

Abstract
Triple-negative breast cancer (TNBC) is the leading cancer in women. Chemotherapeutic agents used for TNBC are mainly associated with dose-dependent toxicities and development of resistance. Hence, novel strategies to overcome resistance and to offer dose reduction are warranted. In this study, we designed a novel dual-functioning agent, conjugate of cholecalciferol with PEG2000 (PEGCCF) which can self-assemble into micelles to encapsulate doxorubicin (DOX) and act as a chemosensitizer to improve the therapeutic potential of DOX. DOX-loaded PEGCCF (PEGCCF-DOX) micelles have particle size, polydispersity index (PDI), and zeta potential of 40 ± 8.7 nm, 0.180 ± 0.051, and 2.39 ± 0.157 mV, respectively. Cellular accumulation studies confirmed that PEGCCF was able to concentration-dependently enhance the cellular accumulation of DOX and rhodamine 123 in MDA-MB-231 cells through its P-glycoprotein (P-gp) inhibition activity. PEGCCF-DOX exhibited 1.8-, 1.5-, and 2.9-fold enhancement in cytotoxicity of DOX in MDA-MB-231, MDA-MB-468, and MDA-MB-231DR (DOX-resistant) cell lines, respectively. Western blot analyses showed that PEGCCF-DOX caused significant reduction in tumor markers including mTOR, c-Myc, and antiapoptotic marker Bcl-xl along with upregulation of preapoptotic marker Bax. Further, reduction in mTOR activity by PEGCCF-DOX indicates reduced P-gp activity due to P-gp downregulation as well and, hence, PEGCCF causes enhanced chemosensitization and induces apoptosis. Substantially enhanced apoptotic activity of DOX (10-fold) in MDA-MB-231(DR) cells confirmed apoptotic potential of PEGCCF. Conclusively, PEGCCF nanomicelles are promising delivery systems for improving anticancer activity of DOX in TNBC, thereby reducing its side effects and may act as a potential carrier for other chemotherapeutic agents.
Keywords: triple-negative breast cancer, cholecalciferol, nanomicelles, doxorubicin, cytotoxicity
INTRODUCTION
Chemotherapy is the primary choice of treatment for early stages of cancer and is also used in conjugation with surgery and radiation for the late stages of cancer. However, chemotherapy is associated with the unavoidable side effects, leading to toxicity to patients and development of drug resistance (1–4). The development of advanced drug delivery systems has shown great potential to yield enhanced therapeutic efficacy and comparatively higher accumulation of drug in tumor cells along with minimal exposure to normal tissues (1,5,6). However, the development of multidrug resistance (MDR) in cancer cells is of grave concern, limiting the efficacy of anticancer agents and, hence, the failure of therapy. Several biochemical changes have been described to be involved for the development of MDR (5–7). Overexpression of ATP-binding cassette transporters causing efflux of drug, overexpression of P-glycoprotein (P-gp), multidrug resistance-associated proteins (MRP1, MRP2), and breast cancer resistance protein (BCRP) are some of the major mechanisms reported to be involved in the development of MDR (6–8).
Triple-negative breast cancer (TNBC) is one such condition offering therapeutic challenge due to its intractable characteristics, lack of well-defined molecular targets, and development of MDR (9,10). It is considered to be one of the most aggressive cancers, lacking estrogen receptor (ER), progesterone receptor (PR), and HER2/Neu receptor expression and accounting for 15% of the breast cancer cases with a high relapse rate (10,11). The absence of ER, PR, and HER2 protein expression makes TNBC highly insensitive to therapies such as tamoxifen and aromatase inhibitors and HER2-targeted therapies such as trastuzumab and lapatinib. Consequently, anthracyclines and taxanes remain a standard therapy for the TNBC patients (9,12). However, long-term doxorubicin (DOX) therapy leads to cytotoxicity in normal tissue, myelosupression, and cardiotoxicity (13).
Application of nanoscale drug delivery carriers has been proved to be a successful strategy to overcome drug resistance (6,14) and enhance therapeutic efficacy of the drugs. The polymeric micelle-based systems have proved to be an efficient inhibitor of P-gp, leading to altered internalization and subcellular localization of drugs (6,15–17). A narrow size distribution also avoids their uptake by the reticuloendothelial system (RES) and promotes their release at the leaky tumor vasculature, leading to passive accumulation (18). The biodistribution of nanomicelles and that of the incorporated drug are largely influenced by the properties of outer shell (19,20). So far, the most frequently used hydrophilic polymer is poly(ethylene glycol) (PEG) due to its biocompatibility and ability to avoid opsonization by serum proteins (21). The P-gp inhibition property of the polymeric micelles has been widely explored in order to enhance the delivery and localization of chemotherapeutics in resistant cancers (6). Lee et al. demonstrated high cytotoxicity in DOX-resistant MCF7 cells via folate-conjugated DOX-loaded micelles by receptor-mediated endocytosis overcoming P-gp (22). Another study by Wei et al. showed significantly lower half maximal inhibitory concentration (IC50) in human lung adenocarcinoma cell lines SPC-A1 and A-549 by paclitaxel-loaded pluronic P123/F127-mixed polymeric micelles (17–19,21–23). Cambón et al. demonstrated enhanced cellular accumulation of DOX by poly(butylene oxide)–poly(ethylene oxide)–poly(butylene oxide) block copolymeric micelles, leading to higher toxicity in ovarian NCIADR-RES cell lines overexpressing P-gp (24).
Several polymeric formulations have been studied in clinical trials. DOX-loaded polymeric micelle (NK911), paclitaxel-loaded radiosensitive nanomicelles (Genexol® PM), cisplatin-loaded polymeric micelles (NC-6004), and epirubicin-loaded nanomicelles (NC-6300) are some of the examples of drug-loaded micelles in clinical trials (6,15). These reports clearly reveal the capability of micelles in overcoming drug resistance and their wide applicability in cancer therapy.
Vitamin D is very well known for its role in calcium and phosphorus metabolism and has also been explored for its inhibition of cell proliferation and induction of cell differentiation). Various studies have shown the impact of vitamin D on reversal of resistance; Yan and Nuriding observed the reversal of MDR on vitamin D-treated Jurkat/ADR and K562/ADR cells, following through reduction in MDR1 and MRP1 mRNA expression, P-gp content on the cell surface, and intracellular GSH level in a dose-dependent manner (25). Vitamin D3 in combination with metformin has been shown to inhibit the proliferation of human breast cancer MDA-MB-231 cells and promoted apoptosis (26). A combination of vitamin D and celecoxib has also been reported to show additive effects along with improved chemotherapeutic efficacy on MCF7 and MDA-MB-231 breast cancer cells (27). Despite all these reports, there has been a very limited effort towards the development of a drug delivery carrier with vitamin D with an aim of simultaneously delivering the payload chemotherapeutic. There has been only one report where PEG–vitamin D succinate conjugate was used as a pharmaceutical additive and has been shown to elicit P-gp inhibitory effect on Caco-2 cells (28). Thus, in the present study, we aim to synthesize vitamin D conjugated with PEG (PEGCCF) and explore its dual role of DOX-loaded PEGCCF (PEGCCF-DOX) micelles, i.e., as an excipient to encapsulate drug in a nanoformulation and to act as a chemosensitizer for enhancing the therapeutic potential of the anticancer drug activity in breast cancer.
MATERIALS AND METHODS
Materials
Cholecalciferol (vitamin D3), poly(ethylene glycol) methyl ether (average Mn ~ 2000) (mPEG), glutaric anhydride, 4-dimethylaminopyridine (DMAP), N-hydroxysuccinimide (NHS), dicyclohexylcarbodiimide (DCC), triethylamine (TEA), and doxorubicin hydrochloride (DOX.HCl) were purchased from Cayman Chemical Company (MI, USA). Rhodamine 123 dichloromethane (DCM), chloroform, methanol, ethyl acetate, and petroleum ether (60:40) were purchased from Sigma-Aldrich, (St. Louis, MO). The cell lines MCF10A, MDA-MB-468, and MDA-MB-231 were purchased from ATCC (Manassas, VA). The cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F-12 and supplemented with 10% fetal bovine serum (FBS) and antibiotic mixture of penicillin streptomycin and neomycin.
Synthesis of Cholecalciferol Glutarate
Cholecalciferol glutarate was synthesized by the reaction of glutaric anhydride with cholecalciferol in dry DCM in the presence of TEA in the dark (29,30). Briefly, about 5 g (12.9 mmol) of cholecalciferol was dissolved in anhydrous chloroform (100 mL) containing 5.4 mL TEA (38.7 mmol) and about 7.4 g (64.9 mmol) of glutaric anhydride was added to it. The reaction was refluxed. Thin-layer chromatography (TLC) with mobile phase methanol/ethyl acetate (1:8) was carried out to observe reaction progress. After 3 days, the chloroform layer was washed with 100 mL of 0.1 N HCl and dried over anhydrous Na2SO4. The chloroform layer was evaporated using a rotary evaporator. The residue thus obtained was dissolved in petroleum ether, and the precipitate of excess glutaric anhydride was filtered. Finally, the petroleum ether was evaporated and brown solid residue (cholecalciferol glutarate) was obtained.
Synthesis of PEGCCF
PEGCCF was synthesized by the reaction of cholecalciferol glutarate with mPEG in the presence of DCC and DMAP using dry DCM as solvent (31,32). Briefly, about 5 g (2.5 mmol) of mPEG was dissolved in anhydrous DCM followed by the addition of 516 mg (2.5 mmol) of DCC and a catalytic amount of DMAP (5 mg). Cholecalciferol glutarate (1.24 g, 2.5 mmol) was added to the reaction mixture, and stirring was done at room temperature for 48 h. The precipitates formed during the reaction were filtered. The filtrate was concentrated, and the residue was dissolved in ethyl acetate. Ethyl acetate was evaporated, and the residue was again dissolved in ethyl ether and filtered. The filtrate was evaporated to obtain the product. The PEGCCF formed in the reaction was dried in a vacuum desiccator.
Characterization of PEGCCF Conjugate
Fourier transform infrared (FTIR) spectra were obtained by the KBr disc method using a Thermo Scientific Nicolet iS5 FT-IR instrument (Waltham, MA, USA). NMR spectra were performed with Varian Mercury 300 MHz NMR Spectrometer using CDCl3 as solvent.
Critical Aggregation Concentration Determination
The critical aggregation concentration (CAC) of PEGCCF micelles was determined by fluorescence spectroscopy using pyrene as a hydrophobic probe according to a procedure reported earlier (28) with slight modifications. Briefly, about 20 μL of a stock solution of pyrene (0.1 mM) in acetone was taken in different glass vials and the solvent was removed by blowing air. To each of these vials, varying volumes of PEGCCF micelle solution were added and the final volume was adjusted with distilled water to get a series of solutions with constant pyrene concentration (1 × 10−6 M) and varying amounts of PEGCCF (0.001 to 0.75 mg/mL). The solutions were allowed to equilibrate overnight at room temperature, and the excitation spectra were recorded from 300 to 350 nm wavelength at an emission wavelength of 390 nm using a fluorescence spectrophotometer (Cary Eclipse, Agilent Technologies). The ratio of the excitation intensities at 338 and 336 nm (I338/I336) was plotted against PEGCCF concentrations, and CAC was obtained from an inflection point in the plot.
Preparation of PEGCCF-DOX Micelles
PEGCCF-DOX micelles were prepared by a pH-induced self-assembly method according to a procedure reported earlier with slight modifications (33,34). Briefly, DOX.HCl was stirred overnight with triethylamine in 2 mol ratio in the presence of DMSO to obtain DOX-free base. PEGCCF (25 mg) and DOX (2 mg) were dissolved in 50 μL of ethanol at 60°C. The solution was then added dropwise to 950 μL of phosphate-buffered saline (PBS) (pH ~ 7.4) at 60°C with constant stirring. The low solubility of DOX in PBS at pH ~ 7.4 led to the incorporation of DOX into the hydrophobic core of the PEGCCF micelles. The particle size of micelles was reduced by ultrasonication (Branson probe sonicator, USA) for 2 min followed by cooling down to room temperature. The measurement of hydrodynamic diameter and zeta potential of micelles was performed using Nicomp 380 ZLS (particle sizing system; Port Richey, FL). The encapsulation efficiency of PEGCCF micelles was obtained by the ratio of experimental drug loading (actual amount of drug encapsulated in the nanomicelles) and by the theoretical drug loading (total amount of DOX initially used for encapsulation). The concentration of DOX was determined by HPLC (Waters e2695 Separation Module; MA, USA) fitted with a Waters 1525 binary pump and Waters 2996 photo diode array detector on a ZORBAX SB-C18 column (4.6 × 250 mm2, 5 μm) at 258 nm wavelength. The mobile phase was acetonitrile and phosphate buffer (pH 3.0; 30:70) at a constant flow rate of 1 mL/min. Blank PEGCCF micelles were prepared following a similar procedure except the addition of DOX. PEGCCF-DOX micelles were stored at 2–8°C for 2 months for stability studies.
In Vitro DOX Release Studies
The DOX release from PEGCCF-DOX micelles was determined by using a dialysis bag technique in PBS (pH 7.4). DOX.HCl aqueous solution (2 mg/mL) was prepared. An aliquot of 1 mL of DOX.HCl solution and 1 mL of PEGCCF-DOX micelles containing 2 mg/mL of DOX were sealed in dialysis tubes (MWCO of 10,000 Da; Sigma-Aldrich Co., MO) and immersed in 250 mL of PBS. The release experiment was conducted in an incubator shaker set at 100 rpm and 37°C temperature. At different time intervals, 1 mL of samples was withdrawn and replaced with an equal amount of fresh PBS. The samples were analyzed on HPLC.
Cellular DOX Accumulation Studies
Cellular uptake studies were conducted for DOX and P-gp substrate rhodamine 123 as mentioned in an earlier report with slight modifications (35). Briefly, MDA-MB-231DR cells were seeded in six-well plates at a density of 50,000 cells/well, washed with PBS after 24 h, and treated with DOX (5 μM), free rhodamine 123 (1 μM), and PEGCCF micelles corresponding to an equivalent amount of free DOX and an equivalent amount of rhodamine 123. After incubation for 30 min, cells were washed with PBS twice and observed using fluorescence microscopy (Olympus, Inc., USA).
Cytotoxicity Evaluation
Cytotoxicity evaluation of DOX, cholecalciferol, and a combination of both was performed at predetermined concentrations on various TNBC cell lines by crystal violet cytotoxic assay according to a procedure reported earlier (36–38). The MCF10A, MDA-MB-231DR, MDA-MB-231, and MDA-MB-468 TNBC cells were plated in 96-well plates, at a density of 10,000 cells/well, and were allowed to incubate for 24 h. After 24 h, the cells were washed with fresh medium and were exposed to different treatments as described above. After 48 h, cells were fixed with 0.5% v/v glutaraldehyde and viability was assessed by crystal violet assay. A microplate reader (Tecan, Tecan Austria GmbH, Austria) was used to measure absorbance at 570 nm. Percentage cytotoxicity was calculated, and the results were expressed as percent cell kill versus concentration of drug. IC50 value was calculated using Sigma Plot software, version 9.0 (Systat Software, San Jose, CA). Combination index (CI) was calculated as per the analytical method of Chougule et al. (39), and drug interaction was analyzed.
Migration Assay
In vitro migration (scratch) assay was performed to analyze the percent inhibition of bridging of migration area by DOX and cholecalciferol alone and in combination. The assay was performed as per the protocol described in our previous reports (37). In short, MDA-MB-231DR cells (1 × 104 cells/well) were seeded in a 96-well plate in complete growth media and allowed to incubate overnight, and a uniform scratch was made in the center of each well using a cell scraper. Cell treatment was done with DOX (10 μM), cholecalciferol (5 μM), and a combination of DOX (10 μM) and cholecalciferol (5 μM). After 48 h of treatment exposure, cells were fixed with 0.5% glutaraldehyde followed by staining with crystal violet for 30 min. An Olympus microscope was used to take images of the scratch area. Gap (scratch) area prior and post the treatment was calculated using ImageJ software.
Western Blot
Western blot analysis was performed according to a procedure standardized in previous report (37). Briefly, MDA-MB 231DR cells were seeded in a culture flask and treated with DOX (0.5 μM), blank PEGCCF micelles, and PEGCCF-DOX micelles. The standard protocol was used to prepare the whole-cell protein lysates. Equivalent amounts of supernatant proteins (50 μg), from all the groups, were denatured, separated using 10% SDS-PAGE gel, and for immunoblotting, transferred onto a nitrocellulose membrane. The nitrocellulose membranes were blocked using PBST containing 5% BSA and probed overnight with primary antibodies against mTOR, Bcl-xl, Bax, and β-actin (1:1000) and c-Myc (1:500), and protein detection was carried out with HRP-conjugated secondary antibodies. Finally, ImageJ software (v1.33u; NIH) was used for quantification and result analysis.
Flow Cytometry
MDA-MB-231DR cells were seeded in a 24-well plate at a density of 1 × 105 cells/well and cultured for 24 h at 37°C in complete media. After 24 h, cells were washed with fresh medium and treated with DOX (0.5 μM), blank PEGCCF micelles, and PEGCCF-DOX micelles containing an equal amount of DOX, respectively. After 48 h of exposure, media were completely discarded and cells were washed with ice-cold PBS twice and incubated with annexin V/FITC. The fluorescence-activated cell sorter (FACS) Calibur flow cytometer (BD, Franklin Lakes, NJ) was used to determine the intracellular fluorescence of apoptotic cells.
Statistical Analysis
The experiments were performed in triplicates. Data are presented as mean ± standard deviation. Comparisons were carried out using Student’s t test. Statistical significance to a level of p < 0.05 was acceptable.
RESULTS
Effect of Cholecalciferol on Cytotoxicity and Cell Migration
Effect of cholecalciferol on cytotoxicity of DOX in MDA-MB-231DR TNBC cell lines was evaluated using crystal violet assay. Different concentrations of cholecalciferol were evaluated, and cholecalciferol (10 μM) was found to be noncytotoxic for the cells (data not shown). Enhanced cytotoxicity of DOX (0.4 μM) was observed in combination with cholecalciferol (10 μM) (Fig. 1a). Further, the effect of cholecalciferol on migratory and invasiveness capacity of MDA-MB-231DR cells was evaluated by scratch assay. About 60% bridging was observed in the control group after 48 h, while cholecalciferol (5 μM) and DOX (10 μM) alone showed 50 and 47% bridging of scratch area, respectively. A significant reduction (p < 0.01) in bridging of scratch area was observed, when cells were treated with a combination of cholecalciferol (5 μM) and DOX (10 μM) as compared to the cells treated with either cholecalciferol or DOX alone (Fig. 1b). Above results indicate that even noncytotoxic concentration of cholecalciferol enhanced cytotoxicity and antimigratory effect of DOX.
Fig. 1.

Effect of cholecalciferol on anticancer activity of DOX. a Cytotoxicity of cholecalciferol (10 μM), DOX (0.4 μM), and a combination of DOX (0.4 μM) and cholecalciferol (10 μM) on MDA-MB-231DR using crystal violet assay. b In vitro migration assay of cholecalciferol (10 μM), DOX (0.4 μM) and a combination of DOX (0.4 μM) and cholecalciferol (10 μM) on MDA-MB-231DR in a 96-well plate for 48 h after scratch formation. *p < 0.05 was considered significant, and p value is represented as **p < 0.01 and *p < 0.05. Error bars refer to SD
Synthesis and Characterization of PEGCCF
The synthesis scheme of PEGCCF is shown in Fig. 2a. Synthesis of PEGCCF was achieved by esterification of PEG and cholecalciferol glutarate and confirmed by 1H NMR spectroscopy (Supplemental Fig. S1) and FTIR (Supplemental Fig. S2). Comparative 1H NMR spectra of cholecalciferol, glutaric anhydride, cholecalciferol glutarate, and PEGCCF are shown in Fig. S1. The characteristic shifts of cholecalciferol H21, H26, and H27 protons were observed at δ 0.97 ppm (40). The stereoisomeric proton H19 was observed at δ 4.8 and δ 5.09 ppm. The proton H3 adjacent to the hydroxyl group was observed as singlet around 3.9 ppm, and the alkene protons H6 and H7 appeared as doublet at δ 6.0 ppm and δ 6.1 ppm. The steroid skeleton shifts were observed as a group of peaks between δ 0.5 ppm and δ 2.9 ppm. The 1H NMR spectrum of cholecalciferol glutarate contained all of the chemical shifts of steroid skeleton except the H3 proton signal shifted downfield to δ 5.0 ppm, indicating that the –OH group adjacent to H3 proton has been converted into ester after reaction with glutaric anhydride. Further, the synthesis of PEGCCF micelles was confirmed by the presence of PEG proton around δ 3.5 to 3.6 ppm besides the proton observed in case of cholecalciferol glutarate. The FTIR spectrum of cholecalciferol glutarate showed the stretching vibration of carboxyl group –OH at 3200 cm−1 and stretching vibration corresponding to the C = O group of ester and carboxylic acid were observed at 1735 and 1710 cm−1, respectively, indicating that glutaric anhydride ring opened during the reaction forming ester with the cholecalciferol hydroxyl group, leaving a free carboxylic group at the other end. In the spectrum of PEGCCF, the appearance of characteristic –C–O–C skeletal stretch at 1104 cm−1 and disappearance of C = O stretching vibration of carboxyl group confirmed a successful synthesis of PEGCCF via esterification of PEG and cholecalciferol glutarate. The particle size, polydispersity index (PDI), and zeta potential of PEGCCF micelles was 40 ± 8.7 nm, 0.180 ± 0.051, and 2.39 ± 0.157 mV, respectively. The encapsulation efficiency was 92.4 ± 2.7%. Further, 2-month stability data demonstrated that micelles were stable at 2–8°C with no significant change in particle size and encapsulation efficiency. As shown in Fig. 2b, the CAC of PEGCCF micelles was calculated using a pyrene probe method and was determined as 0.089 mg/mL.
Fig. 2.
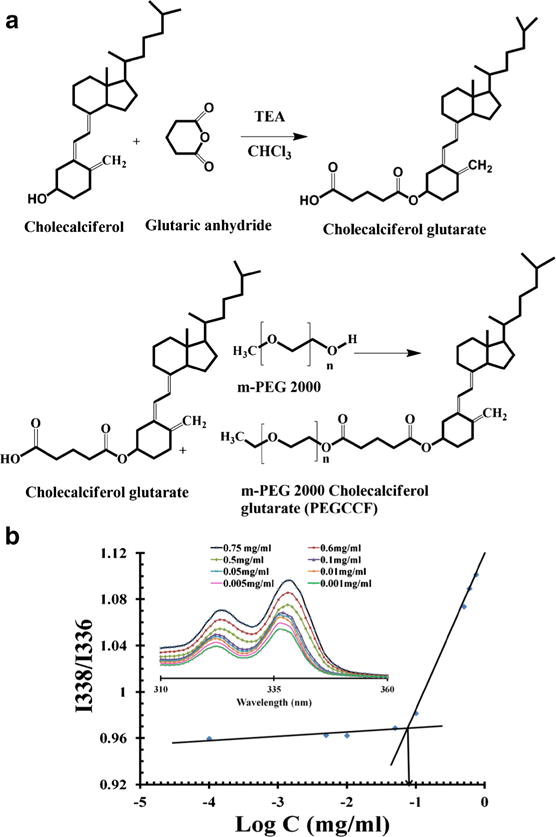
Synthesis scheme and CAC determination of PEGCCF. a Synthesis scheme of mPEG 2000 cholecalciferol glutarate (PEGCCF). b Pyrene excitation spectra as a function of PEGCCF concentration (inset). Plot of intensity ratios I338/I336 versus log C (mg/mL) of PEGCCF
In Vitro DOX Release
Drug release profile of DOX alone and PEGCCF-DOX cells was determined in PBS (pH 7.4). A very high burst release was observed in case of DOX alone with almost 90% release of DOX in first 4 h, whereas PEGCCF-DOX micelles exhibited a better controlled release comparatively with a low initial burst for first 2 h with about 44% drug release in 4 h followed by a longer sustained release phase with 93% drug release in 12 h (Fig. 3a). From this study, it can be concluded that PEGCCF micelles are reducing the rate of release of DOX.
Fig. 3. In vitro.

characterization of PEGCCF-DOX micelles, cellular accumulation of DOX, and IC50 of DOX and PEGCCF-DOX on cancer cells. a In vitro drug release study of DOX and PEGCCF-DOX micelles in PBS (pH 7.4). PEGCCF-DOX micelles showed sustained release of DOX. b Cellular internalization of P-gp substrate DOX and rhodamine 123 in MDA-MB-231(DR) cells. c IC50 value of DOX and PEGCCF-DOX on MDA-MB-231, MDA-MB-231DR, and MDA-MB-468 cells. Experiments were done in triplicate, and average was calculated. Error bars represent SD. *p < 0.05 was considered significant, and p value is represented as **p < 0.01 and *p < 0.05. Error bars refer to SD
Cellular DOX and Rhodamine 123 Accumulation Studies
To determine P-gp inhibition by PEGCCF micelle, rhodamine 123 (fluorescent P-gp substrate) was taken as standard. The cellular uptake of free rhodamine 123, rhodamine 123 + cholecalciferol, and rhodamine 123 loaded in PEGCCF micelles was evaluated. Green fluorescent intensity of rhodamine 123 was highest when loaded in PEGCCF micelles followed by rhodamine 123 + cholecalciferol and was lowest in rhodamine 123 alone. Similar results were observed for uptake studies of DOX (red fluorescence). Intensity of red fluorescence was highest when DOX was loaded in PEGCCF micelles (Fig. 3b). And red fluorescence was lowest when DOX was added alone in the cells. This study distinctly suggests that PEGCCF micelles enhanced the drug accumulation in the cancer cells through P-gp inhibition.
In Vitro Cytotoxicity Studies of PEGCCF-DOX Micelles
Toxicity evaluation of blank micelles was performed on normal breast MCF10A cells and breast cancer MDA-MB-231 and MDA-MB-468 cells. Since PEGCCF micelles did not show any significant cytotoxicity up to 0.062 mM, a concentration of 0.030 mM was utilized to carry out further biological studies. Cytotoxicity evaluation of PEGCCF-DOX micelles was performed on MDA-MB-231, MDA-MB-231DR, and MDA-MB 468 cells (Fig. 3c). Efficacy of DOX was enhanced after encapsulation into PEGCCF micelles. A 2.9-fold, 1.8-fold, and 1.5-fold reduction in IC50 of DOX was observed when evaluated on MDA-MB-231DR, MDA-MB-231, and MDA-MB-468 cell lines, respectively, as compared to free DOX. CI of 0.34, 0.55, and 0.68 was observed in MDA-MB-231DR, MDA-MB-231, and MDA-MB-468 cell lines, respectively.
Western Blot
Consistent with the results from in vitro cytotoxicity and cell uptake studies, western blot analysis of protein mTOR was significantly (p < 0.01) downregulated in PEGCCF-DOX-treated cells when compared to control. Further, western blot bands revealed that c-Myc expressions were reduced by fivefold in PEGCCF-DOX-treated cells, as compared to untreated cells. Also, the expression for proapoptotic marker Bax was upregulated by 3-fold and Bcl-xl expression was downregulated by 2.3-fold (p < 0.01) (Fig. 4).
Fig. 4.

Western blot and densitometry analysis. MDA-MB-231DR cells were treated with PEGCCF, DOX, and PEGCCF-DOX. β-Actin acts as a housekeeping protein. Error bars refer to SD, and p value is represented as **p < 0.01 and *p < 0.05
Flow Cytometry
Flow cytometry analysis was performed to understand the effect of PEGCCF micelles on MDA-MB-231DR cells. Flow cytometry data suggested that PEGCCF-DOX micelles induced apoptosis in MDA-MB-231(DR) cells when incubated for 48 h. PEGCCF-DOX micelles showed significantly enhanced early apoptosis (21.61%) as compared to an equivalent amount of free DOX where merely 2.16% early apoptosis was observed (Fig. 5). All these observations are in agreement with cytotoxicity results of PEGCCF micelles, suggesting chemosensitization and thus enhancing the apoptotic activity of the DOX.
Fig. 5.

Flow cytometry analysis. Flow cytometry analysis of MDA-MB-231(DR) cells after treatment with annexin V/FITC and PI staining for apoptosis. a Control (no treatment). b DOX (0.5 μM). c PEGCCF micelles. d PEGCCF-DOX micelles
DISCUSSION
The absence of therapeutic receptors, metastasis, and drug resistance impede TNBC treatment. Moreover, traditional chemotherapy is associated with dose-related toxicities. Hence, novel delivery systems or combination therapies, which promise higher efficacy and lower toxicities, are being investigated. There are numerous reports in the literature supporting the role of vitamin D and its derivatives in the treatment as well as prevention of various types of cancers (41). Despite huge interest in vitamin D, cholecalciferol and cholecalciferol-based delivery systems have not been explored widely. This is the first study focused on evaluating the role of dual-functioning agent cholecalciferol–PEG conjugate as a nanocarrier and chemosensitizer for the delivery of chemotherapeutic agents in TNBC. We performed studies to investigate the effect of cholecalciferol alone and in combination with the chemotherapeutic agent DOX on TNBC cells.
Our in vitro cytotoxicity studies showed a threefold reduction in IC50 of DOX when used in combination with cholecalciferol against MDA-MB-231(DR) cells. In scratch assay studies, a combination of cholecalciferol and DOX demonstrated 29% bridging of scratch area as compared to 47% bridging by DOX alone. These studies suggested that cholecalciferol has potential to enhance cytotoxicity and antimigration activity of DOX. Our results are supported by a previous study where co-administration of cholecalciferol with cancer DNA vaccine significantly (p < 0.005) suppressed the tumor growth and metastasis (p < 0.05) in lung carcinoma mouse models (42).
Nanomicelle-based drug delivery systems have wide applicability in cancer chemotherapy owing to their enhanced bioavailability, efficacy, and sustained release profiles. Zhao et al. used cholecalciferol–PEG succinate as an adjuvant in DOX-loaded PLGA nanoparticles and demonstrated its P-glycoprotein (P-gp) inhibitory potential (28). It has been reported that carboxylesterases present in tumor cells can efficiently convert various prodrugs into their active metabolites (43). In general, esterase activity and intrinsic clearance increase with an increase in carbon chain length (44,45). Therefore, we chose a glutarate ester linker instead of succinate ester and successfully synthesized PEGCCF. The particle size of the micelles was 40 ± 8.7 nm, and small particle size promotes penetration and accumulation in the tumor cells. Zeta potential is a measure of magnitude of electrostatic repulsion among the particles bearing the same charge and indicates the stability of colloidal dispersion. In general, higher zeta potential value (absolute value) reflects higher stability due to higher repulsion among the particles. However, in PEGylated nanocarriers, often PEG chains on the surface of nanoparticles prevent aggregation and provide stabilization by acting as a steric barrier (46). PEGCCF micelles were sterically stabilized by PEG chains on the surface of micelles. PDI reflects broadness in size distribution of nanoparticles. A high PDI value denotes a broad range of particle sizes, low stability, and aggregation of the particles. PEGCCF had PDI 0.180 ± 0.051, suggesting homogeneity and stability of nanoformulation. Further, stability data of PEGCCF-DOX demonstrated that micelles were stable at 2–8°C with no sign of aggregation and loss of encapsulation efficiency. CAC is a crucial factor for determining the stability of micelles. The CAC of PEGCCF nanomicelles was 0.089 mg/mL and is comparable to that of D-alphatocopheryl–poly(ethylene glycol)–1000 succinate (TPGS) (0.2 mg/mL) (47) showing its stability and significance. Cytotoxicity of PEGCCF micelles with MCF10A and MDA-MB-231 cells revealed that the nanomicelles were safe for the cells at a concentration below 0.062 mM. PEGCCF-DOX exhibited a sustained release pattern with a release of 50% of DOX at 8 h when compared to 90% release of DOX when in the free form. The release study suggests that PEGCCF-DOX release DOX over a longer period of time as compared to free DOX.
It is well known that enhanced expression of plasma membrane-associated protein known as P-gp strongly blocks the transport of several hydrophobic drugs into the cancer cells and thus diminishes their cytotoxicity. To evaluate P-gp inhibitory potential of PEGCCF micelles in MDA-MB 231DR cells, DOX and rhodamine 123 accumulation studies were performed. Both rhodamine 123 and DOX are P-gp activity markers and exhibit green and red fluorescence, respectively. Higher uptake of rhodamine 123 and DOX in the combination group is attributed to P-gp inhibitory activity of cholecalciferol (48). However, the highest uptake was observed via PEGCCF nanomicelles, possibly due to substantial DOX and rhodamine 123 accumulation in the cells by the nanomicelles. Also, due to P-gp inhibitory effect in part by PEGCCF itself which goes in line with other surfactant molecules such as TPGS and in part by hydrolysis of PEGCCF to release cholecalciferol, which also have been shown as a P-gp inhibitor. TPGS has been reported to inhibit P-gp-mediated drug resistance in MDR1 cDNA-transfected cells (G185) having 27–135-fold more resistance to cytotoxic drugs, thereby increasing cytotoxicity of DOX as compared to parental cells. In another study, cholecalciferol succinate has been reported to block P-gp and enhance accumulation of DOX (p < 0.05) (28,49).
We further evaluated the effect of PEGCCF-DOX on various triple-negative breast cancer cell lines. PEGCCF-DOX exhibited enhanced cytotoxicity of DOX by 2.9-fold (p < 0.01), 1.8-fold (p < 0.05), and 1.5-fold against MDA-MB-231(DR), MDA-MB-231, and MDA-MB-468 cells, respectively, as compared to free DOX. The reasons for enhanced cytotoxicity were possibly higher intracellular uptake of DOX via nanomicelles and cholecalciferol released from hydrolysis of PEGCCF in the cells. Cholecalciferol has been reported to inhibit growth, migration, and invasion in prostate cancer cells (50). This observation is supported by another study wherein enhanced uptake of DOX was observed when loaded in dextran and poly(D,L-lactide-co-glycolide) blocks copolymer polymeric micelles as compared to DOX alone in DOX-resistant human cholangiocarcinoma (HuCC-T1) cells (51). Viswanathan et al. observed twofold enhancement in the DOX potency in MDA-MB-231 cells when loaded in di-block copolymer-based micelles (52).
For better understanding of the underlying mechanism, western blot analysis was performed. We observed a significant downregulation in the expression of mTOR (p < 0.01) and c-Myc (p < 0.01). It has been reported that there is a relationship between mTOR signaling and P-gp-associated MDR in cancer cells (53) and mTOR inhibitors have been reported for resistance reversal (54). The decreased mTOR and c-Myc expression supports that PEGCCF-DOX inhibits P-gp-associated drug resistance and enhances DOX cytotoxicity by sensitizing cells. We also observed significant downregulation of Bcl-xl (p < 0.01) and upregulation of Bax. It is well known that Bax and Bcl2 play a role in cell apoptosis; Bax promotes the cell death whereas Bcl2 subfamily including Bcl-xl suppresses cell death (55). Our results suggest that PEGCCF-DOX suppressed Bcl-xl to a greater extent than DOX; thus, it may have a role in enhancing its antiapoptotic activity. The presence of 25-hydroxylase and 25-hydroxyvitamin D3 1α-hydroxylase has been reported in various breast cancer cells (56). These enzymes are capable of converting cholecalciferol to calcitriol. We also believe that the observed effects of PEGCCF in the current study are possibly due to conversion of cholecalciferol released from the PEGCCF conjugate to calcitriol. Calcitriol has been reported to exhibit antiproliferative and antiapoptotic activity on MCF7 and MDA-MB-231 cells by downregulation of mTOR, c-Myc, and Bcl-xl (26,57–59). To further understand the effect of PEGCCF on DOX-induced apoptosis, flow cytometry analysis was performed. PEGCCF-DOX demonstrated 10-fold enhancement in DOX-induced apoptosis on MDA-MB-231(DR) cells. Indeed, flow cytometry data was consistent with apoptotic markers c-Myc, Bcl-xl, and Bax. Cholecalciferol being a precursor of the highly active metabolite calcitriol may exhibit higher anticancer activity in vivo. However, further studies are needed to better understand the mechanism of direct anticancer activity of cholecalciferol and its chemosensitizer action in breast cancer.
CONCLUSION
We have synthesized cholecalciferol–PEG conjugate (PEGCCF) and evaluated it as a carrier for the delivery of anticancer drug DOX. Cytotoxicity and antimigration activity of DOX were significantly enhanced when administered in combination with cholecalciferol. PEGCCF micelles exhibited sustained release profile and enhanced DOX accumulation in cancer cells possibly via inhibition of P-gp-mediated MDR. PEGCCF-DOX micelles reduced IC50 of DOX along with significant downregulation of tumor markers mTOR, c-Myc, and Bcl-xl and upregulation of the apoptotic marker Bax. PEGCCF also increased apoptotic activity of DOX. These effects are essentially important in chemosensitizing and enhancing anticancer activity of DOX and can help in overcoming drug resistance along with reducing side effects. Furthermore, our results have widened the potential use of PEGCCF conjugate as a dual functioning agent, i.e., an excipient to encapsulate anticancer drugs in a nanoformulation and to act as a chemosensitizer for enhancing the therapeutic potential of the anticancer drug. Indeed, further evaluation in animal tumor models is needed to better understand the in vivo therapeutic potential of the proposed strategy.
Supplementary Material
Acknowledgments
This research was supported with funding from the National Institute on Minority Health and Health Disparities (NIMHD) P20 program (Grant no. 1P20 MD006738-03).
Footnotes
Electronic supplementary material: The online version of this article (https://doi.org/10.1208/s12249-017-0885-z) contains supplementary material, which is available to authorized users.
COMPLIANCE WITH ETHICAL STANDARDS
Conflict of Interest: The authors declare that they have no conflict of interest.
References
- 1.Bergh J, Jönsson P-E, Glimelius B, Nygren P. A systematic overview of chemotherapy effects in breast cancer. Acta Oncol. 2001;40(2–3):253–81. doi: 10.1080/02841860151116349. [DOI] [PubMed] [Google Scholar]
- 2.Boehm T, Folkman J, Browder T, O’Reilly MS. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature. 1997;390(6658):404–7. doi: 10.1038/37126. [DOI] [PubMed] [Google Scholar]
- 3.Gottesman MM, Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu Rev Biochem. 1993;62(1):385–427. doi: 10.1146/annurev.bi.62.070193.002125. [DOI] [PubMed] [Google Scholar]
- 4.Litman T, Druley T, Stein W, Bates S. From MDR to MXR: new understanding of multidrug resistance systems, their properties and clinical significance. Cell Mol Life Sci CMLS. 2001;58(7):931–59. doi: 10.1007/PL00000912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang Y, Cole SP, Cai T, Cai Y. Applications of nanoparticle drug delivery systems for the reversal of multidrug resistance in cancer (review) Oncol Lett. 2016;1(12):11–5. doi: 10.3892/ol.2016.4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kapse-Mistry S, Govender T, Srivastava R, Yergeri M. Nanodrug delivery in reversing multidrug resistance in cancer cells. Front Pharmacol. 2014;5:159. doi: 10.3389/fphar.2014.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeng X, Morgenstern R, Nyström AM. Nanoparticle-directed sub-cellular localization of doxorubicin and the sensitization breast cancer cells by circumventing GST-mediated drug resistance. Biomaterials. 2014;35(4):1227–39. doi: 10.1016/j.biomaterials.2013.10.042. [DOI] [PubMed] [Google Scholar]
- 8.Broxterman HJ, Lankelma J, Hoekman K. Resistance to cytotoxic and anti-angiogenic anticancer agents: similarities and differences. Drug Resist Updat. 2003;6(3):111–27. doi: 10.1016/s1368-7646(03)00026-8. [DOI] [PubMed] [Google Scholar]
- 9.O’Reilly EA, Gubbins L, Sharma S, Tully R, Guang MHZ, Weiner-Gorzel K, et al. The fate of chemoresistance in triple negative breast cancer (TNBC) BBA Clin. 2015;3:257–75. doi: 10.1016/j.bbacli.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schneider BP, Winer EP, Foulkes WD, Garber J, Perou CM, Richardson A, et al. Triple-negative breast cancer: risk factors to potential targets. Clin Cancer Res. 2008;14(24):8010–8. doi: 10.1158/1078-0432.CCR-08-1208. [DOI] [PubMed] [Google Scholar]
- 11.Andey T, Sudhakar G, Marepally S, Patel A, Banerjee R, Singh M. Lipid nanocarriers of a lipid-conjugated estrogenic derivative inhibit tumor growth and enhance cisplatin activity against triple-negative breast cancer: pharmacokinetic and efficacy evaluation. Mol Pharm. 2015;12(4):1105–20. doi: 10.1021/mp5008629. [DOI] [PubMed] [Google Scholar]
- 12.Chacón RD, Costanzo MV. Triple-negative breast cancer. Breast Cancer Res. 2010;12(2):1–9. doi: 10.1186/bcr2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biondo PD, Brindley DN, Sawyer MB, Field CJ. The potential for treatment with dietary long-chain polyunsaturated n-3 fatty acids during chemotherapy. J Nutr Biochem. 2008;19(12):787–96. doi: 10.1016/j.jnutbio.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 14.Ayers D, Nasti A. Utilisation of nanoparticle technology in cancer chemoresistance. J Drug Deliv. 2012;2012:1–12. doi: 10.1155/2012/265691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gong J, Chen M, Zheng Y, Wang S, Wang Y. Polymeric micelles drug delivery system in oncology. J Control Release. 2012;159(3):312–23. doi: 10.1016/j.jconrel.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Wang R, Lu X, Lu W, Zhang C, Liang W. Pegylated phospholipids-based self-assembly with water-soluble drugs. Pharm Res. 2010;27(2):361–70. doi: 10.1007/s11095-009-0029-6. [DOI] [PubMed] [Google Scholar]
- 17.Wei Z, Hao J, Yuan S, Li Y, Juan W, Sha X, et al. Paclitaxel-loaded Pluronic P123/F127 mixed polymeric micelles: formulation, optimization and in vitro characterization. Int J Pharm. 2009;376(1):176–85. doi: 10.1016/j.ijpharm.2009.04.030. [DOI] [PubMed] [Google Scholar]
- 18.Jain RK. 1996 Landis Award—delivery of molecular and cellular medicine to solid tumors. Microcirculation. 1997;4(1):1–23. doi: 10.3109/10739689709148314. [DOI] [PubMed] [Google Scholar]
- 19.Jiang W, Wang Y-d, Gan Q, Zhang GJ-z, Zhao X-w, Fei W-y, et al. Preparation and characterization of copolymer micelles formed by poly (ethylene glycol)-polylactide block copolymers as novel drug carriers. Chin J Process Eng. 2006;6(2):289–95. [Google Scholar]
- 20.Singh M, Ferdous AJ, Branham M, Betageri GV. Trends in drug targeting for cancer treatment. Drug Deliv. 1996;3(4):289–304. [Google Scholar]
- 21.Owens DE, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307(1):93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 22.Lee ES, Na K, Bae YH. Doxorubicin loaded pH-sensitive polymeric micelles for reversal of resistant MCF-7 tumor. J Control Release. 2005;103(2):405–18. doi: 10.1016/j.jconrel.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 23.Zhang W, Shi Y, Chen Y, Yu S, Hao J, Luo J, et al. Enhanced antitumor efficacy by paclitaxel-loaded pluronic P123/F127 mixed micelles against non-small cell lung cancer based on passive tumor targeting and modulation of drug resistance. Eur J Pharm Biopharm. 2010;75(3):341–53. doi: 10.1016/j.ejpb.2010.04.017. [DOI] [PubMed] [Google Scholar]
- 24.Cambón A, Rey-Rico A, Mistry D, Brea J, Loza M, Attwood D, et al. Doxorubicin-loaded micelles of reverse poly (butylene oxide)–poly (ethylene oxide)–poly (butylene oxide) block copolymers as efficient “active” chemotherapeutic agents. Int J Pharm. 2013;445(1):47–57. doi: 10.1016/j.ijpharm.2013.01.056. [DOI] [PubMed] [Google Scholar]
- 25.Yan M, Nuriding H. Reversal effect of vitamin D on different multidrug-resistant cells. Genet Mol Res. 2014;13(3):6239–47. doi: 10.4238/2014.August.15.6. [DOI] [PubMed] [Google Scholar]
- 26.Guo L-S, Li H-X, Li C-Y, Zhang S-Y, Chen J, Wang Q-L, et al. Synergistic antitumor activity of vitamin D3 combined with metformin in human breast carcinoma MDA-MB-231 cells involves m-TOR related signaling pathways. Pharmazie Int J Pharm Sci. 2015;70(2):117–22. [PubMed] [Google Scholar]
- 27.Thill M, Reichert K, Woeste A, Polack S, Fischer D, Hoellen F, et al. Combined treatment of breast cancer cell lines with vitamin D and COX-2 inhibitors. Anticancer Res. 2015;35(2):1189–95. [PubMed] [Google Scholar]
- 28.Zhao H, Tan E, Yung L. Potential use of cholecalciferol polyethylene glycol succinate as a novel pharmaceutical additive. J Biomed Mater Res A. 2008;84(4):954–64. doi: 10.1002/jbm.a.31402. [DOI] [PubMed] [Google Scholar]
- 29.Wuts PG, Greene TW. Greene’s protective groups in organic synthesis. 4th. New York: John Wiley & Sons; 2006. [Google Scholar]
- 30.Kazemi M, Kohzadi H, Noori Z. Potassium carbonate: a highly efficient catalyst for the acylation of alcohols, phenols and thiols under mild conditions. Iranian Chem Commun. 2014;2:39–47. [Google Scholar]
- 31.Harris JM. Laboratory synthesis of polyethylene glycol derivatives. J Macromol Sci-Rev Macromol Chem Phys. 1985;25(3):325–73. [Google Scholar]
- 32.Tsakos M, Schaffert ES, Clement LL, Villadsen NL, Poulsen TB. Ester coupling reactions—an enduring challenge in the chemical synthesis of bioactive natural products. Nat Prod Rep. 2015;32(4):605–32. doi: 10.1039/c4np00106k. [DOI] [PubMed] [Google Scholar]
- 33.Gao X, Wang B, Wei X, Rao W, Ai F, Zhao F, et al. Preparation, characterization and application of star-shaped PCL/PEG micelles for the delivery of doxorubicin in the treatment of colon cancer. Int J Nanomedicine. 2013;8:971–82. doi: 10.2147/IJN.S39532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee ES, Na K, Bae YH. Polymeric micelle for tumor pH and folate-mediated targeting. J Control Release. 2003;91(1):103–13. doi: 10.1016/s0168-3659(03)00239-6. [DOI] [PubMed] [Google Scholar]
- 35.Patel K, Doddapaneni R, Chowdhury N, Boakye CH, Behl G, Singh M. Tumor stromal disrupting agent enhances the anticancer efficacy of docetaxel loaded PEGylated liposomes in lung cancer. Nanomedicine. 2016;11(11):1377–92. doi: 10.2217/nnm.16.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chougule MB, Patel AR, Jackson T, Singh M. Antitumor activity of noscapine in combination with doxorubicin in triple negative breast cancer. PLoS One. 2011;6(3):e17733. doi: 10.1371/journal.pone.0017733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patel K, Chowdhury N, Doddapaneni R, Boakye CH, Godugu C, Singh M. Piperlongumine for enhancing oral bioavailability and cytotoxicity of docetaxel in triple-negative breast cancer. J Pharm Sci. 2015;104(12):4417–26. doi: 10.1002/jps.24637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shaik MS, Chatterjee A, Singh M. Effects of monensin liposomes on the cytotoxicity, apoptosis and expression of multidrug resistance genes in doxorubicin-resistant human breast tumour (MCF-7/dox) cell-line. J Pharm Pharmacol. 2004;56(7):899–907. doi: 10.1211/0022357023772. [DOI] [PubMed] [Google Scholar]
- 39.Chougule M, Patel AR, Sachdeva P, Jackson T, Singh M. Anticancer activity of noscapine, an opioid alkaloid in combination with cisplatin in human non-small cell lung cancer. Lung Cancer. 2011;71(3):271–82. doi: 10.1016/j.lungcan.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mizhiritskii MD, Konstantinovskii LE, Vishkautsan R. 2D NMR study of solution conformations and complete 1 H and 13 C chemical shifts assignments of vitamin D metabolites and analogs. Tetrahedron. 1996;52(4):1239–52. [Google Scholar]
- 41.Toffoli G, Corona G, Gigante M, Boiocchi M. Inhibition of Pgp activity and cell cycle-dependent chemosensitivity to doxorubicin in the multidrug-resistant LoVo human colon cancer cell line. Eur J Cancer. 1996;32(9):1591–7. doi: 10.1016/0959-8049(96)00113-x. [DOI] [PubMed] [Google Scholar]
- 42.Zhuravel E, Efanova O, Shestakova T, Glushko N, Mezhuev O, Soldatkina M, et al. Administration of vitamin D3 improves antimetastatic efficacy of cancer vaccine therapy of lewis lung carcinoma. Exp Oncol. 2010;32:33–9. [PubMed] [Google Scholar]
- 43.Yang Y-h, Aloysius H, Inoyama D, Chen Y, Hu L-q. Enzyme-mediated hydrolytic activation of prodrugs. Acta Pharm Sin B. 2011;1(3):143–59. [Google Scholar]
- 44.Hofstee B. Specificity of esterases IV. Behavior of horse liver esterase towards a homologous series of n-fatty acid esters. J Biol Chem. 1954;207(1):219–24. [PubMed] [Google Scholar]
- 45.Williams ET, Bacon JA, Bender DM, Lowinger JJ, Guo W-K, Ehsani ME, et al. Characterization of the expression and activity of carboxylesterases 1 and 2 from the beagle dog, cynomolgus monkey, and human. Drug Metab Dispos. 2011 doi: 10.1124/dmd.111.041335. https://doi.org/10.1124/dmd.111.041335. [DOI] [PubMed]
- 46.Vukovic L, Khatib FA, Drake SP, Madriaga A, Brandenburg KS, Král P, et al. Structure and dynamics of highly PEG-ylated sterically stabilized micelles in aqueous media. J Am Chem Soc. 2011;133(34):13481–8. doi: 10.1021/ja204043b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ismailos G, Reppas C, Macheras P. Enhancement of cyclosporin A solubility by d-alphatocopheryl-polyethylene-glycol-1000 succinate (TPGS) Eur J Pharm Sci. 1994;1(5):269–71. [Google Scholar]
- 48.Chang C, Bahadduri PM, Polli JE, Swaan PW, Ekins S. Rapid identification of P-glycoprotein substrates and inhibitors. Drug Metab Dispos. 2006;34(12):1976–84. doi: 10.1124/dmd.106.012351. [DOI] [PubMed] [Google Scholar]
- 49.Dintaman JM, Silverman JA. Inhibition of P-glycoprotein by D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) Pharm Res. 1999;16(10):1550–6. doi: 10.1023/a:1015000503629. [DOI] [PubMed] [Google Scholar]
- 50.Tokar EJ, Webber MM. Cholecalciferol (vitamin D3) inhibits growth and invasion by up-regulating nuclear receptors and 25-hydroxylase (CYP27A1) in human prostate cancer cells. Clin Exp Metastasis. 2005;22(3):275–84. doi: 10.1007/s10585-005-8393-z. [DOI] [PubMed] [Google Scholar]
- 51.Jeong Y-I, Kim DH, Chung C-W, Yoo J-J, Choi KH, Kim CH, et al. Doxorubicin-incorporated polymeric micelles composed of dextran-b-poly (DL-lactide-co-glycolide) copolymer. Int J Nanomedicine. 2011;6:1415–27. doi: 10.2147/IJN.S19491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Viswanathan G, Hsu YH, Voon SH, Imae T, Siriviriyanun A, Lee HB, et al. A comparative study of cellular uptake and subcellular localization of doxorubicin loaded in self-assemblies of amphiphilic copolymers with pendant dendron by MDA-MB-231 human breast cancer cells. Macromol Biosci. 2016;16(6):882–95. doi: 10.1002/mabi.201500435. [DOI] [PubMed] [Google Scholar]
- 53.Kuo T-C, Chiang P-C, Yu C-C, Nakagawa-Goto K, Bastow KF, Lee K-H, et al. A unique P-glycoprotein interacting agent displays anticancer activity against hepatocellular carcinoma through inhibition of GRP78 and mTOR pathways. Biochem Pharmacol. 2011;81(9):1136–44. doi: 10.1016/j.bcp.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 54.Barančík M, Boháčová V, Sedlák J, Sulová Z, Breier A. LY294, 002, a specific inhibitor of PI3K/Akt kinase pathway, antagonizes P-glycoprotein-mediated multidrug resistance. Eur J Pharm Sci. 2006;29(5):426–34. doi: 10.1016/j.ejps.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 55.Naseri MH, Mahdavi M, Davoodi J, Tackallou SH, Goudarzvand M, Neishabouri SH. Up regulation of Bax and down regulation of Bcl2 during 3-NC mediated apoptosis in human cancer cells. Cancer Cell Int. 2015;15(1):1. doi: 10.1186/s12935-015-0204-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Segersten U, Holm PK, Björklund P, Hessman O, Nordgren H, Binderup L, et al. 25-Hydroxyvitamin D3 1α-hydroxylase expression in breast cancer and use of non-1α-hydroxylated vitamin D analogue. Breast Cancer Res. 2005;7(6):R980. doi: 10.1186/bcr1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lopes N, Paredes J, Costa JL, Ylstra B, Schmitt F. Vitamin D and the mammary gland: a review on its role in normal development and breast cancer. Breast Cancer Res. 2012;14(3):1–7. doi: 10.1186/bcr3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meyer MB, Goetsch PD, Pike JW. VDR/RXR and TCF4/β-catenin cistromes in colonic cells of colorectal tumor origin: impact on c-FOS and c-MYC gene expression. Mol Endocrinol. 2011;26(1):37–51. doi: 10.1210/me.2011-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Salehi-Tabar R, Nguyen-Yamamoto L, Tavera-Mendoza LE, Quail T, Dimitrov V, An B-S, et al. Vitamin D receptor as a master regulator of the c-MYC/MXD1 network. Proc Natl Acad Sci. 2012;109(46):18827–32. doi: 10.1073/pnas.1210037109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.