

Abstract
Based on prior reports that the HIV-1 Tat protein modulates amyloid-beta (Aβ) metabolism, this study aimed to compare CSF neural injury biomarkers between 27 patients with HIV subtype B, 26 patients with HIV subtype C, 18 healthy HIV-negative controls, and 24 patients with Alzheimer’s disease (AD). Immunoassays were used to measure soluble amyloid precursor protein α and β (sAPPα, sAPPβ); Aβ oligomers 38,40,42, and Aβ-total; phosphorylated tau (P-tau181), and total tau (T-tau). Comparisons between HIV(+) and HIV(-) (including AD) were adjusted by linear regression for gender and age; HIV subtype comparisons were adjusted for nadir CD4 and plasma viral load suppression. The p-values were corrected for multiple testing with the Benjamini-Hochberg procedure.
CSF Aβ-42 and Hulstaert (P-tau181) index were lower in HIV1-C than B (p= 0.03, and 0.049 respectively); subtypes did not differ on other CSF biomarkers or ratios. Compared to AD, HIV(+) had lower CSF levels of T-tau, P-tau181 (p <0.001), and sAPPα (p= 0.041); HIV(+) had higher CSF Aβ-42 (p=0.002), and higher CSF indexes: [Aß-42/ (240 + 1.18 T-tau)], P-tau181/Aβ-42, T-tau/Aβ-42, P-tau181/T- tau, sAPPα/β (all p≤0.01) than AD. Compared to HIV(-), HIV(+) had lower CSF Aβ-42, and T-tau (all p≤0.004).
As conclusion, amyloid metabolism was influenced by HIV infection in a subtype-dependent manner. Aß-42 levels were lower in HIV1-C than B, suggesting that there may be greater deposition of Aß-42 in HIV1-C. These findings are supported by CSF Hulstaert (P-tau181) index. Differences between HIV and AD in the patterns of Aß and Tau biomarkers suggest that CNS HIV infection and AD may not share some of same mechanisms of neuronal injury.
Keywords: HIV, amyloid metabolism, CSF, biomarkers, neuronal injury, subtype, central nervous system
The interactions between HIV and aging are an increasingly important topic. Since the introduction of highly active antiretroviral therapy (HAART), patients with HIV (HIV+) have been living longer with an improved quality of life and decreased HIV-related illnesses (Burgoyne and Tan 2008; Raboni et al. 2017). However, HIV and aging-related challenges have emerged and are a clinical, health care, and research priority. Chronic HIV infection results in a 4.9 year increase in biological age due to epigenetic targeting of HLA (Gross et al. 2016).
Aging increases amyloid-beta (Aβ) accumulation and neurocognitive impairment (High et al. 2012); amyloid deposition has been described in the brains of relatively young patients with HIV infection (Achim et al. 2009; Mothapo et al. 2015). Several studies have noted marked increases in diffuse amyloid plaques and intraneuronal amyloid deposition in HIV(+) individuals compared to HIV(-) age-matched controls (Esiri et al. 1998; Green et al. 2005; Rempel and Pulliam 2005; Achim et al. 2009).
HIV1 infected cells actively secrete the viral protein transactivator of transcription (Tat) (Ensoli et al. 1993). Tat was found in the brains of patients with HIV1 infection, and Aβ staining was significantly increased in human brain from individuals with HIV1 infection compared to controls (Green et al. 2005; Achim et al. 2009). Tat inhibits neprilysin (NEP), which degrades brain Aβ deposits, the higher inhibitory activity of NEP depends on the “CC” dimotif (Rempel and Pulliam 2005; Daily et al. 2006). Tat in patients with HIV1-subtype C (HIV1-C) has defective chemokine activity (Satishchandra et al. 2000 ; Ranga et al. 2004) due to replacement of the CC motif with serine, CS or SC (Ranga et al. 2004). There are no existing reports on how HIV1-C Tat interferes with NEP.
The study of aging and neuronal injury proteins will add substantial information to help determine the pathogenesis of HIV neurocognitive disorders. Over a prolonged period of time, HIV-1-induced accumulation of Aβ may lower the threshold for dementia by contributing to the Aβ load (Rempel and Pulliam 2005). The profile of neural injury biomarkers related with amyloid metabolism is not well defined in HIV infected participants. The majority of the previous studies are on HIV-1 subtype B (Brew et al. 2005; Nath and Hersh 2005; Gisslén et al. 2009; Peterson et al. 2013 ; Krut et al. 2013 ; Krut et al. 2014), these biomarkers were not studied up to now on HIV-1 non B subtypes.
The authors hypothesized that HIV+ patients would have lower CSF Aβ levels compared with HIV-subjects, with the lowest levels in patients with Alzheimer’s disease (AD) and highest levels in HIV-healthy controls, and that CSF Aβ levels would be lower in HIV1-B patients than HIV1-C patients because HIV1-B Tat inhibits NEP.
The aim of this study was to compare the CSF levels of biomarkers of Aβ metabolism [Aβ oligomers 38, 40, and 42 and soluble amyloid precursor protein (APP) α (sAPPα) and β (sAPPβ)], neurodegeneration [total tau (T-tau)], and neuronal tangle pathology [tau phosphorylated at threonine 181 (P-tau181)] between HIV1-B and HIV1-C patients, healthy HIV- controls, and patients with AD.
METHODS
This study, which was a cross-sectional survey of stored CSF samples, was approved by the University of California-San Diego (UCSD, San Diego, CA, USA) Institutional Review Board (IRB), Hospital de Clínicas-Universidade Federal do Paraná (HC-UFPR, Curitiba, Paraná, Brazil) IRB, and the National Commission of Ethics in Research (CONEP).
Subjects
All participants signed consent forms approved by the IRBs in the US and Brazil. For patients with AD, the responsible caregiver signed the consent form. Samples of the CSF and serum were collected under a NIMH-funded protocol (R21 MH076651-01). These methods and the demographic and infection characteristics of the HIV+ patients were described previously (de Almeida et al. 2013; de Almeida et al. 2016). A total of 110 CSF samples were analyzed. The demographic characteristics, HIV status, and coinfections, if any, are summarized in Table 1 for HIV+ (n = 68), HIV- (n = 18), and AD (n = 24) patients.
Table 1.
Demographic data, clinical and HIV infection characteristics, and co-morbidities of HIV participants, uninfected volunteers and Alzheimer’s disease group
| HIV+ (n = 68) | HIV−(n = 18) | AD (n=24) | P | |
|---|---|---|---|---|
| Demographics | ||||
| Age, years | 43 (35; 48) | 40 (34; 50) | 76.5(67;79.5) | <0.0001 |
|
| ||||
| Education, years | 8 (5;11) | 12 (11;15.5) | 4 (2;6) | 0.0001 |
|
| ||||
| Gender, n male (%) | 33 (49) | 14 (77.8) | 8(33) | 0.0120 |
|
| ||||
| Clinical | ||||
|
| ||||
| Duration, months | 89 (31; 135)f | - | 36 (24;60)g | - |
|
| ||||
| MEEM | - | - | 14(9.5;20) | - |
|
| ||||
| MoCA, (n=8) | 11.5(10.5;12.5) | - | ||
|
| ||||
| FAQ | - | - | 23.5(15;27.5) | - |
|
| ||||
| GDS | 0.65 (0.30; 1.05) | 0.11(0.0; 0.28) | - | - |
|
| ||||
| Depression | 13 (8, 25)h | - | 1(0.5;3)i | - |
|
| ||||
| HIV status and treatment | ||||
| AIDS, n (%) | 55 (81) | - | - | |
|
| ||||
| Current CD4, cell/mm3 | 369 (201; 534) | - | - | - |
|
| ||||
| Nadir CD4, cell/mm3 | 92 (37; 267) | - | - | - |
|
| ||||
| Log Plasma HIV RNAa | 1.7 (1.7; 3.5) | - | - | - |
|
| ||||
| Plasma HIV RNA <50 copies/mL, n (%) | 38 (56) | - | - | - |
|
| ||||
| Log CSF HIV RNA | 1.7 (1.7; 2.8) | - | - | - |
|
| ||||
| CSF HIV RNA <50 copies/mL, n(%) | 36 (53) | - | - | - |
|
| ||||
| on CARTb, n (%) | 55 (81) | - | - | - |
|
| ||||
| CPEc | 8 (6; 9) | - | - | - |
|
| ||||
| Adherenced, n (%) | 51 (93) | - | - | - |
|
| ||||
| Co-morbidities | ||||
| HCVe, n (%) | 12 (18) | 0 | 0 | - |
|
| ||||
| Log Plasma HCV RNA | 2.9 (1.7; 5.9) | 0 | 0 | - |
Data are presented as median [interquartile range (IQR)] or number of cases (%).
Plasma viral load, log10 c/mL;
CART, combination anti-retroviral therapy;
CPE, anti-retroviral CNS penetration effectiveness (Letendre et al., 2010);
Anti-retroviral treatment adherence was evaluated using AIDS clinical trial (ACTG) adherence questionnaire (4-day recall);
Hepatitis C virus (HCV) status was assessed by antibody testing (Abbott-Architect). Participants co-infected with HCV were not on treatment with interferon-gamma. Clinical duration:
of infection on HIV-positive;
of symptoms on AD. Cognitive impairment evaluated by: global deficit score (GDS) on HIV-positive; mini–mental state examination (MMSE); Montreal Cognitive Assessment (MoCA);Functional Activities Questionnaire (Pfeffer’s FAQ) on AD. Major depression disorder (MDD) diagnosed by
Beck depression inventory (BDI) on HIV-positive;
Geriatric Depression scale (GD scale) on AD.
HIV+ participants
The HIV+ participants, n=68, were recruited at the HC-UFPR. Individuals with opportunistic CNS infections were excluded. All volunteers provided blood and CSF samples and underwent serological testing to confirm HIV status before enrollment in accordance with previously published guidelines (Brasil, Ministério da Saúde 2015). For participants with clinically resistant infection, the infecting HIV strain was genotyped with pol sequences, while env sequences were used for all other participants. Genotyping indicated that 27 individuals were infected with HIV1-B and 40 were infected with non-B HIV-1 subtypes (C, n = 26; BF, 10; BC, 1; CF, 1; and F, 2). In one participant, the HIV-1 subtype could not be genotyped. HIV subtype B- and C-infected individuals were similar in age, gender, and education. CSF samples of HIV subtype B- and C-infected individuals were similar in total protein, WBC, number of cases with pleocitosis (WBC > 5 cell/mm3); albumin; QAlb; CSF HIV RNA Log10; Log CSF HIV RNA <1.7.
HIV- controls
Because Brazilian institutional review boards do not permit lumbar puncture in HIV uninfected volunteers, we recruited a control group of 18 age-matched HIV- individuals at the HIV Neurobehavioral Research Center, University of California San Diego (HNRC-UCSD). They had no neurological comorbidities or cognitive complaints and tested negative on serological tests for hepatitis C and syphilis. The CSF cytochemical criteria for inclusion in the control group was white blood cell (WBC) count ≤5 cells/mm3, total protein ≤45 mg/dL, and glucose ≥55 mg/dL.
AD participants
Twenty-four patients with AD were diagnosed by the Dementia Investigative team from the Cognitive Dysfunction Outclinic (Neurology Unit, HC-UFPR). They underwent detailed anamnesis and clinical examinations and routine blood analyses to rule out treatable causes of the dementia. All patients with AD had negative serum and/or CSF tests for HIV, Hepatitis C, and VDRL. CSF biomarkers are not considered for AD clinical diagnoses.
All AD participants met the dementia criteria of the DSM-V (American Psychiatric Association 2013) and criteria for probable AD of the National Institute on Aging-Alzheimer’s Association (McKhann et al. 2011).
The scales used in the AD group were Brazilian Portuguese versions of the Mini–Mental State Examination (Brucki et al. 2003), Montreal Cognitive Assessment (Memória et al.2013), Geriatric Depression scale (Almeida and Almeida 1999), and Functional Activities Questionnaire (Pfeffer et al.1982). The dementia severity was assessed by the Brazilian version of the Clinical Dementia Rating (CDR) (Bertolucci et al.1998).
The AD participants had probable AD; moderate dementia, CDR median [Interquartile Range (IQR)], 2 (2, 2.5); severely decreased daily activity; and no associated depression (Table 1). Eighteen AD patients had computed tomography or magnetic resonance images, which showed brain volumetric reductions, absent expansive lesions or extra-axial collections, and/or pathological calcifications in the brain. The demographic, clinical and laboratorial characteristics of the groups studied are shown in Tables 1 and 2.
Table 2.
Biochemical, cytological, and virological characteristics of the CSF in HIV-positive, uninfected volunteers and Alzheimer’s disease participants. Significant differences are highlighted in bold.
| HIV+ (n = 68) | HIV- (n = 18) | AD (n=24) | P | |
|---|---|---|---|---|
| WBC, cells/mm3 | 2.1 (0.6; 7.2) | 2 (1;2.5) | 0.6(0.3;1.4) | 0.0025 |
| WBC count > 5 cells/mm3, n(%) | 20 (29) | 0 | 0 | - |
| Glucose, mg/dL | 57 (53; 62) | 63.5 (59; 71) | 60.5(54.0;72.5) | 0.0009 |
| Total protein, mg/dL | 40 (32; 46) | 30.5 (26.5; 38) | 37.35(30.7;49.0) | 0.0129 |
| Total protein > 45 mg/dL, n(%) | 20 (29) | 0 | 7 (29) | 0.0302 |
| Albumin, mg/dL | 22.4 (16.4; 28.9) | 19 (14.5; 24) | - | 0.0885 |
| Albumin quotient, QAlb | 0.0064 (0.0049; 0.0097) | 0.005 (0.003; 0.006) | - | 0.0002 |
| Lactic acid, mmol/L | 1.6 (1.5; 1.8) | - | 1.7(1.5 ; 1.8) | 0.6499 |
| RBC, cells/mm3 | 0.5 (0; 7.5) | 2.0 (1.0;4.0) | 3.4(0.95;53) | 0.0133 |
| Log CSF HIV RNA | 1.7 (1.7; 2.8) | - | - | - |
| Viral load < 50 | 35 | - | - | - |
| HIV RNA CSF > blood, n(%) | 12 (18) | - | - | - |
CSF glucose was significantly higher in HIV-negative volunteers than in HIV participants, although levels in both groups were below reference range.
Data are presented as median (IQR) or number of cases (%).
Laboratory Methods
CSF biomarkers
Neuronal Injury biomarkers were measured in the CSF with immunoassays. T-tau, Aβ (isoforms 38, 40, and 42), sAPPα, and sAPPβ were assayed by electrochemiluminescence (MULTI-ARRAY, Meso Scale Diagnostics, LLC, Rockville, MD, USA). P-tau181 (Thermo Fisher Scientific Inc., Waltham, MA, USA) was assayed by multiplex bead assays (FlexMAP 3D®, Luminex Corporation, Austin, TX, USA). All samples were assayed concurrently in duplicate according to the manufacturers’ instructions. The acceptable coefficient of variation between duplicates was less than 20%. When the results were under the minimum low detection limit determined by the manufacturers, the low-detection-limit value was considered in the statistical analysis.
To enhance the specificity of the neuronal injury biomarkers, combinations of these biomarkers in ratios and indexes were calculated as a single value (Blennow and Vanmechelen 1998), as previously described for AD: Aβ-total = Aβ-38 + Aβ-40 + Aβ-42, Aβ-42/Aβ-total, Aβ-42/Aβ-40, and Aβ-42/Aβ-38 (Anoop et al. 2010); P-tau181/Aβ-42, T-tau/Aβ-42, P-tau181/T-tau, and sAPPα/sAPPβ; Hulstaert (T-tau) index = Aβ- 42/(240 + 1.18T-tau) (Hulstaert et al. 1999); and Hulstaert (P-tau181) index = Aß-42/(240 + 1.18P-tau181) (Molinuevo et al. 2013).
Specimen collection and storage
CSF was collected with lumbar punctures performed with atraumatic spinal needles under aseptic conditions by a trained neurologist. The samples were collected in polypropylene tubes to avoid adherence of the proteins to the tube walls, which is particularly important for Aβ, a sticky protein and centrifuged immediately after the lumbar puncture to separate cells and debris and avoid false increases in T-tau and P-tau181. CSF total protein, glucose, and WBC counts were measured with standard laboratory methods. All HIV+ and AD samples were collected at the same time interval of the day to limit diurnal variability. CSF and serum aliquots were frozen and stored at -80°C at HC-UFPR, Brazil.
Data analyses
Demographic variables (age, sex, and education) were compared between all groups with pairwise Student’s t-tests for continuous variables and Fisher's exact test for binary and categorical variables. Demographic and HIV disease characteristics were compared between HIV1-B and HIV1-C individuals with similar methods. Values were log10-transformed prior to the statistical analyses if their distributions were not approximately normal.
First, a hierarchy of comparisons was performed with the AD versus HIV+ groups as the primary comparison, HIV- versus AD groups as the secondary comparisons, and HIV- versus HIV+ groups as the exploratory comparison, without adjustment for multiple comparisons. Age and gender were included as covariates in multivariable linear regression models if they had p values < 0.2 in the adjusted model. If the age effect was significantly nonlinear, a smooth age effect was used within a generalized additive model (Wood 2006). The p values within each class of biomarkers (CSF, ratio, and index) were then corrected for multiple testing with the Benjamini-Hochberg (BH) procedure.
Second, the CSF biomarkers, biomarker indexes, and biomarker CSF/serum ratios were compared between the HIV1-B and HIV1-C groups. A multivariable model was applied to control for plasma HIV viral load (VL) suppression and nadir CD4 counts. As described above, the p values for the biomarker effects were corrected for multiple testing.
The results were considered statistically significant at the 5% alpha level. The differences between groups are presented as Cohen’s d (and 95% CI). The statistical analyses were performed with R (version 3.2.3, R Foundation for Statistical Computing).
RESULTS
HIV1-B and HIV1-C patients
Neuronal injury biomarker levels in the CSF by HIV-1 subtype are shown in Table 3. CSF Aß-42 levels were decreased in HIV1-C patients compared with HIV1-B patients after adjusting for plasma VL suppression and CD4 nadir count (p = 0.033; after BH correction for multiple comparisons, p = 0.30; Figure 1A).
Table 3.
HIV-1 subtype B and C levels of neuronal injury biomarkers and of indexes in cerebrospinal. Significant differences are in bold typeface.
| Biomarker | HIV-B | HIV-C | Diff (95% CI) | p* | p** |
|---|---|---|---|---|---|
| Aβ-38, pg/mL | 2062 (1652; 2599) | 2045 (1342; 2440) | 0.32 (-0.24, 0.89) | 0.16 | 0.30 |
| Aβ-40, pg/mL | 4140 (3703; 5285) | 3878 (3112; 4646) | 0.43 (-0.13, 1) | 0.068 | 0.30 |
| Aβ-42, pg/mL | 475.8 (409.1; 620.6) | 419.5(303.7; 501.9) | 0.56 (-0.01, 1.13) | 0.033 | 0.30 |
| Aβ-Total, pg/mL | 6742 (5710; 8555) | 6333 (4797; 7705) | 0.41 (-0.16, 0.98) | 0.085 | 0.30 |
| sAPPα, ng/mL | 20.80 (16.10; 29.25) | 19.25(14.45; 25.40) | 0.36 (-0.2, 0.93) | 0.15 | 0.30 |
| sAPPβ, ng/mL | 33.90 (25.80; 52.15) | 31.85(21.50; 41.65) | 0.37 (-0.2, 0.93) | 0.13 | 0.30 |
| T –tau, pg/mL | 298.1 (42.76;600.4) | 194.6(97.10; 385.5) | -0.04 (-0.6, 0.52) | 0.76 | 0.84 |
| P-tau181, pg/mL | 186.0 (120.5; 200.5) | 146.0(122.5; 202.0) | -0.12 (-0.68, 0.44) | 0.48 | 0.58 |
| Aβ-42/Aβ-Total | 0.07 (0.06; 0.08) | 0.07 (0.06; 0.07) | 0.55 (-0.03, 1.12) | 0.08 | 0.25 |
| P-tau181/Aβ-42 | 0.33 (0.25;0.50) | 0.37 (0.25; 0.61) | -0.36 (-0.92, 0.21) | 0.11 | 0.25 |
| T-tau/Aβ-42 | 0.43 (0.09; 1.24) | 0.43 (0.29; 1.07) | -0.2 (-0.76, 0.37) | 0.74 | 0.78 |
| P-tau181/T-tau | 0.41 (0.32; 4.57) | 0.80 (0.30; 1.39) | -0.03 (-0.59, 0.54) | 0.53 | 0.78 |
| Aβ-42/Aβ-38 | 0,24 (0,19;0,29) | 0,22(0,18;0,25) | 0.42(-0.13, 0.96) | 0.18 | 0.41 |
| Aβ-42/Aβ-40 | 0.12 (0.11; 0.12) | 0.11 (0.09; 0.11) | 0.58 (0.01, 1.16) | 0.06 | 0.25 |
| sAPPα/sAPPβ | 0.68 (0.50; 0.86) | 0.58(0.48; 0.88) | -0.06 (-0.62, 0.5) | 0.78 | 0.78 |
| Hulstaert T-tau*** | 0.98 (0.53;1.46) | 0.84 (0.55;1.10) | 0.17 (-0.39, 0.73) | 0.72 | 0.78 |
| Hulstaert P-tau181*** | 1.163(0.921;1.455) | 1.015(0.567;1.279) | 0.47(-0.08, 1.01) | 0.049 | 0.41 |
Values in median (IQR); Diff: Group differences presented as Cohen’s d; CI: confidence interval.
p adjusted for plasma VL suppression and CD4 nadir count;
p adjusted for plasma VL suppresion and CD4 nadir count, corrected for multiple testing with the Benjamini-Hochberg (BH) method.
Aß-42/ (240 + 1.18 T-tau or P-tau181).
Figure 1.
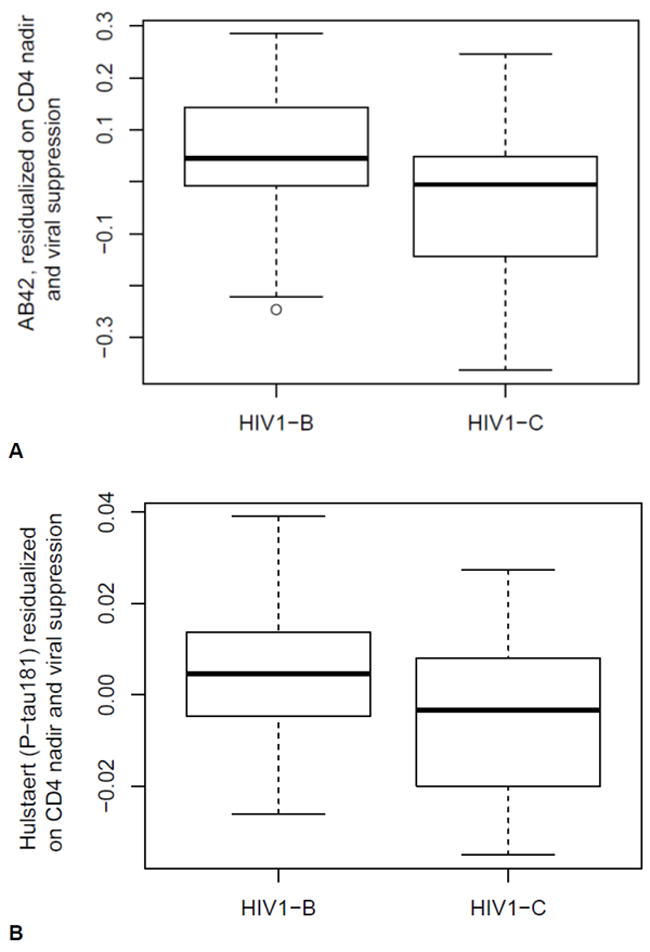
A. CSF Aβ-42 levels adjusted for plasma viral load (VL) suppression and CD4 nadir count in patients with HIV subtypes B and C (p = 0.033). After correcting for multiple testing with the Benjamini-Hochberg (BH) method, the difference was not statistically significant (p = 0.30). B. CSF Hulstaert (P-tau181) = [Aß-42/(240 + 1.18 P-tau181)] index in patients with HIV subtypes B and C adjusted for plasma VL suppression and CD4 nadir count (p = 0.049). After correcting for multiple testing with the BH method, the difference was not statistically significant (p = 0.41).
The Hulstaert (P-tau181) index was higher in HIV1-B patients than in HIV1-C patients, after adjusting for plasma VL suppression and CD4 nadir (p = 0.049; after BH multiple comparisons correction, p = 0.41; Figure 1B). No differences between HIV-1 patient subtypes were found for the other ratios and indexes (Table 3).
Figure 2 plots the CSF Hulstaert (T-tau) index related with CSF P-tau181 values. Data points for AD patients fall in the lower right quadrant as expected. HIV(+) data points were distributed in all four quadrants.
Figure 2. CSF Hulstaert (T-tau) = [Aß-42/(240 + 1.18 T-tau)] and CSF P-tau181 in HIV+ patients according to HIV-1 subtype B or C, AD, and HIV- groups.
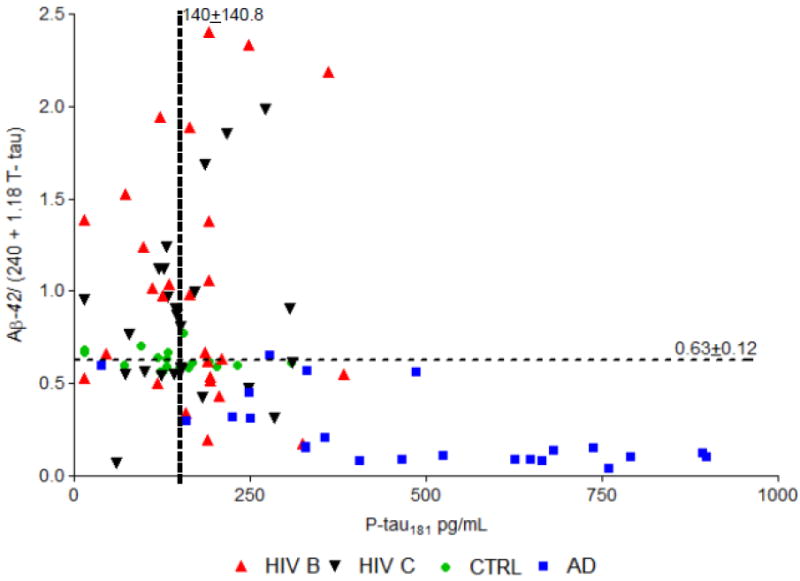
Cutoffs (horizontal and vertical dashed lines) defined as mean ± 2 SD of the HIV- group for the Hulstaert T-tau index was 0.63 ± 0.12 and for P-tau181 was 140 ± 140.8. All participants in the AD group were in the right lower quadrant, which was expected according to previous findings. HIV-positive values were distributed in all four quadrants. Both groups (AD and HIV+) had different distributions from the control group, and controls were distributed close to the horizontal line. The distribution of HIV subtypes B or C patients were on a similar pattern (Fisher’s exact test, p = 1.0).
HIV+, HIV-, and AD patients
Table 4 lists the CSF biomarkers in HIV+, HIV-, and AD patients. In HIV+ patients, CSF Aβ-42 levels were increased compared with AD patients (p = 0.002) and decreased compared with HIV- patients (p = 0.004; Figure 3A), while CSF sAPPα and P-tau181 levels were decreased compared with AD patients (p = 0.041 and < 0.001, respectively) and did not reach significance compared with HIV- patients (Figure 3B and D). T-tau levels were decreased in HIV+ patients compared with AD patients (p < 0.001) and HIV- patients (p < 0.002; Figure 3C).
Table 4.
HIV-positive; HIV-negative and Alzheimer’s disease cerebrospinal fluid levels of neuronal injury biomarkers and indexes. Significant differences are in bold typeface.
| Biomarker | HIV+ | AD | CTRL | (a) Diff (95% CI) | p (a) | (b)Diff (95% CI) | p (b) | (c)Diff (95% CI) | p (c) |
|---|---|---|---|---|---|---|---|---|---|
| Aβ-38, pg/mL | 2101 (1541; 2530) | 1653 (1274; 2589) | 2209 (1840; 2509) | 0.32 (-0.18; 0.8) | 0.39 | 0.17 (-0.36; 0.7) | 0.58 | 0.57(-0.12, 1.26) | 0.65 |
| Aβ-40, pg/mL | 4134 (3499; 4906) | 3937 (3245; 5668) | 4561 (4182; 5116) | 0 (-0.48; 0.48) | 0.84 | 0.34 (-0.2; 0.88) | 0.34 | 0.38(-28,1.04) | 0.65 |
| Aβ-42, pg/mL | 447.9 (328.5; 564.0) | 276.0 (167; 444.5) | 618.3 (548.6;735.0) | 0.87 (0.38;1.37) | 0.002 | 0.91 (0.35; 1.46) | 0.004 | 1.59(0.74, 2.44) | 0.65 |
| Aβ-Total, pg/mL | 6724 (5438; 7930) | 5810 (4737; 8655) | 7357 (6665; 8316) | 0.17 (-0.3; 0.65) | 0.70 | 0.33 (-0.21; 0.87) | 0.34 | 0.57(-0.12, 1.26) | 0.65 |
| sAPPα, ng/mL | 20.25 (15.35; 27.85 | 25.90 (19.00;42.10) | 27.80 (16.40; 35.15) | -0.61(-1.14;-0.08) | 0.041 | 0.48 (-0.06; 1.02) | 0.21 | -0.15(-0.77, 0.48) | 0.65 |
| sAPPβ, ng/mL | 33.50 (22.00; 44.90) | 30.30 (24.00; 50.25) | 35.75 (27.05; 43.35) | 0.15 (-0.38; 0.67) | 0.70 | 0.08 (-0.45; 0.62) | 0.75 | 0.22(-0.42, 0.86) | 0.65 |
| T-tau, pg/mL | 198.5 (66.39; 513.8) | 1201 (864.0; 1538) | 633.7 (531.2; 764.6) | -1.65 (-2.19;-1.11) | <0.001 | 1.03 (0.48; 1.59) | 0.002 | -1.88 (-2.86,-0.9) | <0.001 |
| P-tau181, pg/mL | 164.0 (123.5; 241.0) | 475.0 (302.5;709.0) | 132.0 (106.5; 179.5) | -1.32(-1.84; -0.81) | <0.001 | -0.37(-0.91; 0.16) | 0.34 | -1.69(-2.59, -0.79) | 0.001 |
| Aβ-42/Aβ-38 | 0,224 (0,189; 0,257) | 0,164 (0,122; 0,239) | 0,281(0,267; 0,308) | 0.83(0.35, 1.31) | <0.001 | 1.48(0.91, 2.04) | <0.001 | 1.96(1.2, 2.7) | <0.001 |
| Aβ-42/Aβ-40 | 0.11 (0.10; 0.12) | 0.07(0.05; 0.11) | 0.13 (0.13;0.14) | 1.26 (0.75, 1.78) | 0.70 | 1.69 (1.09, 2.28) | <0.001 | 2.13 (1.26, 2.98) | 0.32 |
| Aβ-42/Aβ-Total | 0.07 (0.06;0.07) | 0.04(0.03;0.07) | 0.08 (0.08; 0.09) | 1.2 (0.7, 1.71) | 0.70 | 1.71 (1.11, 2.3) | <0.001 | 2.17(1.2, 3.13) | 0.25 |
| P-tau181/Aβ-42 | 0.38 (0.27;0.52) | 2.12(0.58;3.67) | 0.22 (0.19;0.29) | -1.57 (-2.1, -1.04) | <0.001 | -0.78 (-1.33, -0.23) | 0.007 | -2.02 (-2.82, -1.2) | <0.001 |
| T-tau/Aβ-42 | 0.43 (0.13; 1.12) | 5.11(2.20;8.02) | 1.05 (0.93;1.13) | -1.93 (-2.49, -1.37) | <0.001 | 0.76 (0.21, 1.31) | 0.007 | -2.27 (-3.25, -1.28) | <0.001 |
| P-tau181/T-tau | 0.71 (0.30;3.08) | 0.39(0.31; 0.48) | 0.23 (0.17;0.28) | 0.61 (0.13, 1.1) | 0.016 | -1.03 (-1.59, - 0.47) | <0.001 | -0.99 (-1.65, -0.31) | 0.100 |
| sAPPα/sAPPβ | 0.69 (0.50; 0.83) | 0.82 (0.80;0.88) | 0.76 (0.60;0.87) | -0.85 (-1.39, -0.31) | 0.003 | 0.4 (-0.14, 0.94) | 0.14 | -0.79 (-1.42, -0.15) | 0.25 |
| Hulstaert T-tau* | 0.78(0.54;1.24) | 0.04(0.05;0.08) | 0.61 (0.59; 0.67) | 2.18 (1.6, 2.76) | <0.001 | -0.39 (-0.93, 0.14) | 0.14 | 2.18(1.21, 3.15) | <0.001 |
| Hulstaert P-tau181* | 1.061(0.781;1.281) | 0.273(0,182; 0.849) | 1.494 (1.332;1.742) | 1.19(0.69, 1.68) | <0.001 | 1.28(0.72, 1.83) | <0.001 | 2.13(1.35, 2.89) | <0.001 |
(a) HIV(+)xAD;(b)HIV(+)xCTRL;(c)ADxCTRL.Values in median (IQR); Diff: Group differences presented as Cohen’s d; CI: confidence interval; all p values
adjusted for multiple testing with the Benjamini-Hochberg (BH) method, (a) and (c) adjusted for gender or age and BH method.
Aβ-42/ (240 + 1.18 T-tau or P-tau181).
Figure 3. CSF neuronal injury biomarkers, all p values are after adjusting for multiple testing with the Benjamini-Hochberg (BH) method.

(A) Aß-42, AD and CTRL unadjusted analysis for age and gender with corrections for multiple testing with BH: p < 0.001; after adjusted for age and gender and corrections for multiple testing with BH, the difference was not significant p = 0.65(B) sAPPα; (C) T-tau; (D) P-tau181; in HIV+, Alzheimer disease (AD), and HIV- (CTRL) groups. HIV(+) and AD and AD and CTRL adjusted for gender or age and BH method.
AD and HIV- patients
CSF Aβ-42 levels were decreased in AD patients compared with HIV- patients (unadjusted for gender and age but corrected for multiple testing with BH, p < 0.001; after adjusting for gender and age and BH multiple comparisons correction, p = 0.65). CSF P-tau181 and T-tau levels were increased in AD patients compared with HIV negative patients (p = 0.001 and < 0.001, respectively). CSF Aβ-38, Aβ-40, Aβ-total, sAPPα, and sAPPβ did not differ between AD and HIV- patients (Table 4 and Figure 3). Across all participants, the levels of Aβ-40 were higher than the levels of the other isoforms, which is the normal pattern of expression. Pairwise comparisons of the ratios and indexes are shown in Table 4.
DISCUSSION
This study aimed to compare Aβ isoforms and related biomarkers of the amyloid pathway between patients with HIV1-B and C. This is the first study to analyze neuronal injury biomarkers in patients with HIV1 non-B subtypes.
Aß-42 levels were decreased in HIV1-C patients compared with HIV1-B patients after adjusting for blood VL suppression and CD4 nadir count. These results were corroborated by similar results for the Hulstaert (P–tau181) index. Combining biomarkers in ratios or indexes enhance their specificity (Blennow and Vanmechelen 1998). These findings were consistent with increased tissue deposition of Aβ-42 or decreased synthesis of Aβ-42 in HIV-1C patients compared with HIV1-B patients. However, against the second possibility the sAPPα and sAPPβ levels did not differ between HIV1-B and HIV1-C patients. These results were no longer significant after BH corrections for multiple testing. Decreased levels of CSF Aß-42 in HIV1-C patients does not support the initial hypothesis that, in HIV1-C, Tat would not inhibit NEP, thus preventing Aß-42 brain deposition and resulting in higher CSF Aß-42 levels. In vitro Tat inhibits NEP activity by 80%, which causes Aβ accumulation (Rempel and Pulliam 2005). Additional studies on NEP and its activity in HIV1-C are needed.
The results of previous studies of neuronal injury biomarkers, especially Aβ-42, T-tau, and P-tau181, vary. The current results showed that HIV, independently of subtype, affected amyloid metabolism differently than it does in AD. Although, similar to findings in AD, Aβ-42 was the main Aβ isoform that was probably deposited in the brains of HIV patients. Compared to AD patients, HIV+ patients had increased CSF Aβ-42 levels and Hulstaert (T-tau), Hulstaert (P-tau181), and P-tau181/T-tau indexes and decreased CSF T-tau, P-tau181, and sAPPα levels and P-tau181/Aβ-42, T-tau/Aβ-42, and sAPPα/β indexes. In HIV+ patients in this study, Aβ-42 was decreased compared with HIV- patients and increased compared with AD patients, which was similar to previous results (Brew et al. 2005; Gisslén et al. 2009; Clifford et al. 2009; Krut et al. 2013; Cysique et al. 2015). However, another study found the opposite: increased CSF Aβ-42 in early HIV infection compared with HIV- controls (Peluso et al. 2013), while other studies found no differences in CSF Aβ-42 in HIV+ individuals compared to HIV- controls (Ances et al. 2012; Steinbrink et al. 2013). Few studies have examined Aβ isoforms other than Aβ-42, such as Aβ-38, Aβ-40, or Aβ-total. A study found no differences between groups for CSF Aβ-38, 40, or 42 (Peterson et al. 2014).
The results of studies of CSF T-tau and P-tau181 in HIV have been more inconsistent. In the current study, CSF T-tau levels were decreased in HIV+ patients compared with HIV- controls, which was similar to previous results (Clifford et al. 2009; Peterson et al. 2014; Cysique et al. 2015). This can be explained by the deposit in brain of tau protein or may be relate to the predominating subcortical pathology of HIV-associated neurognitive disorder (HAND) (Navia et al. 1986), since tau is expressed most prominently in non-myelinated cortical axons (Trojanowski et al. 1989). An AD-like pattern of increased CSF T-tau and P-tau181 has been described (Brew et al. 2005), while normal T-tau and P-tau181 levels have been also reported (Gisslén et al. 2009). Increased P-tau181 in HAND (Brew et al. 2005; Andersson et al. 1999), while others did not confirm these results (Clifford et al. 2009; Ellis et al. 1998; Green et al. 2000). In this study, P-tau181 did not differ between HIV+ and HIV- controls. The observations of normal levels of P-tau181 HIV+ subjects are also consistent with the lack of neurofibrillary tangles in HAND, one of the neuropathological hallmarks of AD (Anthony et al. 2006). Thus, although increased tau deposition has been reported in HIV patients, neuronal tangles are uncommon (Stanley et al. 1994; Anthony et al. 2005). This difference further suggests that HAND and AD have different major mechanisms of neuronal injury.
CSF sAPPα and sAPPβ
sAPPα and sAPPβ are both shed from the cell membrane and diffuse into the CSF. Few HIV studies have examined CSF sAPPα or sAPPβ. In this study, sAPPα and sAPPβ levels were similar to previous reports: decreased sAPPα compared with AD patients but no difference with HIV- patients (Green et al. 2005; Krut et al. 2013), while sAPPβ did not differ between groups. The mechanisms underlying reduced CSF sAPP in HIV are uncertain (Gisslén et al. 2009) but may relate to brain APP deposition in HIV encephalitis (HIVE) (Nebuloni et al. 2001). CSF sAPPα and sAPPβ levels were decreased in patients with CNS HIV infection, most the later (Gisslén et al. 2009; Steinbrink et al. 2013); sAPPα and sAPPβ were decreased in CNS bacterial infections and HSV-1 encephalitis, suggesting a role of neuroinflammation in amyloid metabolism (Krut et al. 2013). APP accumulation has been observed in brain of AIDS patients (Giometto et al. 1997).
Our group described a patient with HIVE who exhibited decreased CSF sAPPα levels compared with the control group (de Almeida et al. 2017), which corroborated the observations of APP axonal accumulation in HIVE (Nebuloni et al. 2001) and the Simian Immunodeficiency Virus encephalitis model (Mankowski et al. 2002), and could explain the low CSF levels of this biomarker on the present study. The HIV viral coat protein Gp120 causes APPβ accumulation in adult rats, resulting in axonal injury (Zhang et al. 2011).
Low CSF Aβ levels resulting from the altered APP metabolism would either decrease sAPPs and Aβ production from APP due to decreased α and β-secretase activity or unspecific proteolysis. Other possible factors could be intracellular APP metabolite deposition in the CNS or increased clearance and elimination of amyloid metabolites during acute inflammation in the CNS (Krut et al. 2013). Aβ CNS concentration is regulated by the rate of production from APP, brain influx across the blood-brain barrier (BBB), mainly through the advanced glycation end product (RAGE) receptor (Deane et al. 2009), rapid clearance across the BBB by transcytosis by the low-density lipoprotein receptor-related protein-1 (LRP1) (Deane et al. 2009), and enzymatic degradation within brain parenchyma mainly by NEP and the insulin-degrading enzyme (IDE) to prevent deleterious Aβ-42 neuronal aggregation (Selkoe 2001; Madani et al. 2006). However, the degradation of free Aβ in brain interstitial fluid has been reported to be insignificant (Shibata et al. 2000).
HIV-1-infected patients
Both animal and human studies suggest that HIV alters amyloid metabolism (Green et al. 2005; Rempel and Pulliam 2005; Aksenov et al. 2010). Previous autopsy studies of HIV+ individuals have demonstrated an increase in diffuse extracellular amyloid plaques before7 and after HAART (Rempel and Pulliam 2005; Anthony et al. 2010). Achim et al. (2009) reported abundant Aβ immunostaining in pyramidal neurons and along axonal tracts in HIV-infected brain. Intraneuronal Aβ immunoreactivity was increased in HIVE patients compared with HIV+ patients without encephalitis. HIV-1 markedly increased the endogenous Aβ levels and accumulation of exogenous Aβ in an HIV-1 exposure model of brain microvascular endothelial cells (András et al. 2010).
Aβ deposition differs in AD and HIV-1 brains. While extracellular amyloid plaques are the major amyloid pathology in AD, intraneuronal amyloid accumulation or diffuse perivascular amyloid depositions are more characteristic for HAND (Xu and Ikezu 2009). The mechanisms underlying the interactions between Aβ and HIV-1 infection are not fully understood but several factors and/or pathways are likely to be involved. It has been hypothesized that aging, HIV-1 infection, and the secondary effects of ART may all contribute to brain Aβ accumulation in neurons and in perivascular space (Green et al. 2005).
HIV disrupts several steps in the amyloid cascade from Aβ biogenesis to clearance (Xu and Ikezu 2009; Pulliam 2009; András and Toborek 2013). Several HIV proteins are amyloidogenic, such as gp120 (Aksenov et al. 2010; Haughey et al. 2010; Zhang et al. 2011), gp41(Mankowski et al. 2002), Tat (Seeger et al. 1997; Apcher et al. 2003; Rempel and Pulliam 2005; Daily et al. 2006; Aksenov et al. 2010; Lan et al. 2011), and Nef (White et al. 2005). Mononuclear phagocytes, which are macrophages and microglia, that are infected by HIV play a pivotal role in Aβ degradation through the expression and execution of two endopeptidases, NEP and IDE (Lan et al. 2011). Multiple studies have described the amyloidogenic interference of ART (Tran et al. 2003; Hamel et al. 2006; Hamel 2007; Lan et al. 2012; Brown et al. 2014).
Tat, which is the main HIV amyloidogenic protein, interferes with Aβ degradation (chiefly NEP) and amyloid peptide reuptake and clearance. HIV-1 Tat protein application increases Aβ levels in cell culture (Rempel and Pulliam 2005). In addition, HIV-1 Tat, specifically HIV-B, increases Aβ-40 levels in neuronal cell cultures by inhibiting NEP (Rempel and Pulliam 2005; Daily et al. 2006; Aksenov et al. 2010; Lan et al. 2011).
In vitro NEP inhibition requires Tat’s cysteine-rich domain, amino acids 22 37 (Daily et al., 2006). Although this domain has been described as highly conserved among different HIV-1 strains, the cysteine residue in position 31 is mutated to a serine in HIV1-C (Ranga et al. 2004). It is unclear how Tat affects HIV1-C.
HIV-1 Tat B and gp120 promote Aβ-42 release and accumulation in primary rat fetal hippocampal cell cultures, while non-neurotoxic variants of HIV-1 Tat (Cys22 Tat 1-86 B [cys22→gly22 substitution] and Tat 1-101 C) do not promote Aβ-42 production in hippocampal cell cultures or cytotoxicity (Aksenov et al. 2010).
Tat interference on Aß-42 clearance
Tat binds to LRP, which is involved in amyloid peptide reuptake and clearance, reduces Aβ42 clearance (András and Toborek 2013; Giunta et al 2009), and attaches to endothelial cell RAGE, increasing blood-brain influx (Xu and Ikezu 2009; András and Toborek 2013). These results suggest that HIV-1 directly contributes to Aβ accumulation at the BBB level (András et al. 2010). In addition, Tat inhibits microglial Aβ phagocytosis (Giunta et al. 2008).
Tat interacts with APP both in vitro and in vivo and increases Aβ-42 levels by recruiting APP into lipid rafts, a site of increased β- and γ-secretase activity. Furthermore, Tat enhances APP cleavage by β-secretase, resulting in higher levels of Aβ-42. These results were consistent with increased β-C-terminal fragment (β-CTF) levels and decreased α-CTF levels and suggest that HIV-1 Tat contributes to HAND by interacting with and modifying APP processing and thereby increasing Aβ production (Kim et al. 2013).
Comparing the results in different studies is very difficult and may reflect heterogeneity in HAND severity, HIV disease duration, patient age, diagnostic criteria, and/or treatment status. Several preanalytical variables can interfere with the results. In particular, Aβ-42 levels are affected by the time of sample collection (Bateman et al. 2007), the type of tube used to collect and store the CSF, and number of thawing and rethawing times. Care was taken to avoid these errors in this study. The lack of standardized cutoff values, even for AD, and the need for more investigation of these biomarkers hampers the widespread application of CSF biomarkers in clinical setting (Molinuevo et al. 2013; Hort et al. 2010) and analysis of the research results. Age-corrected norms have not been defined for sAPPα and β and Aβ isoforms (Peterson et al. 2014). In addition; the analytical platform used for the CSF analysis complicates the comparisons of results because values obtained in one laboratory may differ from those obtained in another laboratory. Even when the same assay is used, considerable variability in the absolute concentrations of AD biomarkers occurs due to preanalytical and analytical factors (Mattsson et al. 2011). Thus, the conflicting CSF Aβ-42 results in HIV+ individuals might reflect the relatively small number of samples used in the analysis. The inconsistency of the T-tau increases and the apparent limited sensitivity of T-tau for detecting this type of injury might also explain conflicting results (Gisslén et al. 2009).
Besides the inconsistent characteristics of the biomarkers studied, the present study had some limitations. The study was limited by its cross-sectional design. A longitudinal study of these markers might be able to predict the development of HAND in patients without clear symptoms. This study did not include a substantial number of older HIV+ patients who are most vulnerable to AD. Finally, we did not assess HIV+ patients with known AD, although we are not aware of reports of patients with this combination of conditions. The HIV- group was younger than the AD group as this group was age- and gender-matched with the HIV+ group, and it was not from Brazil but from the US. Future longitudinal studies that include older HIV+ individuals are necessary for investigating the effects of HIV on the Aβ pathway.
This study had several strengths, this was the first study to examine HIV1-C patients and investigate HIV subtypes effects. Most previous studies of these biomarkers in HIV examined HIV1-B patients or do not describe HIV-1 subtype. HIV+ and AD patients were from the same center and were analyzed at the same period of time. Both healthy and disease controls were used. Neuronal injury biomarkers indexes or rates were described for AD, in order to enhance discrimination power (Hulstaert et al. 1999; Molinuevo et al. 2013). Most of these ratios and indexes were first calculated on HIV+ participants on this study, expanding future studies and clinical relevance. Future studies are necessary to establish the operational characteristics of these indexes on HIV + participants.
The differences in Tat in HIV-1 subtype B and C may be the causative factor for the differences observed, however other factors could be implied as other HIV proteins, as gp120. More studies need to be done to elucidate the impact of HIV and the influence of subtypes on amyloid metabolism. The comparisons of these markers provides very useful information that will lead to expanded studies on neuropathology associated with different HIV subtypes, especially in the aging population.
In conclusion, the results of this study have shown that HIV infection affects amyloid metabolism in a subtype-dependent pattern HIV subtypes B and C differed: Aß-42 levels were decreased in HIV1-C compared with HIV1-B, suggesting that Aß-42 might accumulate more in HIV1-C. These findings were supported by the Hulstaert (P-tau181) index. These findings support the concept that HIV CNS infection and AD may not share some of the same mechanisms of neuronal injury.
Acknowledgments
The authors would like to thank Mr. Gabriel L.O. Salvador for editing figure 3.
Funding
This work was supported by the following grants: National Institute of Health, NIH R21 MH76651 (Ellis, Ronald J; Almeida, Sergio M.), S10 RR31646 (Letendre, Scott), K24 MH097673(Letendre, Scott); University of California, San Diego, Center for AIDS Research (CFAR), an NIH-funded program (P30 AI036214), which is supported by the following NIH Institutes and Centers: NIAID, NCI, NIMH, NIDA, NICHD, NHLBI, NIA, NIGMS, and NIDDK.
The HIV Neurobehavioral Research Center (HNRC) is supported by Center award P30MH062512 from NIMH.The San Diego HIV Neurobehavioral Research Center [HNRC] group is affiliated with the University of California, San Diego, the Naval Hospital, San Diego, and the Veterans Affairs San Diego Healthcare System, and includes: Director: Robert K. Heaton, Ph.D., Co-Director: Igor Grant, M.D.; Associate Directors: J. Hampton Atkinson, M.D., Ronald J. Ellis, M.D., Ph.D., and Scott Letendre, M.D.; Center Manager: Thomas D. Marcotte, Ph.D.; Jennifer Marquie-Beck, M.P.H.; Melanie Sherman; Neuromedical Component: Ronald J. Ellis, M.D., Ph.D. (P.I.), Scott Letendre, M.D., J. Allen McCutchan, M.D., Brookie Best, Pharm.D., Rachel Schrier, Ph.D., Debra Rosario, M.P.H.; Neurobehavioral Component: Robert K. Heaton, Ph.D. (P.I.), J. Hampton Atkinson, M.D., Steven Paul Woods, Psy.D., Thomas D. Marcotte, Ph.D., Mariana Cherner, Ph.D., David J. Moore, Ph.D., Matthew Dawson; Neuroimaging Component: Christine Fennema-Notestine, Ph.D. (P.I.), Monte S. Buchsbaum, M.D., John Hesselink, M.D., Sarah L. Archibald, M.A., Gregory Brown, Ph.D., Richard Buxton, Ph.D., Anders Dale, Ph.D., Thomas Liu, Ph.D.; Neurobiology Component: Eliezer Masliah, M.D. (P.I.), Cristian Achim, M.D., Ph.D.; Neurovirology Component: David M. Smith, M.D. (P.I.), Douglas Richman, M.D.; International Component: J. Allen McCutchan, M.D., (P.I.), Mariana Cherner, Ph.D.; Developmental Component: Cristian Achim, M.D., Ph.D.; (P.I.), Stuart Lipton, M.D., Ph.D.; Participant Accrual and Retention Unit: J. Hampton Atkinson, M.D. (P.I.), Jennifer Marquie-Beck, M.P.H.; Data Management and Information Systems Unit: Anthony C. Gamst, Ph.D. (P.I.), Clint Cushman; Statistics Unit: Ian Abramson, Ph.D. (P.I.), Florin Vaida, Ph.D. (Co-PI), Bin Tang, Ph.D., Anya Umlauf, M.S.
The views expressed in this article are those of the authors and do not reflect the official policy or position of the Department of the Navy, Department of Defense, nor the United States Government.
Footnotes
Part of this work was previously presented at the Conference on Retrovirus and Opportunistic Infections, CROI 2017, February 13-16, 2017, Seattle, USA.
Conflict of Interest:
Sérgio Monteiro de Almeida – the author declare that they have no conflict of interest.
Clea Ribeiro – the author declare that they have no conflict of interest.
Indianara Rotta – the author declare that they have no conflict of interest.
Mauro Piovesan – the author declare that they have no conflict of interest.
Sonia Mara Raboni – the author declare that they have no conflict of interest.
Scott Letendre – the author declare that they have no conflict of interest.
Michael Potter – the author declare that they have no conflict of interest.
Bin Tang – the author declare that they have no conflict of interest.
Meire Silva Batistela – the author declare that they have no conflict of interest.
Florin Vaida – the author declare that they have no conflict of interest.
Ronald Ellis – the author declare that they have no conflict of interest.
References
- Achim CL, Adame A, Dumaop W, Everall IP, Masliah E Neurobehavioral Research Center. Increased accumulation of intraneuronal amyloid beta in HIV-infected patients. J Neuroimmune Pharmacol. 2009;4:190–199. doi: 10.1007/s11481-009-9152-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksenov MY, Aksenova MV, Mactutus CF, Booze RM. HIV-1 protein-mediated amyloidogenesis in rat hippocampal cell cultures. Neurosci Lett. 2010;475:174–178. doi: 10.1016/j.neulet.2010.03.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida OP, Almeida AS. Confiabilidade da versão brasileira da Escala de Depressão em Geriatria (GDS) versão reduzida. Arq Neuro-Psiquiatr. 1999;57:421–426. doi: 10.1590/s0004-282x1999000300013. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-5. Washington, DC: American Psychiatric Association; 2013. [Google Scholar]
- Ances BM, Benzinger TL, Christensen JJ, Thomas J, Venkat R, Teshome M, Aldea P, Fagan AM, Holtzman DM, Morris JC, Clifford DB. 11C-PiB imaging of human immunodeficiency virus-associated neurocognitive disorder. Arch Neurol. 2012;69:72–77. doi: 10.1001/archneurol.2011.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson L, Blennow K, Fuchs D, Svennerholm B, Gisslen M. Increased cerebrospinal fluid protein tau concentration in neuro-AIDS. J Neurol Sci. 1999;171:92–96. doi: 10.1016/s0022-510x(99)00253-1. [DOI] [PubMed] [Google Scholar]
- András IE, Eum SY, Huang W, Zhong Y, Hennig B, Toborek M. HIV-1-induced amyloid beta accumulation in brain endothelial cells is attenuated by simvastatin. Mol Cell Neurosci. 2010;43:232–243. doi: 10.1016/j.mcn.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- András IE, Toborek M. Amyloid beta accumulation in HIV-1- infected brain: The role of the blood brain barrier. IUBMB Life. 2013;65:43–9. doi: 10.1002/iub.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anoop A, Singh PK, Jacob RS, Maji SK. CSF biomarkers for Alzheimer’s disease diagnosis. Int J Alzheimers Dis. 2010;2010:1–12. doi: 10.4061/2010/606802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE. Influence of HAART on HIV-related CNS disease and neuroinflammation. J Neuropathol Exp Neurol. 2005;64:529–536. doi: 10.1093/jnen/64.6.529. [DOI] [PubMed] [Google Scholar]
- Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE. Accelerated Tau deposition in the brains of individuals infected with human immunodeficiency virus-1 before and after the advent of highly active anti-retroviral therapy. Acta Neuropathol. 2006;111:529–538. doi: 10.1007/s00401-006-0037-0. [DOI] [PubMed] [Google Scholar]
- Anthony IC, Norrby KE, Dingwall T, Dingwall T, Carnie FW, Millar T, Arango JC, Robertson R, Bell JE. Predisposition to accelerated Alzheimer-related changes in the brains of human immunodeficiency virus negative opiate abusers. Brain. 2010;133:3685–3698. doi: 10.1093/brain/awq263. [DOI] [PubMed] [Google Scholar]
- Apcher GS, Heink S, Zantopf D, Kloetzel PM, Schmid HP, Mayer RJ, Krüger E. Human immunodeficiency virus-1 Tat protein interacts with distinct proteasomal alpha and beta subunits. FEBS Lett. 2003;553:200–204. doi: 10.1016/s0014-5793(03)01025-1. [DOI] [PubMed] [Google Scholar]
- Bateman RJ, Wen G, Morris JC, Holtzman DM. Fluctuations of CSF amyloid-beta levels: implications for a diagnostic and therapeutic biomarker. Neurology. 2007;68:666–9. doi: 10.1212/01.wnl.0000256043.50901.e3. [DOI] [PubMed] [Google Scholar]
- Bertolucci PH, Okamoto IH, Toniolo Neto J, Ramos LR, Brucki SM. Desempenho da população brasileira na bateria neuropsicológica do Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) Revista de Psiquiatria Clínica. 1998;25:80–83. [Google Scholar]
- Blennow K, Vanmechelen E. Combination of the different biological markers for increasing specificity of in vivo Alzheimer’s testing. J Neural Transm Suppl. 1998;53:223–235. doi: 10.1007/978-3-7091-6467-9_20. [DOI] [PubMed] [Google Scholar]
- Programa Nacional de DST/AIDS. Brasil: Ministério da Saúde; 2015. http://www.aids.gov.br/assistencia/manualdst/item12.htm. [Google Scholar]
- Brew BJ, Pemberton L, Blennow K, Wallin A, Hagberg L. CSF amyloid beta42 and tau levels correlate with AIDS dementia complex. Neurology. 2005;65:1490–1492. doi: 10.1212/01.wnl.0000183293.95787.b7. [DOI] [PubMed] [Google Scholar]
- Brown LAM, Jin J, Ferrell D, Sadic E, Obregon D, Smith AJ, Tan J, Giunta B. Efavirenz Promotes b-Secretase Expression and Increased Ab1-40,42 via Oxidative Stress and Reduced Microglial Phagocytosis: Implications for HIV Associated Neurocognitive Disorders (HAND) PLoS ONE. 2014;9(4):e95500. doi: 10.1371/journal.pone.0095500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brucki SM, Nitrini R, Caramelli P, Bertolucci PHF, Okamoto IH. Sugestões para o uso do Mini-Exame do Estado Mental no Brasil. Arq Neuro-Psiquiatr. 2003;61:777–781. doi: 10.1590/s0004-282x2003000500014. [DOI] [PubMed] [Google Scholar]
- Burgoyne RW, Tan DH. Prolongation and quality of life for HIV-infected adults treated with highly active antiretroviral therapy (HAART): a balancing act. J Antimicrob Chemother. 2008;61:469–473. doi: 10.1093/jac/dkm499. [DOI] [PubMed] [Google Scholar]
- Clifford DB, Fagan AM, Holtzman DM, Morris JC, Teshome M, Shah AR, Kauwe JS. CSF biomarkers of Alzheimer disease in HIV-associated neurologic disease. Neurology. 2009;73(23):1982–7. doi: 10.1212/WNL.0b013e3181c5b445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cysique LA, Hewitt T, Croitoru-Lamoury J, Taddei K, Martins RN, Chew CSN, Davies NNWS, Price P, Brew BJ. APOE ε4 moderates abnormal CSF-abeta-42 levels, while neurocognitive impairment is associated with abnormal CSF tau levels in HIV+ individuals – a cross-sectional observational study. BMC Neurology. 2015;15:51. doi: 10.1186/s12883-015-0298-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daily A, Nath A, Hersh LB. Tat peptides inhibit neprilysin. J Neurovirol. 2006;12:153–160. doi: 10.1080/13550280600760677. [DOI] [PubMed] [Google Scholar]
- de Almeida SM, Ribeiro CE, de Pereira AP, Badiee J, Cherner M, Smith D, Maich I, Raboni SM, Rotta I, Barbosa FJ, Heaton RK, Umlauf A, Ellis RJ. Neurocognitive impairment in HIV-1 clade C- versus B-infected individuals in Southern Brazil. J Neurovirol. 2013;19:550–556. doi: 10.1007/s13365-013-0215-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida SM, Rotta I, Jiang Y, Li X, Raboni SM, Ribeiro CE, Smith D, Potter M, Vaida F, Letendre S, Ellis RJ HNRC Group. Biomarkers of chemotaxis and inflammation in cerebrospinal fluid and serum in individuals with HIV-1 subtype C versus B. J Neurovirol. 2016;22:715–724. doi: 10.1007/s13365-016-0437-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida SM, Rotta I, Ribeiro CE, Oliveira MF, Chaillon A, de Pereira AP, Cunha AP, Zonta M, Bents JF, Raboni SM, Smith D, Letendre S, Ellis RJ. Dynamic of CSF and serum biomarkers in HIV-1 subtype C encephalitis with CNS genetic compartmentalization—case study. J Neurovirol. 2017;23:460–473. doi: 10.1007/s13365-017-0518-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Bell RD, Sagare A, Zlokovic BV. Clearance of amyloid-β peptide across the blood-brain barrier: Implication for therapies in Alzheimer’s disease. CNS Neurol Disord Drug Targets. 2009;8:16–30. doi: 10.2174/187152709787601867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis RJ, Seubert P, Motter R, Galasko D, Deutsch R, Heaton RK, Heyes MP, McCutchan JA, Atkinson JH, Grant I. Cerebrospinal fluid tau protein is not elevated in HIV-associated neurologic disease in humans. Neuroscience Letters. 1998;254(1):1–4. doi: 10.1016/s0304-3940(98)00549-7. [DOI] [PubMed] [Google Scholar]
- Ensoli B, Buonaguro L, Barillari G, Fiorelli V, Gendelman R, Morgan RA, Wingfield P, Gallo RC. Release, uptake, and effects of extracellular human immunodeficiency virus type 1 Tat protein on cell growth and viral transactivation. J Virol. 1993;67:277–87. doi: 10.1128/jvi.67.1.277-287.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esiri MM, Biddolph SC, Morris CS. Prevalence of Alzheimer plaques in AIDS. J Neurol Neurosurg Psychiatry. 1998;65:29–33. doi: 10.1136/jnnp.65.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giometto B, An SF, Groves M, Scaravilli T, Geddes JF, Miller R, Tavolato B, Beckett AA, Scaravilli F. Accumulation of beta-amyloid precursor protein in HIV encephalitis: relationship with neuropsychological abnormalities. Ann Neurol. 1997;42:34–40. doi: 10.1002/ana.410420108. [DOI] [PubMed] [Google Scholar]
- Gisslén M, Krut J, Andreasson U, Blennow K, Cinque P, Brew BJ, Spudich S, Hagberg L, Rosengren L, Price RW, Zetterberg H. Amyloid and tau cerebrospinal fluid biomarkers in HIV infection. BMC Neurology. 2009;9:63. doi: 10.1186/1471-2377-9-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giunta B, Zhou Y, Hou H, Rrapo E, Fernandez F, Tan J. HIV-1 Tat inhibits microglial phagocytosis of Aβ peptide. Int J Clin Exp Pathol. 2008;1:260–275. [PMC free article] [PubMed] [Google Scholar]
- Giunta B, Hou H, Zhu Y, Rrapo E, Tian J, Takashi M, Commins D, Singer E, He J, Fernandez F, Tan J. HIV-1 Tat Contributes to Alzheimer’s Disease-like Pathology in PSAPP Mice. Int J Clin Exp Pathol. 2009;2:433–443. [PMC free article] [PubMed] [Google Scholar]
- Green AJ, Giovannoni G, Hall-Craggs MA, Thompson EJ, Miller RF. Cerebrospinal fluid tau concentrations in HIV infected patients with suspected neurological disease. Sex Transm Infect. 2000;76:443–446. doi: 10.1136/sti.76.6.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DA, Masliah E, Vinters HV, Beizai P, Moore DJ, Achim CL. Brain deposition of beta-amyloid is a common pathologic feature in HIV positive patients. AIDS. 2005;19:407–411. doi: 10.1097/01.aids.0000161770.06158.5c. [DOI] [PubMed] [Google Scholar]
- Gross AM, Jaeger PA, Kreisberg JF, Licon K, Jepsen KL, Khosroheidari M, Morsey BM, Swindells S, Shen H, Ng CT, Flagg K, Chen D, Zhang K, Fox HS, Ideker T. Methylome-wide Analysis of Chronic HIV Infection Reveals Five-Year Increase in Biological Age and Epigenetic Targeting of HLA. Molecular Cell. 2016;62:157–168. doi: 10.1016/j.molcel.2016.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel FG, Fawcett J, Tsui BT, Bennett RG, Duckworth WC. Effect of nelfinavir on insulin metabolism, proteasome activity and protein degradation in HepG2 cells. Diabetes Obes Metab. 2006;8:661–668. doi: 10.1111/j.1463-1326.2005.00546.x. [DOI] [PubMed] [Google Scholar]
- Hamel F. HIV Protease Inhibitors Inhibit Insulin-degrading Enzyme (IDE) Function. 67th Scientific Sessions; American Diabetes Association; 2007. Insulin-degrading enzyme (IDE) is a protease that cleaves insulin and other bioactive peptides such as amyloid-β. [Google Scholar]
- Haughey NJ, Bandaru VVR, Bae M, Mattson M. Roles for dysfunctional sphingolipid metabolism in Alzheimer’s disease neuropathogenesis. Biochim Biophys Acta. 2010;1801:878–86. doi: 10.1016/j.bbalip.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- High KP, Brennan-Ing M, Clifford DB, Cohen MH, Currier J, Deeks SG, Deren S, Effros RB, Gebo K, Goronzy JJ, Justice AC, Landay A, Levin J, Miotti PG, Munk RJ, Nass H, Rinaldo CR, Jr, Shlipak MG, Tracy R, Valcour V, Vance DE, Walston JD, Volberding P OAR Working Group on HIV and Aging. HIV and aging: state of knowledge and areas of critical need for research. A report to the NIH Office of AIDS Research by the HIV and Aging Working Group. J Acquir Immune Defic Syndr. 2012;60(Suppl 1):S1–18. doi: 10.1097/QAI.0b013e31825a3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hort J, O’Brien JT, Gainotti G, Pirttila T, Popescu BO, Rektorova I, Sorbi S, Scheltens P. Scientist Panel on EFNS, Dementia EFNS guidelines for the diagnosis and management of Alzheimer’s disease. Eur J Neurol. 2010;17:1236–1248. doi: 10.1111/j.1468-1331.2010.03040.x. [DOI] [PubMed] [Google Scholar]
- Hulstaert F, Blennow K, Ivanoiu A, Schoonderwaldt HC, Riemenschneider M, DeDeyn PP, Bancher C, Cras P, Wiltfang J, Mehta PD, Iqbal K, Pottel H, Vanmechelen E, Vanderstichele H. Improved discrimination of AD patients using beta-amyloid (1-42) and tau levels in CSF. Neurology. 1999;52:1555–1562. doi: 10.1212/wnl.52.8.1555. [DOI] [PubMed] [Google Scholar]
- Kim J, Yoon JH, Kim YS. HIV-1 Tat interacts with and regulates the localization and processing of amyloid precursor protein. PLoS One. 2013;8(11):e77972. doi: 10.1371/journal.pone.0077972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krut JJ, Zetterberg H, Blennow K, Cinque P, Hagberg L, Price RW, Studahl M, Gisslen M. Cerebrospinal fluid Alzheimer’s biomarker profiles in CNS infections. J Neurol. 2013;260:620–26. doi: 10.1007/s00415-012-6688-y. [DOI] [PubMed] [Google Scholar]
- Krut JJ, Gisslen M, Hagberg L, Zetterberg H, Price RW, Nilsson S, Cinque P. Hyperphosphorylated Tau in Cerebrospinal fluid: a biomarker for neurological aging in HIV? CROI, #453 2014 [Google Scholar]
- Lan X, Xu J, Kiyota T, Peng H, Zheng JC, Ikezu T. HIV-1 reduces Abeta-degrading enzymatic activities in primary human mononuclear phagocytes. J Immunol. 2011;186:6925–32. doi: 10.4049/jimmunol.1100211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan X, Kiyota T, Hanamsagar R, Huang Y, Andrews S, Peng H, Zheng JC, Swindells S, Carlson GA, Ikezu T. The Effect of HIV Protease Inhibitors on Amyloid-β Peptide Degradation and Synthesis in Human Cells and Alzheimer’s Disease Animal Model. J Neuroimmune Pharmacol. 2012;7(2):412–423. doi: 10.1007/s11481-011-9304-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letendre S, Ellis R, Deutsch R, Clifford D, Marra C, McCutchan A, Morgello S, Simpson D, Heaton R, Grant I the CHARTER Group. Correlates of time-to-loss-of-viralresponse in CSF and plasma in the CHARTER cohort. Program and abstracts of the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA. 16-19 February; 2010. poster 430. [Google Scholar]
- Madani R, Poirier R, Wolfer DP, Welzl H, Groscurth P, Lipp HP, Lu B, El Mouedden M, Mercken M, Nitsch RM, Mohajeri MH. Lack of Neprilysin Suffices To Generate Murine Amyloid-Like Deposits in the Brain and Behavioral Deficit In Vivo. J Neurosci Res. 2006;84:1871–1878. doi: 10.1002/jnr.21074. [DOI] [PubMed] [Google Scholar]
- Mankowski JL, Queen SE, Tarwater PM, Fox KJ, Perry VH. Accumulation of beta-Amyloid Precursor Protein in Axons Correlates with CNS Expression of SIV gp41. J Neuropathol Exp Neurol. 2002;61:85–90. doi: 10.1093/jnen/61.1.85. [DOI] [PubMed] [Google Scholar]
- Mattsson N, Andreasson U, Persson S, Arai H, Batish SD, Bernardini S, Bocchio-Chiavetto L, Blankenstein MA, Carrillo MC, Chalbot S, Coart E, Chiasserini D, Cutler N, Dahlfors G, Duller S, Fagan AM, Forlenza O, Frisoni GB, Galasko D, Galimberti D, Hampel H, Handberg A, Heneka MT, Herskovits AZ, Herukka SK, Holtzman DM, Humpel C, Hyman BT, Iqbal K, Jucker M, Kaeser SA, Kaiser E, Kapaki E, Kidd D, Klivenyi P, Knudsen CS, Kummer MP, Lui J, Lladó A, Lewczuk P, Li QX, Martins R, Masters C, McAuliffe J, Mercken M, Moghekar A, Molinuevo JL, Montine TJ, Nowatzke W, O’Brien R, Otto M, Paraskevas GP, Parnetti L, Petersen RC, Prvulovic D, de Reus HP, Rissman RA, Scarpini E, Stefani A, Soininen H, Schröoder J, Shaw LM, Skinningsrud A, Skrogstad B, Spreer A, Talib L, Teunissen C, Trojanowski JQ, Tumani H, Umek RM, Van Broeck B, Vanderstichele H, Vecsei L, Verbeek MM, Windisch M, Zhang J, Zetterberg H, Blennow K. The Alzheimer’s Association external quality control program for cerebrospinal fluid biomarkers. Alzheimers Dement. 2011;7:386–395.e6. doi: 10.1016/j.jalz.2011.05.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memória CM, Yassuda MS, Nakano EY, Forlenza OV. Brief screening for mild cognitive impairment: validation of the Brazilian version of the Montreal Cognitive Assessment. Int J Geriatr Psychiatry. 2013;28:34–40. doi: 10.1002/gps.3787. [DOI] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies B, Weintraub S, Phelps CH. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging -Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–9. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinuevo JL, Gispert JD, Dubois B, Heneka MT, Lleo A, Engelborghs S, Pujol J, de Souza LC, Alcolea D, Jessen F, Sarazind M, Lamari F, Balasa M, Antonell A, Rami L. The AD-CSF-Index Discriminates Alzheimer’s Disease Patients from Healthy Controls: A Validation Study. J Alzheimer’s Disease. 2013;36:67–77. doi: 10.3233/JAD-130203. [DOI] [PubMed] [Google Scholar]
- Mothapo K, Stelma F, Janssen M, Kessels R, Miners S, Verbeek M, Koopmans P, van der Ven A. Amyloid beta-42 (Ab-42): neprilysin and cytokine levels. A pilot study in patients with HIV related cognitive impairments. J Neuroimmunol. 2015;282:73–79. doi: 10.1016/j.jneuroim.2015.03.017. [DOI] [PubMed] [Google Scholar]
- Nath A, Hersh LB. Tat and amyloid: multiple interactions. AIDS. 2005;19:203–204. doi: 10.1097/00002030-200501280-00013. [DOI] [PubMed] [Google Scholar]
- Navia BA, Cho ES, Petito CK, Price RW. The AIDS dementia complex: II. Neuropathology. Ann Neurol. 1986;19(6):525–535. doi: 10.1002/ana.410190603. [DOI] [PubMed] [Google Scholar]
- Nebuloni M, Pellegrinelli A, Ferri A, Bonetto S, Boldorini R, Vago L, Grassi MP, Costanzi G. Beta amyloid precursor protein and patterns of HIV p24 immunohistochemistry in different brain areas of AIDS patients. AIDS. 2001;15(5):571–5. doi: 10.1097/00002030-200103300-00005. [DOI] [PubMed] [Google Scholar]
- Peluso MJ, Meyerhoff DJ, Price RW, Peterson J, Lee E, Young AC, Walter R, Fuchs D, Brew BJ, Cinque P, Robertson K, Hagberg L, Zetterberg H, Gisslén M, Spudich S. Cerebrospinal fluid and neuroimaging biomarker abnormalities suggest early neurological injury in a subset of individuals during primary HIV infection. J Infect Dis. 2013;207(11):1703–12. doi: 10.1093/infdis/jit088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson J, Zetterberg H, Hagberg L, Spudich S, Gisslen M, Price R. Changing CSF concentrations of neurofilament light chain, tau and amyloid proteins, characterize evolving CNS injury in HIV-1 infection. 20th Conference on Retroviruses and Opportunistic Infections; Atlanta. 2013. [Google Scholar]
- Peterson J, Gisslen M, Zetterberg H, Fuchs D, Shacklett BL, Hagberg L, Constantin T, Yiannoutsos CT, Spudich SS, Price RW. Cerebrospinal Fluid (CSF) Neuronal Biomarkers across the Spectrum of HIV Infection: Hierarchy of Injury and Detection. PLoS ONE. 2014;9(12):e116081. doi: 10.1371/journal.pone.0116081. https://doi.org/10.1371/journal.pone.0116081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer RI, Kurosaki TT, Harrah CH, Jr, Chance JM, Filos S. Measurement of functional activities in older adults in the community. J Gerontol. 1982;37:323–329. doi: 10.1093/geronj/37.3.323. [DOI] [PubMed] [Google Scholar]
- Pulliam L. HIV regulation of amyloid beta production. J Neuroimmune Pharmacol. 2009;4:213–217. doi: 10.1007/s11481-009-9151-9. [DOI] [PubMed] [Google Scholar]
- Raboni SM, Ribeiro CE, Almeida SM, Telles JP, Azevedo M, Schaitza GA. Impact of public health strategies on reducing AIDS mortality in southern Brazil. Int J STD AIDS. 2017;28:54–62. doi: 10.1177/0956462415624075. [DOI] [PubMed] [Google Scholar]
- Ranga U, Shankarappa R, Siddapa NB, Ramakrishna L, Nagendran R, Mahalingam M, Mahadevan A, Jayasuryan N, Satishchandra P, Shankar SK, Prasad VR. Tat protein of human immunodeficiency virus type 1 subtype C strains is a defective chemokine. J Virology. 2004;78:2586–90. doi: 10.1128/JVI.78.5.2586-2590.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempel HC, Pulliam L. HIV-1Tat inhibits neprilysin and elevates amyloid beta. AIDS. 2005;19:127–135. doi: 10.1097/00002030-200501280-00004. [DOI] [PubMed] [Google Scholar]
- Satishchandra P, Nalini A, Gourie-Devi M, Khanna N, Santosh V, Rasvi V, Desai A, Chandramuki A, Jayakumar PN, Shankar SK. Profile of neurologic disorders associated with HIV/AIDS from Bangalore, South India (1989-96) Indian J Med Res. 2000;111:14–23. [PubMed] [Google Scholar]
- Seeger M, Ferrell K, Frank R, Dubiel W. HIV-1 tat inhibits the 20 S proteasome and its 11 S regulator-mediated activation. J Biol Chem. 1997;272:8145–8148. doi: 10.1074/jbc.272.13.8145. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Clearing the brain’s amyloid cobwebs. Neuron. 2001;32:177–180. doi: 10.1016/s0896-6273(01)00475-5. [DOI] [PubMed] [Google Scholar]
- Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV. Clearance of Alzheimer’s amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106(12):1489–99. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley LC, Mrak RE, Woody RC, Perrot LJ, Zhang S, Marshak DR, Nelson SJ, Griffin WS. Glial cytokines as neuropathogenic factors in HIV infection: pathogenic similarities to Alzheimer’s disease. J Neuropathol Exp Neurol. 1994;53(3):231–238. doi: 10.1097/00005072-199405000-00003. [DOI] [PubMed] [Google Scholar]
- Steinbrink F, Evers S, Buerke B, Young P, Arendt G, Koutsilieri E, Reichelt D, Lohmann H, Husstedt IW. Cognitive impairment in HIV infection is associated with MRI and CSF pattern of neurodegeneration. Eur J Neurol. 2013;20:420–428. doi: 10.1111/ene.12006. [DOI] [PubMed] [Google Scholar]
- Tran H, Robinson S, Mikhailenko I, Strickland DK. Modulation of the LDL receptor and LRP levels by HIV protease inhibitors. J Lipid Res. 2003;44:1859–1869. doi: 10.1194/jlr.M200487-JLR200. [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, Schuck T, Schmidt ML, Lee VM. Distribution of tau proteins in the normal human central and peripheral nervous system. J Histochem Cytochem. 1989;37:209–215. doi: 10.1177/37.2.2492045. [DOI] [PubMed] [Google Scholar]
- White JA, Manelli AM, Holmberg KH, Van Eldik LJ, Ladu MJ. Differential effects of oligomeric and fibrillar amyloid-beta 1–42 on astrocyte-mediated inflammation. Neurobiol Dis. 2005;18:459–65. doi: 10.1016/j.nbd.2004.12.013. [DOI] [PubMed] [Google Scholar]
- Wood SN. Generalized Additive Models: An Introduction with R. Boca Raton, FL: CRC Press, LLC; 2006. [Google Scholar]
- Xu J, Ikezu T. The comorbidity of HIV-associated neurocognitive disorders and Alzheimer’s disease: a foreseeable medical challenge in post-HAART era. J Neuroimmune Pharmacol. 2009;4:200–212. doi: 10.1007/s11481-008-9136-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Liu J, Katafiasz B, Fox H, Xiong H. HIV-1 gp120-induced axonal injury detected by accumulation of β-amyloid precursor protein in adult rat corpus callosum. J Neuroimmune Pharmacol. 2011;6:650–657. doi: 10.1007/s11481-011-9259-6. [DOI] [PMC free article] [PubMed] [Google Scholar]