

Abstract
Chemotherapy-induced peripheral neuropathy (CIPN) is a common adverse effect experienced by cancer patients receiving treatment with paclitaxel. The voltage-gated sodium channel 1.7 (Nav1.7) plays an important role in multiple preclinical models of neuropathic pain and in inherited human pain phenotypes, and its gene expression is increased in dorsal root ganglia (DRGs) of paclitaxel-treated rats. Hence, the potential of change in the expression and function of Nav1.7 protein in DRGs from male rats with paclitaxel-related CIPN and from male and female humans with cancer-related neuropathic pain was tested here. Double immunofluorescence in CIPN rats showed that Nav1.7 was upregulated in small DRG neuron somata, especially those also expressing calcitonin gene-related peptide (CGRP), and in central processes of these cells in the superficial spinal dorsal horn. Whole-cell patch-clamp recordings in rat DRG neurons revealed that paclitaxel induced an enhancement of ProTx II (a selective Nav1.7 channel blocker)-sensitive sodium currents. Bath-applied ProTx II suppressed spontaneous action potentials in DRG neurons occurring in rats with CIPN, while intrathecal injection of ProTx II significantly attenuated behavioral signs of CIPN. Complementarily, DRG neurons isolated from segments where patients had a history of neuropathic pain also showed electrophysiological and immunofluorescence results indicating an increased expression of Nav1.7 associated with spontaneous activity. Nav1.7 was also colocalized in human cells expressing transient receptor potential vanilloid 1 and CGRP. Furthermore, ProTx II decreased firing frequency in human DRGs with spontaneous action potentials. This study suggests that Nav1.7 may provide a potential new target for the treatment of neuropathic pain, including chemotherapy (paclitaxel)-induced neuropathic pain.
SIGNIFICANCE STATEMENT This work demonstrates that the expression and function of the voltage-gated sodium channel Nav1.7 are increased in a preclinical model of chemotherapy-induced peripheral neuropathy (CIPN), the most common treatment-limiting side effect of all the most common anticancer therapies. This is key as gain-of-function mutations in human Nav1.7 recapitulate both the distribution and pain percept as shown by CIPN patients. This work also shows that Nav1.7 is increased in human DRG neurons only in dermatomes where patients are experiencing acquired neuropathic pain symptoms. This work therefore has major translational impact, indicating an important novel therapeutic avenue for neuropathic pain as a class.
Keywords: cancer, pain, patch clamp, spontaneous activity
Introduction
Paclitaxel is a chemotherapeutic agent originally derived from the bark of the Western yew tree, Taxus brevifolia, with activity against several of the most common cancers, including those of the breast, ovary, and lung (Hagiwara and Sunada, 2004). Paclitaxel exerts its antitumor effects by promoting hyperstabilized microtubule assemblies (Scripture et al., 2006). Chemotherapy-induced peripheral neuropathy (CIPN) is the most commonly reported neurotoxic and dose-limiting side effect of paclitaxel, thereby affecting survival in many cancer patients; and its persistence in survivors negatively affects quality of life and rehabilitation (Chaudhry et al., 1994; Kosturakis et al., 2014; Li et al., 2015). The mechanism of paclitaxel-related CIPN remains to be fully defined, and this has limited the identification of effective therapies for its relief or prevention. However, the recent observations that CIPN is associated with the development of ectopic spontaneous activity (SA) in dorsal root ganglion (DRG) somata that itself appears to be related to changes in the expression of multiple cell membrane ion channels may indicate potential therapeutic avenues (Zhang and Dougherty, 2014; Li et al., 2017). Among these, real-time PCR ion channel microarrays in CIPN rats revealed a significant increase in the expression of the SCN9A gene, which encodes voltage-gated sodium channel 1.7 (Nav1.7) in the DRG 14 d following paclitaxel treatment (Zhang and Dougherty, 2014).
Voltage-gated sodium channels, including Nav1.7, are expressed in peripheral sensory neurons associated with pain sensation, and the expression and functional properties of some of these sodium channels in peripheral sensory neurons can be dynamically regulated after axonal injury or peripheral inflammation (Gold et al., 1996; Cox et al., 2006; Cummins et al., 2007). Nav1.7, one of seven sodium channel subtypes distinctly found in the peripheral nervous system, is tetrodotoxin sensitive (TTX-S) and is expressed in both large- and small-diameter DRG neurons including most of those functionally identified as nociceptors (Black et al., 2012). Nav1.7 has the potential to be a primary target for CIPN due to several associations that have been found in relation to pain phenotypes. Loss-of-function mutations in SCN9A result in congenital insensitivity to pain (Cox et al., 2006; Goldberg et al., 2007). In contrast, gain-of-function mutations underlie chronic pain in inherited erythromelalgia, paroxysmal extreme pain disorder, and chronic painful peripheral neuropathy (Yang et al., 2004; Cox et al., 2006; Fertleman et al., 2006; Han et al., 2006; Faber et al., 2012; Huang et al., 2014a). Furthermore, animal studies have revealed that Nav1.7 expression in DRG neurons, and accordingly pain, can be increased and decreased, respectively, with various stimulation and knock-down strategies (Dib-Hajj et al., 2013). The goal in this project, therefore, was to test the hypothesis that changes in the expression and/or function of Nav1.7 in DRG neurons contributes to paclitaxel CIPN in rodents. As well, given that a number of potential analgesics identified in preclinical studies have failed to translate into effective and safe clinical treatments (Lázár et al., 2009), it was tested whether an increase in the expression and/or function of Nav1.7 accompanies the development of hyperexcitability and spontaneous activity in human DRG neurons from patients with a history of painful neuropathy.
Materials and Methods
Animals.
All experimental protocols were approved by the Institutional Animal Care and Use Committee at The University of Texas MD Anderson Cancer Center and were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All procedures were designed to minimize animal discomfort and to use the fewest animals possible for statistical analysis.
A total of 140 Male Sprague Dawley (Harlan) rats were housed in temperature- and light-controlled (12 h light/dark cycle) conditions with food and water available ad libitum. A total of 132 young adult rats weighing 250–300 g were used in the behavioral studies followed by biochemical, immunohistochemical, or neurophysiological studies, while eight 2-week-old rats were used for cell culture experiments.
Paclitaxel-induced neuropathy model.
The paclitaxel-induced neuropathy model was produced as previously described by our group (Zhang et al., 2013), based on the protocol described by Polomano et al. (2001). In the most basic model, pharmaceutical-grade paclitaxel (Taxol, Teva Pharmaceuticals) was diluted with sterile saline from the original stock concentration of 6 mg/ml (in 1:1 Cremophor EL/ethanol) to 1 mg/ml and given to the rats at 2 mg/kg, i.p., every other day for a total of four injections (days 1, 3, 5, and 7), resulting in a final cumulative dose of 8 mg/kg. Equal numbers of control animals received an equivalent volume of vehicle only, which consisted of Cremophor EL/ethanol diluted with saline to reach a concentration equivalent to the paclitaxel solution. This model was varied as described below to test for the effects of interventions to prevent or reverse change in mechanical withdrawal threshold.
Mechanical withdrawal threshold was tested before, during, and after treatment with paclitaxel by an individual blinded to the treatment groups during the mid-light hours (10:00 A.M. to 5:00 P.M.). The 50% paw withdrawal threshold in response to a series of eight Von Frey hairs (0.41–15.10 g) was examined using the up-down method, as described previously (Chaplan et al., 1994), beginning with a filament having a bending force of 2 g. The animals were placed in clear acrylic cages atop a wire mesh floor and allowed to acclimate for 30 min. The filaments were applied to a paw just below the pads with no acceleration at a force just sufficient to produce a bend and held for 6–8 s. A quick flick or full withdrawal of the paw was considered to be a response.
For studies wherein intrathecal agents were administered (see below), motor performance was also assessed using rotarod performance as the endpoint (Norreel et al., 2003; O'Connor et al., 2003). The rotation velocity was set to increase from 4 to 40 rpm over 2 min. The period of time, in seconds, at which the animal fell from the rod was recorded. For those animals that did not fall, a cutoff limit of 120 s was used, after which they were removed from the rotating wheel.
Dosing regimen for ProTx II and lipopolysaccharide from Rhodobacter sphaeroides to prevent/reverse CIPN.
The tarantula venom peptide ProTx II, a selective Nav1.7 channel blocker (Schmalhofer et al., 2008), was used to test whether this may have an effect on preventing and/or reversing paclitaxel-induced CIPN. In testing whether ProTxII could prevent CIPN, 30 rats (6 per group) were treated with ProTx II beginning 2 d before and then daily through 2 d after paclitaxel treatment. ProTx II (15 or 60 μg in 20 μl of PBS, Tocris Bioscience) or PBS alone (InvivoGen) was given by intrathecal injection. To assess the role of Nav1.7 in maintaining paclitaxel-induced neuropathic pain, 20 rats (5 per group) were given ProTx II (60 μg in 20 μl of PBS) or PBS was given by intrathecal injection on day 14 after confirmation of paclitaxel-induced mechanical hypersensitivity. The ProTx II dose was chosen on the basis of previously published studies (Schmalhofer et al., 2008). Lipopolysaccharide from Rhodobacter sphaeroides (LPS-RS) was administered as previously described, and then the L4 and L5 DRGs and lumbar dorsal horn of spinal cord were collected at days 7 and 14 (Li et al., 2014b).
Immunohistochemical analysis.
Ten rats (5 for each group) were deeply anesthetized with Beuthanasia-D® (130 mg/kg pentobarbital plus 17 mg/kg phenytoin) and perfused through the ascending aorta with warm saline followed by cold 4% paraformaldehyde in 0.1 m phosphate buffer. The L4 and L5 DRGs were removed, fixed in 4% paraformaldehyde for 6 h, and then cryoprotected in a 20% then a 30% sucrose solution. The L4 and L5 spinal cord segments and a portion of cerebellar cortex were also removed, fixed in 4% paraformaldehyde for 12 h, and then cryoprotected in a 20% and then a 30% sucrose solution. Transverse spinal cord sections (15 μm), longitudinal DRG sections (8 μm), and longitudinal cerebellum sections (30 μm) were cut in a cryostat. DRG sections were mounted on gelatin-coated glass slides (Southern Biotech), while spinal cord and cerebellum sections were collected in wells with PBS solution. After blocking in 5% normal donkey serum and 0.2% Triton X-100 in PBS for 1 h at room temperature, the sections were incubated overnight at 4°C in 1% normal donkey serum and 0.2% Triton X-100 in PBS containing primary antibodies against the following targets: Nav1.7 (rabbit anti-rat, 1:500; catalog #ASC-008, Alomone Labs); IB4 (1:1000; BS-Isolectin B4 FITC Conjugate, Sigma-Aldrich); calcitonin gene-related peptide (CGRP; mouse anti-rat, 1:1000; Abcam); NeuN (mouse anti-rat, 1:1000; Millipore); glial fibrillary acid protein (GFAP; mouse anti-rat, 1:1000; Cell Signaling Technology); and OX-42 (mouse anti-rat, 1:1000; Serotec). A subset of DRG and spinal cord sections had an excess of the Nav1.7 epitope control protein added for absorption control. After the sections were washed, they were incubated with Cy3-, Cy5-, or FITC-conjugated secondary antibodies overnight at 4°C. The total numbers of cells, the numbers of cells positive for Nav1.7 alone, and the numbers of cells showing colocalization with IB4 or CGRP were counted in three sections of DRGs from each rat. For a given experiment, all images were taken using identical acquisition parameters by experimenters blinded to treatment groups.
Western blot analysis.
L4 and L5 DRGs were collected from 4 rats in each treatment group (56 rats total) that had been deeply anesthetized with sodium pentobarbital plus phenytoin as described above. The samples were snap frozen in liquid nitrogen. Tissues were later disrupted in neuronal protein extraction reagent (Thermo Fisher Scientific) mixed with 1 mm dithiothreitol, protease inhibitor cocktail (P8340, Sigma-Aldrich), and phosphatase inhibitor cocktails (P0044 and P5726, Sigma-Aldrich) on ice for 1 h, and then centrifuged at 4°C, 12,000 × g for 10 min. The supernatant was then transferred into new tubes and denatured with sample buffer (Laemmli 2× Concentrate, Sigma-Aldrich) for 10 min at 70°C. Lysates (total protein, 20 μg) were separated using SDS-PAGE and transferred to polyvinylidene fluoride membranes (Bio-Rad). After blocking with 5% fat-free milk in Tris-buffered saline with Tween (TBST; 137 mm sodium chloride, 20 mm Tris, 0.1% Tween-20) for 1 h at room temperature, the membranes were incubated with Nav1.7 (rabbit anti-rat, 1:2000; catalog #ASC-008, Alomone Labs) or β-actin antisera (mouse anti-rat, 1:10,000; Sigma-Aldrich) in 5% fat-free milk in TBST overnight at 4°C. After washing with TBST, the membranes were incubated with goat anti-rabbit or goat anti-mouse antibody labeled with horseradish peroxidase (EMD Millipore) diluted with 5% fat-free milk in TBST for 1 h at room temperature, and then the reactive bands were detected with enhanced chemiluminescence reagents (GE Healthcare). The blots were scanned, and the band densities were detected and compared with the use of ImageJ (NIH, Bethesda, MD). Statistical comparisons were made using densities of the individual Nav1.7 bands normalized to their respective matching β-actin bands.
Acute dissociation of rat DRG neurons.
Sixteen rats were deeply anesthetized with pentobarbital/phenytoin, as described above, and the bilateral L4 and L5 ganglia were surgically exposed and removed. The ganglia were placed in a flask containing trypsin (0.0625 mg/ml; Hyclone) and type IA collagenase (1 mg/ml, Sigma-Aldrich) in DMEM and shaken for 50 min in a heated (37°C) bath. After the cells were washed and mechanically dispersed with a fire-polished Pasteur pipette, they were plated on poly-l-lysine-coated glass sheets and held in culture dishes with DMEM (10% FBS) until use within 6 h after plating (Li et al., 2014a).
Whole-cell recording of sodium currents in acutely dissociated DRG neurons.
Six paclitaxel- and six vehicle-treated rats (12 of the 16 noted above) were used in whole-cell patch-clamp recording to assess sodium current function using previously described protocols (Gold et al., 2003; Zhang et al., 2017). Two hours after plating, dissociated DRG neurons were transferred to a recording chamber perfused with an extracellular solution containing the following (in mm): 35 NaCl, 30 tetraethylammonium-Cl, 65 choline-Cl, 0.1 CaCl2, 5 MgCl2, 10 HEPES, and 10 glucose, with pH adjusted to 7.4 with Tris-base, and osmolarity adjusted to 325 mOsm with sucrose. The recording electrode was filled with a solution containing the following (in mm): 100 Cs methanesulfonate, 40 tetraethylammonium-Cl, 5 NaCl, 1 CaCl2, 2 MgCl2, 11 EGTA, 10 HEPES, 2 Mg-ATP, and 1 Li-GTP, with pH adjusted to 7.2 with Tris-base, and osmolarity adjusted to 310 mOsm with sucrose. Series resistance (Rs) was compensated to >70% for all recorded neurons. A P/4 protocol was used for on-line leak subtractions. Capacitance and series resistance were calculated after subtraction of the capacitive transients. Data were excluded in which the holding current was >500 pA. Nav1.7 current was isolated from total Na+ currents by subtraction of the ProTx II-resistant Na+ currents from total current using a previously published subtraction protocol (Gold et al., 2003; Zhang et al., 2017). Neurons were held at −90 mV, and activation was evoked with a 15 ms step to potentials ranging from −90 through +45 mV in 5 mV increments before and after ProTx II administration. Current density was calculated by normalizing maximal peak currents with cell capacitance. The steady-state inactivation current was assessed with a 1 s prepulse from −100 to +10 mV followed by a voltage step to 0 mV. The voltage dependencies of activation and voltage dependence of inactivation were recorded and calculated using single Boltzmann distributions (Gold et al., 2003; Zhang et al., 2017).
Whole-cell recording of spontaneous activity and treatment with ProTx II in acutely dissociated DRG neurons.
Whole-cell patch clamp to record SA was performed in four paclitaxel-treated rats (those remaining that were used to prepare dissociated DRG neurons), as previously described (Li et al., 2017). Briefly, 2 h after plating, dissociated DRG neurons were transferred to a recording chamber perfused with extracellular solution containing 140 mm NaCl, 5 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 10 mm HEPES, and 11 mm glucose adjusted to pH 7.4 with NaOH. Glass micropipettes were filled with an internal solution of 135 mm K-gluconate, 5 mm KCl, 5 mm Mg-ATP, 0.5 mm Na2GTP, 5 mm HEPES, 2 mm MgCl2, 5 mm EGTA, and 0.5 mm CaCl2 adjusted to pH 7.4 with KOH. DRG neurons were held at 0 pA, and the action potentials were evoked using a series of 300 ms depolarizing current injections in 10 pA steps from −50 pA. The current that induced the first action potential was defined as 1× rheobase. Only neurons with a resting membrane potential of at least −40 mV, stable baseline recordings, and evoked spikes that overshot 0 mV were used for further experiments and analysis. Rs was compensated to >70%. All recordings were made at room temperature. Recordings began 5 min after whole-cell access was established, and the condition of cells had reached a steady state. DRG neurons were monitored for spontaneous action potentials for at least 2 min. If SA was detected, an infusion of ProTx II was added to the extracellular bath to achieve a concentration of 5 nm. The infusion time to reach the target concentration based on the volume of the recording dish and infusion rate was ∼2 min. ProTx II was continually infused for 10 min or until cessation of SA for >2 min, whichever occurred first, and then the infusion was stopped. Neurons were then monitored for the return of SA during the washout of ProTx II.
Primary culture of rat DRG neurons.
Eight 2-week-old rats were decapitated after anesthesia with Beuthanasia-D (see above, Immunohistochemical analysis). The spinal column was removed, and the DRGs of both sides were removed from cervical, thoracic, and lumbar levels. The ganglia were placed in DMEM with type I collagenase (3 mg/ml; Sigma-Aldrich) in a heated (37°C) chamber for 45 min. After washing in PBS, the DRG were placed in fresh DMEM with trypsin (0.25%; Sigma-Aldrich) for 10 min in the same heated chamber. Digestion was extinguished by the addition of fetal bovine serum, and the cells were spun at 180 × g for 5 min. The cells were resuspended in fresh DMEM containing fetal bovine serum and triturated using a polished Pasteur pipette then plated on poly-l-lysine-coated 35 mm culture dishes (Li et al., 2014a). The cells from two rats were combined and then distributed to six wells, one for each treatment group, and four replicates were performed. Pharmaceutical-grade paclitaxel (or vehicle) was diluted with culture medium from the original stock concentration and added to the DRG cultures at a final concentration of 300 nm (or equivalent vehicle dilution). The toll-like receptor 4 (TLR4) antagonist ultrapure LPS-RS (InvivoGen) was diluted in PBS and added 12 h ahead of paclitaxel at a concentration of 2 μg/ml. PBS alone was added as the negative control. Following either 2 h in culture with paclitaxel or at 6 h after culture with paclitaxel, the culture medium was discarded, and then the cells were scraped from the dish and collected into centrifuge tubes. Primary cultured DRG neurons were disrupted in lysis buffer on ice for 30 min and then centrifuged at 4°C and 12,000 × g for 10 min. The supernatant was then transferred into new tubes and denatured with sample buffer for 10 min at 70°C. Western blot was then run as described above.
Human DRG neuron preparation.
Six human DRGs were collected from three patient donors (two male, one female) at MD Anderson Cancer Center who had provided legal written consent. Each donor was undergoing spinal surgery for disease treatment wherein a spinal nerve root was to be sacrificed as the standard of care. The human subjects' protocol was reviewed and approved by the MD Anderson Cancer Center Institutional Review Board.
Immediately after each ganglion was excised in the operating room, it was immersed into cold (4°C), sterile BSS containing nutrients and transported to the laboratory in a sterile, sealed 50 ml centrifuge tube. On arrival at the laboratory, ganglia were cut with a sterile scalpel into parts and then either immersed into 4% paraformaldehyde and cryoprotected in sucrose solution for immunohistochemistry (IHC) or digested in mixed enzyme solution as detailed below and used in whole-cell neurophysiology studies.
Human DRGs were processed for immunohistochemistry as described above for the rat tissues, but here using antibodies directed at Nav1.7 (rabbit anti-human, 1:250; Alomone Labs), CGRP (mouse anti-human, 1:1000; Abcam), transient receptor potential vanilloid 1 (TRPV1; guinea pig anti-human, 1:250; Abcam), and GFAP (mouse anti-human, 1:1000; Cell Signaling Technology). Sections were viewed with use of a fluorescent microscope (Eclipse E600; Nikon). For a given experiment, all images were taken using identical acquisition parameters, and experimenters were blinded to pain condition in the associated dermatome. To measure cell size, each individual neuron, including the nuclear region, was graphically highlighted. The total numbers of cells, the numbers of cells positive for Nav1.7 alone, and the numbers of cells showing colocalization with TRPV1 or CGRP were counted in three sections of all DRG from all donors.
DRG tissue for physiological recordings was dissociated with a cocktail of enzymes (w/v, final concentration): 0.1% trypsin (catalog #T9201, Sigma-Aldrich), 0.1% collagenase (catalog #C1764, Sigma-Aldrich), and 0.01% DNase (catalog #D5025, Sigma-Aldrich) prepared by mixing 1 aliquot each of the individual stock solution in a 15 ml centrifuge tube and diluting to a final volume of 8 ml with DMEM/F12. The pieces of tissue were transferred to a vial containing 2 ml of enzyme cocktail. The vial was placed in a 37°C rotator at a shake speed of 124–128/min. To minimize mechanical and enzymatic damage, cells released from the tissue were harvested every 20 min. This is accomplished by allowing the undissociated tissue fragments to settle by gravity, collecting the supernatant, and transferring to a centrifuge tube that contains an enzyme inhibitor solution and then reimmersing the remaining tissue fragments in fresh dissociation solution. The dissociation procedure is continued until the majority of the tissue fragments disintegrate. The cells are washed free of enzyme by centrifugation at 180 × g for 5 min and removal of the supernatant. The cells are resuspended in fresh DMEM/F12 (10% horse serum) culture solution to the desired density. Neurons were placed on coverslips that had been precoated with poly-l-lysine. Neurons were cultured at 37°C with 5% CO2 in DMEM/F12 supplemented with 10% horse serum, 2 mm glutamine, and 25 ng/ml human nerve growth factor (NGF). Only neurons with a stable resting membrane potential of at least −40 mV and evoked spikes that overshot 0 mV were used for further recording and analysis. Series resistance was compensated to >70%. All recordings were made at room temperature. Other recording protocols were as described above.
Experimental design and statistical analysis.
In the animal experiments, all rats were evenly distributed into either a vehicle-treated negative control group or a paclitaxel-treated positive control group; or, where appropriate, into a vehicle–vehicle negative control group, a paclitaxel–vehicle positive control group, and a paclitaxel–test intervention group. The endpoint for the behavioral studies was a change in mechanical paw withdrawal threshold. These rats were then evenly distributed into follow-up biochemical, immunohistochemical, or neurophysiological studies to define the effects of chemotherapy treatment on the expression, localization, and function of Nav1.7. As well, the role of Nav1.7 in contributing to SA following chemotherapy treatment was tested. The human DRG was collected to determine whether the occurrence of pain affects the expression, localization, and function of Nav1.7. All data were acquired and analyzed by investigators blinded to treatment conditions. The rat data were grouped by treatment, while the patient data were grouped by the presence or absence of pain for statistical analysis. Data are expressed as the mean ± SEM and analyzed with GraphPad Prism 6. Behavioral mechanical withdrawal, sodium current neurophysiology, and human DRG intracellular current injection neurophysiology data were analyzed with two-way ANOVA (treatment × time, voltage, or current, respectively) followed by Bonferroni post hoc tests. Cell counts in the immunohistochemical experiments, Western blot, and human DRG neuron membrane physiological data in Figure 8 were analyzed with the Mann–Whitney U test. The effects of ProTx II on dissociated DRG SA (rat and human firing rate) were analyzed using the paired t test.
Figure 8.

The expression of Nav1.7 is increased in human DRGs that are hyperexcitable. A, B, Representative immunohistochemical images are shown in A (DRG with no SA) and B (DRG with SA). The expression of Nav1.7 in the DRG without SA (A) is lower compared with that of the DRG (B) where pronounced hyperexcitability was observed. D, The bar graph shows the summarized mean intensity of Nav1.7 staining from 10 sections of each ganglion and indicates that the increases in expression were significant in the DRG with SA. C, Representative action potential responses evoked at 1×, 2×, and 3× rheobase in DRG without SA and DRG with SA are shown. As summarized in E, neurons from DRG with SA showed more depolarized resting membrane potential than DRG without SA (n = 12, each). The summary graphs in F and G show that the mean current threshold at rheobase was significantly reduced in DRG with SA and that the mean numbers of action potentials evoked at 4× and 5× rheobase was significantly higher. *p < 0.05, ***p < 0.001. Scale bar, 100 μm.
Results
Increased expression and function change of Nav1.7 in DRG neurons in rats with paclitaxel-induced peripheral neuropathy
Expression of Nav1.7 was markedly increased in L4–L5 DRGs at days 7 and 14 after paclitaxel treatment compared with that in DRGs after vehicle treatment, but this increased expression was not sustained at day 21 (Fig. 1A,B; day 7, U = 0, p = 0.0286; day 14, U = 0, p = 0.0286; day 21, U = 0, p = 0.0159; Mann–Whitney unpaired test). Hence, the onset of CIPN is paralleled by increased expression in Nav1.7. The model of paclitaxel CIPN used here persists between 28 and 60 d after chemotherapy (Cata et al., 2004; Boyette-Davis et al., 2011a; Zhang et al., 2013); and so, increased expression of Nav1.7 may not contribute to the ongoing maintenance of paclitaxel CIPN. However, as shown below, increased function of Nav1.7 may contribute during this phase of the model.
Figure 1.

Increased expression and function of Nav1.7 in DRG neurons in rats with paclitaxel-induced peripheral neuropathy. A, The representative Western blot images demonstrate that the expression of Nav1.7 was increased in DRGs by day 7 of chemotherapy through day 14, whereas the expression of Nav1.7 then fell below the baseline level at day 21. The bar graphs in B summarize the grouped data and indicate that the level of expression of Nav1.7 in the dorsal root ganglia in the paclitaxel-treated rats (black bars) was significantly higher than in the vehicle-treated rats (white bars). C, Representative voltage-clamp recordings of Nav1.7 current in acute dissociated DRG neurons of day 7 paclitaxel-treated rats collected using a subtraction protocol with the administration of Nav1.7-selective blocker ProTx II, as described in Results, the mean current density–voltage relationships are plotted in D. Shown in E is a comparison of the voltage dependence of activation for Nav1.7 channels in vehicle- and paclitaxel-treated animals. In F, the comparison of steady-state inactivation for Nav1.7 channels is shown. β-act, β-actin; V, vehicle; P, paclitaxel. *p < 0.05, ***p < 0.001.
Using whole-cell patch-clamp, small dissociated neurons (<35 μm in diameter) were recorded from vehicle- and paclitaxel-treated animals. Figure 1C shows representative whole-cell DRG currents of a paclitaxel-treated rat at day 7 after initial treatment. Current–voltage relationships were measured by using a series of 15 ms depolarizing steps (−90 to 45 mV in 5 mV increments at 5 s intervals) from a holding potential of −90 mV. Nav1.7 current was isolated using a subtraction protocol (Gold et al., 2003; Zhang et al., 2017). The top trace in Figure 1C is total Na+ current, the middle trace is Na+ currents after ProTx II administration, while the current obtained following subtraction of the residual current following ProTx II administration from the total is shown in the bottom trace. Current density was calculated by normalizing the maximal peak current to cell capacitance. The current densities recorded from cells in the paclitaxel treatment group (6 rats, 15 neurons) were greater than were those in the vehicle treatment group (6 rats,12 neurons) across voltages of −15 to −10 mV (F(7,175) = 2.326, p = 0.0271, two-way ANOVA; Fig. 1D). Comparison of the voltage dependence of activation for Nav1.7 channel in vehicle- and paclitaxel-treated animals showed that this was more hyperpolarized in cells from paclitaxel-treated rats than in vehicle-treated rats (F(11,264) = 1.962, p = 0.0324, two-way ANOVA; Fig. 1E). There was no difference in steady-state inactivation of Nav1.7 between vehicle- and paclitaxel-treated rat DRG neurons (Fig. 1F).
Cellular localization of Nav1.7 in DRG and the spinal cord
Nav1.7 has been suggested to be preferentially expressed in DRG neurons (Black et al., 1996), and to a lesser extent in olfactory sensory neuron processes (Ahn et al., 2011). IHC was used to determine the cellular localization of Nav1.7 in rat DRGs and spinal cord on day 7 after paclitaxel treatment. Consistent with Western blot analysis, representative IHC images of DRGs from vehicle-treated rats (Fig. 2A) and paclitaxel-treated rats (Fig. 2B) show that the expression of Nav1.7 was increased following paclitaxel treatment. Double IHC demonstrated that Nav1.7 was colocalized in both IB4-positive (Fig. 2C,D) and CGRP-positive [Fig. 2E,F (with all data combined in Fig. 2G,H)] small DRG neurons. The bar graphs in Figure 2I–K indicate that the increase in Nav1.7 expression was statistically significant (t(22) = 5.085, p < 0.0001, unpaired t test; Fig. 2I), and that Nav1.7 expression was especially concentrated in small-diameter (<30 μm) and medium-diameter (30–45 μm) DRG neurons with little in large-diameter (>45 μm) DRG neurons (F(2,65) = 3.437, p = 0.0381, two-way ANOVA; Fig. 2J); the increased expression was most pronounced in the CGRP-positive neurons (t(22) = 6.339, p < 0.0001, unpaired t test; Fig. 2K).
Figure 2.

The expression of Nav1.7 is increased in nociceptive DRG neurons in rats with paclitaxel-treated CIPN. Representative IHC images are shown in A–H. A, B, The expression of Nav1.7 (red) in DRGs is normally low in naive (data not shown) and vehicle-treated rats (A) but becomes greatly increased by day 7 after paclitaxel treatment (B). C–H, Nav1.7 is colocalized in subsets of IB4-positive (green) neurons (C, D, colocalization indicated in yellow) and CGRP-positive (blue) neurons (E, F, colocalization indicated in purple), as well as in a percentage of neurons negative for both IB4 and CGRP (G, H, overall merged). I–K, The bar graphs summarize the grouped IHC data and show that the overall number of Nav1.7-positive neurons (I) was significantly higher in the paclitaxel-treated rats (black bars) than in the vehicle-treated rats (white bars); and these increases were significantly higher in neurons with diameters <30 μm and to a lesser extent in medium-sized (30–45 μm) DRG neurons (J); and in IB4-positive and CGRP-positive neurons, as well as neurons negative for both markers (K). Scale bar, 100 μm. *p < 0.05, ***p < 0.001.
IHC on spinal dorsal horn showed that the expression of Nav1.7 was low in vehicle-treated rats, largely confined to the most superficial layers in the zone of small fiber terminals (Fig. 3A). However, this expression was markedly elevated at day 7 after paclitaxel treatment both in the superficial dorsal horn and now also extending into deeper lamina (Fig. 3B). Consistent with DRG staining, Nav1.7 in fiber profiles of the superficial lamina were found to colocalize with CGRP (Fig. 3E) and less so with IB4 (Fig. 3D), and this strongly suggests that Nav1.7 is localized within presynaptic central terminals of nociceptive DRG neurons (Fig. 3F, merged picture for all ligands). Double IHC on spinal tissue showed that Nav1.7 was expressed in NeuN-positive spinal cells in deeper lamina (Fig. 3G–I) but not in GFAP-positive or OX42-positive cell profiles (data not shown). Absorption control with an excess of Nav1.7 protein confirmed the specificity of the antibody (Fig. 3C); as a further test of specificity, no positive staining was found in the cerebellum (Fig. 3J–L), consistent with previous reports (Schaller and Caldwell, 2003; Ahn et al., 2011).
Figure 3.
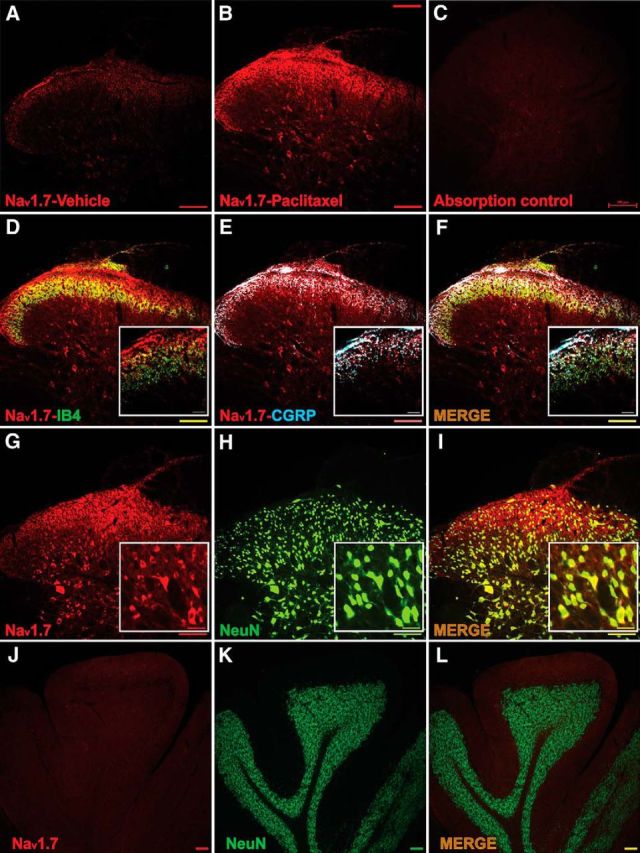
The expression of Nav1.7 is increased and colocalized with IB4, CGRP, and NeuN in spinal cord segments L4–L5 at day 7 after treatment with paclitaxel. A, B, Representative immunohistochemical (IHC) images show that expression of Nav1.7 (red) in spinal cord was quite pronounced compared with vehicle (A) by day 7 after treatment with paclitaxel (B). In C, the absorption control shows no staining. D, E, Double IHC shows that Nav1.7 (red) was expressed in some IB4-positive (D, green, with colocalization shown in yellow) and especially were expressed in CGRP-positive (blue) neuron terminals (E, colocalization indicated in purple). In F, the merged image indicates that Nav1.7 expression was upregulated in a large percentage of peripheral afferent terminals positive for CGRP. G–I show that Nav1.7 was expressed in NeuN-positive spinal cells in deeper lamina following paclitaxel treatment. J–L are a further test of antibody specificity as no expression of Nav1.7 was found in the cerebellum. Scale bars: lower-scale images, 200 μm; higher-magnification images, 50 μm.
Paclitaxel-induced hyperalgesia is prevented and reversed by the Nav1.7 channel blocker ProTx II
The effects of ProTx II, given both during the induction of and in pre-established paclitaxel hyperalgesia, were assessed in behavioral studies. Figure 4A shows the results of intrathecal administration of ProTx II in preventing paclitaxel-induced hyperalgesia. ProTx II at doses of 15 and 60 μg in 20 μl of PBS or vehicle alone (20 μl of PBS) was given daily beginning 2 d before and continuing until 2 d after chemotherapy. ProTx II had no effect on baseline mechanical withdrawal threshold and showed no interaction with the paclitaxel vehicle (Cremophor EL) over time. The paclitaxel–PBS-treated rats showed the expected decrease in mechanical withdrawal threshold that was significantly different from that of the vehicle-treated rats (both the vehicle-PBS and vehicle-ProTx II groups) by day 7, consistent with previous studies by our group (Boyette-Davis et al., 2011a; Zhang et al., 2013; Li et al., 2014b). In contrast, the paclitaxel-treated rats also given 60 μg of ProTx II showed only a partial development of mechanical hypersensitivity that was significantly less than that of the paclitaxel-PBS-treated rats at days 7 through 14. The paclitaxel-treated rats that were given 15 μg of ProTx II showed no significant difference in withdrawal threshold compared with the paclitaxel–PBS-treated rats (F(16,125) = 11.19, p < 0.0001, two-way ANOVA; n = 6 each group).
Figure 4.

Prevention and reversal of paclitaxel-induced mechanical hypersensitivity in rats and reduced spontaneous action potentials in DRG neurons by application of the Nav1.7 blocker ProTx II. A, B, The baseline (BL) behavioral test in A and B were collected before paclitaxel (Pac) or vehicle (Veh) treatments. Rats that yielded the data in A received daily intrathecal treatment with 15 or 60 μg of ProTx II or PBS beginning 2 d before and continuing to 2 d after intraperitoneal treatment with paclitaxel or the vehicle. The paclitaxel-ProTxII 60 μg group (black filled circles) showed only partial development of mechanical hypersensitivity compared with that in the paclitaxel-PBS (open circles) and paclitaxel-ProTx II 15 μg mg/kg groups (filled gray circles). Rats receiving paclitaxel vehicle (Cremophor-EL) and PBS (ProTx II vehicle, open squares) or Cremophor-ProTx II 60 μg (filled black squares) showed no changes from baseline measurements. In B, paclitaxel-induced mechanical hypersensitivity was confirmed at 14 d after treatment; rats were then treated with 60 μg ProTx II (filled black circles) or PBS (open circles). ProTx II transiently reversed the mechanical hyper-responsiveness. A single injection of 60 μg ProTx II had no effect on mechanical withdrawal in Cremophor-treated rats (filled black squares). ***p < 0.001 vs paclitaxel-PBS; +++p < 0.001 vs BL. C shows analog recordings of a representative DRG neuron with spontaneous activity isolated from segment L5 at day 7 after paclitaxel treatment that was reduced by bath application of ProTx II (5 nm) and resumed with drug discontinuation. The bar graphs in D summarize results from eight cells from four paclitaxel-treated animals. #p < 0.05 vs baseline.
In the second experiment, ProTx II was tested as a treatment for pre-established paclitaxel-induced hyperalgesia (Fig. 4B). Two groups were treated with paclitaxel, and mechanical hyperalgesia was confirmed in each group at day 14 after treatment. Rats in both groups were given a single intrathecal dose of either ProTx II (60 μg) in 20 μl of PBS or just 20 μl of PBS. The ProTx II-treated group showed a significant increase in mechanical withdrawal threshold that was evident within the first hour after treatment, compared with the PBS group, and this effect persisted for 24 h after treatment (F(24,144) = 11.91, p < 0.0001, two-way ANOVA; n = 5 each group). None of the animals that received any intrathecal injections showed any change in motor performance, as assessed using the rotarod performance test (data not shown).
ProTx II suppresses ectopic spontaneous activity in DRG neurons from rats with paclitaxel-induced hyperalgesia
Previous work showed that 35% of small L4–L5 DRG neuron somata from rats with paclitaxel-treated CIPN exhibit ectopic spontaneous activity (action potentials) at day 7 after treatment compared with only 3% in vehicle-treated rats (Li et al., 2017). SA was found in 8 of 26 L4–L5 DRG neurons (31%) sampled in this study at day 7 after paclitaxel treatment recorded in four rats (vehicle-treated rats were not sampled). All eight neurons showed a significant reduction in SA by bath application of ProTx II (5 nm). A representative cell is shown in Figure 4C, and the summary data for this are shown in Figure 4D (t(7) = 3.239, p = 0.0143, paired t test).
LPS-RS prevents the increased expression of Nav1.7 induced by paclitaxel both in vivo and in vitro
Previous work has shown that TLR4 expression is increased after paclitaxel treatment in both IB4-positive and CGRP-positive DRG neurons; and that myeloid differentiation primary response gene protein 88 (MyD88), a key immediate downstream signal of TLR4 activation, is expressed only in CGRP-positive DRG neurons (Li et al., 2014b). Given that the increase in Nav1.7 expression after paclitaxel treatment was most prominently shown in CGRP-positive DRG neurons, we tested whether the expression of Nav1.7 is increased by activation of the TLR4–MyD88–MAPK (mitogen-activated protein kinase) signaling pathway (Li et al., 2014b, 2015). As shown in the representative gel in Figure 5A, the expression of Nav1.7 in the DRG was significantly lower at both day 7 and day 14 in the paclitaxel-treated rats cotreated with the TLR4 antagonist LPS-RS in vivo compared with rats cotreated with PBS during chemotherapy. As shown in the representative gel in Figure 5B, the same effect was also observed using in vitro primary DRG culture at 48 h after the addition of 300 nm paclitaxel plus LPS-RS compared with the addition of vehicle. The bar graphs in Figure 5C show that grouped data for the in vivo experiments revealed a significant effect of LPS-RS in preventing an increase in the expression of Nav1.7 (U = 0, p = 0.0286, Mann–Whitney test, four rats in each group), while the bar graphs in Figure 5D indicate that a significant reduction was also observed in the in vitro studies (U = 0, p = 0.0286, Mann–Whitney test; four wells/treatment group, eight rats total).
Figure 5.

A, B, The representative Western blots show that LPS-RS cotreatment with paclitaxel prevented the increased expression of Nav1.7 in DRG neurons both in vivo (A) and in vitro (B). The level of Nav1.7 in vehicle-PBS (V)-treated rats is in the left column, that of paclitaxel-PBS (P)-treated rats is in the middle column, and that in paclitaxel-LPS-RS (P+L)-treated rats is in the right-hand column of each set of representative blots. C, D, The bar graphs show the summarized mean level of expression of Nav1.7 in the DRGs at day 7 and day 14 in vehicle-PBS-treated (open bars), paclitaxel-PBS-treated (filled black bars), and paclitaxel-LPS-RS-treated (filled gray bars) rats following in vivo treatments (C) and in the primary DRG culture at 2 and 48 h after incubation with 300 nm paclitaxel or vehicle (D). Nav1.7 was significantly lower in the paclitaxel-treated rats cotreated with LRS-RS (gray bars) compared with the paclitaxel-treated rats cotreated with PBS (black bars) at both day 7 and day 14 after paclitaxel intraperitoneal injection but only at 48 h after 300 nm paclitaxel incubation in vitro culture, while there is no difference compared with vehicle-PBS-treated (open bars) animal. *p < 0.05, **p < 0.01.
Studies of Nav1.7 in human DRGs
Tissue was donated by three patients in whom standard-of-care surgical resection of metastatic oncological disease involved ligation of spinal nerve roots. Each patient had a history of cancer-related neuropathic pain that appeared to be multifactorial in etiology, including direct neural compression and physiological effects of peritumoral environment, and possibly were due to radiation therapy involving the spine. One patient had received cisplatin/paclitaxel therapy for 5 months before tissue donation but had no symptoms of CIPN. IHC and neurophysiological data were obtained from a total of six DRGs from these patients.
IHC was used to determine the localization of Nav1.7 in subsets of human DRG neurons. As shown in the representative images in Fig. 6A,B, Nav1.7 staining was strongly positive in approximately half of human DRG neurons (red). Figure 6, C and D, shows the localization of TRPV1 and CGRP (top row and bottom row, respectively; in green) in the same sections, while the merged images in Figure 6, E and F, show that many neurons were positive for both Nav1.7 and TRPV1 or Nav1.7 and CGRP (Fig. 6E,F, top row and bottom row, respectively; in yellow). The bar graphs in Figure 6, G and H, show the group summaries of the IHC data. Human DRG neurons ranged from 20 to 95 μm in diameter, with an average diameter of 45 μm. Surprisingly, both TRPV1 and CGRP were found throughout the size range of neurons observed. Similarly, Nav1.7 was found across all sizes of neurons and was found in approximately half of both the CGRP and TRPV1 neuron populations. Nav1.7-positive staining was also found in ring-shaped cellular profiles around DRG neurons and colocalized with GFAP, consistent with localization to satellite glial cells. Sodium channel α subunit expression was previously reported in cultured Schwann cells (Belcher et al., 1995). As shown in Figure 7A–C, this immunoreactivity was not found in all cells, and it is distinct from that shown in nearby neurons. The annular appearance in many of the neurons suggests that Nav1.7 is highly concentrated in the cell membrane.
Figure 6.

Nav1.7 is expressed in human DRG neurons. Representative IHC images show that Nav1.7 colocalizes with TRPV1 and CGRP. A–F, Nav1.7 alone (red; A, B), TRPV1 (C), and CGRP (D) are shown alone in green, and E and F show the colocalization (yellow/orange). The bar graphs in G and H show the group summaries of the IHC data. Scale bar, 100 μm.
Figure 7.

Nav1.7 is expressed in human DRG satellite cells and neurons and contributes to ectopic spontaneous activity. A–C, Representative immunohistochemical images show labeling for Nav1.7 (A; red), GFAP (B; green), and merged (C; yellow indicates double label). A representative analog recording of a human DRG neuron with spontaneous activity is shown in D that is suppressed by bath application of Pro-TX II (5 nm), while the bar graphs in E show the combined results from eight separate whole-cell patch-clamp experiments in human DRGs. Application of ProTx II significantly decreased firing frequency in all neurons. *p < 0.05. Scale bar, 100 μm.
Neurophysiological activity was recorded from 39 acutely dissociated neurons (diameter, 27.87–75.0 μm; mean, 43.92 ± 1.57 μm) using the same electrophysiological methods as described for acutely dissociated rat DRG neurons. Ectopic SA was found in human DRG neurons, but only when these were collected from dermatomes where patients had a history of radiating pain and/or radiographic evidence of nerve root compression. SA was found in 8 of 27 neurons (30%) from the four ganglia from dermatomes associated with pain, but in 0 of 12 neurons when isolated from the two ganglia in dermatomes without pain. Figure 7D shows a representative example of whole-cell recording with SA. This SA is reduced by bath application of ProTx II (5 nm), suggesting that Nav1.7 has a functional role in human DRGs like those observed in rats. After washout of ProTx II and switching to voltage clamp, the same cell was confirmed to be responsive to bath application of capsaicin (data not shown). Summary data for ProTX II treatment on SA for human DRG neurons is shown in Figure 7E and indicates that the results were statistically significant (t(5) = 2.894, p = 0.034, paired t test; n = 8).
One patient donated two dorsal root ganglia, one from an area of pain (3 of 12 neurons positive for SA) and one from outside an area of pain (0 of 12 neurons positive for SA). This patient had a history of locally invasive squamous cell carcinoma involving the right upper lobe of the lung and adjacent thoracic vertebral bodies T2–T5 with the most extensive involvement at the T4 level. His clinical symptoms included progressive upper back pain with radiation into the right shoulder with associated sensory loss and tenderness in the T4 dermatome. The patient had undergone concurrent fractionated radiation and chemotherapy with cisplatin and paclitaxel finishing ∼5 months before surgical treatment/donation of DRGs, but had no symptom complaints consistent with CIPN. IHC comparing these DRGs, right T4 DRG versus right T3 DRG, as shown in Figure 8, A and B, reveals that the overall expression of Nav1.7 was markedly increased in the DRG where ectopic SA was observed in the right T4 DRG (U = 23, p = 0.0433, Mann–Whitney test; Fig. 8D).
Representative analog recordings testing intrinsic excitability of DRG neurons from ganglia with and without SA are shown in Figure 8C, with bar graph summaries of this and other neurophysiology data shown in Figure 8E–G (n = 12 each group). Evoked action potential frequencies (spikes/500 ms) at 1×, 3×, and 5× rheobase were higher for neurons from the T4 DRG with SA compared with those from the T3 DRG without SA (F(4,83) = 3.962, p = 0.0054, two-way ANOVA). As summarized in Figure 8E, neurons from the DRGs associated with SA showed significantly more depolarized resting membrane potential (−55.12 ± 1.92 mV) compared with cells from the DRG without SA (−62.2 ± 1.42 mV; U = 36, p = 0.0211, Mann–Whitney test). Rheobase, as shown in Figure 8F, was lower for neurons from the DRG with SA compared with the DRG without SA (653.1 ± 270 and 1192 ± 306.9 pA, respectively; U = 38, p = 0.0298, Mann–Whitney test).
Discussion
It was previously reported that DRG neurons develop ectopic SA, decreased rheobase, a more depolarized membrane potential, and increased discharge frequency to suprathreshold stimuli after paclitaxel treatment in rats (Li et al., 2017). In addition, unbiased microarray screening indicated changes in gene expression of multiple membrane ion channels in the DRGs after paclitaxel treatment, with that for Nav1.7, in particular, among those that were increased (Zhang and Dougherty, 2014). Here, these observations are extended wherein protein expression of Nav1.7 is confirmed as increased in small- and medium-sized IB4- and CGRP-positive DRG neurons. In parallel, the current density for Nav1.7 is also increased, and a Nav1.7 channel blocker prevents the development of as well as alleviates established paclitaxel-related mechanical behavioral hypersensitivity. These effects appear to be related to the suppression of ectopic SA that occurs in DRG neurons following paclitaxel treatment. Finally, findings in DRGs from human donors show colocalization like that observed in rats and support a role for Nav1.7 in human pain conditions.
Nav1.7 is a key determinant of action potential threshold in DRG neurons (Dib-Hajj et al., 2007, 2009; Rush et al., 2007), and a change in function of these channels results in significant pain disorders (Dib-Hajj et al., 2007). Mutations of human Nav1.7 leading to loss of function result in congenital insensitivity to pain (Cox et al., 2006; Fertleman et al., 2006; Dib-Hajj et al., 2007), while, in marked contrast, gain-of-function mutations leading to enhanced TTX-S currents result in the occurrence of spontaneous pain disorders (Yang et al., 2004; Cox et al., 2006; Fertleman et al., 2006; Han et al., 2006; Faber et al., 2012). Importantly, the symptoms of two of these disorders, inherited erythromelalgia and chronic painful peripheral neuropathy, involve burning sensations in the hands and feet (Yang et al., 2004; Cox et al., 2006; Fertleman et al., 2006; Han et al., 2006; Faber et al., 2012) similar to those described by CIPN patients (Boyette-Davis et al., 2011b, 2013).
Preclinical studies indicate that Nav1.7 expression and function are increased in models of inflammatory, diabetic, bone cancer and neuropathic pain (Hong et al., 2004; Chattopadhyay et al., 2008; Huang et al., 2014b). The function of Nav1.7 in turn is regulated by the MAPKs, phospho-extracellular signal-related kinase 1/2 (pERK1/2), and phospho-P38 (Stamboulian et al., 2010). More specifically, pERK1/2 phosphorylates Nav1.7, resulting in a depolarizing shift in activation threshold. This in turn impacts the resting membrane potential and firing properties of adult DRG neurons, resulting in increased neuronal excitability. In contrast, the inhibition of pERK1/2 results in hyperpolarized resting membrane potentials, increased threshold for action potential firing, and reduced firing frequency of small DRG neurons (Stamboulian et al., 2010).
These findings dovetail well with previous results showing that TLR4 signaling is important in the generation of paclitaxel-related CIPN. TLR4 expression is increased in IB4- and CGRP-positive DRG neurons, while MyD88, a key immediate downstream signal of TLR4, is increased only in CGRP+ DRG neurons after paclitaxel treatment (Li et al., 2014b). As well, MAPK signaling downstream to TLR4, notably ERK1/2 and P38, but not c-Jun N-terminal kinase or phosphoinositide 3 kinase-Akt, are also increased in DRGs after paclitaxel treatment (Li et al., 2015). The MAPK inhibitors PD98059 (MEK1/2), U0126 (MEK1/2), and SB203580 (P38) given alone partially prevented paclitaxel-induced behavioral hypersensitivity (Li et al., 2015). Like MyD88, ERK1/2 was found to be increased primarily in CGRP-positive neurons, which supports the conclusion that the increased expression and function of Nav1.7 after paclitaxel treatment is due to the activation of a TLR4–MyD88–ERK1/2 signaling pathway that in turn leads to behavioral hypersensitivity. Presumably, the P38 pathway engages the contribution of the IB4+ neurons in CIPN, with the combined effects of both MAPKs on both populations of cells, resulting in the full CIPN phenotype.
Although there have been reports of Nav1.7 expression in spinal cord motor neurons (Fukuoka et al., 2010, 2015; Black et al., 2012), no publications have reported plasticity in the expression of Nav1.7 in the dorsal horn in CIPN or other types of preclinical pain models. Naive animals here, as in the study by Black et al. (2012), showed dense staining for Nav1.7 in the afferent terminal zone in the superficial dorsal horn and only very faint staining in indistinct profiles in the deeper part of the dorsal and ventral horn border. Although sciatic nerve injury did not influence this pattern (Fukuoka et al., 2015), CIPN animals showed a marked, but transient, increase at day 7 after chemotherapy in cellular profiles consistent with neurons and that colocalized with NeuN. Interestingly, Nav1.3, also not normally expressed at high levels in spinal cord, similarly showed an increase in expression in the spinal cord following overt traumatic sciatic nerve injury (Hains et al., 2004). Increased expression of spinal Nav1.3 was also observed following spinal cord injury (Hains et al., 2003). The question that arises in the context of the additional observations provided here is why a differential effect on sodium channel expression occurs with CIPN versus direct trauma to peripheral nerves or the spinal cord. Perhaps this is in accord with the differential responsiveness that is also shown by spinal glial cells in CIPN versus following trauma. Astrocytes alone show signs of activation following CIPN regardless of whether this is induced using paclitaxel, oxaliplatin, or bortezomib (Zhang et al., 2012; Robinson et al., 2014). In contrast, a prominent role for microglia in contributing to both spinal cord injury and peripheral nerve injury-related pain are well documented (Milligan and Watkins, 2009). Thus, CIPN would seem not to engage the brain-derived neurotrophic factor-mediated change in neuronal chloride gradient that appears to play a key role in neuropathic pain produced by surgical nerve injury (Tsuda et al., 2013), and would presumably also not show the sexual dimorphism proposed in trauma-related neuropathic pain (Sorge et al., 2015). In contrast, paclitaxel treatment is associated with increased levels of NGF in both the DRG and dorsal horn (Nakahashi et al., 2014) as well as increased levels of the proinflammatory cytokine tumor necrosis factor α (TNFα) in the DRG and dorsal horn (Janes et al., 2014; Zhang et al., 2016). Increased expression of NGF suppresses the expression of Nav1.3 following nerve injury and so could account for the lack of change in that channel here. As well, TNFα increases the expression of Nav1.7 in diabetic neuropathy and so could play the same role in CIPN (Huang et al., 2014b).
The studies conducted here on the human DRG validate many of the findings observed in rat tissues as well as reveal important new fundamental data. As in animals, the expression of Nav1.7 is increased in human DRGs that show hyperexcitability and ectopic spontaneous activity that were both linked to the presence of ongoing pain. This hyperexcitability could be reduced by the presumptive Nav1.7-specific blocker ProTxII. The distribution of Nav1.7 was similar to that in rodent tissue as the channel was found colocalized with both TRPV1 and CGRP, further implicating the channel as being important in pain processing. On the other hand, many Nav1.7+ neurons also showed no colocalization, leaving the possibility open that the channel is also important in the signaling of non-noxious sensory information. Perhaps more striking is the observation of all three markers, Nav1.7, TRPV1, and CGRP, across the gamut of somal diameters that were observed, clearly indicating that diameter alone is not an adequate approach for the selection of nociceptors versus mechanoreceptors in studies of human DRG neurons. It is unlikely that the sample here from thoracic dermatomes is biased in its cell diameter profile because the data closely match data from other studies using cervical (Anand et al., 2006) and lumbar (L5) ganglia (Holford et al., 1994). As well, previous physiological studies in human cervical DRGs revealed capsaicin-sensitive neurons as large as 100 μm in diameter. Thus, although somal size has been used as a means to target nociceptive neurons in animal models, this approach is not suitable in the study of human DRG neurons.
A final issue concerns the impact of ProTxII on human Nav1.7 and the observed suppression here of ectopic SA in human DRG neurons. ProTxII was shown to have high selectivity for heterologously expressed human Nav1.7 in HEK cells versus other human Nav1 subtypes in detailed whole-cell patch-clamp studies (Schmalhofer et al., 2008). ProTxII and derivatives with heightened selectivity to human Nav1.7 again assayed using detailed whole-cell neurophysiology generated an apparent loss of pain sensitivity when given by intrathecal injection in rats (Flinspach et al., 2017). Yet, an examination of the effects of ProTxII on TTX-S currents in whole-cell studies in DRGs from human donors revealed no effects in any neurons tested (Zhang et al., 2017), bringing into question whether the suppression of ectopic SA observed here was in fact mediated by the inhibition of Nav1.7. Perhaps it is important that there was an apparent increase in the expression of Nav1.7 in a DRG from a dermatome associated with neuropathic pain versus a DRG not associated with pain from a single human donor, raising the possibility that Nav1.7 plays a significant role in human DRGs only with the onset of a pain condition.
Footnotes
This work was supported by Grant CA-200263 from the National Institutes of Health and the H.E.B. Professorship in Cancer Research.
The authors declare no competing financial interests.
References
- Ahn HS, Black JA, Zhao P, Tyrrell L, Waxman SG, Dib-Hajj SD (2011) Nav1.7 is the predominant sodium channel in rodent olfactory sensory neurons. Mol Pain 7:32. 10.1186/1744-8069-7-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand U, Otto WR, Casula MA, Day NC, Davis JB, Bountra C, Birch R, Anand P (2006) The effect of neurotrophic factors on morphology, TRPV1 expression and capsaicin responses of cultured human DRG sensory neurons. Neurosci Lett 399:51–56. 10.1016/j.neulet.2006.01.046 [DOI] [PubMed] [Google Scholar]
- Belcher SM, Zerillo CA, Levenson R, Ritchie JM, Howe JR (1995) Cloning of a sodium channel alpha subunit from rabbit Schwann cells. Proc Natl Acad Sci U S A 92:11034–11038. 10.1073/pnas.92.24.11034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JA, Dib-Hajj S, McNabola K, Jeste S, Rizzo MA, Kocsis JD, Waxman SG (1996) Spinal sensory neurons express multiple sodium channel alpha-subunit mRNAs. Brain Res Mol Brain Res 43:117–131. 10.1016/S0169-328X(96)00163-5 [DOI] [PubMed] [Google Scholar]
- Black JA, Frézel N, Dib-Hajj SD, Waxman SG (2012) Expression of Nav1.7 in DRG neurons extends from peripheral terminals in the skin to central preterminal branches and terminals in the dorsal horn. Mol Pain 8:82. 10.1186/1744-8069-8-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyette-Davis JA, Cata JP, Zhang H, Driver LC, Wendelschafer-Crabb G, Kennedy WR, Dougherty PM (2011b) Follow-up psychophysical studies in bortezomib-related chemoneuropathy patients. J Pain 12:1017–1024. 10.1016/j.jpain.2011.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyette-Davis JA, Cata JP, Driver LC, Novy DM, Bruel BM, Mooring DL, Wendelschafer-Crabb G, Kennedy WR, Dougherty PM (2013) Persistent chemoneuropathy in patients receiving the plant alkaloids paclitaxel and vincristine. Cancer Chemother Pharmacol 71:619–626. 10.1007/s00280-012-2047-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyette-Davis J, Xin W, Zhang H, Dougherty PM (2011a) Intraepidermal nerve fiber loss corresponds to the development of Taxol-induced hyperalgesia and can be prevented by treatment with minocycline. Pain 152:308–313. 10.1016/j.pain.2010.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cata JP, Weng HR, Dougherty PM (2004) Cyclooxygenase inhibitors and thalidomide ameliorate vincristine-induced hyperalgesia in rats. Cancer Chemother Pharmacol 54:391–397. 10.1007/s00280-004-0809-y [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL (1994) Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53:55–63. 10.1016/0165-0270(94)90144-9 [DOI] [PubMed] [Google Scholar]
- Chattopadhyay M, Mata M, Fink DJ (2008) Continuous delta-opioid receptor activation reduces neuronal voltage-gated sodium channel (NaV1.7) levels through activation of protein kinase C in painful diabetic neuropathy. J Neurosci 28:6652–6658. 10.1523/JNEUROSCI.5530-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry V, Rowinsky EK, Sartorius SE, Donehower RC, Cornblath DR (1994) Peripheral neuropathy from taxol and cisplatin combination chemotherapy: clinical and electrophysiological studies. Ann Neurol 35:304–311. 10.1002/ana.410350310 [DOI] [PubMed] [Google Scholar]
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al-Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM, Woods CG (2006) An SCN9A channelopathy causes congenital inability to experience pain. Nature 444:894–898. 10.1038/nature05413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Sheets PL, Waxman SG (2007) The roles of sodium channels in nociception: implications for mechanisms of pain. Pain 131:243–257. 10.1016/j.pain.2007.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib-Hajj SD, Cummins TR, Black JA, Waxman SG (2007) From genes to pain: Na v 1.7 and human pain disorders. Trends Neurosci 30:555–563. 10.1016/j.tins.2007.08.004 [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Binshtok AM, Cummins TR, Jarvis MF, Samad T, Zimmermann K (2009) Voltage-gated sodium channels in pain states: role in pathophysiology and targets for treatment. Brain Res Rev 60:65–83. 10.1016/j.brainresrev.2008.12.005 [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Yang Y, Black JA, Waxman SG (2013) The Na(V)1.7 sodium channel: from molecule to man. Nat Rev Neurosci 14:49–62. 10.1038/nrc3640 [DOI] [PubMed] [Google Scholar]
- Faber CG, Hoeijmakers JG, Ahn HS, Cheng X, Han C, Choi JS, Estacion M, Lauria G, Vanhoutte EK, Gerrits MM, Dib-Hajj S, Drenth JP, Waxman SG, Merkies IS (2012) Gain of function Nav1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol 71:26–39. 10.1002/ana.22485 [DOI] [PubMed] [Google Scholar]
- Fertleman CR, Baker MD, Parker KA, Moffatt S, Elmslie FV, Abrahamsen B, Ostman J, Klugbauer N, Wood JN, Gardiner RM, Rees M (2006) SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron 52:767–774. 10.1016/j.neuron.2006.10.006 [DOI] [PubMed] [Google Scholar]
- Flinspach M, Xu Q, Piekarz AD, Fellows R, Hagan R, Gibbs A, Liu Y, Neff RA, Freedman J, Eckert WA, Zhou M, Bonesteel R, Pennington MW, Eddinger KA, Yaksh TL, Hunter M, Swanson RV, Wickenden AD (2017) Insensitivity to pain induced by a potent selective closed-state Nav1.7 inhibitor. Sci Rep 7:39662. 10.1038/srep39662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuoka T, Kobayashi K, Noguchi K (2010) Laminae-specific distribution of alpha-subunits of voltage-gated sodium channels in the adult rat spinal cord. Neuroscience 169:994–1006. 10.1016/j.neuroscience.2010.05.058 [DOI] [PubMed] [Google Scholar]
- Fukuoka T, Miyoshi K, Noguchi K (2015) De novo expression of Nav1.7 in injured putative proprioceptive afferents: multiple tetrodotoxin-sensitive sodium channels are retained in the rat dorsal root after spinal nerve ligation. Neuroscience 284:693–706. 10.1016/j.neuroscience.2014.10.027 [DOI] [PubMed] [Google Scholar]
- Gold MS, Reichling DB, Shuster MJ, Levine JD (1996) Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proc Natl Acad Sci U S A 93:1108–1112. 10.1073/pnas.93.3.1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS, Weinreich D, Kim CS, Wang R, Treanor J, Porreca F, Lai J (2003) Redistribution of NaV1.8 in uninjured axons enables neuropathic pain. J Neurosci 23:158–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg YP, MacFarlane J, MacDonald ML, Thompson J, Dube MP, Mattice M, Fraser R, Young C, Hossain S, Pape T, Payne B, Radomski C, Donaldson G, Ives E, Cox J, Younghusband HB, Green R, Duff A, Boltshauser E, Grinspan GA, Dimon JH, Sibley BG, Andria G, Toscano E, Kerdraon J, Bowsher D, Pimstone SN, Samuels ME, Sherrington R, Hayden MR (2007) Loss-of-function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clin Genet 71:311–319. 10.1111/j.1399-0004.2007.00790.x [DOI] [PubMed] [Google Scholar]
- Hagiwara H, Sunada Y (2004) Mechanism of taxane neurotoxicity. Breast Cancer 11:82–85. 10.1007/BF02968008 [DOI] [PubMed] [Google Scholar]
- Hains BC, Klein JP, Saab CY, Craner MJ, Black JA, Waxman SG (2003) Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J Neurosci 23:8881–8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hains BC, Saab CY, Klein JP, Craner MJ, Waxman SG (2004) Altered sodium channel expression in second-order spinal sensory neurons contrbutes to pain after peripheral nerve injury. J Neurosci 24:4832–4839. 10.1523/JNEUROSCI.0300-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C, Rush AM, Dib-Hajj SD, Li S, Xu Z, Wang Y, Tyrrell L, Wang X, Yang Y, Waxman SG (2006) Sporadic onset of erythermalgia: a gain-of-function mutation in Nav1.7. Ann Neurol 59:553–558. 10.1002/ana.20776 [DOI] [PubMed] [Google Scholar]
- Holford LC, Case P, Lawson SN (1994) Substance P, neurofilament, peripherin and SSEA4 immunocytochemistry of human dorsal root ganglion neurons obtained from post-mortem tissue: a quantitative morphometric analysis. J Neurocytol 23:577–589. 10.1007/BF01262058 [DOI] [PubMed] [Google Scholar]
- Hong S, Morrow TJ, Paulson PE, Isom LL, Wiley JW (2004) Early painful diabetic neuropathy is associated with differential changes in tetrodotoxin-sensitive and -resistant sodium channels in dorsal root ganglion neurons in the rat. J Biol Chem 279:29341–29350. 10.1074/jbc.M404167200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Yang Y, Dib-Hajj SD, van Es M, Zhao P, Salomon J, Drenth JP, Waxman SG (2014a) Depolarized inactivation overcomes impaired activation to produce DRG neuron hyperexcitability in a Nav1.7 mutation in a patient with distal limb pain. J Neurosci 34:12328–12340. 10.1523/JNEUROSCI.2773-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Zang Y, Zhou L, Gui W, Liu X, Zhong Y (2014b) The role of TNF-alpha/NK-kappa B pathway on the up-regulation of voltage-gated sodium channel Nav1.7 in DRG neurons of rats with diabetic neuropathy. Neurochem Int 75:112–119. 10.1016/j.neuint.2014.05.012 [DOI] [PubMed] [Google Scholar]
- Janes K, Esposito E, Doyle T, Cuzzocrea S, Tosh DK, Jacobson KA, Salvemini D (2014) A3 adenosine receptor agonist prevents the development of paclitaxel-induced neuropathic pain by modulating spinal glial-restricted redox-dependent signaling pathways. Pain 155:2560–2567. 10.1016/j.pain.2014.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosturakis AK, He Z, Li Y, Boyette-Davis JA, Shah N, Thomas SK, Zhang H, Vichaya EG, Wang XS, Wendelschafer-Crabb G, Kennedy WR, Simone DA, Cleeland CS, Dougherty PM (2014) Subclinical peripheral neuropathy in patients with multiple myeloma before chemotherapy is correlated with decreased fingertip innervation density. J Clin Oncol 32:3156–3162. 10.1200/JCO.2013.54.5418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lázár J, Gharat L, Khairathkar-Joshi N, Blumberg PM, Szallasi A (2009) Screening TRPV1 antagonists for the treatment of pain: lessons learned over a decade. Expert Opin Drug Discov 4:159–180. 10.1517/17460440802681300 [DOI] [PubMed] [Google Scholar]
- Li Y, Cai J, Han Y, Xiao X, Meng XL, Su L, Liu FY, Xing GG, Wan Y (2014a) Enhanced function of TRPV1 via up-regulation by insulin-like growth factor-1 in a rat model of bone cancer pain. Eur J Pain 18:774–784. 10.1002/j.1532-2149.2013.00420.x [DOI] [PubMed] [Google Scholar]
- Li Y, Zhang H, Zhang H, Kosturakis AK, Jawad AB, Dougherty PM (2014b) Toll-like receptor 4 signaling contributes to paclitaxel-induced peripheral neuropathy. J Pain 15:712–725. 10.1016/j.jpain.2014.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhang H, Kosturakis AK, Cassidy RM, Zhang H, Kennamer-Chapman RM, Jawad AB, Colomand CM, Harrison DS, Dougherty PM (2015) MAPK signaling downstream to TLR4 contributes to paclitaxel-induced peripheral neuropathy. Brain Behav Immun 49:255–266. 10.1016/j.bbi.2015.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Tatsui CE, Rhines LD, North RY, Harrison DS, Cassidy RM, Johansson CA, Kosturakis AK, Edwards DD, Zhang H, Dougherty PM (2017) Dorsal root ganglion neurons become hyperexcitable and increase expression of voltage-gated T-type calcium channels (Cav3.2) in paclitaxel-induced peripheral neuropathy. Pain 158:417–429. 10.1097/j.pain.0000000000000774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Watkins LR (2009) Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci 10:23–36. 10.1038/nrn2533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahashi Y, Kamiya Y, Funakoshi K, Miyazaki T, Uchimoto K, Tojo K, Ogawa K, Fukuoka T, Goto T (2014) Role of nerve growth factor-tyrosine kinase receptor A signaling in paclitaxel-induced peripheral neuropathy in rats. Biochem Biophys Res Comm 444:415–419. 10.1016/j.bbrc.2014.01.082 [DOI] [PubMed] [Google Scholar]
- Norreel JC, Vinay L, Fontes M, Clarac F (2003) Close relationship between motor impairments and loss of functional motoneurons in a Charcot-Marie-Tooth type 1A model. Neuroscience 116:695–703. 10.1016/S0306-4522(02)00741-8 [DOI] [PubMed] [Google Scholar]
- O'Connor C, Heath DL, Cernak I, Nimmo AJ, Vink R (2003) Effects of daily versus weekly testing and pre-training on assessment of neurologic impairment following diffuse traumatic brain injury. J Neurotrauma 20:985–993. 10.1089/089771503770195830 [DOI] [PubMed] [Google Scholar]
- Polomano RC, Mannes AJ, Clark US, Bennett GJ (2001) A painful peripheral neuropathy in the rat produced by the chemotherapeutic drug, paclitaxel. Pain 94:293–304. 10.1016/S0304-3959(01)00363-3 [DOI] [PubMed] [Google Scholar]
- Robinson CR, Zhang H, Dougherty PM (2014) Astrocytes, but not microglia, are activated in oxaliplatin and bortezomib-induced peripheral neuropathy. Neuroscience 274:308–317. 10.1016/j.neuroscience.2014.05.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AM, Cummins TR, Waxman SG (2007) Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J Physiol 579:1–14. 10.1113/jphysiol.2006.121483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller KL, Caldwell JH (2003) Expression and distribution of voltage-gated sodium channels in the cerebellum. Cerebellum 2:2–9. 10.1080/14734220309424 [DOI] [PubMed] [Google Scholar]
- Schmalhofer WA, Calhoun J, Burrows R, Bailey T, Kohler MG, Weinglass AB, Kaczorowski GJ, Garcia ML, Koltzenburg M, Priest BT (2008) ProTx-II, a selective inhibitor of NaV1.7 sodium channels, blocks action potential propagation in nociceptors. Mol Pharmacol 74:1476–1484. 10.1124/mol.108.047670 [DOI] [PubMed] [Google Scholar]
- Scripture CD, Figg WD, Sparreboom A (2006) Peripheral neuropathy induced by paclitaxel: recent insights and future perspectives. Curr Neuropharmacol 4:165–172. 10.2174/157015906776359568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorge RE, Mapplebeck JC, Rosen S, Beggs S, Taves S, Alexander JK, Martin LJ, Austin JS, Sotocinal SG, Chen D, Yang M, Shi XQ, Huang H, Pillon NJ, Bilan PJ, Tu Y, Klip A, Ji RR, Zhang J, Salter MW, Mogil JS (2015) Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci 18:1081–1083. 10.1038/nn.4053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamboulian S, Choi JS, Ahn HS, Chang YW, Tyrrell L, Black JA, Waxman SG, Dib-Hajj SD (2010) ERK1/2 mitogen-activated protein kinase phosphorylates sodium channel Na(v)1.7 and alters its gating properties. J Neurosci 30:1637–1647. 10.1523/JNEUROSCI.4872-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Beggs S, Salter MW, Inoue K (2013) Microglia and intractable chronic pain. Glia 61:55–61. 10.1002/glia.22379 [DOI] [PubMed] [Google Scholar]
- Yang Y, Wang Y, Li S, Xu Z, Li H, Ma L, Fan J, Bu D, Liu B, Fan Z, Wu G, Jin J, Ding B, Zhu X, Shen Y (2004) Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet 41:171–174. 10.1136/jmg.2003.012153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Dougherty PM (2014) Enhanced excitability of primary sensory neurons and altered gene expression of neuronal ion channels in dorsal root ganglion in paclitaxel-induced peripheral neuropathy. Anesthesiology 120:1463–1475. 10.1097/ALN.0000000000000176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Yoon SY, Zhang H, Dougherty PM (2012) Evidence that spinal astrocytes but not microglia contribute to the pathogenesis of paclitaxel-induced painful neuropathy. J Pain 13:293–303. 10.1016/j.jpain.2011.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Boyette-Davis JA, Kosturakis AK, Li Y, Yoon SY, Walters ET, Dougherty PM (2013) Induction of monocyte chemoattractant protein-1 (MCP-1) and its receptor CCR2 in primary sensory neurons contributes to paclitaxel-induced peripheral neuropathy. J Pain 14:1031–1044. 10.1016/j.jpain.2013.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Li Y, de Carvalho-Barbosa M, Kavelaars A, Heijnen CJ, Albrecht PJ, Dougherty PM (2016) Dorsal root ganglion infiltration by macrophages contributes to paclitaxel chemotherapy-induced peripheral neuropathy. J Pain 17:775–786. 10.1016/j.jpain.2016.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Priest BT, Belfer I, Gold MS (2017) Voltage-gated Na+ currents in human dorsal root ganglion neurons. eLife 6:e23235. 10.7554/eLife.23235 [DOI] [PMC free article] [PubMed] [Google Scholar]