

Abstract
Alterations of the gamma-aminobutyric acid (GABA) signaling system has been strongly linked to the pathophysiology of autism spectrum disorder (ASD). Genetic associations of common variants in GABA-receptor subunits, in particular GABRA4 on chromosome 4p12, with ASD have been replicated by several studies. Moreover, molecular investigations have identified altered transcriptional and translational levels of this gene and protein in brains of ASD individuals. Since the genotyped common variants are likely not the functional variants contributing to the molecular consequences or underlying ASD phenotype, this study aims to examine rare sequence variants in GABRA4, including those outside the protein coding regions of the gene. We comprehensively re-sequenced the entire protein coding and noncoding portions of the gene and putative regulatory sequences in 82 ASD individuals and 55 developmentally typical pediatric controls, all homozygous for the most significant previously associated ASD risk allele (G/G at rs1912960). We identified only a single common, coding variant and no association of any single marker or set of variants with ASD. Functional annotation of non-coding variants identified several rare variants in putative regulatory sites. Finally, a rare variant unique to ASD cases, in an evolutionary conserved site of the 3′ UTR, shows a trend toward decreasing gene expression. Hence, GABRA4 rare variants in noncoding DNA may be variants of modest physiological effects in ASD etiology.
Keywords: Autism, GABRA4, sequencing, noncoding
Introduction
Autism spectrum disorder (ASD) comprises a range of neurodevelopmental conditions characterized by difficulties in social relatedness, communication, and behavior [1] and are diagnosed at a rate of approximately 1 in 68 children over eight years old in the United States[2]. There is a strong genetic component to risk for ASD. Among identified genetic contributors to ASD are large chromosomal irregularities, [3], copy number variations (CNVs), [4, 5], and recurrent de novo protein altering variants[6–9].
Genome-wide attempts to identify common genetic variants contributing to ASD risk have been moderately successful implicating a number of genes and genomic regions. These include the region between Cadherin 9 (CDH9) and Cadherin 10 (CDH10) on chromosome 5p14.1 [10, 11], MACROD2 on 20p12.1 [12], and the intergenic region between SEMA5A and TAS2R1 on 5p15.31 [13]. Though these studies have identified several positive associations, they have not been widely replicated. One possibility is that hundreds of factors impact ASD risk [14] and a complex architecture of many rare disease causing variants make it difficult to replicate single association signals in studies using current sample sizes [15].
Though genome-wide common variant association results have not widely replicated, there have been several candidate gene studies that have implicated the GABAergic system in ASD. GABA is the primary inhibitory neurotransmitter in the adult brain and directs neuronal development, a process which is altered in ASD [16–18]. Cytogenetic [19–21] and genotyping based linkage and association studies [22–24] have repeatedly implicated GABA receptors in ASD. Furthermore, candidate gene association tests for GABA receptor genes have revealed common polymorphisms in GABRA4 on chromosome 4p12 with association to ASD in multiple datasets [25–27]. Beyond these genetic findings, functional evidence for the role of GABRA4 in ASD is supported by biochemical studies which have shown reduced protein and mRNA levels in the cerebella of patients with autism [28–30].
Since the common SNPs associated with ASD in GABRA4 may not be the functional variants contributing to ASD phenotypes, it is necessary to follow-up association with comprehensive re-sequencing to uncover the complete catalogue of potentially functional rare genetic variants. To identify rare potentially functional variants we sequenced an 89kb region of chromosome 4 including the entire GABRA4 transcriptional region, 5kb 5′ and 10kb 3′ of the gene, in 82 White European ASD individuals and 55 age and ethnicity matched controls. We identify novel variants in evolutionarily conserved and predicted functional regions of GABRA4 and demonstrate that these can function by altering transcription of the gene.
Materials and Methods
Sample Ascertainment
The 82 ASD individuals sequenced in this study were recruited and enrolled as part of a previous study of GABA receptor genes [25]. The 55 controls consisted of white individuals aged 4 to 21 years sampled through the John. P. Hussman Institute for Human Genomics (Miami, FL). Control individuals were screened to determine whether they have been diagnosed with or have a parent or sibling with a developmental, behavioral, neurological or other disability. Parents of young children or the participants were informed and signed the informed consent, completed the Social Communication Questionnaire [31] to screen for potential ASDs, and donated saliva. In order to enrich for rare variants in possible linkage disequilibrium (LD) with a previously identified risk allele we chose only homozygous risk ASD individuals and controls (G/G at rs1912960). Genomic DNA was purified from whole blood from ASD individuals and saliva from controls using Puregene chemistry on the Qiagen Autopure LS according to standard automated Qiagen protocols (Qiagen, Valencia, CA, USA).
Targeted Region Amplification
We targeted chr4.hg19:46918163-47005187 encompassing 74kb of GABRA4 (NM_000809) and 10kb upstream and 5kb downstream of the gene with long-range PCR. Ten tiled primer pairs each amplifying ~8.7kb were designed using Primer3 software (http://frodo.wi.mit.edu) [32, 33] (Online Resource 1) and synthesized by IDT (Integrated DNA Technologies, Coralville, IA, USA). PCR was performed using the Expand Long Range dNTPack PCR kit (Roche Applied Science, Indianapolis, IN, USA) on Veriti 96-Well Fast Thermal Cyclers (Life Technologies, Carlsbad, CA, USA). PCR products were visualized on 0.75% agarose-ethidium bromide gel and primer dimer and nonspecific products removed with the NucleoFast 96 PCR kit (Machery-Nagel, Bethlehem, PA, USA) following standard manufacturer protocol for a vacuum manifold.
Massively Parallel Sequencing
Each of the 10 PCR products for each individual were quantified in a high-throughput microplate format on the SpectraMax M5 (Molecular Devices, Sunnyvale, CA, USA) using the Quant-iT PicoGreen dsDNA assay kit (Life Technologies, Carlsbad, CA, USA). The products were equimolar pooled, sequencing libraries prepared using reagents from New England Biolabs (NEB, Ipswich, Massachusetts, USA) according to Illumina manufacturer instructions, and 12 libraries pooled together into a single multiplexed sequencing library. These used for paired end cluster generation on the Illumina v2 flow cell (Illumina, San Diego, CA, USA) and sequenced on the Illumina GAIIx with a paired-end run of 52 cycles, with a 7-cycle index read.
Sequence Analysis
Raw image analysis and base calling were performed with Illumina Pipeline v1.5 and Sequence Control Software v2.5, and individual sequencing reads binned using a split.on.index.pl script supplied by Illumina. Subsequent data processing was done through an in-house pipeline including: alignment to the hg19 human reference genome with the Burrows-Wheeler Aligner (BWA) [34], generation of coverage metrics for quality control with PICARD, and genotype calling with the Genome Analysis Toolkit (GATK) Universal Genotyper [35]. As these reads result from PCR and coverage was extremely high, duplicates were not removed before variant calling. Only variant positions with a read depth greater than 50×, a call-rate of at least 95% across all samples, normalized Phred-scaled likelihood (PL) of a reference genotype ≥ 100, and at least 30% alternate allele reads were analyzed. Variant annotation was performed with ANNOVAR software [36, 37] and an in house non-coding variant annotation pipeline utilizing available ENCODE data [38]. For this pipeline, DNase hypersensitivity and transcription factor binding sites identified in any neuronal lineage cell line (SK-N-SH, BE2_C, Gliobla, HA-sp, HAc, NH-A, PFSK-1, SH-SY5Y, SK-N-MC, SK-N-SH_RA, and U87) were acquired from the UCSC table browser and compared against variant positions with BEDtools[39].
Rare variant association analyses
Single variant association was determined using the Fisher’s exact test implemented in PLINK [40] with power calculations performed with the Genetic Association Study (GAS) Power Calculator[41, 42]. To identify sets of variants associated with ASD, the optimized Sequence Kernal Association Test (SKAT-O) [43, 44] was performed with resampling with 10,000 iterations. This analysis was performed for all variants, for minor allele frequency cutoffs of 0.1, 0.05, 0.01, and 0.005, and for variants within discrete functional units. We also implemented a sliding window approach in each of the minor allele cut offs in groups of ten markers, overlapping by five, and used SKAT-O to determine association of variants within any window.
Functional assessment of 3′UTR variants by luciferase assay
The 9.3kb 3′UTR of GABRA4 was amplified from a control individual with Q5 Hot Start High-Fidelity DNA Polymerase (New England BioLabs, Ipswich, MA, USA) and cloned into 5′PmeI and 3′XhoI sites of the pmirGLO vector (Promega, Madison, WI, USA) using Blunt/TA Ligase Master Mix (New England BioLabs, Ipswich, MA, USA). Mutations were induced with the QuickChange Lightning Site-Directed Mutagenesis Kit (Agilent, Santa Clara, CA, USA). Confirmation of correct orientation, presence of the mutagenized base, and absence of off-target variants in all clones were determined by tiled PCR and Sanger sequencing of the entire insert on the ABI3730xl. Luciferase activity was measured in Human embryonic kidney (HEK293) transfected with 4ug of plasmid with Lipofectamine (Life Technologies, Carlsbad, CA, USA). We utilized the Dual-Glo® Luciferase Assay (Promega, Madison, WI, USA) in seven independent transfections for biological replication and two technical replicates for each experiment. We performed two-tailed, unequal variance Student’s t-tests across the seven biological replicates to compare luciferase expression from constructs with induced mutations compared to the control 3′UTR sequence.
Results
PCR Amplification
PCR optimization resulted in ideal thermocycling conditions for each primer set to yield a specific, single product represented by a band at the expected ~8.7kb size when visualized on an agarose gel (Figure 1). We successfully amplified the entire targeted region of GABRA4 in 82 ASD cases and 55 controls using ten tiled long range PCR reactions. Quantitation of the products after PCR cleanup showed DNA concentrations of ~35ng/μl and total yields of ~1.75μg of purified PCR products. We pooled 2 × 10−15 moles (~100ng) of each product into a single library per individual to be used in the sequencing sample preparation procedure.
Figure 1.
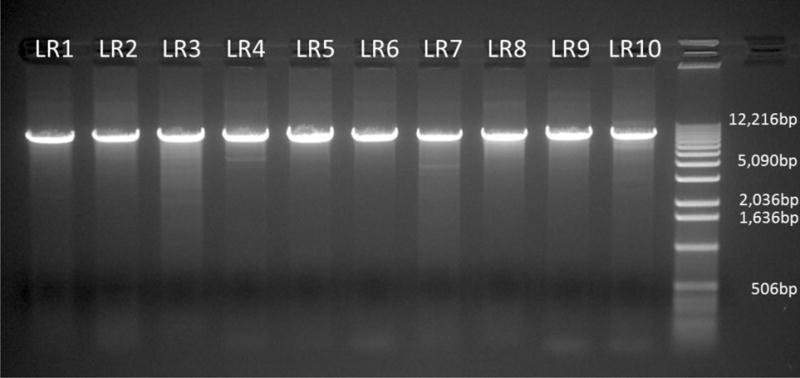
Agarose gel electrophoresis of PCR amplicons. 50ng of PCR products prior to NuceloFast clean-up were run on a 0.75% agarose gel for 1 hour at 90V, stained with ethidium bromide, and imaged under ultraviolet light. At the right is a 1kb ladder with bands at indicated sizes to estimate amplicon size.
Sequence Alignment and Variant Detection
Sequencing of the 137 samples in 2×52 paired end sequencing runs of the Illumina GAIIx and subsequent splitting of reads based on indexes, resulted in 1.8million ± 0.3million reads passing Illumina quality filters per sample. Of these, BWA aligned 97.8 ± 0.8% to the 87kb targeted region. This resulted in an average depth of coverage of 872 ± 180× at each targeted base position. Coverage demonstrated fluctuation across the targeted region but more than 97% of all targeted bases across all samples were covered at least 50× to allow for confident variant calling (Figure 2).
Figure 2.
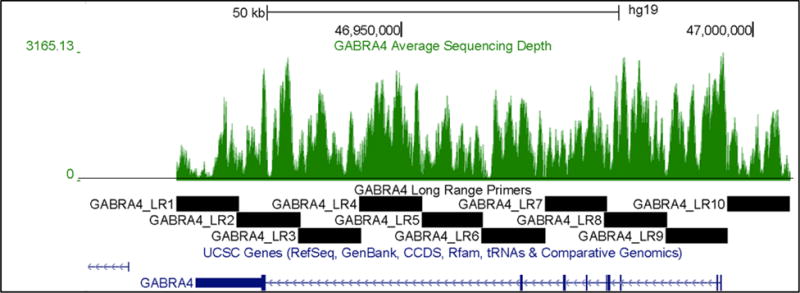
Sequence Depth Coverage Across Targeted Region. The average depth of coverage at each position across all samples is indicated in a green histogram with a black line representing 50× coverage on a UCSC Genome Browser track. The black bars represent the 10 amplicons used to target the region.
We identified 387 single nucleotide variants (SNVs) (Online Resource 2) and 52 short insertion-deletions (indels) (Online Resource 3) across all individuals. All individuals sequenced were genotyped in a previous GWAS study [10] and 36 markers from that study are in targeted region. Overall concordance of genotype calls across all samples was 98.6% suggesting highly accurate genotype calling at common variant positions as well as sample identity integrity. Of the total variants, 87 of 387 SNVs (22.4%) and 18 of 52 indels (34.6%) were not present in dbSNP138 or 1000 Genomes release 3. The average minor allele frequency of these novel variants was 0.004% while those in dbSNP were 14.1% in our sequenced population suggesting that these novel variants are novel or very rare in the overall and sequenced population. There was no difference in the average number of variants between cases and controls t-test p = 0.48 suggesting both not an overall increase in variation based on status nor by sample source (blood or saliva).
Variant Annotation
In order to assess potential functional effects of the variants identified, we annotated each variant using genomic features databases with ANNOVAR and publically available ENCODE databases. Only one variant was identified within the 1,665bp open reading frame of the gene at chr4.hg19:46995366G>T (rs2229940). It is a common variant with a MAF of 43.8% in our sequenced cases, 40% of sequenced controls, and 36.7% in the Exome Variant Server suggesting this coding variant is unlikely to be a risk allele for ASD. Therefore, we sought to determine the functional potential of the remaining noncoding variants using fetal CNS or neuronal cell-line data from the ENCODE Consortium. Overall, 25 total variants (14 rare variants with 1000 Genomes frequency < 0.05) overlapped with any ENCODE DNAse Hypersenstivity, transcription factor binding site (all were CTCF sites), or putative enhancer region as defined by H3K4me1 or H3K9ac (Online Resource 4).
Association analysis
Though at this sample size we are only powered to detect variants of large effects, odds ratio >4 at minor allele frequency of 0.2 and odds ratio > 6 at minor allele frequency 0.01, we analyzed each individual variant for association with ASD via a Fisher’s Exact test of the number of variant alleles, but none survived multiple testing correction for overall significance across all variants tested (threshold p<1.2×10−4) with a lowest p-value of 0.03.
Because aggregation of rare variants could be associated with ASD, we performed analyses with SKAT-O on sets of rare variants. First, we ran a series of analyses including all 387 SNVs and subsequently only SNVs with MAF cut-offs of 0.1 (272 SNVs), 0.05 (266 SNVs), 0.01 (198 SNVs), and 0.005 (150 SNVs). No sets demonstrated significance, with the lowest p-value being 0.118 in the set of rare SNVs with a MAF cut-off of 0.005. Second, we adapted this approach to identify potential regions within GABRA4 with sets of rare variants associated with ASD, by dividing our targeted region into windows consisting of ten SNVs overlapping by five positions. We created these windows at the same MAF cut-offs for a total of 80 (all variants), 56 (0.1 MAF cut-off), 54 (0.05 MAF), 39 (0.01MAF) and 30 (0.005 MAF) windows, but found no region of significance. Finally, because rare variants within specific functional units rather than spatially distributed could be important, we performed SKAT-O across the eight introns, the 3′UTR, 5′UTR, the proximal promoter (5kb from the transcription start site), and the 3′ regulatory region (5kb downstream of the gene). Again, no regions achieved significance.
Functional analysis of 3′UTR variants
In order to assess potential functional individual variants, we focused on those in the 3′UTR. First we utilized the most recent version of TargetScan[45] and the software MicroSNiPer[46] to identify potential disruptions or creation of miRNA binding sites, but none were found. Therefore, we identified four variants in the 3′UTR with GERP scores greater than one, suggesting evolutionary and potential conservation, and unique to our ASD cases at chr4.hg19:46929991A>G (GERP = 2.59), chr4.hg19:46926840T>C (GERP = 1.06), chr4.hg19:46922930G>T (GERP = 1.06) and chr4.hg19:46929433G>T (GERP = 1.16). Since our hypothesis is that rare variants can have a functional effect, we tested whether these variants could alter the expression of genes by cloning the GABRA4 3′UTR to a luciferase reporter. We created five constructs one containing the wild type 3′UTR and one each with the four case unique mutations. Transfection of the wild type into HEK293 cells demonstrated a significant decrease in overall firefly luminescence in comparison with transfection of pmirGLO alone. However, none of the other four constructs showed a significant difference from the wild-type in expression with only the A to C variant at chr4:46922930 showing a slight trend toward significance (p < 0.16 and p < 0.2 in two biological replicates, each of four technical replicates) (Figure 3).
Figure 3.
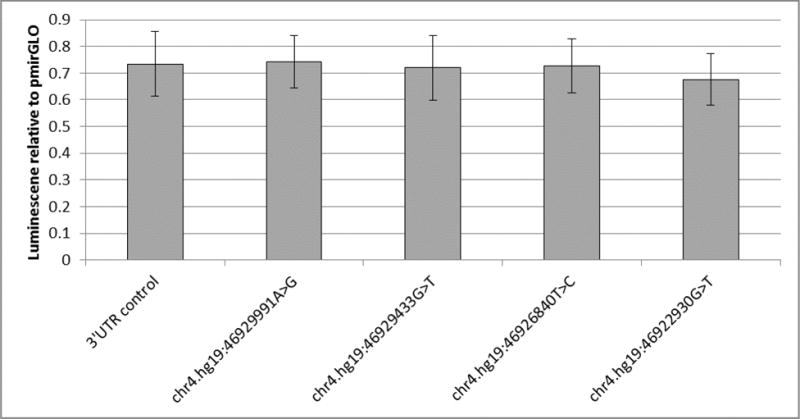
Luciferase assay for four case unique conserved variants in the 3′UTR. Four variants unique to cases and with GERP>1 were cloned into pMirGLO and their luciferase in HEK293 cells measured. Columns represent percentage of luciferase fluorescent units compared to pmirGLO transfected alone. 3′UTR control is a clone of the wild type 3′UTR and each position label is the induced variation tested. Error bars represent SEM across 8 technical replicates.
Discussion
The technological advances of massively parallel sequencing allow for comprehensive variant discovery across large and targeted genomic regions [47]. Sequencing of candidate regions has been touted as a logical follow-up to common variant association studies be they GWAS or candidate gene association approaches in complex diseases [48]. Identification and characterization of all genetic polymorphisms within an associated gene or genomic region, as done in this study for GABRA4, has the potential to identify all common genetic variants that might explain a greater proportion of contributable risk than previously genotyped markers and reveal rare variants which are not sufficiently covered by GWAS. Several post-association analysis sequencing studies have been performed on candidate genes and genomic regions for complex disorders ranging from cancer [49] to sick sinus syndrome [50] to schizophrenia [51]. Association of the G/G genotype of rs1912960 in GABRA4 with ASD by the genotype-pedigree disequilibrium test (geno-PDT) has been demonstrated in two separate family based studies with p=0.003[25] and p=0.0046[27]. As such, sequencing of individuals homozygous at chr4.hg19:g.46953881C>G (G/G at rs1912960) should enrich for discovery or rare ASD risk variants in GABRA4.
The most recent sequencing efforts in ASD have focused on exome and whole genome sequencing to identify rare and de novo mutations in several genes which can contribute to autism etiology [6–9, 52–56]. GABRA4 was not implicated by these studies as having a de novo gene disrupting mutation, nor did the whole genome studies particularly investigate the gene. As in the whole genome studies, our targeted approach offers the benefit of uncovering genetic variation beyond the exome by identifying the complete compendium of variants from exons, introns, untranslated regions, and potential up and downstream regulatory elements. In addition, this experimental design allows focus on a single gene with evidence for genetic association [25, 27], a function that could potentially modulate autism behaviors [57], and biochemical evidence for variability in expression and function in autistic patients [28–30, 58]. Moreover, this candidate gene approach allows for greater depths of coverage, greater sequencing completion rates, and a lower cost when compared to larger scale sequencing studies [59].
We identified variants at 439 sites (387 SNVs and 52 indels) across the GABRA4 region with about 22% of those variants not previously reported in large scale sequencing efforts such as the 1000 Genomes Project. These variants do not occur disproportionately in cases or controls nor are there any regions of the gene that have an association of rare variants with ASD. Therefore, we attempted a single variant analysis to identify rare functional variation in ASD cases. As only a single of these variants, and a common one, was in the coding portion of the gene, we annotated all of the variants using available genomic feature databases to identify potential noncoding functional variants. Because ASD is likely to alter neuronal development or function, we annotated our variants against functional regions identified by the ENCODE project in neuronal cell lines. These included regions of open chromatin (DNase hypersensitivity sites) where variation could alter chromatin conformation and therefore gene expression, and transcription factor binding sites, where variation could alter gene regulatory mechanisms. While 14 rare variants occurred in at least one predicted ENCODE functional domain, again there was no trend for elements specific to ASD. We identify novel variants in evolutionarily conserved and potentially functional regions of GABRA4 including those overlapping transcription factor binding sites and in the 3′UTR. We also identified four variants unique to cases with high evolutionary conservation, but none showed statistical difference in affecting gene transcription in a luciferase assay. Candidate gene analysis of genes similarly associated with ASD, NLGN3 and NLGN4X [60], have arrived at similar conclusions as those seen in this study.
In summary, we have comprehensively re-sequenced the entire autism candidate gene GABRA4 gene region, including all exons, intron, UTRs, and up and downstream potential regulatory regions, in 82 autism cases and 55 controls. While we identify novel and rare variation in the gene, there is no statistical association with any individual variant or any cluster of rare variants in the gene. While association with GABRA4 and ASD has been replicated and functional evidence points toward a role, these data suggest that individual rare variants in the gene may not play a role. Rather, there is a possibility that combinations of rare variants in this gene with others may induce risk. Alternatively, the effect of each rare variant may be undetectable in the assays used here, and future research is warranted. Overall, it is important to sequence candidate genes in larger cohorts to identify the entire spectrum of variation, including in noncoding portions of genes, to point toward the subtle functional effects each of these rare risk variants might play in a complex disease etiology of ASD.
Supplementary Material
Acknowledgments
We thank all participants and their family members in this study and personnel at the John P. Hussman Institute for Human Genomics (HIHG) specifically, Joseph Rantus, Natalia Hofmann, and Patrice Whitehead, the staff at the HIHG Biorepository, the autism ascertainment team, and the HIHG Center for Genome Technology. We also thank John P. Hussman for helpful review and discussion of this manuscript. A subset of the participants was ascertained while Dr Pericak-Vance was a faculty member at Duke University. This work was supported by the National Institutes of Health (R01MH080647, P01NS026630); and a gift from the John P. Hussman Foundation.
References
- 1.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th. American Psychiatric Publishing; Arlington, VA: 2013. [Google Scholar]
- 2.Developmental Disabilities Monitoring Network Surveillance Year 2010 Principal Investigators, Centers for Disease Control and Prevention (CDC) Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill Summ. 2014;63:1–21. [PubMed] [Google Scholar]
- 3.Kusenda M, Sebat J. The role of rare structural variants in the genetics of autism spectrum disorders. Cytogenet Genome Res. 2008;123:36–43. doi: 10.1159/000184690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Griswold AJ, Ma D, Cukier HN, Nations LD, Schmidt MA, Chung RH, Jaworski JM, Salyakina D, Konidari I, Whitehead PL, Wright HH, Abramson RK, Williams SM, Menon R, Martin ER, Haines JL, Gilbert JR, Cuccaro ML, Pericak-Vance MA. Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Hum Mol Genet. 2012;21:3513–3523. doi: 10.1093/hmg/dds164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Almeida J, Bacchelli E, Bader GD, Bailey AJ, Baird G, Battaglia A, Berney T, Bolshakova N, Bolte S, Bolton PF, Bourgeron T, Brennan S, Brian J, Bryson SE, Carson AR, Casallo G, Casey J, Chung BH, Cochrane L, Corsello C, Crawford EL, Crossett A, Cytrynbaum C, Dawson G, de Jonge M, Delorme R, Drmic I, Duketis E, Duque F, Estes A, Farrar P, Fernandez BA, Folstein SE, Fombonne E, Freitag CM, Gilbert J, Gillberg C, Glessner JT, Goldberg J, Green A, Green J, Guter SJ, Hakonarson H, Heron EA, Hill M, Holt R, Howe JL, Hughes G, Hus V, Igliozzi R, Kim C, Klauck SM, Kolevzon A, Korvatska O, Kustanovich V, Lajonchere CM, Lamb JA, Laskawiec M, Leboyer M, Le Couteur A, Leventhal BL, Lionel AC, Liu XQ, Lord C, Lotspeich L, Lund SC, Maestrini E, Mahoney W, Mantoulan C, Marshall CR, McConachie H, McDougle CJ, McGrath J, McMahon WM, Merikangas A, Migita O, Minshew NJ, Mirza GK, Munson J, Nelson SF, Noakes C, Noor A, Nygren G, Oliveira G, Papanikolaou K, Parr JR, Parrini B, Paton T, Pickles A, Pilorge M, Piven J, Ponting CP, Posey DJ, Poustka A, Poustka F, Prasad A, Ragoussis J, Renshaw K, Rickaby J, Roberts W, Roeder K, Roge B, Rutter ML, Bierut LJ, Rice JP, Salt J, Sansom K, Sato D, Segurado R, Sequeira AF, Senman L, Shah N, Sheffield VC, Soorya L, Sousa I, Stein O, Sykes N, Stoppioni V, Strawbridge C, Tancredi R, Tansey K, Thiruvahindrapduram B, Thompson AP, Thomson S, Tryfon A, Tsiantis J, Van Engeland H, Vincent JB, Volkmar F, Wallace S, Wang K, Wang Z, Wassink TH, Webber C, Weksberg R, Wing K, Wittemeyer K, Wood S, Wu J, Yaspan BL, Zurawiecki D, Zwaigenbaum L, Buxbaum JD, Cantor RM, Cook EH, Coon H, Cuccaro ML, Devlin B, Ennis S, Gallagher L, Geschwind DH, Gill M, Haines JL, Hallmayer J, Miller J, Monaco AP, Nurnberger JI, Jr, Paterson AD, Pericak-Vance MA, Schellenberg GD, Szatmari P, Vicente AM, Vieland VJ, Wijsman EM, Scherer SW, Sutcliffe JS, Betancur C. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C, Rieder MJ, Nickerson DA, Bernier R, Fisher SE, Shendure J, Eichler EE. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, Malig M, Baker C, Reilly B, Akey JM, Borenstein E, Rieder MJ, Nickerson DA, Bernier R, Shendure J, Eichler EE. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, Dilullo NM, Parikshak NN, Stein JL, Walker MF, Ober GT, Teran NA, Song Y, El-Fishawy P, Murtha RC, Choi M, Overton JD, Bjornson RD, Carriero NJ, Meyer KA, Bilguvar K, Mane SM, Sestan N, Lifton RP, Gunel M, Roeder K, Geschwind DH, Devlin B, State MW. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, Kendall J, Grabowska E, Ma B, Marks S, Rodgers L, Stepansky A, Troge J, Andrews P, Bekritsky M, Pradhan K, Ghiban E, Kramer M, Parla J, Demeter R, Fulton LL, Fulton RS, Magrini VJ, Ye K, Darnell JC, Darnell RB, Mardis ER, Wilson RK, Schatz MC, McCombie WR, Wigler M. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma DQ, Salyakina D, Jaworski JM, Konidari I, Whitehead PL, Andersen A, Hoffman JD, Slifer SH, Hedges DJ, Cukier H, Beecham GW, 4d321F92AWright HHA,R.K. Martin ER, Hussman JP, Gilbert JR, Cuccaro ML, Haines JL, Pericak-Vance MA. A genome-wide association study of autism reveals a common novel risk locus at 5p14.1. Annals of Human Genetics. 2009;73:263–73. doi: 10.1111/j.1469-1809.2009.00523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS, Salyakina D, Imielinski M, Bradfield JP, Sleiman PM, Kim CE, Hou C, Frackelton E, Chiavacci R, Takahashi N, Sakurai T, Rappaport E, Lajonchere CM, Munson J, Estes A, Korvatska O, Piven J, Sonnenblick LI, Alvarez Retuerto AI, Herman EI, Dong H, Hutman T, Sigman M, Ozonoff S, Klin A, Owley T, Sweeney JA, Brune CW, Cantor RM, Bernier R, Gilbert JR, Cuccaro ML, McMahon WM, Miller J, State MW, Wassink TH, Coon H, Levy SE, Schultz RT, Nurnberger JI, Haines JL, Sutcliffe JS, Cook EH, Minshew NJ, Buxbaum JD, Dawson G, Grant SF, Geschwind DH, Pericak-Vance MA, Schellenberg GD, Hakonarson H. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009;459:528–533. doi: 10.1038/nature07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anney R, Klei L, Pinto D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Sykes N, Pagnamenta AT, Almeida J, Bacchelli E, Bailey AJ, Baird G, Battaglia A, Berney T, Bolshakova N, Bolte S, Bolton PF, Bourgeron T, Brennan S, Brian J, Carson AR, Casallo G, Casey J, Chu SH, Cochrane L, Corsello C, Crawford EL, Crossett A, Dawson G, de Jonge M, Delorme R, Drmic I, Duketis E, Duque F, Estes A, Farrar P, Fernandez BA, Folstein SE, Fombonne E, Freitag CM, Gilbert J, Gillberg C, Glessner JT, Goldberg J, Green J, Guter SJ, Hakonarson H, Heron EA, Hill M, Holt R, Howe JL, Hughes G, Hus V, Igliozzi R, Kim C, Klauck SM, Kolevzon A, Korvatska O, Kustanovich V, Lajonchere CM, Lamb JA, Laskawiec M, Leboyer M, Le Couteur A, Leventhal BL, Lionel AC, Liu XQ, Lord C, Lotspeich L, Lund SC, Maestrini E, Mahoney W, Mantoulan C, Marshall CR, McConachie H, McDougle CJ, McGrath J, McMahon WM, Melhem NM, Merikangas A, Migita O, Minshew NJ, Mirza GK, Munson J, Nelson SF, Noakes C, Noor A, Nygren G, Oliveira G, Papanikolaou K, Parr JR, Parrini B, Paton T, Pickles A, Piven J, Posey DJ, Poustka A, Poustka F, Prasad A, Ragoussis J, Renshaw K, Rickaby J, Roberts W, Roeder K, Roge B, Rutter ML, Bierut LJ, Rice JP, Salt J, Sansom K, Sato D, Segurado R, Senman L, Shah N, Sheffield VC, Soorya L, Sousa I, Stoppioni V, Strawbridge C, Tancredi R, Tansey K, Thiruvahindrapduram B, Thompson AP, Thomson S, Tryfon A, Tsiantis J, Van Engeland H, Vincent JB, Volkmar F, Wallace S, Wang K, Wang Z, Wassink TH, Wing K, Wittemeyer K, Wood S, Yaspan BL, Zurawiecki D, Zwaigenbaum L, Betancur C, Buxbaum JD, Cantor RM, Cook EH, Coon H, Cuccaro ML, Gallagher L, Geschwind DH, Gill M, Haines JL, Miller J, Monaco AP, Nurnberger JI, Jr, Paterson AD, Pericak-Vance MA, Schellenberg GD, Scherer SW, Sutcliffe JS, Szatmari P, Vicente AM, Vieland VJ, Wijsman EM, Devlin B, Ennis S, Hallmayer J. A genome-wide scan for common alleles affecting risk for autism. Hum Mol Genet. 2010;19:4072–82. doi: 10.1093/hmg/ddq307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weiss LA, Arking DE, Gene Discovery Project of Johns Hopkins & the Autism Consortium. Daly MJ, Chakravarti A. A genome-wide linkage and association scan reveals novel loci for autism. Nature. 2009;461:802–808. doi: 10.1038/nature08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Betancur C. Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res. 2011;1380:42–77. doi: 10.1016/j.brainres.2010.11.078. [DOI] [PubMed] [Google Scholar]
- 15.Hirschhorn JN, Gajdos ZK. Genome-wide association studies: results from the first few years and potential implications for clinical medicine. Annu Rev Med. 2011;62:11–24. doi: 10.1146/annurev.med.091708.162036. [DOI] [PubMed] [Google Scholar]
- 16.Barker JL, Behar TN, Ma W, Maric D, Maric I. GABA emerges as a developmental signal during neurogenesis of the rat central nervous system. In: Martin DL, Olsen RW, editors. GABA in the Nervous System: The view at fifty years. Lippincott Williams and Wilkins; Philadelphia: 2000. pp. 245–263. [Google Scholar]
- 17.Hussman JP. Suppressed GABAergic inhibition as a common factor in suspected etiologies of autism. J Autism Dev Disord. 2001;31:247–248. doi: 10.1023/a:1010715619091. [DOI] [PubMed] [Google Scholar]
- 18.Blatt GJ, Fitzgerald CM, Guptill JT, Booker AB, Kemper TL, Bauman ML. Density and distribution of hippocampal neurotransmitter receptors in autism: an autoradiographic study. J Autism Dev Disord. 2001;31:537–543. doi: 10.1023/a:1013238809666. [DOI] [PubMed] [Google Scholar]
- 19.Bundey S, Hardy C, Vickers S, Kilpatrick MW, Corbett JA. Duplication of the 15q11-13 region in a patient with autism, epilepsy and ataxia. Dev Med Child Neurol. 1994;36:736–742. doi: 10.1111/j.1469-8749.1994.tb11916.x. [DOI] [PubMed] [Google Scholar]
- 20.Baker P, Piven J, Schwartz S, Patil S. Duplication of chromosome 15q11-13 in two additional individuals with Autistic Disorder. J Autism Dev Disord. 1994;24:529–535. doi: 10.1007/BF02172133. [DOI] [PubMed] [Google Scholar]
- 21.Repetto GM, White LM, Bader PJ, Johnson D, Knoll JH. Interstitial duplications of chromosome region 15q11q13: Clinical and molecular characterization. Am J Med Genet. 1998;79:82–89. doi: 10.1002/(sici)1096-8628(19980901)79:2<82::aid-ajmg2>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 22.Martin ER, Menold MM, Wolpert CM, Bass MP, Donnelly SL, Ravan SA, Zimmerman A, Gilbert JR, Vance JM, Maddox LO, Wright HH, Abramson RK, DeLong GR, Cuccaro ML, Pericak-Vance MA. Analysis of linkage disequilibrium in gamma-aminobutyric acid receptor subunit genes in autistic disorder. Am J Med Genet. 2000;96:43–48. doi: 10.1002/(sici)1096-8628(20000207)96:1<43::aid-ajmg9>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 23.Menold MM, Shao Y, Kim SJ, Wolpert CM, Donnelly SL, Ravan SA, Abramson RK, Wright HH, DeLong GR, Cuccaro ML, Pericak-Vance MA, Gilbert JR. Evidence for linkage but not association to teh GABR3 region of chromosome 15 in a subset of autistic disorder (AutD) families characterized by an increase in restricted and repetitive behaviors. Am J Hum Gen. 2002;71:488. [Google Scholar]
- 24.Buxbaum JD, Silverman JM, Smith CJ, Greenberg DA, Kilifarski M, Reichert J, Cook EH, Jr, Fang Y, Song CY, Vitale R. Association between a GABRB3 polymorphism and autism. Mol Psychiatry. 2002;7:311–316. doi: 10.1038/sj.mp.4001011. [DOI] [PubMed] [Google Scholar]
- 25.Ma DQ, Whitehead PL, Menold MM, Martin ER, Ashley-Koch AE, Mei H, Ritchie MD, DeLong GR, Abramson RK, Wright HH, Cuccaro ML, Hussman JP, Gilbert JR, Pericak-Vance MA. Identification of Significant Association and Gene-Gene Interaction of GABA Receptor Subunit Genes in Autism. Am J Hum Genet. 2005;77:377–388. doi: 10.1086/433195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Collins AL, Ma DQ, Whitehead PL, Martin ER, Wright HH, Abramson RK, Hussman JP, Gilbert JR, Cuccaro ML, Pericak-Vance MA. Association of GABRA4 and GABRB1 in African American autism families. American Society of Human Genetics 55th Annual Meeting; Salt Lake City, Utah. 2005. [Google Scholar]
- 27.Collins AL, Ma D, Whitehead PL, Martin ER, Wright HH, Abramson RK, Hussman JP, Haines JL, Cuccaro ML, Gilbert JR, Pericak-Vance MA. Investigation of autism and GABA receptor subunit genes in multiple ethnic groups. Neurogenetics. 2006;7:167–174. doi: 10.1007/s10048-006-0045-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fatemi SH, Folsom TD, Reutiman TJ, Thuras PD. Expression of GABA(B) receptors is altered in brains of subjects with autism. Cerebellum. 2009;8:64–69. doi: 10.1007/s12311-008-0075-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fatemi SH, Reutiman TJ, Folsom TD, Thuras PD. GABA(A) receptor downregulation in brains of subjects with autism. J Autism Dev Disord. 2009;39:223–230. doi: 10.1007/s10803-008-0646-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fatemi SH, Reutiman TJ, Folsom TD, Rooney RJ, Patel DH, Thuras PD. mRNA and protein levels for GABAAalpha4, alpha5, beta1 and GABABR1 receptors are altered in brains from subjects with autism. J Autism Dev Disord. 2010;40:743–750. doi: 10.1007/s10803-009-0924-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rutter M, Bailey A, Berument SK, LeCouteur A, Lord C, Pickles A. Social Communication Questionnaire (SCQ) Western Psychological Services; Los Angeles, CA: 2003. [Google Scholar]
- 32.Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007;23:1289–1291. doi: 10.1093/bioinformatics/btm091. [DOI] [PubMed] [Google Scholar]
- 33.Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. Primer3–new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang X, Wang K. wANNOVAR: annotating genetic variants for personal genomes via the web. J Med Genet. 2012;49:433–436. doi: 10.1136/jmedgenet-2012-100918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.ENCODE Project Consortium. Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, Khatun J, Lajoie BR, Landt SG, Lee BK, Pauli F, Rosenbloom KR, Sabo P, Safi A, Sanyal A, Shoresh N, Simon JM, Song L, Trinklein ND, Altshuler RC, Birney E, Brown JB, Cheng C, Djebali S, Dong X, Dunham I, Ernst J, Furey TS, Gerstein M, Giardine B, Greven M, Hardison RC, Harris RS, Herrero J, Hoffman MM, Iyer S, Kelllis M, Khatun J, Kheradpour P, Kundaje A, Lassman T, Li Q, Lin X, Marinov GK, Merkel A, Mortazavi A, Parker SC, Reddy TE, Rozowsky J, Schlesinger F, Thurman RE, Wang J, Ward LD, Whitfield TW, Wilder SP, Wu W, Xi HS, Yip KY, Zhuang J, Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M, Pazin MJ, Lowdon RF, Dillon LA, Adams LB, Kelly CJ, Zhang J, Wexler JR, Green ED, Good PJ, Feingold EA, Bernstein BE, Birney E, Crawford GE, Dekker J, Elinitski L, Farnham PJ, Gerstein M, Giddings MC, Gingeras TR, Green ED, Guigo R, Hardison RC, Hubbard TJ, Kellis M, Kent WJ, Lieb JD, Margulies EH, Myers RM, Snyder M, Starnatoyannopoulos JA, Tennebaum SA, Weng Z, White KP, Wold B, Khatun J, Yu Y, Wrobel J, Risk BA, Gunawardena HP, Kuiper HC, Maier CW, Xie L, Chen X, Giddings MC, Bernstein BE, Epstein CB, Shoresh N, Ernst J, Kheradpour P, Mikkelsen TS, Gillespie S, Goren A, Ram O, Zhang X, Wang L, Issner R, Coyne MJ, Durham T, Ku M, Truong T, Ward LD, Altshuler RC, Eaton ML, Kellis M, Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, Xue C, Marinov GK, Khatun J, Williams BA, Zaleski C, Rozowsky J, Roder M, Kokocinski F, Abdelhamid RF, Alioto T, Antoshechkin I, Baer MT, Batut P, Bell I, Bell K, Chakrabortty S, Chen X, Chrast J, Curado J, Derrien T, Drenkow J, Dumais E, Dumais J, Duttagupta R, Fastuca M, Fejes-Toth K, Ferreira P, Foissac S, Fullwood MJ, Gao H, Gonzalez D, Gordon A, Gunawardena HP, Howald C, Jha S, Johnson R, Kapranov P, King B, Kingswood C, Li G, Luo OJ, Park E, Preall JB, Presaud K, Ribeca P, Risk BA, Robyr D, Ruan X, Sammeth M, Sandu KS, Schaeffer L, See LH, Shahab A, Skancke J, Suzuki AM, Takahashi H, Tilgner H, Trout D, Walters N, Wang H, Wrobel J, Yu Y, Hayashizaki Y, Harrow J, Gerstein M, Hubbard TJ, Reymond A, Antonarakis SE, Hannon GJ, Giddings MC, Ruan Y, Wold B, Carninci P, Guigo R, Gingeras TR, Rosenbloom KR, Sloan CA, Learned K, Malladi VS, Wong MC, Barber GP, Cline MS, Dreszer TR, Heitner SG, Karolchik D, Kent WJ, Kirkup VM, Meyer LR, Long JC, Maddren M, Raney BJ, Furey TS, Song L, Grasfeder LL, Giresi PG, Lee BK, Battenhouse A, Sheffield NC, Simon JM, Showers KA, Safi A, London D, Bhinge AA, Shestak C, Schaner MR, Kim SK, Zhang ZZ, Mieczkowski PA, Mieczkowska JO, Liu Z, McDaniell RM, Ni Y, Rashid NU, Kim MJ, Adar S, Zhang Z, Wang T, Winter D, Keefe D, Birney E, Iyer VR, Lieb JD, Crawford GE, Li G, Sandhu KS, Zheng M, Wang P, Luo OJ, Shahab A, Fullwood MJ, Ruan X, Ruan Y, Myers RM, Pauli F, Williams BA, Gertz J, Marinov GK, Reddy TE, Vielmetter J, Partridge EC, Trout D, Varley KE, Gasper C, Bansal A, Pepke S, Jain P, Amrhein H, Bowling KM, Anaya M, Cross MK, King B, Muratet MA, Antoshechkin I, Newberry KM, McCue K, Nesmith AS, Fisher-Aylor KI, Pusey B, DeSalvo G, Parker SL, Balasubramanian S, Davis NS, Meadows SK, Eggleston T, Gunter C, Newberry JS, Levy SE, Absher DM, Mortazavi A, Wong WH, Wold B, Blow MJ, Visel A, Pennachio LA, Elnitski L, Margulies EH, Parker SC, Petrykowska HM, Abyzov A, Aken B, Barrell D, Barson G, Berry A, Bignell A, Boychenko V, Bussotti G, Chrast J, Davidson C, Derrien T, Despacio-Reyes G, Diekhans M, Ezkurdia I, Frankish A, Gilbert J, Gonzalez JM, Griffiths E, Harte R, Hendrix DA, Howald C, Hunt T, Jungreis I, Kay M, Khurana E, Kokocinski F, Leng J, Lin MF, Loveland J, Lu Z, Manthravadi D, Mariotti M, Mudge J, Mukherjee G, Notredame C, Pei B, Rodriguez JM, Saunders G, Sboner A, Searle S, Sisu C, Snow C, Steward C, Tanzer A, Tapanan E, Tress ML, van Baren MJ, Walters N, Washieti S, Wilming L, Zadissa A, Zhengdong Z, Brent M, Haussler D, Kellis M, Valencia A, Gerstein M, Raymond A, Guigo R, Harrow J, Hubbard TJ, Landt SG, Frietze S, Abyzov A, Addleman N, Alexander RP, Auerbach RK, Balasubramanian S, Bettinger K, Bhardwaj N, Boyle AP, Cao AR, Cayting P, Charos A, Cheng Y, Cheng C, Eastman C, Euskirchen G, Fleming JD, Grubert F, Habegger L, Hariharan M, Harmanci A, Iyenger S, Jin VX, Karczewski KJ, Kasowski M, Lacroute P, Lam H, Larnarre-Vincent N, Leng J, Lian J, Lindahl-Allen M, Min R, Miotto B, Monahan H, Moqtaderi Z, Mu XJ, O’Geen H, Ouyang Z, Patacsil D, Pei B, Raha D, Ramirez L, Reed B, Rozowsky J, Sboner A, Shi M, Sisu C, Slifer T, Witt H, Wu L, Xu X, Yan KK, Yang X, Yip KY, Zhang Z, Struhl K, Weissman SM, Gerstein M, Farnham PJ, Snyder M, Tenebaum SA, Penalva LO, Doyle F, Karmakar S, Landt SG, Bhanvadia RR, Choudhury A, Domanus M, Ma L, Moran J, Patacsil D, Slifer T, Victorsen A, Yang X, Snyder M, White KP, Auer T, Centarin L, Eichenlaub M, Gruhl F, Heerman S, Hoeckendorf B, Inoue D, Kellner T, Kirchmaier S, Mueller C, Reinhardt R, Schertel L, Schneider S, Sinn R, Wittbrodt B, Wittbrodt J, Weng Z, Whitfield TW, Wang J, Collins PJ, Aldred SF, Trinklein ND, Partridge EC, Myers RM, Dekker J, Jain G, Lajoie BR, Sanyal A, Balasundaram G, Bates DL, Byron R, Canfield TK, Diegel MJ, Dunn D, Ebersol AK, Ebersol AK, Frum T, Garg K, Gist E, Hansen RS, Boatman L, Haugen E, Humbert R, Jain G, Johnson AK, Johnson EM, Kutyavin TM, Lajoie BR, Lee K, Lotakis D, Maurano MT, Neph SJ, Neri FV, Nguyen ED, Qu H, Reynolds AP, Roach V, Rynes E, Sabo P, Sanchez ME, Sandstrom RS, Sanyal A, Shafer AO, Stergachis AB, Thomas S, Thurman RE, Vernot B, Vierstra J, Vong S, Wang H, Weaver MA, Yan Y, Zhang M, Akey JA, Bender M, Dorschner MO, Groudine M, MacCoss MJ, Navas P, Stamatoyannopoulos G, Kaul R, Dekker J, Stamatoyannopoulos JA, Dunham I, Beal K, Brazma A, Flicek P, Herrero J, Johnson N, Keefe D, Lukk M, Luscombe NM, Sobral D, Vaquerizas JM, Wilder SP, Batzoglou S, Sidow A, Hussami N, Kyriazopoulou-Panagiotopoulou S, Libbrecht MW, Schaub MA, Kundaje A, Hardison RC, Miller W, Giardine B, Harris RS, Wu W, Bickel PJ, Banfai B, Boley NP, Brown JB, Huang H, Li Q, Li JJ, Noble WS, Bilmes JA, Buske OJ, Hoffman MM, Sahu AO, Kharchenko PV, Park PJ, Baker D, Taylor J, Weng Z, Iyer S, Dong X, Greven M, Lin X, Wang J, Xi HS, Zhuang J, Gerstein M, Alexander RP, Balasubramanian S, Cheng C, Harmanci A, Lochovsky L, Min R, Mu XJ, Rozowsky J, Yan KK, Yip KY, Birney E. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson JL, Abecasis GR. GAS Power Calculator: web-based power calculator for genetic association studies. bioRxiv. 2017 doi: 10.1101/164343. [DOI] [Google Scholar]
- 42.Skol AD, Scott LJ, Abecasis GR, Boehnke M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet. 2006;38:209–213. doi: 10.1038/ng1706. [DOI] [PubMed] [Google Scholar]
- 43.Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. 2011;89:82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee S, Emond MJ, Bamshad MJ, Barnes KC, Rieder MJ, Nickerson DA, NHLBI GO Exome Sequencing Project-ESP Lung Project Team. Christiani DC, Wurfel MM, Lin X. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet. 2012;91:224–237. doi: 10.1016/j.ajhg.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barenboim M, Zoltick BJ, Guo Y, Weinberger DR. MicroSNiPer: a web tool for prediction of SNP effects on putative microRNA targets. Hum Mutat. 2010;31:1223–1232. doi: 10.1002/humu.21349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 48.Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet. 2010;11:415–425. doi: 10.1038/nrg2779. [DOI] [PubMed] [Google Scholar]
- 49.Yeager M, Deng Z, Boland J, Matthews C, Bacior J, Lonsberry V, Hutchinson A, Burdett LA, Qi L, Jacobs KB, Gonzalez-Bosquet J, Berndt SI, Hayes RB, Hoover RN, Thomas G, Hunter DJ, Dean M, Chanock SJ. Comprehensive resequence analysis of a 97 kb region of chromosome 10q11.2 containing the MSMB gene associated with prostate cancer. Hum Genet. 2009;126:743–750. doi: 10.1007/s00439-009-0723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Holm H, Gudbjartsson DF, Sulem P, Masson G, Helgadottir HT, Zanon C, Magnusson OT, Helgason A, Saemundsdottir J, Gylfason A, Stefansdottir H, Gretarsdottir S, Matthiasson SE, Thorgeirsson GM, Jonasdottir A, Sigurdsson A, Stefansson H, Werge T, Rafnar T, Kiemeney LA, Parvez B, Muhammad R, Roden DM, Darbar D, Thorleifsson G, Walters GB, Kong A, Thorsteinsdottir U, Arnar DO, Stefansson K. A rare variant in MYH6 is associated with high risk of sick sinus syndrome. Nat Genet. 2011;43:316–320. doi: 10.1038/ng.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dow DJ, Huxley-Jones J, Hall JM, Francks C, Maycox PR, Kew JN, Gloger IS, Mehta NA, Kelly FM, Muglia P, Breen G, Jugurnauth S, Pederoso I, St Clair D, Rujescu D, Barnes MR. ADAMTSL3 as a candidate gene for schizophrenia: gene sequencing and ultra-high density association analysis by imputation. Schizophr Res. 2011;127:28–34. doi: 10.1016/j.schres.2010.12.009. [DOI] [PubMed] [Google Scholar]
- 52.Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, Smith JD, Paeper B, Nickerson DA, Dea J, Dong S, Gonzalez LE, Mandell JD, Mane SM, Murtha MT, Sullivan CA, Walker MF, Waqar Z, Wei L, Willsey AJ, Yamrom B, Lee YH, Grabowska E, Dalkic E, Wang Z, Marks S, Andrews P, Leotta A, Kendall J, Hakker I, Rosenbaum J, Ma B, Rodgers L, Troge J, Narzisi G, Yoon S, Schatz MC, Ye K, McCombie WR, Shendure J, Eichler EE, State MW, Wigler M. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, Singh T, Klei L, Kosmicki J, Shih-Chen F, Aleksic B, Biscaldi M, Bolton PF, Brownfeld JM, Cai J, Campbell NG, Carracedo A, Chahrour MH, Chiocchetti AG, Coon H, Crawford EL, Curran SR, Dawson G, Duketis E, Fernandez BA, Gallagher L, Geller E, Guter SJ, Hill RS, Ionita-Laza J, Jimenz Gonzalez P, Kilpinen H, Klauck SM, Kolevzon A, Lee I, Lei I, Lei J, Lehtimaki T, Lin CF, Ma’ayan A, Marshall CR, McInnes AL, Neale B, Owen MJ, Ozaki N, Parellada M, Parr JR, Purcell S, Puura K, Rajagopalan D, Rehnstrom K, Reichenberg A, Sabo A, Sachse M, Sanders SJ, Schafer C, Schulte-Ruther M, Skuse D, Stevens C, Szatmari P, Tammimies K, Valladares O, Voran A, Li-San W, Weiss LA, Willsey AJ, Yu TW, Yuen RK, DDD Study, Homozygosity Mapping Collaborative for Autism, UK10K Consortium. Cook EH, Freitag CM, Gill M, Hultman CM, Lehner T, Palotie A, Schellenberg GD, Sklar P, State MW, Sutcliffe JS, Walsh CA, Scherer SW, Zwick ME, Barett JC, Cutler DJ, Roeder K, Devlin B, Daly MJ, Buxbaum JD. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, Goldberg AP, Jinlu C, Keaney JF, 3rd, Klei L, Mandell JD, Moreno-De-Luca D, Poultney CS, Robinson EB, Smith L, Solli-Nowlan T, Su MY, Teran NA, Walker MF, Werling DM, Beaudet AL, Cantor RM, Fombonne E, Geschwind DH, Grice DE, Lord C, Lowe JK, Mane SM, Martin DM, Morrow EM, Talkowski ME, Sutcliffe JS, Walsh CA, Yu TW, Autism Sequencing Consortium. Ledbetter DH, Martin CL, Cook EH, Buxbaum JD, Daly MJ, Devlin B, Roeder K, State MW. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron. 2015;87:1215–1233. doi: 10.1016/j.neuron.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yuen RK, Thiruvahindrapuram B, Merico D, Walker S, Tammimies K, Hoang N, Chrysler C, Nalpathamkalam T, Pellecchia G, Liu Y, Gazzellone MJ, D’Abate L, Deneault E, Howe JL, Liu RS, Thompson A, Zarrei M, Uddin M, Marshall CR, Ring RH, Zwaigenbaum L, Ray PN, Weksberg R, Carter MT, Fernandez BA, Roberts W, Szatmari P, Scherer SW. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat Med. 2015;21:185–191. doi: 10.1038/nm.3792. [DOI] [PubMed] [Google Scholar]
- 56.C Yuen RK, Merico D, Bookman M, L Howe J, Thiruvahindrapuram B, Patel RV, Whitney J, Deflaux N, Bingham J, Wang Z, Pellecchia G, Buchanan JA, Walker S, Marshall CR, Uddin M, Zarrei M, Deneault E, D’Abate L, Chan AJ, Koyanagi S, Paton T, Pereira SL, Hoang N, Engchuan W, Higginbotham EJ, Ho K, Lamoureux S, Li W, MacDonald JR, Nalpathamkalam T, Sung WW, Tsoi FJ, Wei J, Xu L, Tasse AM, Kirby E, Van Etten W, Twigger S, Roberts W, Drmic I, Jilderda S, Modi BM, Kellam B, Szego M, Cytrynbaum C, Weksberg R, Zwaigenbaum L, Woodbury-Smith M, Brian J, Senman L, Iaboni A, Doyle-Thomas K, Thompson A, Chrysler C, Leef J, Savion-Lemieux T, Smith IM, Liu X, Nicolson R, Seifer V, Fedele A, Cook EH, Dager S, Estes A, Gallagher L, Malow BA, Parr JR, Spence SJ, Vorstman J, Frey BJ, Robinson JT, Strug LJ, Fernandez BA, Elsabbagh M, Carter MT, Hallmayer J, Knoppers BM, Anagnostou E, Szatmari P, Ring RH, Glazer D, Pletcher MT, Scherer SW. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci. 2017;20:602–611. doi: 10.1038/nn.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, Ekker M, Rubenstein JL, Noebels JL, Rosenmund C, Zoghbi HY. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oblak AL, Gibbs TT, Blatt GJ. Decreased GABA(B) Receptors in the Cingulate Cortex and Fusiform Gyrus in Autism. J Neurochem. 2010;114:1414–1423. doi: 10.1111/j.1471-4159.2010.06858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Johansson H, Isaksson M, Sorqvist EF, Roos F, Stenberg J, Sjoblom T, Botling J, Micke P, Edlund K, Fredriksson S, Kultima HG, Ericsson O, Nilsson M. Targeted resequencing of candidate genes using selector probes. Nucleic Acids Res. 2011;39:e8. doi: 10.1093/nar/gkq1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Steinberg KM, Ramachandran D, Patel VC, Shetty AC, Cutler DJ, Zwick ME. Identification of rare X-linked neuroligin variants by massively parallel sequencing in males with autism spectrum disorder. Mol Autism. 2012;3:8-2392-3-8. doi: 10.1186/2040-2392-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.