

Pyruvate kinase deficiency (PKD) is the most frequent glycolytic enzyme defect causing hereditary non-spherocytic hemolytic anemia.1 PKD leads to energy deprivation of the red cell, ultimately resulting in premature red cell death. Premature red cell death causes clinical symptoms of hemolytic anemia. The degree of hemolysis can vary widely, from very mild and fully compensated forms to life-threatening anemia with transfusion dependency.2 The treatment for PKD is mainly supportive, and consists of regular red blood cell transfusions, splenectomy and chelation therapy for iron overload.3 Hematopoietic allogeneic stem cell transplantation (HSCT) has the potential to cure PKD. However, there is little experience of applying HSCT in PKD. The current knowledge of HSCT in PKD is predominantly based on animal studies, and guidelines are not available.4,5 To date, only four human cases of HSCT have been published in the literature.6–8 The total number of cases transplanted worldwide is unknown.
The aim of this study is to make a worldwide inventory of PKD cases that have been treated by HSCT, and to evaluate indication, procedures employed and outcome as a first step towards the establishment of guidelines for HSCT in PKD. In order to achieve this goal queries were sent to national and international databanks, including the European Society for Blood and Marrow Transplantation (EBMT), the Center for International Blood and Marrow Transplant Research (CIBMTR), and the National Institute of Health (NIH), as well as to physicians known to be involved in HSCT in PKD patients. For each case found, a specifically designed questionnaire was sent to the physician involved. The questionnaire contained questions on disease characteristics, pre-transplant condition, transplant regimen and post-transplant outcome.9 All data were evaluated by an experienced physician, and institutions were contacted in case of inconsistencies. An adapted EBMT score (i.e., age, donor type and donor-recipient sex combination) was calculated based on the answers provided.10 In addition, data from two additional cases, published recently, were extracted from the literature and included.8 To the best of our knowledge, we have included all cases worldwide.
In total, 16 cases were found to be treated by HSCT between 1996 and 2015. Patient characteristics are summarized in Table 1. Patients had all been treated in either European or Asian centers. No cases resulted as being transplanted in the USA. Patient’s median age at transplantation was 6.5 years. All patients were transfusion-dependent before transplantation, with median transfusion needs of 13 units of packed red blood cells per year (range: 6 to 34 units).
Table 1.
Patient characteristics.
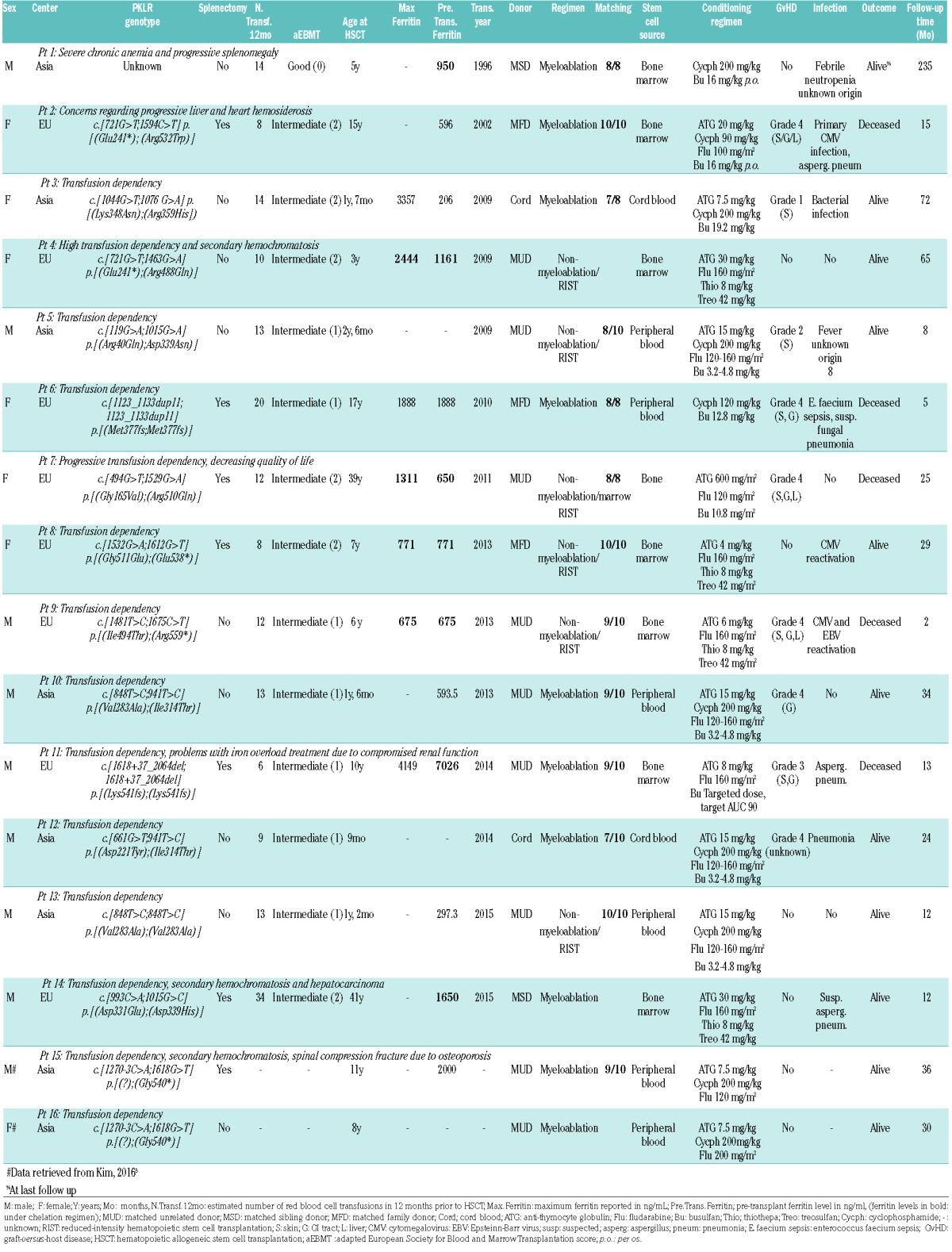
Conditioning and prophylaxis characteristics are summarized in Table 1. All patients received graft-versus-host disease (GvHD) prophylaxis. Ex vivo T-cell depletion was performed in one transplant. In another, red cell depletion was performed. Five transplants were sex-matched, four were female receiver-male donor and four were male receiver-female donor; this information was not available for three cases.
Median follow-up time after transplantation was 2.3 years (range: 2 months to 19 years). Fifteen patients showed engraftment. The sixteenth patient initially showed pancytopenia and mixed chimerism. Following splenectomy six months post-transplantation, this patient’s cell count spontaneously transitioned to normal with full donor chimerism. Two patients suffered from secondary graft loss; in one there was recovery to 91% donor chimerism after donor lymphocyte infusion. The outcome in the second patient was unknown.
Infectious complications and occurrence of GvHD are summarized in Table 1. The most significant infectious complications were aspergillus pneumonia (two patients), suspected aspergillus pneumonia (one patient), suspected fungal pneumonia (one patient), pneumonia (one patient), sepsis (one patient) and bacterial infection e causa ignota (one patient). GvHD grade 4 was reported in 6/16 cases (38%). Seven out of 16 cases (44%) did not show symptoms of GvHD. There was no correlation between GvHD prophylaxis or any other clinical factors and the occurrence of GvHD grade 2-4 in these patients.
Five out of 16 patients (31%) did not survive. All died of transplant-related causes. They had a median survival time of 13 months (range: 2–25 months). The two-year cumulative survival was 74%. Two patients had not yet reached the two-year milestone at the time of the questionnaire. The three-year cumulative survival rate was 65% (Figure 1); seven patients had not yet reached the three-year milestone.
Figure 1.

Overall survival, according to age.
Patients who did not survive differed significantly from surviving patients. (Figure 1, Table 2). They were significantly older (P=0.036). Nine out of ten patients (90%) <10 years of age survived transplantation, whereas two out of six (33%) ≥10 survived. Patients <10 years were less often splenectomized (P=0.001) and had lower pre-transfusion hemoglobin levels prior to HSCT (P=0.04). Patients who did not survive had all been treated in European centers. All patients treated in Asian centers survived transplantation (8/8). Patients treated in Asian centers were younger (P=0.001), less often splenectomized (P=0.041), and had lower ferritin levels prior to HSCT (P=0.048). In addition, they were more often transplanted using peripheral blood stem cells as a source (P=0.014) and more often conditioned on a cyclophosphamide regimen (P=0.007). Furthermore, patients who did not survive had frequently suffered from GvHD grade 2-4 (P=0.031). Notably, four out of five deceased patients had suffered from both GvHD grade 3-4 and infection or viral reactivation.
Table 2.
Statistical differences between surviving and non-surviving patients.

There were no significant differences in sex, plasma ferritin level, use of pre-transplant chelation therapy, transfusion burden in the 12 months prior to HSCT, adapted EBMT-score, conditioning regimen, relation to donor, graft type, donor-recipient sex combination, or transplant source.
In conclusion, herein we discuss the first global study on the outcome of all patients known to have undergone HSCT in PKD. Since guidelines for HSCT in PKD are lacking, this report may be a helpful first step toward future protocols. Compared to published survival rates for other forms of hereditary anemias, cohorts that are otherwise comparable in age, time period and transplant hospital, the overall survival rate after HSCT in PKD is relatively low.11–13 The present analysis of all 16 PKD patients known to be transplanted to date showed a three-year overall survival of 65%. Significantly better survival was observed for patients transplanted before the age of ten. A negative effect of age on survival is also reported for other forms of hereditary anemia.11,12 Concurrently, we noticed a striking difference in survival between patients treated in Asian and European centers, which could possibly be explained by the difference in age at which patients were transplanted. In addition, Asian patients were non-splenectomized in many instances, and had lower pre-transplantation ferritin levels, which could also be related to the young age at which HSCT was performed.
Asian patients were more frequently transplanted with peripheral blood stem cells as opposed to bone marrow-derived stem cells. Peripheral blood stem cells are easier to collect from the donor, but reportedly increase the risk of chronic GvHD.14 Our cohort, however, was too small to analyze the specific effect of stem cell source on the occurrence of chronic GvHD.
An important limitation of this study is its retrospective character, and the fact that the small sample size did not allow us to perform post hoc correction for multiple testing. Therefore, the quantitative analysis of this data should be interpreted with care. Other limitations include the heterogeneity of conditioning regimens, and heterogeneity in the pre-transplant risk classification systems used. However, we did observe a better survival for patients transplanted prior to age ten. This effect of age might also play a role in the observed differences in survival between patients treated in European centers and those treated in Asian centers.
Although HSCT should be considered an investigational treatment, the strong decline in survival of treated patients over the age of ten suggests the need to evaluate HSCT as a treatment option early in life. However, since the rate of grade 3-4 GvHD was relatively high (7/16 = 44%), and death resulting from GvHD was likewise high (5/16 = 31%), transfusion dependency alone should not be an indication for performing HSCT in PKD.
Supplementary Material
Footnotes
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Miwa S, Fujii H. Molecular basis of erythroenzymopathies associated with hereditary hemolytic anemia: tabulation of mutant enzymes. Am J Hematol. 1996;51(2):122–132. [DOI] [PubMed] [Google Scholar]
- 2.Zanella A, Fermo E, Bianchi P, et al. Red cell pyruvate kinase deficiency: molecular and clinical aspects. Br J Haematol. 2005;130(1):11–25. [DOI] [PubMed] [Google Scholar]
- 3.Grace RF, Zanella A, Neufeld EJ, et al. Erythrocyte pyruvate kinase deficiency: 2015 Status report. Am J Hematol. 2015;90(9):825–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morimoto M, Kanno H, Asai H, et al. Pyruvate kinase deficiency of mice associated with nonspherocytic hemolytic anemia and cure of the anemia by marrow transplantation without host irradiation. Blood. 1995;86(11):4323–4330. [PubMed] [Google Scholar]
- 5.Weiden PL, Hackman RC, Deeg HJ, et al. Long-term survival and reversal of iron overload after marrow transplantation in dogs with congenital hemolytic anemia. Blood. 1981;57(1):66–70. [PubMed] [Google Scholar]
- 6.Tanphaichitr VS, Suvatte V, Issaragrisil S, et al. Successful bone marrow transplantation in a child with red blood cell pyruvate kinase deficiency. Bone Marrow Transplant. 2000;26(6):689–690. [DOI] [PubMed] [Google Scholar]
- 7.Akiyoshi K, Sekiguchi K, Okamoto T, et al. Cord blood transplantation in a young child with pyruvate kinase deficiency. Pediatr Int. 2016;58(7):634–636. [DOI] [PubMed] [Google Scholar]
- 8.Kim M, Park J, Lee J, et al. Hemolytic anemia with null PKLR mutations identified using whole exome sequencing and cured by hematopoietic stem cell transplantation combined with splenectomy. Bone Marrow Transplant. 2016;51(12):1605–1608 [DOI] [PubMed] [Google Scholar]
- 9.Sorror ML, Maris MB, Storb R, et al. Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood. 2005;106(8):2912–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gratwohl A, Hermans J, Goldman JM, et al. Risk assessment for patients with chronic myeloid leukaemia before allogeneic blood or marrow transplantation. Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Lancet. 1998;352(9134):1087–1092. [DOI] [PubMed] [Google Scholar]
- 11.Baronciani D, Angelucci E, Potschger U, et al. Hemopoietic stem cell transplantation in thalassemia: a report from the European Society for Blood and Bone Marrow Transplantation Hemoglobinopathy Registry, 2000–2010. Bone Marrow Transplant. 2016;51(4):536–541. [DOI] [PubMed] [Google Scholar]
- 12.Fagioli F, Quarello P, Zecca M, et al. Haematopoietic stem cell transplantation for Diamond Blackfan anaemia: a report from the Italian Association of Paediatric Haematology and Oncology Registry. Br J Haematol. 2014;165(5):673–681. [DOI] [PubMed] [Google Scholar]
- 13.Smetsers SE, Smiers FJ, Bresters D, et al. Four decades of stem cell transplantation for Fanconi anaemia in the Netherlands. Br J Haematol. 2016;174(6):952–961. [DOI] [PubMed] [Google Scholar]
- 14.Adhikari J, Sharma P, Bhatt VR. Optimal graft source for allogeneic hematopoietic stem cell transplant: bone marrow or peripheral blood? Future Oncol. 2016;12(15):1823–1832. [DOI] [PubMed] [Google Scholar]
- 15.Przepiorka D, Weisdorf D, Martin P, et al. 1994 Consensus conference on acute GVHD grading. Bone Marrow Transplant. 1995;15(6):825–828. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.