activity was evaluated by the 2-stage amidolytic assay (Coamatic Factor VIII; Chromogenix, Lexington, MA, USA) and the 1-stage coagulation assay as described.11
Ectopic mRNA was isolated from fresh leukocytes21 and evaluated by RT-PCR using primers in F8 exon 17 and 22.
Studies of patients’ sample were approved by the Review Board of the Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico of Milan, and patients gave their written informed consent.
Sequences of all primers used are listed in the Online Supplementary Appendix.
Results
The impact of the F8 c.6046c>t/p.R2016W mutation on FVIII protein and F8 mRNA splicing were extensively analyzed using recombinant technologies.
The R2016W substitution impairs both FVIII secretion and function
So far, few HA-causing missense variants have been characterized22–26 because of the very low secretion efficiency of recombinant full-length rFVIII, which makes the evaluation of the residual levels associated with variants very difficult. To overcome this limitation, we exploited the lentiviral-mediated delivery of the expression cassette consisting of the codon-optimized FVIII cDNA lacking the B domain (Figure 1A).14 In this experimental system, the rFVIII-2016W missense variant (Figure 1B) was secreted in medium at reduced levels (11.0±0.4% of rFVIIIwt) and, in chromogenic assays, displayed an activity of 6.0±2.9%. Although reduced, these FVIII activity values were slightly higher than those measured in patients. The activity levels from1-stage clotting assays ranged from 1 to 6% [mean±Standard Deviation (SD): 3.6±1.8%; median: 4%] whereas those from chromogenic assays ranged from less than 1 to 5%. The mean value of the latter cannot be estimated because several patients display undefined values below 1%. When the comparison was conducted for each single patient, the chromogenic activity was, with the above mentioned limitation, roughly half that of the clotting one.
The c.6046c>t change promotes skipping of the poorly defined F8 exon 19
The availability of RNA from leukocytes from a panel (n=20) of HA patients affected by the F8 c.6046c>t mutation prompted us to investigate the F8 mRNA splicing patterns.27 The RT-PCR analysis showed, with the limitation of studying ectopic expression, that the c.6046c>t change alters splicing and decreases the proportion of correct transcripts (74.1±14.5%) by inducing exon 19 skipping (Figure 3A and B, upper panels). To experimentally demonstrate the causative nature of the c.6046c>t change and investigate splicing mechanisms, we exploited F8 minigenes preserving the authentic exon 19 nucleotide sequence (Figure 2, lower panel). Splicing assays in two different human cell lines, HepG2 and HEK293, produced overlapping results indicating that 93±2% of transcripts were correctly spliced in the wild-type context (Figure 3A, lower panel). Computational analysis showed that the 5′ and 3′ splice sites (ss) defining exon 19 diverge significantly from the consensus sequence, as witnessed by the low scores (5′ss, 0.55; 3′ss 0.14 vs. 1 of the consensus) (Figure 2, lower panel), which might explain the observed exon skipping rate (~7%). In this nucleotide context, the introduction of the c.6046c>t mutation further decreased exon 19 inclusion to 70% (Figure 3A, lower panel). To test the mechanistic hypothesis that exon 19 is inherently prone to skipping, we artificially strengthened exon 19 definition by acting on the recognition of the 5′ss by the spliceosomal U1snRNA, which is crucial in exon definition in the earliest splicing step.28 First, we optimized the authentic exon 19 5′ss by mutagenesis (high complementary 5′ss, 5′ssHC) to make it fully-complementary to the wild-type U1snRNA binding sequence, mimicking a perfect 5′ss. Conversely, we challenged the F8 minigene with an engineered U1snRNA (U1 F8ex19) designed to recognize the authentic exon 19 5′ss (Figure 2, lower part). Both approaches counteracted the effects of the c.6046c>t change (Figure 3A, lower panel), indicating that the poorly defined exon 19 strongly favored the variant’s pathogenic role.
Figure 3.

The c.6046c>t mutation as well as other adjacent missense changes promote exon 19 skipping differentially. (A) (Top) F8 splicing patterns in white blood cells from a normal control (N) and from 6 representative hemophilia A (HA) patients (P1-6) affected by the c.6046c>t mutation. The RT-PCR was conducted with primers in exons 17 and 22, and amplified fragments were separated on 2% agarose gel. (Bottom) Splicing assays with the wild-type (wt) and the mutated (c.6046c>t) minigenes. Effects of the antisense U7 F8ex19 (+) or the compensatory U1 F8ex19 (+) and of the strengthened 5′ss (5′ssHC) are shown. The RT-PCR was conducted with primers alpha 2,3 and Bra2 in the pTB construct (see arrows in Figure 2). Amplified fragments were separated on 2% agarose gel. Numbers report the percentage of exon 19 inclusion measured in at least three independent experiments; expressed as mean ± Standard Deviation. The percentage of exon 19 inclusion was estimated by densitometric analysis of bands. n: normal; s: exon 19 skipped. (B) (Top) Comparison of the exon 19 inclusion rates measured in 20 HA patients affected by the c.6046c>t mutation (light gray column) and in HepG2 cells expressing F8 minigene variants (dark gray columns). (Bottom) Representative example of splicing patterns of the selected missense variants. RT-PCR and analysis was conducted as in (A).
The c.6046c>t mutation marks a splicing regulatory region that is affected by several HA-causing missense changes
To investigate whether the c.6046c>t change affects a splicing regulatory element we took advantage of the potent antisense approach based on the U7snRNA,29 which we designed (U7 F8exon19) to mask by base-pairing the region including the variant (Figure 2, upper part). Co-expression of the U7 F8exon19 (Figure 3A, lower panel) induced complete exon skipping even in the wild-type context, strongly suggesting the presence of a positive splicing regulatory element.
We therefore selected five missense variants reported as causing HA clustered in the region covered by the U7 F8exon19 (Figure 2). Minigene assays demonstrated that two of them affected splicing (Figure 3B) and reduced exon inclusion to 41±3% (p.G2013R/c.6037g>a)30 and 28±2% (p.E2018G/c.6053a>g).31–33 We extended the investigation to missense variants close to (p.V2035A/c.6104t>c) or at (p.N2038S/c.6113a>g; -3 position)32,34,35 the 5′ss, and only the latter reduced exon inclusion (26±2%).
The splicing-defective F8 missense mutations differentially impair FVIII protein secretion and function
To detail the impact of the underlying amino acid changes on FVIII protein we expressed and characterized the splicing-defective rFVIII missense variants. Results from expression studies (Figure 1B) demonstrated a spectrum of secreted levels, from virtually normal (rFVIII-2018G and rFVIII-2038S) to poor (rFVIII-2013R, 7.0±0.9% of rFVIIIwt). Activity levels ranged from normal (rFVIII-2038S) to significantly reduced (rFVIII-2018G, 19.4±2.3%; rFVIII-2013R, 8.4±0.8%). The specific co-factor activity (activity/antigen ratio) ranged from normal (rFVIII-2013R) to 50% (rFVIII-2016W) or was significantly (rFVIII-2018G) reduced, indicating dysfunctional features of the latter FVIII variants.
The study at the protein level was completed with the expression of the in-frame transcript deriving from exon 19 skipping, clearly detectable in patients’ mRNA and associated with all splicing-defective variants. The rFVIIIΔ19 variant was not appreciably secreted, and, therefore, did not contribute to the FVIII protein levels.
Discussion
This study stems from the notion that amino acid and splicing codes overlap36 and thus the frequent missense changes, commonly considered only for the impact of the amino acid substitution on protein biology, might also have a detrimental effect on mRNA splicing. To provide qualitative and quantitative insights into the potential pleiotropic effect of missense mutations we chose to analyze the frequent p.R2016W/c.6046c>t variant reported to cause HA.11,12 It has previously been suggested to alter splicing13 and the amino change affects a partially (R2016) conserved (Figure 1A, lower panel) residue that in the FVIII structure (PDB: 2R7E)37 is surface exposed on the A3 domain at the interface with A1 domain (Figure 1C). Previous studies have shown that amino acid substitutions in this region have been shown to destabilize the FVIIIa structure and impair FVIII function,38 with a discrepancy in the functional activity values that were higher when measured by 1-stage clotting assays than with the chromogenic method.39–41 This activity profile was consistent with that observed in plasma from several HA patients bearing the F8 p.R2016W variant, which strongly supported an impact on the FVIII protein.
Lentiviral-mediated expression of rFVIII led us to demonstrate that the R2016W substitution differentially impairs secreted (~11% of FVIIIwt) and functional (~6%) protein levels, a finding that confirms the dysfunctional features of the molecule. On the other hand, investigation at the ectopic F8 mRNA level in several HA patients, and through minigene assays, indicated that the corresponding nucleotide change promotes exon 19 skipping. Interestingly, the c.6046c>t change manifested the negative impact because of the weak context of exon 19, as witnessed by the observation that its effect was removed when placed in an artificially strengthened exon 19. Moreover, the observation that masking the region by an antisense U7snRNA induced exon-skipping revealed the presence of a splicing regulatory element with enhancer features not predicted by computational analysis.42 This was further supported by the identification of two adjacent missense changes causing HA that remarkably reduced exon inclusion (p.G2013R/c.6037g>a and p.E2018G/c.6053a>g). Altogether these findings suggest that splicing impairment by missense changes is largely under-estimated.
The identified splicing-defective missense variants occur at residues that in the FVIII sequence alignment are completely (G2013, E2018) conserved (Figure 1A, lower panel), and in the FVIII structure37 are buried (G2013, E2018) within the A3 domain at the interface with A1 domain (Figure 1C). Recombinant FVIII protein expression demonstrated that they alter FVIII secretion and function differentially. We also identified the p.N2038S/c.6113a>g at the 5′ss (-3 position) that, as predicted, affected splicing. However, the amino acid change at the poorly conserved N2038 residue (Figure 1A) did not impact on FVIII protein biology, a finding that supports the relevance of aberrant splicing triggered by missense mutations.
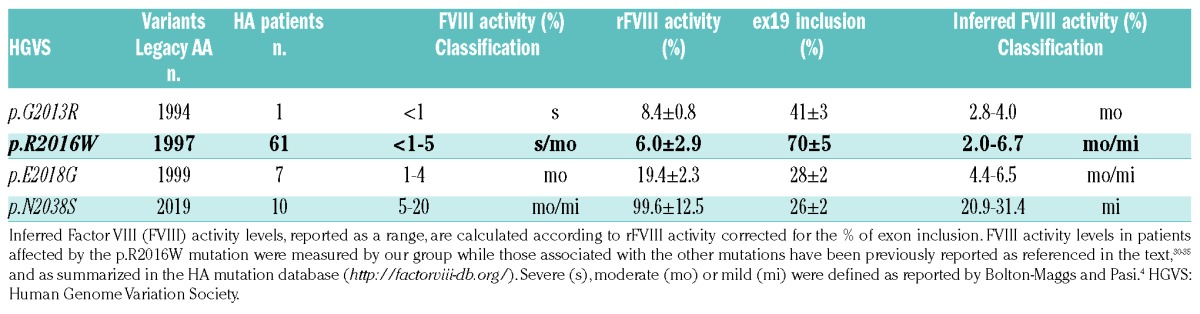
The combination of detrimental effects on splicing and protein observed in vitro (Figure 4) led us to infer FVIII functional levels (Table 1) that are consistent, albeit slightly over-estimated, with the FVIII activity measured in patients affected by the mutations, in spite of the considerable biological variation in patients and some limitations of the study. In particular, the exploitation of hybrid mini- genes and the expression of FVIII molecules lacking the B domain, necessary to appreciate the residual FVIII levels. However, the ‘improved’ B-domainless features could minimize the detrimental impact of amino acid substitutions on intracellular FVIII biosynthesis, potentially relevant for FVIII variants poorly secreted and functionally normal (e.g. rFVIII-2013R).
Figure 4.

Pleiotropic effects of F8 variants. Detrimental effects of variants on F8 splicing and on Factor VIII (FVIII) protein expressed as % reduction extent of wild-type (wt) of correct transcripts (top: splicing impairment) or of co-factor activity (bottom: protein impairment).
Table 1.
Comprehensive overview of detrimental effects of in vivo and in vitro on F8 mRNA and FVIII protein expression.
In summary, data clearly identify an F8 exonic region that has evolved in relation to both mRNA maturation (nucleotide splicing code) and protein (amino acid code) constraints, a feature potentially shared with other exons and with implications for the HA pathogenic mechanisms and mutation pattern.
For the first time, the combination of differentially altered mRNA processing, FVIII biosynthesis and co-factor activity was experimentally evaluated for clustered variants, and showed to substantially contribute to variably significant FVIII deficiency. This accounts for a gradient of residual FVIII activity that provides valuable information as to how to interpret the HA coagulation phenotypes.
Supplementary Material
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/103/2/344
Funding
The authors would like to thank Novo Nordisk (“Access to Insight Basic Research Grant 2014” to MP), Telethon-Italy (GGP14190 to MP) for the financial support and Prof. Shumperli for U7snRNA expressing vector.
References
- 1.Branchini A, Rizzotto L, Mariani G, et al. Natural and engineered carboxy-terminal variants: decreased secretion and gain-of-function result in asymptomatic coagulation factor VII deficiency. Haematologica. 2012;97(5):705–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Branchini A, Campioni M, Mazzucconi MG, et al. Replacement of the Y450 (c234) phenyl ring in the carboxyl-terminal region of coagulation factor IX causes pleiotropic effects on secretion and enzyme activity. FEBS Lett. 2013;587(19):3249–3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Margaglione M, Castaman G, Morfini M, et al. The Italian AICE-Genetics hemophilia A database: results and correlation with clinical phenotype. Haematologica. 2008; 93(5):722–728. [DOI] [PubMed] [Google Scholar]
- 4.Bolton-Maggs PH, Pasi KJ. Haemophilias A and B. Lancet. 2003; 361(9371):1801–1809. [DOI] [PubMed] [Google Scholar]
- 5.Peyvandi F, Kunicki T, Lillicrap D. Genetic sequence analysis of inherited bleeding diseases. Blood. 2013;122(20):3423–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lenting PJ, van Mourik JA, Mertens K. The life cycle of coagulation factor VIII in view of its structure and function. Blood. 1998; 92(11):3983–3996. [PubMed] [Google Scholar]
- 7.Furlan Freguia C, Toso R, Pollak ES, Arruda VR, Pinotti M, Bernardi F. Characterization of mild coagulation factor VII deficiency: activity and clearance of the Arg315Trp and Arg315Lys variants in the Cys310-Cys329 loop (c170s). Haematologica. 2004; 89(12):1504–1509. [PubMed] [Google Scholar]
- 8.Balestra D, Barbon E, Scalet D, et al. Regulation of a strong F9 cryptic 5′ss by intrinsic elements and by combination of tailored U1snRNAs with antisense oligonucleotides. Hum Mol Genet. 2015; 24(17):4809–4916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tajnik M, Rogalska ME, Bussani E, et al. Molecular Basis and Therapeutic Strategies to Rescue Factor IX Variants That Affect Splicing and Protein Function. PLoS Genet. 2016;12(5):e1006082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goodeve AC, Reitsma PH, McVey JH, Working Group on Nomenclature of the S, Standardisation Committee of the International Society on T, Haemostasis. Nomenclature of genetic variants in hemostasis. J Thromb Haemost. 2011;9(4):852–855. [DOI] [PubMed] [Google Scholar]
- 11.Santagostino E, Mancuso ME, Tripodi A, et al. Severe hemophilia with mild bleeding phenotype: molecular characterization and global coagulation profile. J Thromb Haemost. 2010;8(4):737–743. [DOI] [PubMed] [Google Scholar]
- 12.Garagiola I, Seregni S, Mortarino M, et al. A recurrent F8 mutation (c.6046C>T) causing hemophilia A in 8% of northern Italian patients: evidence for a founder effect. Mol Genet Genomic Med. 2016;4(2):152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Theophilus BD, Enayat MS, Williams MD, Hill FG. Site and type of mutations in the factor VIII gene in patients and carriers of haemophilia A. Haemophilia. 2001; 7(4):381–391. [DOI] [PubMed] [Google Scholar]
- 14.Ward NJ, Buckley SM, Waddington SN, et al. Codon optimization of human factor VIII cDNAs leads to high-level expression. Blood. 2011;117(3):798–807. [DOI] [PubMed] [Google Scholar]
- 15.Casari C, Pinotti M, Lancellotti S, et al. The dominant-negative von Willebrand factor gene deletion p.P1127_C1948delinsR: molecular mechanism and modulation. Blood. 2010;116(24):5371–5376. [DOI] [PubMed] [Google Scholar]
- 16.Oberbek A, Matasci M, Hacker DL, Wurm FM. Generation of stable, high-producing CHO cell lines by lentiviral vector-mediated gene transfer in serum-free suspension culture. Biotechnol Bioeng. 2011; 108(3):600–610. [DOI] [PubMed] [Google Scholar]
- 17.Pagani F, Stuani C, Tzetis M, et al. New type of disease causing mutations: the example of the composite exonic regulatory elements of splicing in CFTR exon 12. Hum Mol Genet. 2003;12(10):1111–1120. [DOI] [PubMed] [Google Scholar]
- 18.Pinotti M, Rizzotto L, Balestra D, et al. U1-snRNA-mediated rescue of mRNA processing in severe factor VII deficiency. Blood. 2008;111(5):2681–2684. [DOI] [PubMed] [Google Scholar]
- 19.Pagani F, Buratti E, Stuani C, Bendix R, Dork T, Baralle FE. A new type of mutation causes a splicing defect in ATM. Nat Genet. 2002;30(4):426–429. [DOI] [PubMed] [Google Scholar]
- 20.Kudaravalli R, Tidd T, Pinotti M, et al. Polymorphic changes in the 5′ flanking region of factor VII have a combined effect on promoter strength. Thromb Haemost. 2002;88(5):763–767. [PubMed] [Google Scholar]
- 21.Lunghi B, Pinotti M, Maestri I, Batorova A, Bernardi F. Evaluation of factor V mRNA to define the residual factor V expression levels in severe factor V deficiency. Haematologica. 2008;93(3):477–478. [DOI] [PubMed] [Google Scholar]
- 22.O’Brien DP, Pattinson JK, Tuddenham EG. Purification and characterization of factor VIII 372-Cys: a hypofunctional cofactor from a patient with moderately severe hemophilia A. Blood. 1990;75(8):1664–1672. [PubMed] [Google Scholar]
- 23.Pipe SW, Eickhorst AN, McKinley SH, Saenko EL, Kaufman RJ. Mild hemophilia A caused by increased rate of factor VIII A2 subunit dissociation: evidence for nonproteolytic inactivation of factor VIIIa in vivo. Blood. 1999;93(1):176–183. [PubMed] [Google Scholar]
- 24.Pipe SW, Saenko EL, Eickhorst AN, Kemball-Cook G, Kaufman RJ. Hemophilia A mutations associated with 1-stage/2-stage activity discrepancy disrupt protein-protein interactions within the triplicated A domains of thrombin-activated factor VIIIa. Blood. 2001;97(3):685–691. [DOI] [PubMed] [Google Scholar]
- 25.Nogami K, Zhou Q, Wakabayashi H, Fay PJ. Thrombin-catalyzed activation of factor VIII with His substituted for Arg372 at the P1 site. Blood. 2005;105(11):4362–4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jourdy Y, Nougier C, Roualdes O, et al. Characterization of five associations of F8 missense mutations containing FVIII B domain mutations. Haemophilia. 2016; 22(4):583–589. [DOI] [PubMed] [Google Scholar]
- 27.Liang Q, Xiang M, Lu Y, et al. Characterisation and quantification of F8 transcripts of ten putative splice site mutations. Thromb Haemost. 2015;113(3):585–592. [DOI] [PubMed] [Google Scholar]
- 28.Roca X, Krainer AR, Eperon IC. Pick one, but be quick: 5′ splice sites and the problems of too many choices. Genes Dev. 2013;27(2):129–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marquis J, Meyer K, Angehrn L, Kampfer SS, Rothen-Rutishauser B, Schumperli D. Spinal muscular atrophy: SMN2 pre-mRNA splicing corrected by a U7 snRNA derivative carrying a splicing enhancer sequence. Mol Ther. 2007;15(8):1479–1486. [DOI] [PubMed] [Google Scholar]
- 30.Repessé Y, Slaoui M, Ferrandiz D, et al. Factor VIII (FVIII) gene mutations in 120 patients with hemophilia A: detection of 26 novel mutations and correlation with FVIII inhibitor development. J Thromb Haemost. 2007;5(7):1469–1476. [DOI] [PubMed] [Google Scholar]
- 31.Hill M, Deam S, Gordon B, Dolan G. Mutation analysis in 51 patients with haemophilia A: report of 10 novel mutations and correlations between genotype and clinical phenotype. Haemophilia. 2005;11(2):133–141. [DOI] [PubMed] [Google Scholar]
- 32.Green PM, Bagnall RD, Waseem NH, Giannelli F. Haemophilia A mutations in the UK: results of screening one-third of the population. Br J Haematol. 2008; 143(1):115–128. [DOI] [PubMed] [Google Scholar]
- 33.Markoff A, Gerke V, Bogdanova N. Combined homology modelling and evolutionary significance evaluation of missense mutations in blood clotting factor VIII to highlight aspects of structure and function. Haemophilia. 2009;15(4):932–941. [DOI] [PubMed] [Google Scholar]
- 34.Liu M, Murphy ME, Thompson AR. A domain mutations in 65 haemophilia A families and molecular modelling of dysfunctional factor VIII proteins. Br J Haematol. 1998;103(4):1051–1060. [DOI] [PubMed] [Google Scholar]
- 35.Schwaab R, Oldenburg J, Schwaab U, et al. Characterization of mutations within the factor VIII gene of 73 unrelated mild and moderate haemophiliacs. Br J Haematol. 1995;91(2):458–464. [DOI] [PubMed] [Google Scholar]
- 36.Pagani F, Buratti E, Stuani C, Baralle FE. Missense, nonsense and neutral mutations define juxtaposed regulatory elements of splicing in CFTR Exon 9. J Biol Chem. 2003;278(29):26580–26588. [DOI] [PubMed] [Google Scholar]
- 37.Shen BW, Spiegel PC, Chang CH, et al. The tertiary structure and domain organization of coagulation factor VIII. Blood. 2008;111(3):1240–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Armstrong E, Hillarp A. Assay discrepancy in mild haemophilia A. Eur J Haematol Suppl. 2014;76:48–50. [DOI] [PubMed] [Google Scholar]
- 39.Pavlova A, Delev D, Pezeshkpoor B, Müller J, Oldenburg J. Haemophilia A mutations in patients with non-severe phenotype associated with a discrepancy between one-stage and chromogenic factor VIII activity assays. Thromb Haemost. 2014;111(5):851–861. [DOI] [PubMed] [Google Scholar]
- 40.Duncan EM, Rodgers SE, McRae SJ. Diagnostic testing for mild hemophilia a in patients with discrepant one-stage, two-stage, and chromogenic factor VIII:C assays. Semin Thromb Hemost. 2013; 39(3):272–282. [DOI] [PubMed] [Google Scholar]
- 41.Trossaërt M, Boisseau P, Quemener A, et al. Prevalence, biological phenotype and genotype in moderate/mild hemophilia A with discrepancy between one-stage and Chromogenic factor VIII activity. J Thromb Haemost. 2011;9(3):524–530. [DOI] [PubMed] [Google Scholar]
- 42.Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31(13):3568–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.