

Genetic predisposition due to germline mutations in a number of genes (e.g., ETV6, GATA2, and RUNX1) has been recognized as an important cause of myeloid malignancies. In the 2016 update of the World Health Organization classification of hematological malignancies, a new section, ‘myeloid neoplasms with germline predisposition’, has been introduced. Genetically-defined hereditary myeloid malignancy syndromes (HMMSs) can clinically present with preceding hematological findings and/or syndromic features. There is remarkable intrafamilial and interfamilial variability. The identification of the underlying genetic defect has an impact on the clinical care of patients and their families.1 Herein, we report on the first familial germline mutation in MDS1 and EVI1 complex locus (MECOM). Our finding extends MECOM germline mutation associated phenotypes and proposes MECOM as a novel HMMS candidate gene.
In the family investigated (Figure 1), HMMS was suspected due to: (i) myeloid malignancies observed in individual I:1 and II:3, (ii) congenital thrombocytopenia in individual I:1, and (iii) the autosomal dominant trait of limb dysmorphisms, including radioulnar synostosis, and hearing impairment in all affected family members (Online Supplementary Table S1 and Online Supplementary Table S2). In individual I:1, myelodysplastic syndrome with excess blasts 2 (MDS-EB-2) was diagnosed at age 73 following several months of progressive neutropenia and anemia. In the bone marrow, hypercellularity, dysplasia and 10–15% blasts were observed. Cytogenetic investigations detected an uncommon interstitial deletion in the long arm of chromosome 9. A fluorescence in situ hybridization panel addressing frequent aberrations in MDS revealed normal results. The patient died during the first treatment cycle of a 5-azacytidine-based chemotherapy following a systemic infection with fever and multiple organ failure. In individual II:3, following a period of 6 years with anemia and neutropenia, MDS/myeloproliferative neoplasm-unclassifiable (MDS/MPN-U) was diagnosed at age 48. An uncommon balanced translocation between the long arm of chromosome 1 and the IGH locus at chromosome 14q32 was observed. Moderate hypercellularity, dysplasia, megakaryocytopenia, and 6–8% blasts were observed in the bone marrow. Following myeloablative conditioning, individual II:3 underwent allogenic peripheral blood stem cell transplantation from a matched unrelated donor; four years post transplantation, there is no sign of a recurrence or of graft-versus-host disease. For individuals I:1 and II:3, no data was available regarding their platelet count, morphology or function prior to the malignancies observed. Samples for further investigations in the myeloid malignancies observed (e.g., expression of MECOM or molecular genetic characterization) were not available. In individuals III:3 and III:2 (peripheral blood cell count at age 18: erythrocytes 4.2 × 1012/l, leukocytes 9.0 × 109/l, thrombocytes 240 × 109/l), no cytopenia or overt leukemia have been reported at their current age of 24. Bone marrow biopsies were not performed in either individual.
Figure 1.

Family with MECOM:p.Cys766Gly missense mutation. Individual identifiers are given below the symbols. For additional information on the phenotype and an overview regarding the hematological malignancies in individual I:1 and II:3, please refer to Online Supplementary Table S1 and Online Supplementary Table S2. Symbol patterns refer to the following features: black upper left third, limb defects including radioulnar synostosis and hearing impairment, grey upper right third, congenital thrombocytopenia, and black-striped lower third, myeloid malignancy. Cys766Gly: heterozygous MECOM missense variant p.Cys766Gly; wt: wild-type; - signifies that no genetic analysis was performed.
To screen for the causative genetic alteration, whole exome sequencing of individuals II:3, III:2, and III:3 was performed. Variants were filtered for those that are (i) called in all individuals, (ii) predicted to be damaging, (iii) reported to have an allele frequency of ≤0.1%, and (iv) not listed in our in house database of recurrent variants (Online Supplementary Table S3). Following this examination, the novel heterozygous MECOM variant NM_001105078.3:c.2296T>G p.(Cys766Gly) was detected and confirmed by Sanger sequencing. The variant was not reported in sequencing databases. No pathogenic variants were observed in HMMSs-associated genes1 (Online Supplementary Table S4) and HOXA11, in which a single base pair deletion was reported to cause an inherited bone marrow failure syndrome designated as radioulnar synostosis and amegakaryocytic thrombocytopenia (RUSAT).2 The variant was also identified in individual I:1 via Sanger sequencing of DNA extracted from fixed nuclei stored after cytogenetic investigations. It was absent in the DNA extracted from the peripheral blood of healthy relatives I:3, I:4, II:1 and III:1.
The novel MECOM missense mutation affects a conserved cysteine (Figure 2A), which is crucial for the tetrahedral coordination of the zinc atom in C2H2-type zinc finger motif 9 (Figure 2B). Due to the non-conservative substitution at this crucial site, the C2H2-type zinc finger motif 9 matching Pfam motif zf-C2H2 (PF00096) is no longer recognized by in silico analyses (Figure 2C). Motif 9 has been shown to be crucial for DNA-binding of zinc finger domain 2 (ZF2) targeting the consensus sequence GAAGATGAG.3 The mutation, in all likelihood, hinders DNA interaction via ZF2, and may alter protein-protein interactions.4,5 In silico prediction tools indicated its probable damaging effect (Online Supplementary Table S5). Missense mutations in C2H2-type zinc finger motif 9 of MECOM have already been reported (Figure 2D). None of these previously reported mutations hinder recognition of the C2H2-type zinc finger motif 9 (Figure 2C). The missense mutation R769C, being equivalent to p.Arg778Cys, affects one of the DNA contact residues3 (Figure 2D). This missense mutant failed to bind the consensus sequence while maintaining its transforming capability.6 Mice of the mutant strain Junbo are susceptible to otitis media and develop chronic inflammation and deafness. The causative mutation affects MECOM and corresponds to human p.Asn782Ile (Figure 2D). In contrast to the middle ear bone dysplasia in the family we studied, no ear bone defects were reported in Junbo mice. However, they showed limb defects (i.e., brachydactyly and polydactyly) and defects in neutrophil differentiation (MecomJbo, MGI: 2158381).7 Based on these careful theoretical presumptions, MECOM:c.2296T>G was assumed to be the causative genetic alteration in the family presented herein. We suppose that the missense variant disturbs the secondary structure of zinc finger motif 9 and that this leads to an altered and/or lost DNA-binding specificity and protein-protein interaction of ZF2. Thus, expression of the MECOM missense variant may dysregulate signaling networks and can thereby lead to congenital malformations, thrombocytopenia and/or bone marrow failure, and predisposition to myeloid malignancies.
Figure 2.

MECOM C2H2-type zinc finger motif 9. (A) Multiple sequence alignment of C2H2-zinc finger motif 9 of MECOM protein sequences using ClustalW2. Individual sequences are headed by the UniProtKB database identifier|species information. The alignment encompasses amino acid Q03112|758-787, of which amino acids 761-784 form ZF2, C2H2-zinc finger motif 9 of MECOM. The open arrows and cylinder depicted above the sequence alignment indicate β sheets and a helix, respectively. The blue and orange triangles below the alignment mark the amino acids that are essential for the folding stability of the zinc finger motif and potential DNA contact residues, respectively. Cys766 is highlighted in red. Alignment mismatches are highlighted in yellow. (B) SWISS-MODELL of Q03112, MDS and EVI1 complex locus protein EVI1 showing the four conserved amino acids of C2H2-type zinc finger motif 9 (i.e., Cys763, Cys766, His 779, and His784) which are crucial for the tetrahedral coordination of the zinc atom, shown in green. Cys766 is highlighted in red. (C) Screening of MECOM wild-type protein sequence (UniprotKB Q03112) for Pfam motif matches identifies zinc finger motifs zf-C2H2_6 (PF13912) and zf-C2H2 (PF00096), schematically displayed as grey and black boxes, respectively. The Pfam zinc finger motif zf-C2H2, matching C2H2-type zinc finger motif 9 of MECOM, is highlighted in blue. Below the wild-type sequence, results of reported mutations are shown and grouped with regard to C2H2-type zinc finger motif 8 and 9. The MECOM missense mutant p.Cys766Gly is highlighted in red. (D) At the bottom of the figure, a domain scheme of MECOM (Q03112) is given. C2H2-zinc finger motifs are shown in orange. Motif 1–7 and motif 8–10 form the N-terminal (ZF1) and C-terminal zinc finger domain 2 (ZF2), respectively. The nuclear localization signal, CTBP-binding motif 1/2, and an acidic domain are depicted in red, blue, and green, respectively. Above this outline, a schematic view of C2H2-zinc finger motif 9 is shown. The amino acids of the C2H2-zinc finger motif 9 refer to UniprotKB Q03112-1|760-787. Residues crucial for the tetrahedral coordination of the central zinc atom and potential DNA contact residues are highlighted in blue and orange, respectively. The germline variant p.Cys766Gly identified in the present study is highlighted in red. Somatic variants reported in COSMIC are shown in black. A mutant mouse strain and an in vitro variant are depicted in green and blue, respectively.
MECOM (3q26) was initially identified as a common ecotropic viral integration site 1 (Evi1) in murine myeloid leukemia.8 Its isoforms act as transcription factors. They contain an N-terminal (ZF1) and C-terminal (ZF2) zinc finger domain formed by seven and three C2H2-type zinc finger motifs, respectively (Figure 2D). Evi1 is involved in hematopoietic stem cell maintenance, differentiation and leukemogenesis.9 De novo microdeletions affecting MECOM (i.e., the entire locus or MDS1-encoding parts) have been described in two patients with congenital thrombocytopenia.10,11 Reports that these patients had no skeletal defects provided the first evidence regarding the impact of MECOM germline alterations. Recently, three simplex patients showing a RUSAT phenotype were reported to carry de novo missense variants in ZF2, C2H2-type zinc finger motif 8 of MECOM (i.e., p.Arg750Trp, p.His751Arg, and p.Thr756Ala), which affect MDS1-EVI1 and EVI1 transcripts. The observed variants showed diminished target sequence binding and altered transcriptional regulation,12 which is in line with our theoretical assumptions regarding the MECOM missense variant p.Cys766Gly. Herein, we report on the first family with autosomal dominant inheritance of a MECOM germline mutation in four patients spanning three generations. The clinical findings resemble the clinical observations in those patients reported previously (Table 1). Limb defects, including radioulnar synostosis, were seen in all affected individuals with missense variants in ZF2, but not in the patients with deletions. Congenital thrombocytopenia was reported in only four out of seven probands, including individual I:1 of the family we investigated, for whom congenital thrombocytopenia, but no amegakaryocytosis, was observed. No bone marrow failure was seen in II:3, III:2 and III:3. In HOXA11-associated RUSAT, incomplete penetrance of thrombocytopenia or bone marrow failure was also reported.2
Table 1.
Patients with constitutional MECOM alterations.
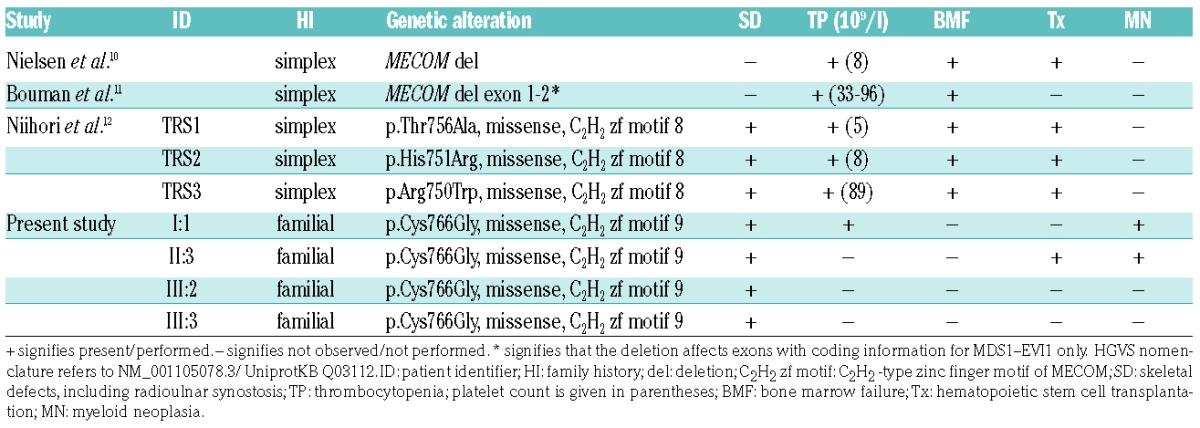
Four out of the five previously reported patients with MECOM germline mutations received hematopoietic stem cell transplantation within the first two years of life due to progressive pancytopenia. The fifth patient died at 28 days of age. No hematological malignancy was described in the affected children.10–12 However, we observed, for the first time, that two individuals with MECOM:p.(Cys766Gly) developed myeloid malignancies. In sporadic myeloid leukemia, recurrent chromosomal aberrations targeting MECOM are known: inversion inv(3;3) and translocation t(3;3) leading to EVI1 overexpression, and translocation t(3;21), and t(3;12) leading to RUNX1-EVI1 and ETV6-EVI1 fusion genes, respectively.13 Aberrant EVI1 expression in patients with inv(3;3)/t(3;3) is due to the repositioning of a distal GATA2 enhancer which causes the activation of MECOM transcription while simultaneously conferring GATA2 haploinsufficiency.14 Haploinsufficiency of GATA2 due to germline mutations is associated with an increased risk for MDS. In particular, adolescents with MDS and monosomy 7 showed a high prevalence of GATA2 germline mutations.15 Leukemias with inv(3;3)/t(3;3) are frequently accompanied by monosomy 7, and acute myeloid leukemia with monosomy 7 as the sole abnormality showed significant upregulation of MECOM.14 Inherited thrombocytopenia caused by mutation of ANKRD26, ETV6, and RUNX1, these two latter genes being involved in the recurrent translocations t(3;12) and t(3;21),13 are also associated with an increased risk for hematological malignancies.1
In conclusion, we report on a novel MECOM germline mutation within the C2H2-zinc finger motif 9. Our observations reconfirm previous findings and extend the phenotypic spectrum, since two of the four patients of the family presented herein developed myeloid malignancies. This association has not been heretofore reported. Thus, one should be aware of a probable increased risk for hematological neoplasia and, as recommended for several bone marrow failure syndromes and HMMSs,1 surveillance should be critically considered in affected individuals. Explorations of additional individuals and/or families with MECOM germline mutations are required in order to: (i) confirm the present conclusions, (ii) investigate the clinical variability and genotype-phenotype correlations, (iii) gain more insights into the probable increased risk for hematological neoplasia, and (iv) elucidate its molecular path.
Supplementary Material
Acknowledgments
The authors would like to thank the family, described herein, for supporting this study and Michaela Losch and Bernd Haermeyer for their expert technical assistance. TR is supported by a John Goldman Clinical Research grant of the European Hematology Association and an intramural HiLF grant of the Hannover Medical School. This study was supported in part by research funding from the German Research Foundation (DFG), Cluster of Excellence REBIRTH to B.S. and D.S.
Footnotes
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Churpek JE, Godley LA. How I diagnose and manage individuals at risk for inherited myeloid malignancies. Blood. 2016;128(14):1800–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thompson AA, Nguyen LT. Amegakaryocytic thrombocytopenia and radio-ulnar synostosis are associated with HOXA11 mutation. Nat Genet. 2000;26(4):397–398. [DOI] [PubMed] [Google Scholar]
- 3.Funabiki T, Kreider BL, Ihle JN. The carboxyl domain of zinc fingers of the Evi-1 myeloid transforming gene binds a consensus sequence of GAAGATGAG. Oncogene. 1994;9(6):1575–1581. [PubMed] [Google Scholar]
- 4.Brayer KJ, Segal DJ. Keep your fingers off my DNA: protein-protein interactions mediated by C2H2 zinc finger domains. Cell Biochem Biophys. 2008;50(3):111–131. [DOI] [PubMed] [Google Scholar]
- 5.Pavletich NP, Pabo CO. Zinc finger-DNA recognition: crystal structure of a Zif268-DNA complex at 2.1 A. Science. 1991;252(5007):809–817. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Sicot G, Cui X, et al. Targeting a DNA binding motif of the EVI1 protein by a pyrrole-imidazole polyamide. Biochemistry. 2011; 50(48):10431–10441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parkinson N, Hardisty-Hughes RE, Tateossian H, et al. Mutation at the Evi1 locus in Junbo mice causes susceptibility to otitis media. PLoS Genet. 2006;2(10):e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kustikova O, Fehse B, Modlich U, et al. Clonal dominance of hematopoietic stem cells triggered by retroviral gene marking. Science. 2005;308(5725):1171–1174. [DOI] [PubMed] [Google Scholar]
- 9.Kustikova OS, Schwarzer A, Stahlhut M, et al. Activation of Evi1 inhibits cell cycle progression and differentiation of hematopoietic progenitor cells. Leukemia. 2013;27(5):1127–1138. [DOI] [PubMed] [Google Scholar]
- 10.Nielsen M, Vermont CL, Aten E, et al. Deletion of the 3q26 region including the EVI1 and MDS1 genes in a neonate with congenital thrombocytopenia and subsequent aplastic anaemia. J Med Genet. 2012;49(9):598–600. [DOI] [PubMed] [Google Scholar]
- 11.Bouman A, Knegt L, Groschel S, et al. Congenital thrombocytopenia in a neonate with an interstitial microdeletion of 3q26.2q26.31. Am J Med Genet A. 2016;170A(2):504–509. [DOI] [PubMed] [Google Scholar]
- 12.Niihori T, Ouchi-Uchiyama M, Sasahara Y, et al. Mutations in MECOM, encoding Oncoprotein EVI1, cause radioulnar synostosis with amegakaryocytic thrombocytopenia. Am J Hum Genet. 2015; 97(6):848–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hinai AA, Valk PJ. Review: Aberrant EVI1 expression in acute myeloid leukaemia. Br J Haematol. 2016;172(6):870–878. [DOI] [PubMed] [Google Scholar]
- 14.Groschel S, Sanders MA, Hoogenboezem R, et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 2014;157(2):369–381. [DOI] [PubMed] [Google Scholar]
- 15.Wlodarski MW, Hirabayashi S, Pastor V, et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood. 2016;127(11):1387–1397. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.