

Abstract
The intercalated cells (ITCs) of the amygdala have been shown to be critical regulatory components of amygdalar circuits, which control appropriate fear responses. Despite this, the molecular processes guiding ITC development remain poorly understood. Here we establish the zinc finger transcription factor Tshz1 as a marker of ITCs during their migration from the dorsal lateral ganglionic eminence through maturity. Using germline and conditional knock-out (cKO) mouse models, we show that Tshz1 is required for the proper migration and differentiation of ITCs. In the absence of Tshz1, migrating ITC precursors fail to settle in their stereotypical locations encapsulating the lateral amygdala and BLA. Furthermore, they display reductions in the ITC marker Foxp2 and ectopic persistence of the dorsal lateral ganglionic eminence marker Sp8. Tshz1 mutant ITCs show increased cell death at postnatal time points, leading to a dramatic reduction by 3 weeks of age. In line with this, Foxp2-null mutants also show a loss of ITCs at postnatal time points, suggesting that Foxp2 may function downstream of Tshz1 in the maintenance of ITCs. Behavioral analysis of male Tshz1 cKOs revealed defects in fear extinction as well as an increase in floating during the forced swim test, indicative of a depression-like phenotype. Moreover, Tshz1 cKOs display significantly impaired social interaction (i.e., increased passivity) regardless of partner genetics. Together, these results suggest that Tshz1 plays a critical role in the development of ITCs and that fear, depression-like and social behavioral deficits arise in their absence.
SIGNIFICANCE STATEMENT We show here that the zinc finger transcription factor Tshz1 is expressed during development of the intercalated cells (ITCs) within the mouse amygdala. These neurons have previously been shown to play a crucial role in fear extinction. Tshz1 mouse mutants exhibit severely reduced numbers of ITCs as a result of abnormal migration, differentiation, and survival of these neurons. Furthermore, the loss of ITCs in mouse Tshz1 mutants correlates well with defects in fear extinction as well as the appearance of depression-like and abnormal social interaction behaviors reminiscent of depressive disorders observed in human patients with distal 18q deletions, including the Tshz1 locus.
Keywords: brain development, telencephalon, transcription factor
Introduction
The amygdala is a diverse collection of nuclei located in the lateral base of the telencephalon involved in the regulation of emotions (Zola-Morgan et al., 1991; Phelps and LeDoux, 2005). Projections from the prefrontal cortex (PFC) transmit signals encoding emotionally relevant stimuli to amygdalar inputs in the lateral amygdala (LA) (Iwata et al., 1986; Mascagni et al., 1993; Vertes, 2004; Gabbott et al., 2005; Likhtik et al., 2005). Signals are subsequently processed by circuits linking the LA to amygdalar outputs in the central amygdala (CeA) (Veening et al., 1984; Pitkänen et al., 1997). Studies in animal models have shown that disruption of amygdalar circuitry leads to abnormalities in fear-, anxiety-, and depression-related behaviors (Wellman et al., 2007; Alò et al., 2014; Gafford and Ressler, 2016). Furthermore, studies in human patients have associated mental illnesses, such as anxiety disorders and major depressive disorders with amygdalar abnormalities (Savitz and Drevets, 2009; Taylor and Whalen, 2015). Recently, a specialized class of amygdalar interneurons, the intercalated cells (ITCs), has been established as a critical regulator of amygdala circuitry (Royer et al., 1999; Marowsky et al., 2005; Likhtik et al., 2008). ITCs comprise three distinct clusters of GABAergic neurons along the medial and lateral boarders of the basolateral complex as well as in the main intercalated nucleus (IA), each with unique functions (Nitecka and Ben-Ari, 1987; McDonald and Augustine, 1993; Paré and Smith, 1993; Geracitano et al., 2007; Zikopoulos et al., 2016). Lateral clusters have been shown to regulate activity of neurons in the LA and BLA, whereas medial ITC clusters gate signaling from the basolateral complex to the CeA, which serves as the output of the amygdala (Marowsky et al., 2005; Ehrlich et al., 2009; Palomares-Castillo et al., 2012; Duvarci and Pare, 2014). Moreover, the medial ITCs have been shown to play a crucial role in fear extinction (Jüngling et al., 2008; Likhtik et al., 2008). Whereas the role of ITCs in fear extinction is well established, their role in other amygdalar functions remains relatively uncharacterized.
In mice, ITCs originate at embryonic time points in the dorsal lateral ganglionic eminence (dLGE) and subsequently migrate to the amygdala via the lateral migratory stream (LMS) (Carney et al., 2009; Waclaw et al., 2010; Cocas et al., 2011). The zinc finger transcription factor Tshz1 is expressed in a subpopulation of dLGE cells as well as in a subset of mature dLGE-derived olfactory bulb interneurons and mature ITCs (Caubit et al., 2005). In addition to a role in soft palate, middle ear, and skeletal development (Coré et al., 2007), Tshz1 has recently been shown to play a key role in the migration and development of olfactory bulb interneuron subtypes (Ragancokova et al., 2014). Interestingly, these neurons are also derived from the dLGE (Stenman et al., 2003a). However, the role Tshz1 plays in ITC development and function remains unexplored.
Here we use germline and conditional mutant mice to investigate the function of Tshz1 in the development of ITCs. We show that Tshz1 is first expressed in cells of the dLGE, which exit into the LMS and that its expression persists in mature ITCs. In Tshz1 mutant mice, ITCs displayed abnormal migration and increased cell death. Additionally, Tshz1 mutant ITCs display ectopic expression of the dLGE gene Sp8 and a loss of the ITC marker Foxp2. Moreover, Foxp2 homozygous mutant mice displayed impaired ITC survival at postnatal stages, suggesting that Foxp2 may play a critical role downstream of Tshz1 in the survival of ITCs. Interestingly, ventral forebrain-specific Tshz1 conditional mutant (cKO) mice showed behavioral deficits related to fear, depression, and abnormal socialization reminiscent of depressive disorders in human patients with distal 18q deletions, including the Tshz1 locus (Daviss et al., 2013), suggesting a potential role for ITCs in the regulation of these behaviors. Overall, our results establish a critical role for Tshz1 in the development of ITCs and the assembly of neural circuitry regulating fear, depression-like and social behaviors.
Materials and Methods
Animals
Animal protocols were conducted in accordance with guidelines set forth by the Cincinnati Children's Hospital Medical Center Institutional Animal Care and Use Committee and the National Institutes of Health. All mice used in this study were maintained on an outbred background. Dlx1-Cre mice (RRID:MMRRC_036076-UCD) were obtained from GENSAT (Gong et al., 2007; Gerfen et al., 2013) and were genotyped with the following primers: Dlx1-Cre5 (5′-ATGCAAGAGAGCCGACCAAT-3′) and Dlx1-Cre3 (5′-GGCAAACGGACAGAAGCATT-3′). Sp8-GFP BAC (RRID:MMRRC_034608-UCD) mice were obtained from GENSAT (Gong et al., 2003) and genotyped with the primers GFP57-5′ (5′-AGCAAAGACCCCAACGAGAAGC-3′) and GFP57-3′ (5′-CCAACAACAGATGGCTGGCAAC-3′). Tshz1GFP mice (Ragancokova et al., 2014) were genotyped with either of the following two primer pairs: Tshz1GFP5 (5′-GTTGAGGTGGCCTTGTAAGC-3′) and Tshz1GFP-GFP3 (5′-AAGTCGTGCTGCTTCATGTG-3′) or EGFP5 (5′-GACGTAAACGGCCACAAGTTC) and EGFP3 (5′-CTTCAGCTCGATGCGGTTCA-3′). The Tshz1Flox allele (Ragancokova et al., 2014) was genotyped with the following primers: Tshz1RA5 (ATCAGGGGTCTTGGTGTCCT) and Tshz1RA-WT3 (5′-AGTTCAGTCCTTCCGTGGTG-3′). The Tshz1Flox mice were crossed with EIIa-cre mice (The Jackson Laboratory; RRID:IMSR_JAX:003724) to generate the recombined null allele Tshz1RA and genotyped with the following primers: Tshz1RA5 (5′-ATCAGGGGTCTTGGTGTCCT-3′) and Tshz1RA-RA3 (5′-TCCCCACAGCCTCTAACCATA-3′). The Tshz1WT allele was genotyped with the primer set: Tshz1GFP5: (5′-GTTGAGGTGGCCTTGTAAGC-3′) and Tshz1GFP-WT3 (5′-ATTCGCTCTCCTGAATGTCC-3′). The Gsx2RA allele (RRID:MGI:4412087) (Waclaw et al., 2009) was genotyped with the primers: Gsx2RA5: (5′-ACGGAGATTCCACTGCCTCT-3′) and Gsx2RA3 (5′-CTCCCAGACACAGATCCAGAC-3′). The Gsx2WT allele was genotyped with the primers Gsx2–1437 (5′-GCATCCACCCCAAATCTCAGTC-3′) and Gsx2-Int5b (5′-CCACGGAGATTCCACTGCC-3′). Foxp2S321X mice (RRID:MGI:3795717) were genotyped as described previously (Gaub et al., 2010).
For staging of embryos, the day of vaginal plug detection was considered embryonic day 0.5 (E0.5). Brains were collected at the time point indicated in the figures. Brains of embryos E15.5 and older were dissected from the skull before fixation, whereas brains of embryos E14.5 and younger were fixed with the forming skull intact. Tissues were fixed in 4% PFA overnight. Brains at P3 and younger were cryoprotected in 30% sucrose, and 12 μm sections were collected with a cryostat and stored at −20°C. Brains that were P12 and older were cryoprotected in 12% sucrose and sectioned on a sliding microtome at 35 μm. Sections were stored at −20°C in a solution of 30% glycerol/30% ethylene glycol in PBS.
Immunohistochemistry
Sections from brains P12 and older were stained free-floating and subsequently mounted on slides, whereas staining of brains that were P3 and younger was performed on slides. Immunohistochemistry was performed as described by Olsson et al. (1997). Immunofluorescence staining was performed as described by Qin et al. (2016). Primary antibodies were used at the following concentrations: guinea pig anti-μ opioid receptor (1:1000, Millipore, RRID:AB_177511) rabbit anti-cleaved Caspase-3 (1:200, Cell Signaling Technology, RRID:AB_2341188), guinea pig anti-doublecortin (1:3000, Millipore, RRID:AB_1586992), rabbit anti-Er81 (1:1000) (Arber et al., 2000), rabbit anti-ErbB4 (1:1000) (Zhu et al., 1995), rabbit anti-Foxp1 (1:5000, Abcam, RRID:AB_732428) rabbit anti-Foxp2 (1:5000, Abcam, RRID:AB_2107107), goat anti-Foxp2 (1:1000, Abcam, RRID:AB_1268914), chicken anti-GFP (1:1000, Aves Laboratories, RRID:AB_10000240), rabbit anti-Gsx2 (1:5000) (Toresson et al., 2000), rabbit anti-Ki67 (1:1000, Novacastra, RRID:AB_442102), rabbit anti-Mef2c (1:2000, Proteintech, RRID:AB_513447), rabbit anti-Meis2 (1:500, Atlas Antibodies, RRID:AB_611953), rabbit anti-Pax6 (1:1000, Biolegend, RRID:AB_291612), and goat anti-Sp8 (1:5000, Santa Cruz Biotechnology, RRID:AB_2194626). Secondary antibodies used were as follows: donkey anti-chicken conjugated with Alexa-488 (1:400, Jackson ImmunoResearch Laboratories, RRID:AB_2340375); donkey anti-goat conjugated with Alexa-594 (1:400, Jackson ImmunoResearch Laboratories, RRID:AB_2340434); donkey anti-guinea pig conjugated with Alexa-594 (1:400, Jackson ImmunoResearch Laboratories, RRID:AB_2340475); and donkey anti-rabbit conjugated with Alexa-594, Cy3, or Alexa-647 (1:400, Jackson ImmunoResearch Laboratories, RRID:AB_2340622, RRID:AB_2307443, and AB_2340625, respectively). Donkey anti-chicken conjugated with biotin (1:200, Jackson ImmunoResearch Laboratories, RRID:AB_2340355) followed by ABC HRP kit (both reagents 1:200, Vectastain, RRID:AB_2336827) was used for immunohistochemistry.
Digital micrographs of immunohistochemical stains were acquired with a Nikon 90i upright microscope. For fluorescent stains, Z stacks were acquired with either a Nikon A1R LUN-V laser scanning inverted confocal microscope or a Nikon A1 LUN-A laser scanning inverted confocal microscope. Z stacks were converted into maximum intensity projections using NIS-elements software. Brightness and contrast or color adjustments were made equally to both control and mutant images using either GIMP 2 or Adobe Photoshop CS6 software.
In situ hybridization
In situ hybridization was performed at 65°C on 12 μm cryosections as described by Toresson et al. (1999). Tshz1 coding domain antisense probe was generated using the primer pair Tshz15 (5′-GCATCAAGAAGCAACCGGAC-3′) and T3-Tshz13 (5′-AATTAACCCTCACTAAAGGGAGACTTGGGAGTCAGACGACCTG-3′). Adora2a antisense probe was generated using the primer pair Adora2a5 (5′-GGTTTGAGTGGGTACACGGC-3′) and T3-Adora2a3 (5′-AATTAACCCTCACTAAAGGGAGAGCAGTTGATGATGTGCAGGG-3′). Cyp26b1 antisense probe was generated with the primer pair Cyp26b15 (5′-GGGTGGAAGACGAGGGATTC-3′) and T3-Cyp26b13 (5′-AATTAACCCTCACTAAAGGCAACGAGACACACGAACACG-3′). Digital micrographs were obtained with a Nikon 90i Upright microscope. To generate overlays, in situ images were pseudocolored red using Adobe Photoshop CS6 software and superimposed onto micrographs of GFP immunohistochemical staining (pseudocolored green) from immediately adjacent sections. Images were rotated and resized to align the two images using the hippocampus and edge of the cortex as landmarks.
RNA sequencing
E16.5 Embryos were harvested and stored on ice while tail tissue samples were used for genotyping. Brains from Tshz1GFP/+ or Tshz1GFP/RA embryos were dissected, quickly embedded in low melting agarose at 36°C, and hardened on ice. Brains were cut into 700 μm coronal sections using a vibratome. The ventrolateral portions of caudal telencephalic sections containing the amygdala were dissected as depicted in Figure 6A. Tissue from embryos of the same genotype was pooled in PBS and dissociated. Cell suspensions were diluted to a concentration of ∼1.5 × 106 cells/ml, and GFP-expressing cells were isolated by FACS sorting. Sorted cells were collected in 350 μl buffer RLT (QIAGEN) with 1% (v/v) 2-mercaptoethanol, and RNA was isolated using the QIAGEN RNeasy Micro Kit. Double-stranded cDNA was generated and amplified with the NuGEN Ovation RNA-Seq System version 2. The Nextera XT DNA Sample Preparation Kit was used to create DNA library templates from the double-stranded cDNA. The size of the libraries for each sample was measured using the Agilent HS DNA chip. The samples were placed in a pool, and the concentration of the pool was optimized to acquire at least 30–35 million reads per sample. Paired 75 bp reads were obtained with the Illumina HiSeq 2500 platform. Sequencing data have been deposited in NCBI's Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE99164 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE99164). Reads were mapped to the mm10 transcriptome using RSEM (Li and Dewey, 2011).
Figure 6.

Increased postnatal apoptosis in Tshz1 conditional mutants. A–F, Immunostaining for cleaved Caspase-3 reveals a 1.3-fold increase in apoptotic cells in Tshz1 conditional mutants (D–F) relative to controls (A–C). G, Quantification of total cleaved Caspase-3 cells found within the amygdala, the region occupied by the ectopic clumped cells, and the LMS in mutants (n = 3) and the corresponding region in controls (n = 3). Quantifications are displayed as mean ± SEM. **p ≤ 0.01. Scale bars, 100 μm.
Behavioral testing procedures
Male mice were weaned at postnatal day 28 and housed with littermates. Behavioral testing was performed between 9:00 A.M. and 3:00 P.M. by an experimenter who was blind to genotype. All mice were sequentially tested in the behaviors listed below during the light phase of the light/dark cycle with a 3 d intertest interval between each test.
Elevated zero maze test.
Mice were placed in the maze consisting of two open quadrants and two closed quadrants elevated 24 inches off the floor and left undisturbed for 5 min. Mice were video recorded for the duration of testing. Time spent in the open quadrants and number of entries into the open quadrants were measured by a trained observer blind to experimental groups (Zarrindast et al., 2012).
Social interaction test.
The social interaction test was performed as described by Spencer et al. (2011). Briefly, individual mice were housed for 2 d and 2 nights in one side of a partitioned cage divided in half by a clear perforated (0.6 cm diameter holes) partition with a partner mouse of either the same or different genotype. On the following day (10:00 A.M. to 2:00 P.M.), the partition was removed and mice were acclimated for 5 min before interaction was video recorded for the next 10 min. Social behavior was later scored using the video recordings by an observer (Spencer et al., 2011). Scored behaviors were grouped into three main categories: (I) active social behavior, which is any behavior initiated by the experimental mouse toward the partner mouse (categorized as either investigative or aggressive), including the following: (1) anogenital sniffing, (2) nonanogenital sniffing, (3) direct aggressive attacks, (4) lateral threats, (5) tail rattling, (6) chasing, (7) aggressive grooming, and (8) wrestling/boxing; (II) passive social behavior, defined as behavior of the experimental mouse responding to behavior initiated by the partner mouse, including (1) freezing, (2) fleeing, (3) defeat postures, (4) acceptance of the partner mouse investigation without defensive behavior, and (5) active defense; and (III) nonsocial behaviors, including (1) cage exploration, (2) rest, (3) self-grooming, and (4) eating.
Forced swim test (FST).
As previously described (Boyle et al., 2005), mice were placed in a 2 L beaker with 1.3 L of water (18°C–20°C). The level of the water prevented the animals from escaping or from reaching the bottom of the container. Mice were continuously monitored for immobility behavior from 1 to 6 min of a 6 min trial. Immobility was defined as the lack of all motion, except respiration, and the minimal movement required to keep the mouse afloat. At the end of the trial, the mouse was removed from the water, dried, and returned to its home cage.
Open field test.
Following the above described test battery, mice were assessed in an automated locomotor activity chamber (Photobeam Activity System, San Diego Instruments) for 1 h as described by Stottmann et al. (2017). Activity chambers were 41 cm (width) × 41 cm (depth) × 38 cm (height) with 16 photobeams spaced 2.5 cm apart in the x and y planes.
Fear conditioning and extinction.
A separate cohort of male mice, between 6 and 11 months of age, was used for fear conditioning and extinction and did not undergo the above behavioral test battery. Testing was conducted over 5 d. Mice were placed in 25 × 25 cm conditioned fear boxes (San Diego Instruments) with grid floors, speakers, and light mounted in the ceiling, and infrared photocells in the x and y planes. On day 1, mice were habituated to the arena for 20 min. On day 2, conditioning consisted of a 3 min habituation followed by 6 tone/light (conditioned stimuli [CS])-footshock (unconditioned stimulus [US]) pairings. The tone (82 dB, 2 kHz) and light were on for 8 s followed by a footshock that lasted 2 s (1 mA) through the grid floor. There was a 100 s intertrial interval between pairings. On day 3, contextual fear was tested by placing mice in the same chamber for 6 min with no CS present. On day 4, cued fear and extinction were tested by placing mice in a different chamber (black triangular boxes with solid floor), with no tone for the first 3 min followed by light and tone for another 3 min. This sequence was then repeated 20 times to extinguish the CS-US association. On day 5, the extinction sequence was repeated 11 times as a test of extinction recall.
Experimental design and statistical analysis
Both male and female mice were included in all anatomical analyses. For observations not accompanied by cell counts, at least 3 mice/embryos were analyzed for each condition and time point described. For cell counts, all cells expressing the indicated markers within the indicated areas of interest were counted from three consecutive sections from 3 or 4 mice/embryos (n values, ages, and regions of interest indicated in figures and figure legends) using either Imaris or ImageJ (Schneider et al., 2012) software. Statistics comparing cell numbers or percentage coexpression between controls and mutants were performed using a two-tailed t test (Microsoft Excel) with the variance parameter determined by the result of an F test. Marker coexpression between three distinct regions (see Fig. 3F) were compared using a single-factor ANOVA. Significance was set at p ≤ 0.05. Bar graphs represent mean ± SEM. Bar graphs depicting cell numbers indicate total cells counted across three sections.
Figure 3.
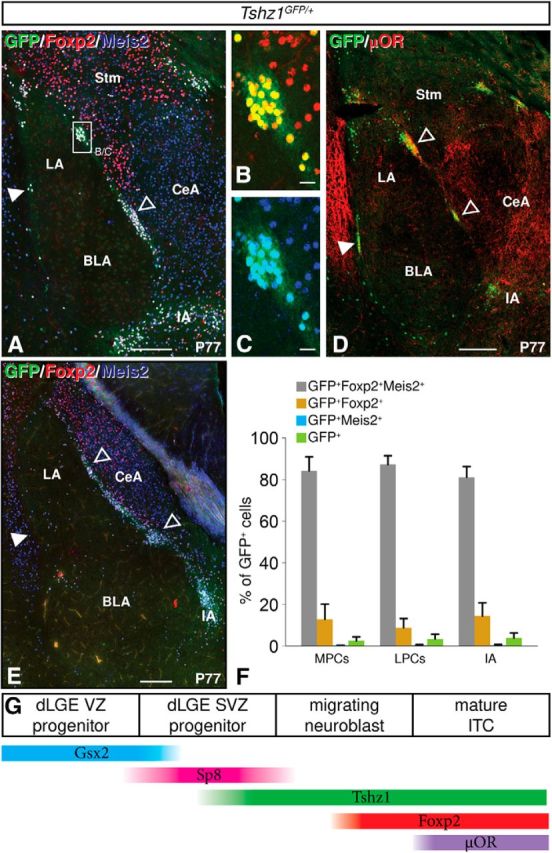
GFP expression in postnatal Tshz1GFP/+ mice. A–D, Adult ITCs maintain robust expression of GFP driven by the Tshz1 allele, Foxp2, and Meis2 (A–C) as well as μOR (D). E, Robust GFP expression colocalizing with Foxp2 and Meis2 is apparent in ITCs at posterior levels as well. F, Quantification of colocalization of GFP with ITC markers Foxp2 and Meis2 in postnatal mice (n = 4). G, Schematic depicting changes in gene expression that occur in the ITC lineage. Solid arrowheads indicate lateral paracapsular intercalated cell clusters. Open arrowheads indicate medial paracapsular intercalated cell clusters. LPCs, Lateral paracapsular clusters; MPCs, medial paracapsular clusters; Stm, striatum. Quantifications are displayed as mean ± SEM. Scale bars: A, D, E, 200 μm; B, C, 20 μm.
Four mice of each genotype were sequenced for RNA-Seq experiments. Differential expression and significance testing were determined by pairwise comparison of controls and mutants from each litter using the GLM functionality of the EdgeR package following TMM normalization (Robinson et al., 2010). Multiple hypothesis-corrected significance measures were obtained using the Benjamini–Hochberg method, and significance threshold was set at false discovery rate ≤0.1 (Benjamini and Hochberg, 1995). Gene ontology (GO) and KEGG pathway gene sets were obtained from Bioconductor, and enrichment within our dataset was determined by paired analysis in the GAGE R package with the same.dir argument set to FALSE (Luo et al., 2009). Pathways and GO terms were considered significant if the q value following Benjamini–Hochberg adjustment was <0.05. To analyze differential expression of olfactory bulb Tshz1 targets, microarray data from E18.5 embryo olfactory bulbs were obtained from GEO (accession no. GSE51761), and enrichment of genes differentially expressed at p ≤ 0.01 was analyzed with EdgeR's “camera” function.
Male mice were used for all behavioral studies. One cohort of mice ∼6–7 months in age underwent the following tests in order: elevated zero maze, social interaction test, FST, open field test. A second cohort of mice between 6 and 11 months was analyzed for fear conditioning and extinction. The number of mice analyzed for each assay are indicated in the figure legends. Significance for all behavioral tests was set at p ≤ 0.05. All fold changes reported are calculated as (mutant value − control value)/control value.
In the elevated zero maze, differences in the amount of time spent in the open quadrants between controls and mutants were compared with a two-tailed t test assuming equal variance, whereas differences in the number of entries into the open quadrants were compared using a two-tailed t test assuming unequal variance. In the FST, differences between control and mutant mice in the percentage time spent immobile were compared with a two-tailed t test assuming unequal variance.
Four groups of mice were tested in the social interaction test: controls paired with control partners, mutants paired with control partners, controls paired with mutant partners, and mutants paired with mutant partners. Dependent measures were latency to enter partner's portion of the cage, time engaged in active social behavior, and numbers of aggressive, investigative, passive, and nonsocial behaviors. The effects of subject genotype, partner genotype, and the interaction of the two were modeled with a two-way ANOVA, and p values for between-group comparisons were calculated with post hoc Tukey HSD tests.
In the open field test, dependent measures were the total number of infrared photobeam interruptions (beam breaks) and the number of beam breaks in the peripheral and central regions of the apparatus, as well as repetitive breaks of the same photocell beam as an index of fine motor movement. Measures were recorded across twelve 5 min intervals. Effects of genotype, interval, and genotype × interval interaction on distance traveled and time spent in the central region were analyzed using mixed linear factorial ANOVA where interval was a repeated-measures factor (SAS version 9.2, SAS Institute). Degrees of freedom were calculated using the Kenward–Roger method. Significance was set at p ≤ 0.05.
In fear conditioning and extinction experiments, the dependent measure was the number of infrared photobeam interruptions (inverse of freezing). Effects of genotype, test interval, and genotype × interval interaction were analyzed for each day using mixed linear ANOVA models (SAS Proc Mixed, SAS Institute, version 9.3 TS Level 1M2) with an autoregressive-1 covariance structure and interval as a repeated-measures factor. Kenward–Rogers first-order degrees of freedom were used. Significant interactions were analyzed using slice-effect ANOVAs at each level of the repeated-measures factor. The effect of extinction training was tested by comparing the first cued interval on day 5 (extinction testing) to the first cued interval on day 4 (extinction training) using one-tailed paired t tests.
Results
Tshz1 expression characterization
Previous studies have described Tshz1 gene expression in the dLGE and intercalated cell masses of the amygdala as well as in the LMS linking these structures during development (Caubit et al., 2005; Carney et al., 2009; Cocas et al., 2011). To analyze further this Tshz1-expressing population of cells, we investigated the timing and location of GFP expression in Tshz1GFP/+ mice, in which one allele coding for Tshz1 protein was replaced with a GFP-encoding cassette (Ragancokova et al., 2014). Immunostaining for GFP protein (Fig. 1E) recapitulated the expression pattern of the Tshz1 gene (Fig. 1A) within the ventrolateral region of the telencephalon. GFP protein was detectable at E13.5 in cells emerging from the LGE and migrating laterally toward the basolateral mantle region (Fig. 1B). Two gestational days later (i.e., E15.5), Tshz1 expression in the subventricular zone (SVZ) had become restricted to the dLGE, and robust staining was observed in the LMS as well as in clusters in the forming amygdalar complex (Fig. 1C). By E18.5, several distinct clusters of GFP+ cells were observed to surround the basolateral amygdalar complex as well as in the main IA (Fig. 1D,F–H). GFP+ cells coexpressed the forkhead transcription factor Foxp2, a previously described ITC marker (Fig. 1F) (Takahashi et al., 2008; Kaoru et al., 2010; Waclaw et al., 2010). These cell clusters encapsulated the BLA, marked by Er81, and the LA as labeled by Mef2c (Fig. 1G,H) (Stenman et al., 2003b; Waclaw et al., 2010). GFP+ cells were largely absent from the LA, BLA, or CeA.
Figure 1.

Tshz1GFP drives GFP in the LGE and ITC clusters. A, In situ hybridization showing Tshz1 gene expression in the LGE, LMS (arrow), and ITC clusters. Solid arrowhead indicates lateral. Open arrowhead indicates medial. B, Immunohistology for GFP (green) in E13.5 Tshz1GFP/+ mice shows GFP protein extending from the dLGE SVZ to the mantle zone. C, D, At E15.5 (C) and E18.5 (D), GFP protein expression refines to a distinct stream (arrow) emerging from the dLGE and several contiguous clusters in the amygdala comprising the lateral paracapsular clusters (solid arrowheads), medial paracapsular clusters (open arrowheads), and IA. E, GFP immunohistochemical staining recapitulates the Tshz1 expression pattern (A). F, Amygdalar GFP staining colocalizes with the ITC marker Foxp2. G, H, GFP-labeled cells in the amygdala surround cells expressing the BLA marker Er81 (G) and the LA marker Mef2c (H). BM, Basomedial amygdala; Ctx, cortex; HPC, hippocampus; LGE, lateral ganglionic eminence; MA, medial amygdala; MGE, medial ganglionic eminence; Stm, striatum; TH, thalamus. Scale bars: A–E, 500 μm; F–H, 100 μm.
The dLGE contains cells representing distinct lineages and at different stages of maturity. To understand further the Tshz1-expressing subpopulation of dLGE cells, we costained Tshz1GFP/+ embryo brains for GFP and known markers of previously characterized populations. GFP-expressing cells were distinct from cells expressing the proliferation marker Ki67 (Gerdes et al., 1984) (Fig. 2A,B) and the dorsally enriched LGE progenitor marker Gsx2 (Yun et al., 2001; Waclaw et al., 2009) (Fig. 2C,D). The GFP+ cells in the dLGE showed limited colabeling with the transcription factor Sp8, which marks the SVZ of the dLGE (Waclaw et al., 2006) (Fig. 2E,F). Moreover, Tshz1-driven GFP showed no coexpression with Pax6, which marks another, largely distinct population of dLGE cells (Yun et al., 2001; Stenman et al., 2003b; Waclaw et al., 2006) (Fig. 2G,H). In the LMS and amygdala, GFP+ cells highly coexpressed the migratory neuroblast marker, doublecortin (Francis et al., 1999) (Fig. 2I,J) as well as the ITC marker Foxp2 (Fig. 2K,L). Previous work has shown reduced ITC numbers in Gsx2 and Sp8 mutants (Waclaw et al., 2010). Consistent with this, we observed a 40% reduction in the total number of GFP-labeled cells in the amygdala (t(4) = 4.07, p = 0.015) in Gsx2-null mutants containing the Tshz1GFP allele compared with Gsx2 heterozygous controls (Fig. 2M,N,Q). Further examination revealed that this reduction was largely driven by a loss of cells in the lateral paracapsular clusters (t(4) = 4.33, p = 0.012) and IA (t(4) = 3.10, p = 0.036), whereas the number of cells in the medial paracapsular clusters was not significantly altered (t(4) = 0.44, p = 0.68). To support further the dLGE origin of ITCs, we used E16.5 Sp8-GFP BAC transgenic mice from GENSAT (Gong et al., 2003) to label dLGE progenitors as well as their neuronal offspring (e.g., ITCs). We found that the Sp8-driven GFP signal persists in the ITCs, many of which are marked by Foxp2 (Fig. 2O,P) despite the fact that few of the Tshz1 GFP+ ITCs express Sp8 protein, suggesting that these cells are derived from Sp8-expressing progenitors (Fig. 2E).
Figure 2.
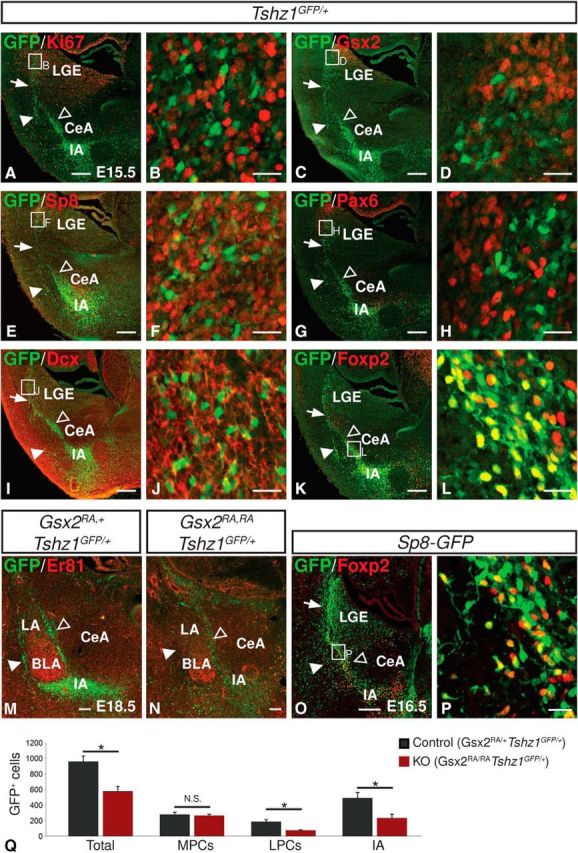
Characterization of ITC lineage progression. A–D, G, H, GFP+ cells in Tshz1GFP/+ embryos do not express Ki67 (A, B), Gsx2 (C, D), or Pax6 (G, H). E, F, GFP+ cells show partial overlap with Sp8. I–L, GFP+ cells highly express doublecortin (I, J) and Foxp2 (K, L). M, N, Gsx2 mutants (N) show reduced amygdala GFP staining compared with heterozygous controls (M). O, P, Sp8-GFP drives robust GFP expression in Foxp2-labeled ITCs. Q, Quantification of amygdalar GFP+ cells in Gsx2 mutants (n = 3) and controls (n = 3). Arrows indicate LMS. Solid arrowheads indicate lateral paracapsular intercalated cell clusters. Open arrowheads indicate medial paracapsular intercalated cell clusters. LGE, Lateral ganglionic eminence; LPCs, lateral paracapsular clusters; MPCs, medial paracapsular clusters. Quantifications are displayed as mean ± SEM. *p ≤ 0.05. Scale bars: A, C, E, G, I, K, M, O, 200 μm; B, D, F, H, J, L, P, 20 μm.
We next assessed GFP expression in Tshz1GFP/+ mice at postnatal time points. GFP protein was found to label ITCs at both rostral (Fig. 3A–D) and caudal (Fig. 3E) portions of the amygdala. As expected, mature Tshz1 GFP+ ITCs in postnatal mice were largely immunopositive for Foxp2 and another ITC marker, Meis2 (Fig. 3A–C) (Stenman et al., 2003b; Takahashi et al., 2008; Kaoru et al., 2010; Waclaw et al., 2010). Among all GFP cells occupying the medial and lateral paracapsular ITC clusters and the IA, 84.0% coexpressed Meis and Foxp2, 12.3% coexpressed Foxp2 only, 2.5% coexpressed Meis2 only, and 2.2% were negative for Foxp2 and Meis2. No significant differences were observed in the proportion of cells coexpressing any of these combinations of markers between the lateral paracapsular clusters, medial paracapsular clusters, and IA (GFP+Foxp2+Meis2+, F(2,9) = 0.32, p = 0.73; GFP+Foxp2+Meis2−, F(2,9) = 0.23, p = 0.80; GFP+Foxp2−Meis2+, F(2,9) = 0.043, p = 0.96; GFP+Foxp2−Meis2−, F(2,9) = 0.099, p = 0.91; Figure 3F). Moreover, μ opioid receptor (μOR), an established marker of ITCs (Jacobsen et al., 2006; Busti et al., 2011; Blaesse et al., 2015), also appears to label the Tshz1 GFP+ ITCs (Fig. 3D). Thus, Tshz1GFP mice appear to be a very useful tool for the early identification of developing ITCs as well as to follow these important amygdalar interneurons into postnatal stages. This is in line with previous reports on Tshz1 expression in other regions of the telencephalon, including the interneurons of the olfactory bulb glomerular layer and granule cell layer as well as the striosomes of the caudate and putamen (Caubit et al., 2005; Ragancokova et al., 2014). Consistent with these reports, we observed robust GFP expression in each of these regions in our mice (data not shown).
Together, our observations led us to propose the ITC differentiation model depicted in Figure 3G in which Gsx2-positive dLGE progenitors give rise to Sp8-positive secondary (i.e., SVZ) progenitors. Sp8 is subsequently downregulated as these progenitors enter the LMS, upregulating Tshz1 and subsequently Foxp2 and μOR in the differentiating ITCs that settle in the amygdala.
ITC abnormalities in Tshz1 mutants
Previous research has demonstrated a requirement for Tshz1 in a subpopulation of olfactory bulb bound neuroblasts that fail to migrate radially once they reach the bulb (Ragancokova et al., 2014). However, no function has been attributed to Tshz1 during development of the other dLGE neuronal subtype (i.e., the ITCs). To determine whether Tshz1 is required for proper ITC development, we first analyzed germline Tshz1 mutants at E18.5 by crossing Tshz1GFP/+ mice with mice containing a Tshz1-null allele (Tshz1RA) to generate Tshz1 mutants (i.e., Tshz1GFP/RA). Examination of the Tshz1GFP/RA mice revealed a complete disruption of the GFP+ ITC distribution pattern with an 82.9% reduction in the number of cells located in the lateral paracapsular clusters (t(2) = 4.95, p = 0.039) and an 82.2% reduction in number of cells located in the IA (t(4) = 10.2, p = 5.19 × 10−4; Fig. 4A–E). There was no change in the expression of the LA/BLA markers Er81 and Mef2c in Tshz1GFP/RA embryos. On the medial side, a large cluster of GFP-labeled cells was observed next to the LA (compare Fig. 4C,D with Fig. 4A,B), although no significant alteration in the total number of cells along the medial boundary of the basolateral complex was detected (t(4) = −1.81, p = 0.14). Further analysis of this population revealed that many of the mutant GFP-expressing cells aberrantly coexpressed Sp8 (compare Fig. 4H,I with Fig. 4F,G; see also Fig. 2E,F). An average of 57.7% of GFP-labeled cells in the amygdala of mutants were observed to coexpress Sp8, whereas only 8.4% of GFP+ cells in the control amygdala expressed Sp8 (t(4) = −8.21, p = 1.20 × 10−3; Fig. 4F–J). Moreover, GFP+ Tshz1 mutant ITCs frequently failed to coexpress Foxp2, with only 62.4% colocalization in contrast to controls in which 95.0% of ITCs were Foxp2-positive (t(4) = 7.46, p = 1.72 × 10−3; Fig. 4K–O). Thus, in the absence of Tshz1, ITCs appear to undergo altered migration and inappropriate differentiation at embryonic stages.
Figure 4.

Disrupted localization and gene expression in Tshz1 mutant ITCs. A, B, GFP+ ITCs are concentrated in the IA and distributed as clusters along the entire extent of the lateral (solid arrowheads) and medial (open arrowheads) borders of the BLA marked by Er81 (A) and LA marked by Mef2c (B) of control embryos (Tshz1GFP/+). C, D, Tshz1 mutants (Tshz1GFP/RA) display a large cluster (open arrowheads) of cells lying dorsal to the BLA (C) and medial to the LA (D), a striking reduction in IA density, and a nearly complete absence of lateral ITCs (solid arrowheads). E, Quantification of ITC numbers in Tshz1 controls (n = 3) and mutants (n = 3). F–I, Mutant ITCs (H, I) show increased Sp8 expression compared with controls (F, G). J, Quantification of the percentage of total GFP+ cells in the amygdala that also coexpress Sp8 (n = 3 for controls and mutants). K–N, Mutant ITCs (M, N) show reduced Foxp2 expression compared with heterozygous controls (M, N). O, Quantification of the percentage of total GFP+ cells in the amygdala that also coexpress Foxp2 (n = 3 for controls and mutants). LPCs, Lateral paracapsular clusters; MPCs, medial paracapsular clusters; Stm, striatum. Quantifications are displayed as mean ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. Scale bars: A–D, F, G, K, M, 100 μm; G, I, L, N, 20 μm.
Tshz1 germline mutants have been shown to die within 24 h of birth (Coré et al., 2007; Ragancokova et al., 2014). To follow the development of Tshz1-null ITCs at postnatal time points, we generated ventral forebrain-specific Tshz1 cKOs. Previous work has shown Dlx2 expression in subpallial germinal zones, and these subpallial regions contribute to the LMS (Panganiban and Rubenstein, 2002; Carney et al., 2006). This expression is governed by a set of shared enhancers located in the intergenic region between Dlx1 and Dlx2 (Ghanem et al., 2003; Park et al., 2004). We obtained a Dlx1-cre BAC transgenic line from GENSAT, reasoning that this intergenic region may be sufficient to drive Cre expression in ITC precursors while sparing Tshz1-expressing populations outside of the basal forebrain (Gong et al., 2007; Gerfen et al., 2013). Indeed, Dlx1-cre;Tshz1GFP/Flox (i.e., ventral forebrain-specific Tshz1 cKO) mice were viable into adulthood and at P21 displayed nearly complete loss of Tshz1 coding mRNA in the dLGE and amygdala, whereas Tshz1 expression in the dorsal thalamus remained largely intact (compare Fig. 5C with Fig. 5A).
Figure 5.
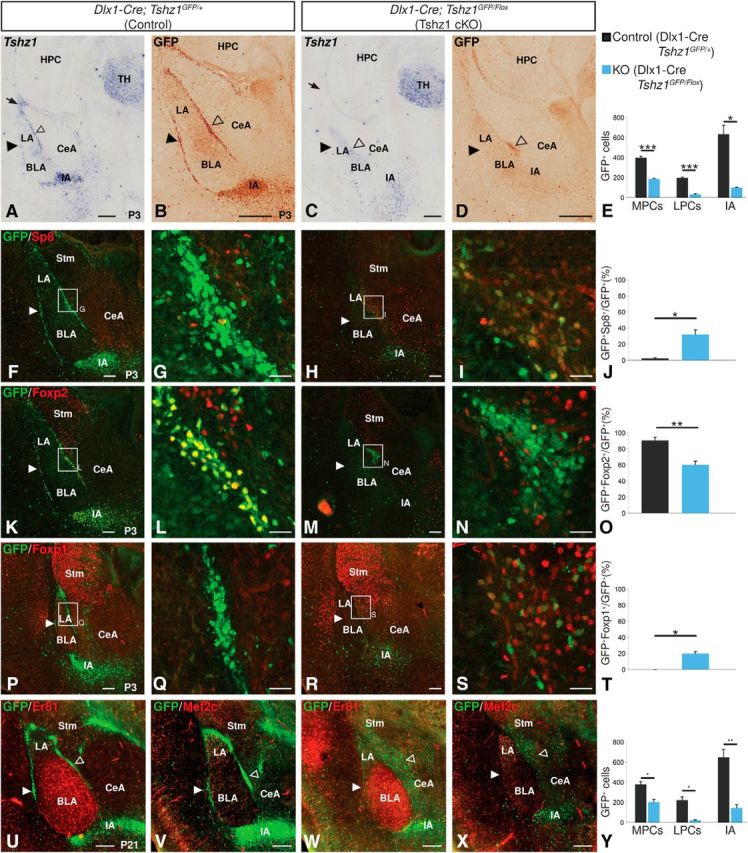
Altered ITCs in postnatal Tshz1 conditional mutants. A, C, In situ hybridization for the coding region of Tshz1 mRNA. Dlx1-Cre efficiently recombines Tshz1 in the LGE (arrows) and ITCs (solid arrowheads indicate lateral paracapsular clusters; open arrowheads indicate medial paracapsular clusters) while sparing Tshz1 in the thalamus of conditional knock-outs. B, D, GFP expression is maintained in the LGE of Tshz1 cKOs and can be seen in a cluster (open arrowhead) of cells medial to the LA and dorsal to the BLA of cKOs. cKOs display a nearly complete loss of GFP-labeled cells in the lateral paracapsular clusters (solid arrowheads). E, Quantification of GFP-labeled ITCs in P3 controls (n = 3) and cKOs (n = 3). F–I, Clustered cells in P3 Tshz1 mutants (H, I) show increased Sp8 expression compared with controls (F, G). J, Quantification of the percentage of total GFP+ cells in the amygdala that also coexpress Sp8 in P3 mice (n = 3 for controls and mutants). K–N, Clustered cells in P3 Tshz1 mutants (M, N) show reduced Foxp2 expression compared with controls (K, L). O, Quantification of the percentage of total GFP+ cells in the amygdala that also coexpress Foxp2 in P3 mice (n = 3 for controls and mutants). P–S, Tshz1 mutant ITCs (R, S) show ectopic Foxp1 expression compared with controls (P, Q). T, Quantification of the percentage of total GFP+ cells in the amygdala that also coexpress Foxp1 in P3 mice (n = 3 for controls and mutants). U–X, In P21 mutants (W, X), ITC numbers are severely reduced compared with controls (U, V), and ITCs are scattered along the medial border of the LA and BLA. Y, Quantification of GFP-labeled ITCs in P21 controls (n = 3) and cKOs (n = 3). HPC, Hippocampus; LPCs, lateral paracapsular clusters; MPCs, medial paracapsular clusters; Stm, striatum; TH, thalamus. Quantifications are displayed as mean ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. Scale bars: A–D, 250 μm; F, H, K, M, P, R, 100 μm; G, I, L, N, Q, S, 25 μm U–X, 200 μm.
Analysis of postnatal Tshz1 cKOs revealed an amygdalar phenotype reminiscent of that seen in the E18.5 germline Tshz1 mutants. Specifically, at P3, Tshz1 cKO mutants displayed a nearly complete loss of ITCs in the lateral paracapsular clusters (83.4% reduction, t(4) = 17.63, p = 6.08 × 10−5) and IA (84.1% reduction, t(2) = 5.89, p = 0.028), and the presence of ectopically located GFP+ cells clustered off the medial border of the lateral amygdala. In contrast to our findings in E18.5 embryos, however, P3 conditional mutants also displayed a 53% reduction in the number of cells observed medial to the basolateral complex compared with controls (t(4) = 12.8, p = 2.12 × 10−4), suggesting a loss of GFP-labeled cells between E18.5 and P3 (Fig. 5B,D,E).
Again, in agreement with the findings at E18.5 (Fig. 4), the clustered mutant GFP+ cells exhibited ectopic Sp8 expression (compare Fig. 5H,I with Fig. 5F,G) with 32.1% of cells observed to coexpress Sp8 in mutants compared with 2.4% of control cells (t(2) = −5.25, p = 0.034; Fig. 5J) and loss of Foxp2 (compare Fig. 5M,N with Fig. 5K,L) with only 60.3% of mutant cells expressing Foxp2 compared with 90.5% of control cells (t(4) = 5.15, p = 6.74 × 10−3; Fig. 5O). Additionally, 19.8% of GFP-labeled cells in cKOs coexpressed Foxp1, a marker of striatal projection neurons (Tamura et al., 2004; Precious et al., 2016) that we only observed in 3.0 × 10−4% of control ITCs. We interpreted this atypical gene expression pattern as an indication that Tshz1-null ITC precursors become stalled in an intermediate, molecularly abnormal, state and fail to differentiate properly into mature ITCs.
GFP-expressing cells were abnormally distributed within the olfactory bulb in a manner similar to that previously reported (Ragancokova et al., 2014). Specifically, GFP+ cells were more prominent within the RMS and were reduced in number in the granule cell layer and glomerular layer (data not shown). Examination of the striosomes by staining for GFP and μOR revealed no alteration in size, morphology, or number (data not shown). Nissl staining revealed no alterations in non–Tshz1-expressing regions known to participate in ITC-containing circuits, such as the LA, BLA, CeA, or mPFC.
Examination of the amygdala in P21 Tshz1 conditional mutants using GFP, Er81, and Mef2c expression revealed a pattern of GFP labeling around the LA/BLA similar to that observed at perinatal stages and suggestive of a perinatal loss of mutant ITCs (compare Fig. 5W,X with Fig. 5U,V). Specifically, mutants exhibited a 46.7% reduction in cells located in the medial paracapsular region (t(4) = 4.48, p = 0.011), an 89.8% reduction in cells located in the lateral paracapsular clusters (t(2) = 5.88, p = 0.028), and a 70.4% reduction in cells observed in the IA (t(4) = 5.89, p = 4.17 × 10−3; Fig. 5Y). Indeed, immunolabeling of P0.5 brains with the apoptosis marker, cleaved Caspase-3, revealed a dramatically increased number of apoptotic cells among the GFP-labeled cKO ITCs relative to controls (compare Fig. 6D–F with Fig. 6A–C). Within the LMS and amygdala of mutant animals, we observed a 1.3-fold increase in cleaved Caspase-3-positive cells compared with controls, indicating that the conditional mutant ITCs are dying already at early postnatal time points (t(4) = −10.5, p = 4.72 × 10−4; Fig. 6G). Apoptotic cells appeared to be concentrated within the clusters of GFP cells located medial to the basolateral complex, suggesting that cell death may underlie the reduction in GFP+ cells in this region between E18.5 (Fig. 4) and P3 (Fig. 5). In total, our results suggest that mutant ITCs in Tshz1 cKOs fail to properly migrate and differentiate and largely undergo apoptosis within the first postnatal week.
We next performed gene-expression profiling of Tshz1 controls and germline mutants to identify perturbations in transcriptional regulation resulting from loss of Tshz1 activity. We chose to collect material from E16.5 embryos because the LMS was most prominent at that time point. The caudal portion of the ventrolateral telencephalon of Tshz1GFP/+ mice and Tshz1GFP/RA mice was dissected, pooled based on genotype, and dissociated for each of four litters. Because GFP+ cells comprise a minority of cells in this region of the brain, we enriched our sample for ITC precursors via FACS isolation of GFP+ cells before library preparation and sequencing (Fig. 7A). Comparison of transcript abundance identified 131 genes upregulated and 85 genes downregulated (Benjamini–Hochberg corrected false discovery rate ≤ 0.1) in Tshz1 mutants compared with controls (Fig. 7B). The list of downregulated genes included known regulators of neuronal migration ErbB4, Prokr2, and Dcc (Hamasaki et al., 2001; Anton et al., 2004; Ng et al., 2005; Prosser et al., 2007; Li et al., 2012). Notably, Prokr2, a gene essential for olfactory bulb development has previously been shown to be downregulated in the olfactory bulb of Tshz1 mutants (Ragancokova et al., 2014). Additionally, Foxp2 expression was found to be reduced, whereas Foxp1 expression was increased, consistent with immunostaining results presented above.
Figure 7.
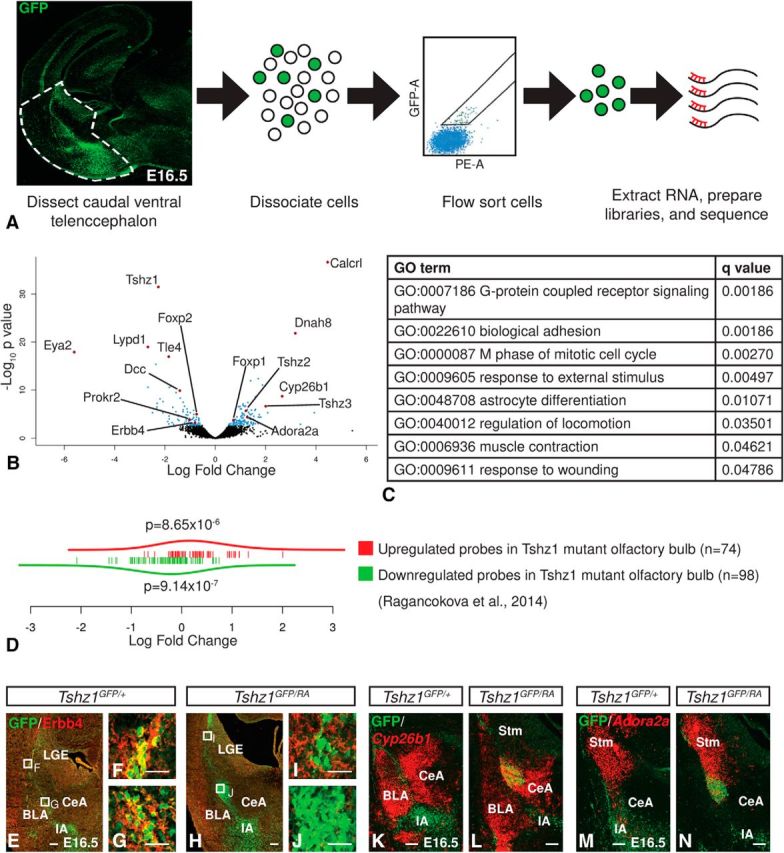
Gene expression profile of Tshz1 mutants. A, Caudal regions of the ventrolateral telencephalon of control (n = 4) and Tshz1 mutant (n = 4) embryos were dissected and GFP+ ITCs were enriched by FACS sorting before RNA extraction and library preparation. B, Volcano plot illustrating global alterations in gene expression in Tshz1 mutants compared with controls with notable genes annotated. C, Significantly disrupted biological process gene ontology terms in Tshz1 mutants (if gene membership of two terms overlapped by 75% or more, only the more significant term is shown). D, Gene set enrichment analysis showed upregulation of genes previously shown to be upregulated in Tshz1 mutant olfactory bulbs and downregulation of genes previously shown to be downregulated in Tshz1 mutant olfactory bulbs. E–J, Immunofluorescence for Erbb4 showed reduced expression in Tshz1 mutant ITCs (H–J) compared with controls (E–G). K, L, Cyp26b1 in situ hybridization and GFP immunohistology pseudocolored and overlaid showing ectopic Cyp26b1 expression in Tshz1 mutant ITCs (L) compared with controls (K). M, N, Adora2a in situ hybridization and GFP immunohistochemistry pseudocolored and overlaid showing ectopic Cyp26b1 expression in Tshz1 mutant ITCs (N) compared with controls (M). Scale bars: E, H, K–N, 100 μm; F, G, I, J, 25 μm.
We performed GO analysis to identify biological processes associated with the altered transcriptional profiles of Tshz1 mutants (Fig. 7C). Among the most significant processes altered in Tshz1 mutants were processes associated with G-protein-coupled receptor signaling, biological adhesion, response to external stimuli, and regulation of locomotion. A similar analysis of differential gene abundance trends associated with molecular functions identified only one altered term, GO:0038023: signaling receptor activity (q = 2.1 × 10−7). Gene set enrichment analysis of KEGG pathways identified only mmu04080: neuroactive ligand–receptor interaction as significantly altered (q = 8.8 × 10−5). These results suggest that a critical function of Tshz1 in ITCs is the regulation of receptors mediating cells' ability to respond appropriately to extracellular migratory and survival cues.
Ragancokova et al. (2014) identified disruptions in the radial migration of Tshz1-null olfactory bulb interneurons at the level of the bulb. To determine whether the migratory deficits detected in the amygdala could be due to misregulation of Tshz1 targets common to both ITCs and olfactory bulb interneurons, we performed gene set enrichment analysis on genes corresponding to microarray probes detected as either upregulated or downregulated (p < 0.01) in olfactory bulbs from Tshz1 mutant embryos by Ragancokova et al. (2014) (Fig. 7D). A set of 74 genes corresponding to probes identified as upregulated in the Tshz1 mutant olfactory bulb demonstrated a significant trend toward upregulation in our amygdala dataset as well (p = 8.65 × 10−6). Likewise, a set of 98 genes corresponding to probes downregulated in the mutant olfactory bulb was also significantly downregulated in the mutant amygdala (p = 9.14 × 10−7), suggesting that Tshz1 may play similar roles during ITC and olfactory bulb interneuron development.
Immunostaining of E16.5 Tshz1 mutants confirmed a reduction of ErbB4, a neuregulin receptor known to play roles in neuronal migration and interneuron activity (compare Fig. 7H–J with Fig. 7E–G) (Anton et al., 2004; Bi et al., 2015). Cyp26b1 (cytochrome P450 subunit 26b1) gene expression can be detected in cells located in the BLA and CeA of wild-type embryos (Fig. 7K) (Abu-Abed et al., 2002). In situ hybridization confirmed robust upregulation in Cyp26b1 in GFP+ Tshz1 mutant ITCs (Fig. 7L). Adora2a is an adenosine receptor that robustly marks the indirect pathway in the striatum of wild-types (Fig. 7M) (Lobo et al., 2006; Heiman et al., 2008), and in situ hybridization demonstrated ectopic Adora2a expression in the mislocated GFP+ ITCs of Tshz1 mutants (Fig. 7N). The ectopic expression of markers of multiple distinct telencephalic regions in Tshz1 mutant ITCs is suggestive of a confused state in these maturing neurons. Indeed, the aberrant expression of markers in mutant ITCs is likely an effect of abnormal responses to local differentiation signals possibly stemming from perturbed Tshz1-dependent receptor expression, which ultimately leads to cell death.
Impaired ITC survival in Foxp2 mutant ITCs
Foxp2 has been shown to play a role in cortical neurogenesis and heterozygous mutations are implicated in human speech disorders (Lai et al., 2001; Fisher and Scharff, 2009; Tsui et al., 2013). Moreover, Foxp2 represents a definitive marker of the ITCs (Takahashi et al., 2008; Kaoru et al., 2010; Waclaw et al., 2010). However, to our knowledge, no role in amygdalar development has been attributed to Foxp2. To determine whether Foxp2 reduction/loss could explain aspects of the Tshz1 mutant ITC phenotype, we next investigated Foxp2S321X mouse mutants, which possess a nonsense mutation leading to a null allele (Gaub et al., 2010). Consistent with previous descriptions of these mutants, Foxp2S321X/S321X; Tshz1GFP/+ mice were runted and died between 2 and 3 weeks of age, whereas Foxp2S321X/+; Tshz1GFP/+ mice were healthy and viable (Groszer et al., 2008; Gaub et al., 2010). Analysis of the amygdala of E18.5 Foxp2S321X/S321X; Tshz1GFP/+ mice revealed no apparent difference in GFP+ ITC number or distribution compared with Foxp2S321X/+; Tshz1GFP/+ controls (Fig. 8A–D). By P12, however, Foxp2S321X/S321X; Tshz1GFP/+ mice exhibited a 34.2% reduction in the number of ITCs compared with controls (t(8) = 3.32, p = 0.0105; Fig. 8E–I), suggestive of a critical role of Foxp2 downstream of Tshz1 for the postnatal survival of ITCs.
Figure 8.

Impaired ITC survival in homozygous Foxp2 mutants carrying the Tshz1GFP allele. A–D, Immunofluorescence analysis of embryonic Foxp2 homozygote mutants (compare C with A) showed loss of Foxp2 and apparently normal numbers of GFP+ ITCs (compare D with B) encapsulating the Er81-positive BLA. E–G, Analysis of postnatal mutants also showed ITCs lacking Foxp2 protein (compare G with E) and revealed a 32% reduction in ITC number in homozygous Foxp2 mutants (H) compared with heterozygous controls (G). I, Quantification of GFP+ ITC numbers in Foxp2 mutants (n = 3) and controls (n = 3). Solid arrowheads indicate lateral paracapsular intercalated cell clusters. Open arrowheads indicate medial paracapsular intercalated cell clusters. Stm, Striatum. Quantifications are displayed as mean ± SEM. Scale bars: A–D, 100 μm; E–H, 200 μm. *p ≤ 0.05.
Tshz1 mutant behavioral abnormalities
Prior studies have associated ITC immunotoxic ablation in rats (Likhtik et al., 2008) or inhibition of excitatory inputs to the ITCs in mice (Jüngling et al., 2008) with an impaired ability to extinguish conditioned fear responses. To assess whether disrupted ITC development results in similar deficits, mice were trained in a fear conditioning paradigm (Laxmi et al., 2003), and movement (as an unbiased assessment of freezing) was measured to assess response to the CS (Jablonski et al., 2017). To simplify our breeding scheme, Dlx1-cre; Tshz1Flox/Flox (cKOs) were compared with Dlx1-cre; Tshz1Flox/+ (controls). Twenty-four hours following habituation, mice were reintroduced to the chamber for 6 min of exploration, followed by six CS-US pairings analyzed in 3 min intervals. A genotype × interval ANOVA showed no effect of genotype and a significant effect of interval (F(3,75.6) = 40.46, p = 0.0001) that reflected the decrease in movement on intervals 3 and 4 following CS-US pairing and showing that Tshz1 cKOs fear condition similarly to controls. There was also a genotype × interval interaction (F(3,75.6) = 5.78, p < 0.0013). Slice-effect ANOVAs on each interval showed a significant effect of genotype on interval 1 (i.e., pre-stim) (F(1,55.21) = 7.41, p = 0.0087) but not thereafter. During interval 1, Tshz1 cKO mice explored less than control mice (Fig. 9A). Twenty-four hours after conditioning, contextual response was assessed over two 3 min intervals in the same chamber. A genotype × interval ANOVA showed no effect of genotype, interval, or genotype × interval (data not shown). On day 4, mice were placed in a novel environment and exposed to 21 unpaired CS presentations to assess cued fear responses as well as to extinguish the conditioned fear response. A genotype × interval ANOVA showed significant effects of genotype (F(1,158) = 9.97, p = 0.0019), interval (F(41,1025) = 6.37, p = 0.0001), and genotype × interval (F(41,1025) = 1.48, p = 0.027; Fig. 9B). Slice-effect ANOVAs on each interval showed an effect on the first non-CS interval (F(1636.2) = 4.80, p = 0.0288), again with the Tshz1 cKO exploring less, but no effects thereafter (Fig. 9B; cued intervals shown are representative). On day 5, we assessed extinction recall over 11 repeated CS-on/CS-off trials. A genotype × interval ANOVA showed no effect of genotype and significant effects of interval (F(21,521) = 3.66, p = 0.0001) and genotype × interval (F(21,521) = 1.68, p = 0.03). Slice-effect ANOVAs on each interval showed an effect on the first CS interval (F(1,289.1) = 4.86, p = 0.0283). As can be seen in Figure 9C, Tshz1 cKO mice showed reduced movement (i.e., increased freezing) compared with controls, indicating greater recall for the CS-US association. The effect size on the recall trial was Cohen's d = 1.09 (large effect). To confirm this, we did paired t tests on each genotype between the first cued trial of day 4 versus the first cued trial of day 5. As predicted, the control mice increased movement (i.e., reduced freezing) on the first cued interval compared with the same interval on the previous day (day 4: 1137 ± 119.8 vs day 5: 1415.7 ± 81.0; t(12) = 2.08, p = 0.03), indicating that they extinguished the fear response (Fig. 9B,C). However, the Tshz1 cKO's response was not different between the days (day 4: 1071.7 ± 80.6 vs day 5: 1111.5 ± 69.2; t(14) = 1.0, not significant). Thus, as is the case in adult rodents (Jüngling et al., 2008; Likhtik et al., 2008), lack of functional ITC signaling in Tshz1 cKOs results in impaired expression of fear extinction.
Figure 9.

Reduced fear extinction recall in Tshz1 cKOs. A, On day 2, Tshz1 cKOs (n = 16) exhibited reduced exploratory behavior before stimulus exposure compared with controls (n = 14). Following paired CS-US exposure, both controls and mutants exhibited reduced movement. B, In a novel environment on day 4, Tshz1 cKOs exhibited reduced exploratory behavior before CS exposure. Both Tshz1 cKOs and controls showed reduced movement following CS exposure. C, On day 5, following the initial CS exposure, control mice activity rate was significantly elevated compared with the first CS exposure on day 4, indicative of an extinguished fear response. Tshz1 cKOs moved less than controls in response to the CS and did not exhibit a significantly different response than they did on day 4, suggesting impaired expression of fear extinction. Data are mean ± SEM. *p ≤ 0.05 (ANOVA). † p ≤ 0.05 (one-tailed paired t test).
In addition to fear, the PFC-amygdala circuit, in which ITCs play an integral function, has been shown to play an important role in regulating anxiety and depression (Jüngling et al., 2008; Price and Drevets, 2010; Palomares-Castillo et al., 2012; Duvarci and Pare, 2014). To determine whether cellular alterations in Tshz1 cKOs lead to abnormalities in these functions, we performed a number of additional behavioral assays on Tshz1 cKOs and controls. We first assessed anxiety-like behavior using the elevated zero maze (Kulkarni et al., 2007). These experiments revealed no significant difference between the Tshz1 cKOs and controls either in the amount of time spent in the open quadrants versus the closed quadrants (t(62) = −0.290, p = 0.77; Fig. 10A) or in the number of entries into the open quadrants (t(42) = −1.85, p = 0.071; Fig. 10B) between controls and Tshz1 conditional mutants. Using the open field test (Hall and Ballachey, 1932; Belzung and Berton, 1997), Tshz1 cKOs were 1.30 times as active as controls (F(1,54) = 4.20, p = 0.045; Fig. 10C). We also observed an effect of interval number (i.e., a reduction in activity over the course of the test; F(11,442) = 3.97, p = 1.0 × 10−4; data not shown), but no effect of genotype-interval interaction (F(11,442) = 1.24, p = 0.26; data not shown). This result is somewhat at odds with the reduced exploratory behavior of the Tshz1 cKOs observed in the fear conditioning paradigm (see Fig. 9A,B); however, the environments in which movement was measured were different; and in the case of the open field test, mice underwent stressful tests (i.e., FST and social interaction) before testing. Tshz1 mutants also displayed a 31.0% reduction in the time spent in the center of the open field chamber compared with the edges, which has been suggested to indicate an anxiety-like phenotype (F(1,94.8) = 10.13, p = 0.002; Fig. 10D). We observed no effect of interval (F(11,426) = 0.79, p = 0.65) or genotype-interval interaction (F(11,426) = 1.24, p = 0.25) on preference for the center versus periphery (data not shown). Depression-like behavior was evaluated with the forced swim test (FST), a routine assay for behavioral despair, in which mice exhibiting depression-like conditions tend to float rather than struggle (Porsolt et al., 1977). Tshz1 cKOs showed an 86% increase in the amount of time spent immobile (i.e., floating) in the FST compared with controls (t(47) = −7.54, p = 1.28 × 10−9; Fig. 10E). Although these tests provide conflicting findings regarding anxiety phenotypes in Tshz1 cKOs, they indicate that these mutants exhibit a depression-like phenotype, which is possibly due to the loss of ITCs in these animals.
Figure 10.
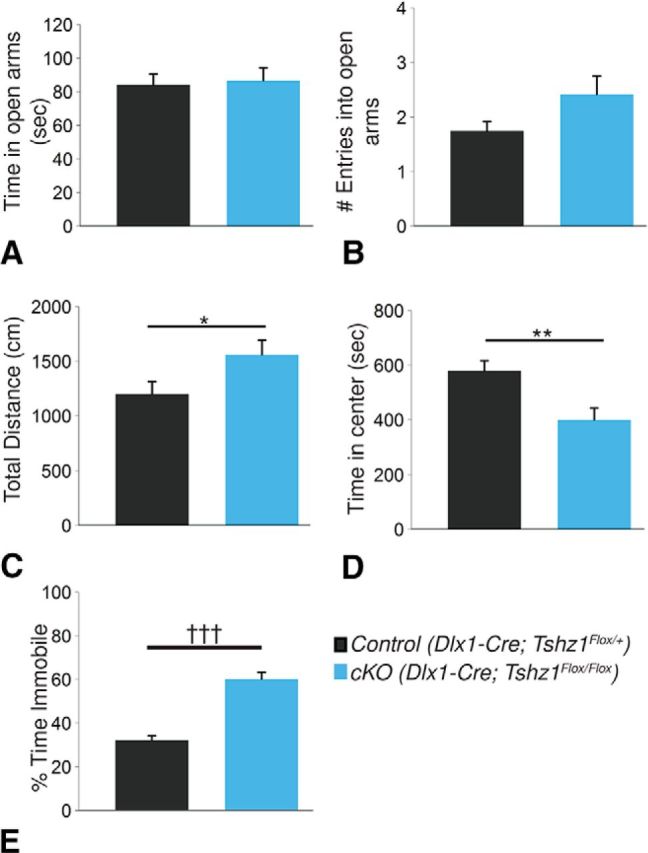
Anxiety and depression-like behaviors examined in Tshz1 cKOs. A, B, In the elevated zero maze, no significant differences were observed between controls (n = 36) and cKOs (n = 29) in either the amount of time spent in the open quadrants of the maze (A) or in the number of entries into the open quadrants (B). C, D, During the open field test, Tshz1 cKOs (n = 18) traveled a greater total distance compared with controls (n = 25; C) and spent less time in the center of the chamber (D). E, In the FST, Tshz1 cKOs (n = 36) spent significantly more time floating compared with controls (n = 29), suggestive of a depression-like phenotype. Data are mean ± SEM. *p ≤ 0.05 (mixed linear factorial ANOVA). **p ≤ 0.01 (mixed linear factorial ANOVA). †††p ≤ 0.001 (two-tailed t test).
Amygdalar circuits have also been implicated in the regulation of social behavior in humans and mice (Adolphs, 2001; Phelps and LeDoux, 2005; Felix-Ortiz and Tye, 2014; Felix-Ortiz et al., 2016). Furthermore, humans with major depressive disorder frequently exhibit impaired social function (Kupferberg et al., 2016), as an inability to perform normal social roles can develop from an underlying depression (Hirschfeld et al., 2000). To determine whether Tshz1 loss perturbs social behavior, Tshz1 cKO mice were analyzed via the direct social interaction test (Spencer et al., 2011). Each mouse was observed for 10 min as it interacted with either a control or cKO stranger. For the latency of the mouse (subject) to enter its partner's portion of the cage (Fig. 11A), we detected significant effects of genotype (F(1,36) = 111.37, p = 2.1 × 10−12), partner's genotype (F(1,36) = 35.17, p = 9.5 × 10−7), and the interaction between-subject genotype and partner genotype (F(1,36) = 30.44, p = 3.3 × 10−6). Regardless of partner, cKOs exhibited significantly longer latency to enter the partner's portion of the cage (fold change = 7.57, p = 1.9 × 10−13 for control partners; fold change = 3.40, p = 1.3 × 10−3 for mutant partners). However, this latency was reduced by 60.4% when the cKO was partnered with another cKO rather than a control (p = 1.7 × 10−12). We also detected effects on the amount of time engaged in active social behavior (Fig. 11B) of subject genotype (F(1,36) = 91.87, p = 1.9 × 10−11), partner genotype (F(1,36) = 17.70, p = 1.64 × 1−4), and the subject genotype-partner genotype interaction (F(1,36) = 17.89, p = 1.5 × 10−4). Conditional mutants spent a significantly shorter portion of the trial engaging in active social behaviors with the partner mouse, regardless of the partner's genotype (67.6% reduction, p = 8.1 × 10−4 for control partner; 83.5% reduction, p = 2.0 × 10−11 for cKO partners). Interestingly, both control and mutant behavior patterns were highly dependent on the genotype of the partner mouse. Control mice spent 95.0% more time engaging in active social behavior with cKO partners than with control partners (p = 4.5 × 10−6). Close examination of the types of behaviors exhibited revealed significant effects of subject genotype (F(1,36) = 11.10, p = 2.0 × 10−3), partner genotype (F(1,36) = 10.57, p = 2.4 × 10−3), and subject genotype-partner genotype interaction (F(1,36) = 12.14, p = 1.3 × 10−3) on the number of aggressive behaviors, such as biting or chasing, that the subject engaged in (Fig. 11C). When paired with cKOs, control mice exhibited a 3.17-fold increase in aggressive behaviors (p = 1.8 × 10−4). The number of investigative behaviors, such as sniffing or grooming, the subject engaged in (Fig. 11D) was similarly affected by subject genotype (F(1,36) = 42.03, p = 1.5 × 10−7), partner genotype (F(1,36) = 25.48, p = 1.3 × 10−5), and subject genotype-partner genotype interaction (F(1,36) = 23.74, p = 2.2 × 10−5). Control mice engaged in 2.41-fold more investigative behaviors when paired with mutant partners than they did when paired with control partners (p = 1.8 × 10−7). The number of passive behaviors (Fig. 11E), such as freezing, fleeing, defensive posturing, or defeat posturing, was also significantly affected by subject genotype (F(1,36) = 101.16, p = 5.3 × 10−12), partner genotype (F(1,36) = 64.94, p = 1.4 × 10−9), and subject genotype-partner genotype interaction (F(1,36) = 82.22, p = 7.9 × 10−11). cKOs paired with control partners displayed a 3.33-fold increase in the number of passive interactions in which they engaged compared with those with cKO partners (p = 1.0 × 10−18). Subject genotype (F(1,36) = 4.97, p = 0.032), partner genotype (F(1,36) = 4.57, p = 0.039), and subject genotype-partner genotype interaction (F(1,36) = 27.71, p = 6.7 × 10−6) also significantly affected the likelihood of mice engaging in nonsocial behaviors, such as digging, cage exploration, or self-grooming (Fig. 11F). cKOs paired with other cKOs engaged in 3.61-fold more nonsocial activities (p = 4.2 × 10−5). Together, these results suggest impaired social interactions on the part of Tshz1 cKOs compared with controls. Conditional mutants rarely engaged in active social behavior. When paired with socially active controls, cKOs tended to respond to their partner's approaches passively. When paired with cKO partners, cKOs showed a preference for nonsocial behavior. Overall, we interpret these findings to indicate that the Tshz1 mutants are exceptionally passive in social situations. Interestingly, the passivity displayed by Tshz1 cKO partners appeared to elicit a significant increase in both aggressive (Fig. 11C) and investigative (Fig. 11D) interactions on the part of control mice, suggesting a complete disinterest by Tshz1 cKOs in establishing a position within the social hierarchy or even self-defense.
Figure 11.
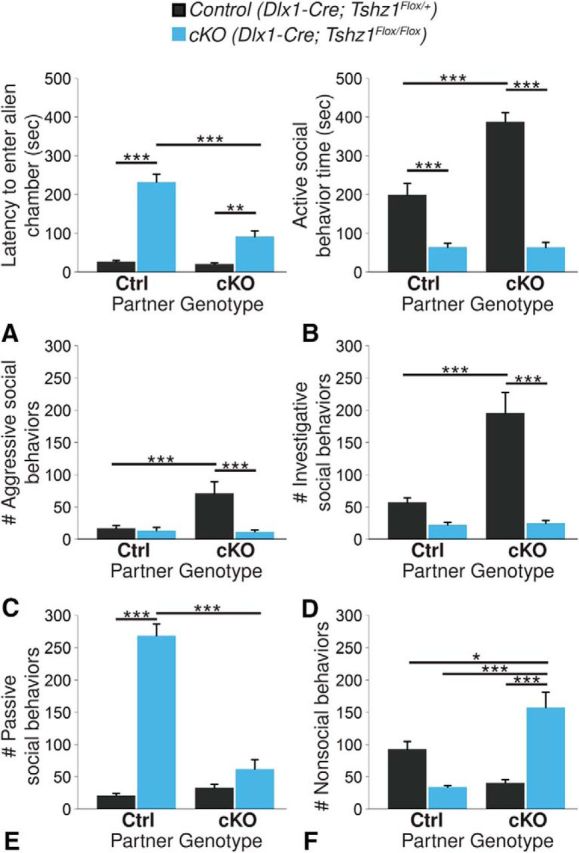
Tshz1 cKOs display impaired social function. Each mouse spent 10 min interacting with an age- and weight-matched partner mouse of either cKO or control genotype (control-control, n = 12; control-cKO, n = 8; cKO-control, n = 8; cKO-cKO, n = 8). A, Tshz1 cKOs were in general more hesitant to enter the partner's portion of the cage than controls. This latency was significantly more pronounced when Tshz1 cKOs were paired with control partners. B, Controls spent more time engaged in active social behaviors than mutants and showed significant increases in the duration of active social behavior when paired with Tshz1 cKO partners. C, D, Control mice paired with Tshz1 cKO partners showed a significantly increased number of both aggressive (C) and investigative (D) social behaviors. E, Tshz1 cKO mice with control partners showed significantly increased numbers of passive social behaviors. F, Tshz1 cKO mice paired with other Tshz1 cKO mice engaged in significantly more nonsocial behaviors. Data are mean ± SEM. *p ≤ 0.05 (two-factor ANOVA). **p ≤ 0.01 (two-factor ANOVA). ***p ≤ 0.001 (two-factor ANOVA).
Discussion
In this study, we have further characterized the molecular cascade that occurs in the lineage of ITCs from their origin as dLGE progenitors, through the LMS, and ultimately within the amygdalar complex. Moreover, we have established that Tshz1GFP mice are a useful tool to mark ITC development as well as placed Tshz1 within the ITC lineage, showing that it first appears as cells leave the dLGE and enter the LMS and remains expressed in mature ITCs of the amygdala. We further established critical roles for Tshz1 and Foxp2 during ITC development. Loss of Tshz1 results in abnormal ITC migration and maturation, leading to impaired neuronal survival at early postnatal time points. The ITC death phenotype is likely mediated, at least in part, by loss of Foxp2, which was consistently reduced in Tshz1 mutant ITCs. ITCs are known to modulate PFC-amygdalar circuitry (Paré et al., 2004; Sotres-Bayon and Quirk, 2010), and these circuits have been implicated in fear, anxiety, and depression (Wellman et al., 2007; Vialou et al., 2014; Tovote et al., 2015). In line with this, we identified behavioral alterations in Tshz1 mutants, which include predicted defects in fear extinction as well as novel phenotypes indicative of depression and impaired social interactions.
Our findings suggest that Tshz1 regulates the molecular code of maturing ITC precursors as evidenced by the maintained expression of the dLGE marker Sp8 in the mutant ITCs within the amygdala. Because Tshz factors are thought to function as repressors (Alexandre et al., 1996; Waltzer et al., 2001; Manfroid et al., 2004), the upregulation of Tshz1 in dLGE cells entering the LMS may be required to downregulate dLGE progenitor identity (i.e., Sp8), allowing for proper migration to the amygdala and differentiation into ITCs. In addition, it appears that the mutant ITCs lose their normal molecular identity (e.g., loss of Foxp2) and become molecularly misspecified. Among the top differentially regulated GO processes from our RNA-Seq analysis of Tshz1 mutant ITCs were G-protein-coupled receptor signaling, biological adhesion, and response to external stimuli. Dysregulation of any of these processes could lead to abnormalities in ITC migration as well as a confused molecular identity. Accordingly, we found a number of genes downregulated that participate in neuronal migration, including ErbB4, Prokr2, and Dcc (Hamasaki et al., 2001; Anton et al., 2004; Ng et al., 2005; Prosser et al., 2007; Li et al., 2012; Ragancokova et al., 2014). Finally, the ultimate fate of Tshz1 mutant ITCs is death, perhaps as a result of altering their ability to interact with the extracellular environment and/or the loss of ITC-specific factors, such as Foxp2, which may more directly regulate cell survival as evidenced by the loss of ITCs in Foxp2 homozygous mutant mice.
It is interesting to note that two separate, but molecularly similar, neuronal populations commonly arise from Gsx2+ (i.e., VZ)-Sp8+ (i.e., SVZ) progenitors in the dLGE: One is the olfactory bulb interneurons that migrate rostrally to the bulb (Stenman et al., 2003a; Waclaw et al., 2006, 2009), and the other is the ITCs that migrate laterally through the LMS (Carney et al., 2009; Waclaw et al., 2010; Cocas et al., 2011). Both neuronal subtypes are dependent of Gsx2 and Sp8 for their normal development (Corbin et al., 2000; Toresson and Campbell, 2001; Yun et al., 2001, 2003; Waclaw et al., 2009, 2010). However, it is currently unknown whether these two neuronal subtypes originate from a common or distinct pool of dLGE progenitors. The migration of dLGE-derived cells toward the ventrolateral telencephalon, including the amygdala, occurs in association with a dense radial glial palisade (Carney et al., 2006). However, Carney et al. (2006) suggested that these neurons undergo chain migration similar to their dLGE counterparts that migrate within the rostral migratory stream to the olfactory bulb (Lois and Alvarez-Buylla, 1994; Wichterle et al., 1999). Ragancokova et al. (2014) showed that migration of olfactory bulb interneuron progenitors within the rostral migratory stream of Tshz1 mutants is largely intact. However, the transition from chain to radial migration, within the bulb, is markedly impaired in Tshz1 mutants. Thus, it may be that, for the normal distribution of ITCs (i.e., lateral and medial clusters), a transition from a chain migration mode to a radial glial-associated migratory mode is required to occur at the apex of the lateral amygdala.
The major function of ITCs relates to their role in the normal expression of fear extinction (Jüngling et al., 2008; Likhtik et al., 2008). While these neurons are not required for the fear conditioning response itself, they play a crucial role in extinguishing a conditioned fear response after multiple nonreinforced presentations of the feared conditioned stimulus. Our findings indicate that Tshz1 cKOs undergo fear conditioning similar to the controls but, unlike the controls, do not extinguish the fear response after extinction training. This is in line with the fact that Tshz1 mutants show severe reductions in ITCs. Importantly, these mutants are compromised in their ITC population from embryonic stages; thus, our results suggest that these neurons are required for the fear extinction learning process from birth. It has been suggested that defects in fear extinction could lead to increased anxiety; thus, ITCs may represent a target for anxiolytic therapies (Jüngling et al., 2008; Likhtik et al., 2008). Indeed, Jüngling et al. (2008) showed that neuropeptide S is able to increase activity of medial ITC clusters and that its application in the amygdala produces anxiolytic effects as demonstrated by a tendency to venture into the center of the testing chamber in the open field test (Belzung and Berton, 1997). Our findings provided mixed results regarding anxiety in Tshz1 mutants. Although they did not show any significant effects in the elevated zero maze, mutants did avoid the center of the open field chamber, compared with controls. It is worth noting, however, that the open field test was performed after the FST and social interaction tests, which are stressful behavioral paradigms.
Despite the above-mentioned requirement for ITCs in conditioned fear responses and anxiety, no role has been attributed to them in the regulation of mood or social behavior. Indeed, ITCs are known to modulate circuits that link the PFC and the amygdala, two structures that play critical roles in depression (Price and Drevets, 2010). To address this, we used the FST, a well-established readout of depressive-like behavior as measured by the time a rodent spends immobile (i.e., floating) (Porsolt et al., 1977). We show here that Tshz1 cKO mice exhibit a significant increase in the time spent immobile in the FST. Interestingly, Andolina et al. (2013) speculated that increased ITC activity may underlie an observed reduction in floating time in the FST following suppression of 5-HT-dependent PFC projections to the BLA. Thus, the severe loss of ITCs observed in the Tshz1 mutants would be consistent with their hypothesis, which would predict an increased floating time in a mouse model lacking ITCs. This depressive-like behavior suggests an interesting parallel between Tshz1 mutant mice and human patients with distal 18q deletions, including the Tshz1 locus who have been reported to frequently suffer from major depressive disorders and abnormal social interactions (Daviss et al., 2013).
Human patients with major depressive disorders frequently show abnormal social function (Kupferberg et al., 2016). Despite this, no link between ITCs and social behavior has been postulated. In this respect, Felix-Ortiz et al. (2016) have demonstrated that BLA projections to the PFC and ventral hippocampus (Felix-Ortiz and Tye, 2014) are able to modify social behaviors. Because lateral ITCs modulate the activity of BLA neurons (Marowsky et al., 2005), the loss of these neurons in the Tshz1 mutants may account for the altered social behavior observed. It bears mentioning, however, that Tshz1 mutants have previously been shown to exhibit olfactory deficits (Ragancokova et al., 2014), and we have observed similar olfactory bulb defects in Tshz1 cKOs generated with Dlx1-cre. Thus, aspects of the social interaction phenotypes observed here (i.e., reduced investigative behavior) could be due to olfactory bulb defects. However, it is unlikely that the observed immobility in the FST and lack of self-defense in the social interaction are due to olfactory deficits but more likely as a result of the fear extinction defects for which the ITC phenotype is central.
In conclusion, our findings show that Tshz1 is essential for the correct development of ITCs; and in its absence, ITC precursors migrate abnormally within the amygdalar complex and ultimately die in the early postnatal period. This leaves the PFC-amygdala circuit without ITCs to modulate either the cortical input to the basolateral complex or the output from the basolateral complex. This anatomical phenotype correlates well with the observed defects in expression of fear extinction as well as the appearance of depression-like and social interaction behaviors in the Tshz1 cKOs, suggesting that ITCs play a role in modulating these behaviors. Future studies using chemogenetic manipulations (i.e., designer receptor exclusively activated by designer drugs-DREADDs) (Roth, 2016) of ITC neuronal activity in wild-type animals, may help to uncover the specific role of ITCs in the depressive and social behavioral abnormalities observed in Tshz1 mutants.
Footnotes
This work was supported in part by National Institutes of Health Grant R01 NS044080 to K.C. and National Institute of General Medical Sciences Grant T32 GM063483-14. We thank Cary Lai for the ErbB4 antibody; and Tom Jessell for the Er81 antibody.
The authors declare no competing financial interests.
References
- Abu-Abed S, MacLean G, Fraulob V, Chambon P, Petkovich M, Dollé P (2002) Differential expression of the retinoic acid-metabolizing enzymes CYP26A1 and CYP26B1 during murine organogenesis. Mech Dev 110:173–177. 10.1016/S0925-4773(01)00572-X [DOI] [PubMed] [Google Scholar]
- Adolphs R. (2001) The neurobiology of social cognition. Curr Opin Neurobiol 11:231–239. 10.1016/S0959-4388(00)00202-6 [DOI] [PubMed] [Google Scholar]
- Alexandre E, Graba Y, Fasano L, Gallet A, Perrin L, De Zulueta P, Pradel J, Kerridge S, Jacq B (1996) The Drosophila Teashirt homeotic protein is a DNA-binding protein and modulo, a HOM-C regulated modifier of variegation, is a likely candidate for being a direct target gene. Mech Dev 59:191–204. 10.1016/0925-4773(96)00594-1 [DOI] [PubMed] [Google Scholar]
- Alò R, Avolio E, Mele M, Storino F, Canonaco A, Carelli A, Canonaco M (2014) Excitatory/inhibitory equilibrium of the central amygdala nucleus gates anti-depressive and anxiolytic states in the hamster. Pharmacol Biochem Behav 118:79–86. 10.1016/j.pbb.2014.01.007 [DOI] [PubMed] [Google Scholar]
- Andolina D, Maran D, Valzania A, Conversi D, Puglisi-Allegra S (2013) Prefrontal/amygdalar system determines stress coping behavior through 5-HT/GABA connection. Neuropsychopharmacology 38:2057–2067. 10.1038/npp.2013.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anton ES, Ghashghaei HT, Weber JL, McCann C, Fischer TM, Cheung ID, Gassmann M, Messing A, Klein R, Schwab MH, Lloyd KC, Lai C (2004) Receptor tyrosine kinase ErbB4 modulates neuroblast migration and placement in the adult forebrain. Nat Neurosci 7:1319–1328. 10.1038/nn1345 [DOI] [PubMed] [Google Scholar]
- Arber S, Ladle DR, Lin JH, Frank E, Jessell TM (2000) ETS gene Er81 controls the formation of functional connections between group Ia sensory afferents and motor neurons. Cell 101:485–498. 10.1016/S0092-8674(00)80859-4 [DOI] [PubMed] [Google Scholar]
- Belzung C, Berton F (1997) Further pharmacological validation of the BALB/c neophobia in the free exploratory paradigm as an animal model of trait anxiety. Behav Pharmacol 8:541–548. 10.1097/00008877-199711000-00012 [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57:289–300. [Google Scholar]
- Bi LL, Sun XD, Zhang J, Lu YS, Chen YH, Wang J, Geng F, Liu F, Zhang M, Liu JH, Li XW, Mei L, Gao TM (2015) Amygdala NRG1-ErbB4 is critical for the modulation of anxiety-like behaviors. Neuropsychopharmacology 40:974–986. 10.1038/npp.2014.274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaesse P, Goedecke L, Bazelot M, Copogna M, Pape HC, Jüngling K (2015) μ-Opioid receptor-mediated inhibition of intercalated neurons and effect on synaptic transmission to the central amygdala. J Neurosci 35:7317–7325. 10.1523/JNEUROSCI.0204-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle MP, Brewer JA, Funatsu M, Wozniak DF, Tsien JZ, Izumi Y, Muglia LJ (2005) Acquired deficit of forebrain glucocorticoid receptor produces depression-like changes in adrenal axis regulation and behavior. Proc Natl Acad Sci U S A 102:473–478. 10.1073/pnas.0406458102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busti D, Geracitano R, Whittle N, Dalezios Y, Mańko M, Kaufmann W, Sätzler K, Singewald N, Capogna M, Ferraguti F (2011) Different fear states engage distinct networks within the intercalated cell clusters of the amygdala. J Neurosci 31:5131–5144. 10.1523/JNEUROSCI.6100-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney RS, Alfonso TB, Cohen D, Dai H, Nery S, Stoica B, Slotkin J, Bregman BS, Fishell G, Corbin JG (2006) Cell migration along the lateral cortical stream to the developing basal telencephalic limbic system. J Neurosci 26:11562–11574. 10.1523/JNEUROSCI.3092-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney RS, Cocas LA, Hirata T, Mansfield K, Corbin JG (2009) Differential regulation of telencephalic pallial-subpallial boundary patterning by Pax6 and Gsh2. Cereb Cortex 19:745–759. 10.1093/cercor/bhn123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caubit X, Tiveron MC, Cremer H, Fasano L (2005) Expression patterns of the three Teashirt-related genes define specific boundaries in the developing and postnatal mouse forebrain. J Comp Neurol 486:76–88. 10.1002/cne.20500 [DOI] [PubMed] [Google Scholar]
- Cocas LA, Georgala PA, Mangin JM, Clegg JM, Kessaris N, Haydar TF, Gallo V, Price DJ, Corbin JG (2011) Pax6 is required at the telencephalic pallial-subpallial boundary for the generation of neuronal diversity in the postnatal limbic system. J Neurosci 31:5313–5324. 10.1523/JNEUROSCI.3867-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin JG, Gaiano N, Machold RP, Langston A, Fishell G (2000) The Gsh2 homeodomain gene controls multiple aspects of telencephalic development. Dev Camb Engl 127:5007–5020. [DOI] [PubMed] [Google Scholar]
- Coré N, Caubit X, Metchat A, Boned A, Djabali M, Fasano L (2007) Tshz1 is required for axial skeleton, soft palate and middle ear development in mice. Dev Biol 308:407–420. 10.1016/j.ydbio.2007.05.038 [DOI] [PubMed] [Google Scholar]
- Daviss WB, O'Donnell L, Soileau BT, Heard P, Carter E, Pliszka SR, Gelfond JA, Hale DE, Cody JD (2013) Mood disorders in individuals with distal 18q deletions. Am J Med Genet B Neuropsychiatr Genet 162B:879–888. 10.1002/ajmg.b.32197 [DOI] [PubMed] [Google Scholar]
- Duvarci S, Pare D (2014) Amygdala microcircuits controlling learned fear. Neuron 82:966–980. 10.1016/j.neuron.2014.04.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE (2002) Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30:207–210. 10.1093/nar/30.1.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Lüthi A (2009) Amygdala inhibitory circuits and the control of fear memory. Neuron 62:757–771. 10.1016/j.neuron.2009.05.026 [DOI] [PubMed] [Google Scholar]
- Felix-Ortiz AC, Tye KM (2014) Amygdala inputs to the ventral hippocampus bidirectionally modulate social behavior. J Neurosci 34:586–595. 10.1523/JNEUROSCI.4257-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix-Ortiz AC, Burgos-Robles A, Bhagat ND, Leppla CA, Tye KM (2016) Bidirectional modulation of anxiety-related and social behaviors by amygdala projections to the medial prefrontal cortex. Neuroscience 321:197–209. 10.1016/j.neuroscience.2015.07.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher SE, Scharff C (2009) FOXP2 as a molecular window into speech and language. Trends Genet 25:166–177. 10.1016/j.tig.2009.03.002 [DOI] [PubMed] [Google Scholar]
- Francis F, Koulakoff A, Boucher D, Chafey P, Schaar B, Vinet MC, Friocourt G, McDonnell N, Reiner O, Kahn A, McConnell SK, Berwald-Netter Y, Denoulet P, Chelly J (1999) Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron 23:247–256. 10.1016/S0896-6273(00)80777-1 [DOI] [PubMed] [Google Scholar]
- Gabbott PL, Warner TA, Jays PR, Salway P, Busby SJ (2005) Prefrontal cortex in the rat: projections to subcortical autonomic, motor, and limbic centers. J Comp Neurol 492:145–177. 10.1002/cne.20738 [DOI] [PubMed] [Google Scholar]
- Gafford GM, Ressler KJ (2016) Mouse models of fear-related disorders: cell-type-specific manipulations in amygdala. Neuroscience 321:108–120. 10.1016/j.neuroscience.2015.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaub S, Groszer M, Fisher SE, Ehret G (2010) The structure of innate vocalizations in Foxp2-deficient mouse pups. Genes Brain Behav 9:390–401. 10.1111/j.1601-183X.2010.00570.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geracitano R, Kaufmann WA, Szabo G, Ferraguti F, Capogna M (2007) Synaptic heterogeneity between mouse paracapsular intercalated neurons of the amygdala. J Physiol 585:117–134. 10.1113/jphysiol.2007.142570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H (1984) Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol 133:1710–1715. [PubMed] [Google Scholar]
- Gerfen CR, Paletzki R, Heintz N (2013) GENSAT BAC Cre-recombinase driver lines to study the functional organization of cerebral cortical and basal ganglia circuits. Neuron 80:1368–1383. 10.1016/j.neuron.2013.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanem N, Jarinova O, Amores A, Long Q, Hatch G, Park BK, Rubenstein JL, Ekker M (2003) Regulatory roles of conserved intergenic domains in vertebrate Dlx bigene clusters. Genome Res 13:533–543. 10.1101/gr.716103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N (2003) A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature 425:917–925. 10.1038/nature02033 [DOI] [PubMed] [Google Scholar]
- Gong S, Doughty M, Harbaugh CR, Cummins A, Hatten ME, Heintz N, Gerfen CR (2007) Targeting Cre recombinase to specific neuron populations with bacterial artificial chromosome constructs. J Neurosci 27:9817–9823. 10.1523/JNEUROSCI.2707-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groszer M, Keays DA, Deacon RM, de Bono JP, Prasad-Mulcare S, Gaub S, Baum MG, French CA, Nicod J, Coventry JA, Enard W, Fray M, Brown SD, Nolan PM, Pääbo S, Channon KM, Costa RM, Eilers J, Ehret G, Rawlins JN, et al. (2008) Impaired synaptic plasticity and motor learning in mice with a point mutation implicated in human speech deficits. Curr Biol 18:354–362. 10.1016/j.cub.2008.01.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall C, Ballachey EL (1932) A study of the rat's behavior in a field: a contribution to method in comparative psychology. Univ Calif Publ Psychol 6:1–12. [Google Scholar]
- Hamasaki T, Goto S, Nishikawa S, Ushio Y (2001) A role of Netrin-1 in the formation of the subcortical structure striatum: repulsive action on the migration of late-born striatal neurons. J Neurosci 21:4272–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suárez-Fariñas M, Schwarz C, Stephan DA, Surmeier DJ, Greengard P, Heintz N (2008) A translational profiling approach for the molecular characterization of CNS cell types. Cell 135:738–748. 10.1016/j.cell.2008.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschfeld RM, Montgomery SA, Keller MB, Kasper S, Schatzberg AF, Möller HJ, Healy D, Baldwin D, Humble M, Versiani M, Montenegro R, Bourgeois M (2000) Social functioning in depression: a review. J Clin Psychiatry 61:268–275. 10.4088/JCP.v61n0405 [DOI] [PubMed] [Google Scholar]
- Iwata J, LeDoux JE, Meeley MP, Arneric S, Reis DJ (1986) Intrinsic neurons in the amygdaloid field projected to by the medial geniculate body mediate emotional responses conditioned to acoustic stimuli. Brain Res 383:195–214. 10.1016/0006-8993(86)90020-X [DOI] [PubMed] [Google Scholar]
- Jablonski SA, Williams MT, Vorhees CV (2017) Learning and memory effects of neonatal methamphetamine exposure in rats: role of reactive oxygen species and age at assessment. Synapse 71:11. 10.1002/syn.21992 [DOI] [PubMed] [Google Scholar]
- Jacobsen KX, Höistad M, Staines WA, Fuxe K (2006) The distribution of dopamine D1 receptor and μ-opioid receptor 1 receptor immunoreactivities in the amygdala and interstitial nucleus of the posterior limb of the anterior commissure: relationships to tyrosine hydroxylase and opioid peptide terminal systems. Neuroscience 141:2007–2018. 10.1016/j.neuroscience.2006.05.054 [DOI] [PubMed] [Google Scholar]
- Jüngling K, Seidenbecher T, Sosulina L, Lesting J, Sangha S, Clark SD, Okamura N, Duangdao DM, Xu YL, Reinscheid RK, Pape HC (2008) Neuropeptide S-mediated control of fear expression and extinction: role of intercalated GABAergic neurons in the amygdala. Neuron 59:298–310. 10.1016/j.neuron.2008.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaoru T, Liu FC, Ishida M, Oishi T, Hayashi M, Kitagawa M, Shimoda K, Takahashi H (2010) Molecular characterization of the intercalated cell masses of the amygdala: implications for the relationship with the striatum. Neuroscience 166:220–230. 10.1016/j.neuroscience.2009.12.004 [DOI] [PubMed] [Google Scholar]
- Kulkarni SK, Singh K, Bishnoi M (2007) Elevated zero maze: a paradigm to evaluate antianxiety effects of drugs. Methods Find Exp Clin Pharmacol 29:343–348. 10.1358/mf.2007.29.5.1117557 [DOI] [PubMed] [Google Scholar]
- Kupferberg A, Bicks L, Hasler G (2016) Social functioning in major depressive disorder. Neurosci Biobehav Rev 69:313–332. 10.1016/j.neubiorev.2016.07.002 [DOI] [PubMed] [Google Scholar]
- Lai CS, Fisher SE, Hurst JA, Vargha-Khadem F, Monaco AP (2001) A forkhead-domain gene is mutated in a severe speech and language disorder. Nature 413:519–523. 10.1038/35097076 [DOI] [PubMed] [Google Scholar]
- Laxmi TR, Stork O, Pape HC (2003) Generalisation of conditioned fear and its behavioural expression in mice. Behav Brain Res 145:89–98. 10.1016/S0166-4328(03)00101-3 [DOI] [PubMed] [Google Scholar]
- Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. 10.1186/1471-2105-12-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Chou SJ, Hamasaki T, Perez-Garcia CG, O'Leary DD (2012) Neuregulin repellent signaling via ErbB4 restricts GABAergic interneurons to migratory paths from ganglionic eminence to cortical destinations. Neural Dev 7:10. 10.1186/1749-8104-7-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Likhtik E, Pelletier JG, Paz R, Paré D (2005) Prefrontal control of the amygdala. J Neurosci 25:7429–7437. 10.1523/JNEUROSCI.2314-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Likhtik E, Popa D, Apergis-Schoute J, Fidacaro GA, Paré D (2008) Amygdala intercalated neurons are required for expression of fear extinction. Nature 454:642–645. 10.1038/nature07167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo MK, Karsten SL, Gray M, Geschwind DH, Yang XW (2006) FACS-array profiling of striatal projection neuron subtypes in juvenile and adult mouse brains. Nat Neurosci 9:443–452. 10.1038/nn1654 [DOI] [PubMed] [Google Scholar]
- Lois C, Alvarez-Buylla A (1994) Long-distance neuronal migration in the adult mammalian brain. Science 264:1145–1148. 10.1126/science.8178174 [DOI] [PubMed] [Google Scholar]
- Luo W, Friedman MS, Shedden K, Hankenson KD, Woolf PJ (2009) GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics 10:161. 10.1186/1471-2105-10-161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfroid I, Caubit X, Kerridge S, Fasano L (2004) Three putative murine Teashirt orthologues specify trunk structures in Drosophila in the same way as the Drosophila teashirt gene. Development 131:1065–1073. 10.1242/dev.00977 [DOI] [PubMed] [Google Scholar]
- Marowsky A, Yanagawa Y, Obata K, Vogt KE (2005) A specialized subclass of interneurons mediates dopaminergic facilitation of amygdala function. Neuron 48:1025–1037. 10.1016/j.neuron.2005.10.029 [DOI] [PubMed] [Google Scholar]
- Mascagni F, McDonald AJ, Coleman JR (1993) Corticoamygdaloid and corticocortical projections of the rat temporal cortex: a Phaseolus vulgaris leucoagglutinin study. Neuroscience 57:697–715. 10.1016/0306-4522(93)90016-9 [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Augustine JR (1993) Localization of GABA-like immunoreactivity in the monkey amygdala. Neuroscience 52:281–294. 10.1016/0306-4522(93)90156-A [DOI] [PubMed] [Google Scholar]
- Ng KL, Li JD, Cheng MY, Leslie FM, Lee AG, Zhou QY (2005) Dependence of olfactory bulb neurogenesis on prokineticin 2 signaling. Science 308:1923–1927. 10.1126/science.1112103 [DOI] [PubMed] [Google Scholar]
- Nitecka L, Ben-Ari Y (1987) Distribution of GABA-like immunoreactivity in the rat amygdaloid complex. J Comp Neurol 266:45–55. 10.1002/cne.902660105 [DOI] [PubMed] [Google Scholar]
- Olsson M, Campbell K, Turnbull DH (1997) Specification of mouse telencephalic and mid-hindbrain progenitors following heterotopic ultrasound-guided embryonic transplantation. Neuron 19:761–772. 10.1016/S0896-6273(00)80959-9 [DOI] [PubMed] [Google Scholar]
- Palomares-Castillo E, Hernández-Pérez OR, Pérez-Carrera D, Crespo-Ramírez M, Fuxe K, Pérez de la Mora M (2012) The intercalated paracapsular islands as a module for integration of signals regulating anxiety in the amygdala. Brain Res 1476:211–234. 10.1016/j.brainres.2012.03.047 [DOI] [PubMed] [Google Scholar]
- Panganiban G, Rubenstein JL (2002) Developmental functions of the Distal-less/Dlx homeobox genes. Development 129:4371–4386. [DOI] [PubMed] [Google Scholar]
- Paré D, Smith Y (1993) The intercalated cell masses project to the central and medial nuclei of the amygdala in cats. Neuroscience 57:1077–1090. 10.1016/0306-4522(93)90050-P [DOI] [PubMed] [Google Scholar]
- Paré D, Quirk GJ, Ledoux JE (2004) New vistas on amygdala networks in conditioned fear. J Neurophysiol 92:1–9. 10.1152/jn.00153.2004 [DOI] [PubMed] [Google Scholar]
- Park BK, Sperber SM, Choudhury A, Ghanem N, Hatch GT, Sharpe PT, Thomas BL, Ekker M (2004) Intergenic enhancers with distinct activities regulate Dlx gene expression in the mesenchyme of the branchial arches. Dev Biol 268:532–545. 10.1016/j.ydbio.2004.01.010 [DOI] [PubMed] [Google Scholar]
- Phelps EA, LeDoux JE (2005) Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron 48:175–187. 10.1016/j.neuron.2005.09.025 [DOI] [PubMed] [Google Scholar]
- Pitkänen A, Savander V, LeDoux JE (1997) Organization of intra-amygdaloid circuitries in the rat: an emerging framework for understanding functions of the amygdala. Trends Neurosci 20:517–523. 10.1016/S0166-2236(97)01125-9 [DOI] [PubMed] [Google Scholar]
- Porsolt RD, Le Pichon M, Jalfre M (1977) Depression: a new animal model sensitive to antidepressant treatments. Nature 266:730–732. 10.1038/266730a0 [DOI] [PubMed] [Google Scholar]
- Precious SV, Kelly CM, Reddington AE, Vinh NN, Stickland RC, Pekarik V, Scherf C, Jeyasingham R, Glasbey J, Holeiter M, Jones L, Taylor MV, Rosser AE (2016) FoxP1 marks medium spiny neurons from precursors to maturity and is required for their differentiation. Exp Neurol 282:9–18. 10.1016/j.expneurol.2016.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JL, Drevets WC (2010) Neurocircuitry of mood disorders. Neuropsychopharmacology 35:192–216. 10.1038/npp.2009.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser HM, Bradley A, Caldwell MA (2007) Olfactory bulb hypoplasia in Prokr2 null mice stems from defective neuronal progenitor migration and differentiation. Eur J Neurosci 26:3339–3344. 10.1111/j.1460-9568.2007.05958.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S, Madhavan M, Waclaw RR, Nakafuku M, Campbell K (2016) Characterization of a new Gsx2-cre line in the developing mouse telencephalon. Genesis 54:542–549. 10.1002/dvg.22980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragancokova D, Rocca E, Oonk AM, Schulz H, Rohde E, Bednarsch J, Feenstra I, Pennings RJ, Wende H, Garratt AN (2014) TSHZ1-dependent gene regulation is essential for olfactory bulb development and olfaction. J Clin Invest 124:1214–1227. 10.1172/JCI72466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth BL. (2016) DREADDs for neuroscientists. Neuron 89:683–694. 10.1016/j.neuron.2016.01.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer S, Martina M, Paré D (1999) An inhibitory interface gates impulse traffic between the input and output stations of the amygdala. J Neurosci 19:10575–10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitz JB, Drevets WC (2009) Imaging phenotypes of major depressive disorder: genetic correlates. Neuroscience 164:300–330. 10.1016/j.neuroscience.2009.03.082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotres-Bayon F, Quirk GJ (2010) Prefrontal control of fear: more than just extinction. Curr Opin Neurobiol 20:231–235. 10.1016/j.conb.2010.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer CM, Alekseyenko O, Hamilton SM, Thomas AM, Serysheva E, Yuva-Paylor LA, Paylor R (2011) Modifying behavioral phenotypes in Fmr1KO mice: genetic background differences reveal autistic-like responses. Autism Res 4:40–56. 10.1002/aur.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenman J, Toresson H, Campbell K (2003a) Identification of two distinct progenitor populations in the lateral ganglionic eminence: implications for striatal and olfactory bulb neurogenesis. J Neurosci 23:167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenman J, Yu RT, Evans RM, Campbell K (2003b) Tlx and Pax6 co-operate genetically to establish the pallio-subpallial boundary in the embryonic mouse telencephalon. Development 130:1113–1122. 10.1242/dev.00328 [DOI] [PubMed] [Google Scholar]
- Stottmann RW, Driver A, Gutierrez A, Skelton MR, Muntifering M, Stepien C, Knudson L, Kofron M, Vorhees CV, Williams MT (2017) A heterozygous mutation in tubulin, beta 2B (Tubb2b) causes cognitive deficits and hippocampal disorganization. Genes Brain Behav 16:250–259. 10.1111/gbb.12327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Liu FC, Oishi T, Mori T, Higo N, Hayashi M, Hirokawa K, Takahashi H (2008) Expression of FOXP2 in the developing monkey forebrain: comparison with the expression of the genes FOXP1, PBX3, and MEIS2. J Comp Neurol 509:180–189. 10.1002/cne.21740 [DOI] [PubMed] [Google Scholar]
- Tamura S, Morikawa Y, Iwanishi H, Hisaoka T, Senba E (2004) Foxp1 gene expression in projection neurons of the mouse striatum. Neuroscience 124:261–267. 10.1016/j.neuroscience.2003.11.036 [DOI] [PubMed] [Google Scholar]
- Taylor JM, Whalen PJ (2015) Neuroimaging and anxiety: the neural substrates of pathological and non-pathological anxiety. Curr Psychiatry Rep 17:49. 10.1007/s11920-015-0586-9 [DOI] [PubMed] [Google Scholar]
- Toresson H, Campbell K (2001) A role for Gsh1 in the developing striatum and olfactory bulb of Gsh2 mutant mice. Development 128:4769–4780. [DOI] [PubMed] [Google Scholar]
- Toresson H, Mata de Urquiza A, Fagerström C, Perlmann T, Campbell K (1999) Retinoids are produced by glia in the lateral ganglionic eminence and regulate striatal neuron differentiation. Development 126:1317–1326. [DOI] [PubMed] [Google Scholar]
- Toresson H, Potter SS, Campbell K (2000) Genetic control of dorsal-ventral identity in the telencephalon: opposing roles for Pax6 and Gsh2. Development 127:4361–4371. [DOI] [PubMed] [Google Scholar]
- Tovote P, Fadok JP, Lüthi A (2015) Neuronal circuits for fear and anxiety. Nat Rev Neurosci 16:317–331. 10.1038/nrn3945 [DOI] [PubMed] [Google Scholar]
- Tsui D, Vessey JP, Tomita H, Kaplan DR, Miller FD (2013) FoxP2 regulates neurogenesis during embryonic cortical development. J Neurosci 33:244–258. 10.1523/JNEUROSCI.1665-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veening JG, Swanson LW, Sawchenko PE (1984) The organization of projections from the central nucleus of the amygdala to brainstem sites involved in central autonomic regulation: a combined retrograde transport-immunohistochemical study. Brain Res 303:337–357. 10.1016/0006-8993(84)91220-4 [DOI] [PubMed] [Google Scholar]
- Vertes RP. (2004) Differential projections of the infralimbic and prelimbic cortex in the rat. Synapse 51:32–58. 10.1002/syn.10279 [DOI] [PubMed] [Google Scholar]
- Vialou V, Bagot RC, Cahill ME, Ferguson D, Robison AJ, Dietz DM, Fallon B, Mazei-Robison M, Ku SM, Harrigan E, Winstanley CA, Joshi T, Feng J, Berton O, Nestler EJ (2014) Prefrontal cortical circuit for depression- and anxiety-related behaviors mediated by cholecystokinin: role of ΔFosB. J Neurosci 34:3878–3887. 10.1523/JNEUROSCI.1787-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waclaw RR, Allen ZJ 2nd, Bell SM, Erdélyi F, Szabó G, Potter SS, Campbell K (2006) The zinc finger transcription factor Sp8 regulates the generation and diversity of olfactory bulb interneurons. Neuron 49:503–516. 10.1016/j.neuron.2006.01.018 [DOI] [PubMed] [Google Scholar]
- Waclaw RR, Wang B, Pei Z, Ehrman LA, Campbell K (2009) Distinct temporal requirements for the homeobox gene Gsx2 in specifying striatal and olfactory bulb neuronal fates. Neuron 63:451–465. 10.1016/j.neuron.2009.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waclaw RR, Ehrman LA, Pierani A, Campbell K (2010) Developmental origin of the neuronal subtypes that comprise the amygdalar fear circuit in the mouse. J Neurosci 30:6944–6953. 10.1523/JNEUROSCI.5772-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltzer L, Vandel L, Bienz M (2001) Teashirt is required for transcriptional repression mediated by high Wingless levels. EMBO J 20:137–145. 10.1093/emboj/20.1.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellman CL, Izquierdo A, Garrett JE, Martin KP, Carroll J, Millstein R, Lesch KP, Murphy DL, Holmes A (2007) Impaired stress-coping and fear extinction and abnormal corticolimbic morphology in serotonin transporter knock-out mice. J Neurosci 27:684–691. 10.1523/JNEUROSCI.4595-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichterle H, Garcia-Verdugo JM, Herrera DG, Alvarez-Buylla A (1999) Young neurons from medial ganglionic eminence disperse in adult and embryonic brain. Nat Neurosci 2:461–466. 10.1038/8131 [DOI] [PubMed] [Google Scholar]
- Yun K, Potter S, Rubenstein JL (2001) Gsh2 and Pax6 play complementary roles in dorsoventral patterning of the mammalian telencephalon. Development 128:193–205. [DOI] [PubMed] [Google Scholar]
- Yun K, Garel S, Fischman S, Rubenstein JL (2003) Patterning of the lateral ganglionic eminence by the Gsh1 and Gsh2 homeobox genes regulates striatal and olfactory bulb histogenesis and the growth of axons through the basal ganglia. J Comp Neurol 461:151–165. 10.1002/cne.10685 [DOI] [PubMed] [Google Scholar]
- Zarrindast MR, Aghamohammadi-Sereshki A, Rezayof A, Rostami P (2012) Nicotine-induced anxiogenic-like behaviours of rats in the elevated plus-maze: possible role of NMDA receptors of the central amygdala. J Psychopharmacol 26:555–563. 10.1177/0269881111412094 [DOI] [PubMed] [Google Scholar]
- Zhu X, Lai C, Thomas S, Burden SJ (1995) Neuregulin receptors, erbB3 and erbB4, are localized at neuromuscular synapses. EMBO J 14:5842–5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zikopoulos B, John YJ, García-Cabezas MÁ, Bunce JG, Barbas H (2016) The intercalated nuclear complex of the primate amygdala. Neuroscience 330:267–290. 10.1016/j.neuroscience.2016.05.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zola-Morgan S, Squire LR, Alvarez-Royo P, Clower RP (1991) Independence of memory functions and emotional behavior: separate contributions of the hippocampal formation and the amygdala. Hippocampus 1:207–220. 10.1002/hipo.450010208 [DOI] [PubMed] [Google Scholar]