

Abstract
Maintenance of a low intracellular Cl− concentration ([Cl−]i) is critical for enabling inhibitory neuronal responses to GABAA receptor-mediated signaling. Cl− transporters, including KCC2, and extracellular impermeant anions ([A]o) of the extracellular matrix are both proposed to be important regulators of [Cl−]i. Neurons of the reticular thalamic (RT) nucleus express reduced levels of KCC2, indicating that GABAergic signaling may produce excitation in RT neurons. However, by performing perforated patch recordings and calcium imaging experiments in rats (male and female), we find that [Cl−]i remains relatively low in RT neurons. Although we identify a small contribution of [A]o to a low [Cl−]i in RT neurons, our results also demonstrate that reduced levels of KCC2 remain sufficient to maintain low levels of Cl−. Reduced KCC2 levels, however, restrict the capacity of RT neurons to rapidly extrude Cl− following periods of elevated GABAergic signaling. In a computational model of a local RT network featuring slow Cl− extrusion kinetics, similar to those we found experimentally, model RT neurons are predisposed to an activity-dependent switch from GABA-mediated inhibition to excitation. By decreasing the activity threshold required to produce excitatory GABAergic signaling, weaker stimuli are able to propagate activity within the model RT nucleus. Our results indicate the importance of even diminished levels of KCC2 in maintaining inhibitory signaling within the RT nucleus and suggest how this important activity choke point may be easily overcome in disorders such as epilepsy.
SIGNIFICANCE STATEMENT Precise regulation of intracellular Cl− levels ([Cl−]i) preserves appropriate, often inhibitory, GABAergic signaling within the brain. However, there is disagreement over the relative contribution of various mechanisms that maintain low [Cl−]i. We found that the Cl− transporter KCC2 is an important Cl− extruder in the reticular thalamic (RT) nucleus, despite this nucleus having remarkably low KCC2 immunoreactivity relative to other regions of the adult brain. We also identified a smaller contribution of fixed, impermeant anions ([A]o) to lowering [Cl−]i in RT neurons. Inhibitory signaling among RT neurons is important for preventing excessive activation of RT neurons, which can be responsible for generating seizures. Our work suggests that KCC2 critically restricts the spread of activity within the RT nucleus.
Keywords: chloride, epilepsy, GABAA, KCC2, reticular thalamic nucleus
Introduction
Proper inhibitory neurotransmission in the CNS is critical for many neural processes (Wong et al., 2003; Kaila et al., 2014) and is primarily mediated by the influx of chloride (Cl−) through the GABAA receptor. However, if the concentration of intracellular Cl− ([Cl−]i) is sufficiently elevated, then GABAA receptor activation enables Cl− efflux and neuronal depolarization (Cherubini et al., 1990; Rohrbough and Spitzer, 1996; Staley and Smith, 2001). Thus, depending on the [Cl−]i, the actions of GABA can be either inhibitory or excitatory.
The low [Cl−]i required for GABAergic inhibition is primarily achieved by active Cl− extrusion driven by the K+-Cl− cotransporter KCC2 (Payne et al., 1996; Rivera et al., 1999). However, negatively charged, fixed macromolecules known as impermeant anions can also promote a low [Cl−]i (Donnan, 1911), perhaps even more prominently than KCC2 (Delpire and Staley, 2014; Glykys et al., 2014a). To date, the relative contribution of KCC2 and impermeant anions to [Cl−]i remains an open debate (Glykys et al., 2014b; Kaila et al., 2014; Luhmann et al., 2014; Voipio et al., 2014; Doyon et al., 2016).
Evaluating the contributions of KCC2 and impermeant anions to GABAergic processes that regulate global brain excitability is particularly pertinent for understanding seizure propagation. The reticular thalamic (RT) nucleus envelops and critically regulates the nuclei of the dorsal thalamus (Jones, 2007). Importantly, local circuitry within the RT nucleus operates as an inhibitory choke point that prevents the propagation of seizures (Huntsman et al., 1999; Sohal and Huguenard, 2003; Paz and Huguenard, 2015; Makinson et al., 2017). Notably, the RT nucleus is one of the few adult brain regions with greatly reduced KCC2 expression (Kanaka et al., 2001; Barthó et al., 2004; Sun et al., 2012). Indeed, the paucity of KCC2 in the RT nucleus suggests that GABAergic neurotransmission within the nucleus is excitatory (Sun et al., 2012), thus presenting a conundrum regarding the role of the RT nucleus as an inhibitory seizure choke point.
Reciprocal GABAergic connections among RT neurons form the basis of the thalamic seizure choke point (Ahlsén and Lindström, 1982; Yen et al., 1985; Pinault et al., 1997). Eliminating these connections increases thalamic excitability (Huntsman et al., 1999; Makinson et al., 2017) and possibly causes seizures (Homanics et al., 1997; DeLorey et al., 1998). Computational modeling suggests that local GABAergic signaling among RT neurons prevents the simultaneous activation of nearby neurons through an activity-vetoing mechanism, thereby restricting seizure propagation (Sohal and Huguenard, 2003). This model assumes that GABA is inhibitory, an effect mediated by low [Cl−]i (Ulrich and Huguenard, 1997). However, if GABA is excitatory due to low KCC2 expression, it then remains unclear how the RT nucleus can function as a seizure choke point.
In this study, we aim to better understand GABAergic regulation of excitability within the RT nucleus. Using immunohistochemistry, we confirm a reduced KCC2 expression in the RT nucleus and further demonstrate that it remains low throughout development. However, despite low KCC2 immunoreactivity, intracellular, perforated patch-clamp recordings of RT neurons indicate that the [Cl−]i is low, and that GABAergic signaling inhibits RT neurons. We then show that impermeant anions, abundantly expressed in the RT nucleus, only moderately contribute to a low RT [Cl−]i. Surprisingly, despite low RT immunoreactivity, KCC2 effectively regulates basal [Cl−]i in RT neurons. However, diminished KCC2 leaves RT neurons more susceptible to activity-dependent Cl− accumulation, likely weakening the capacity of the RT inhibitory choke point to prevent seizures.
Materials and Methods
Subjects.
Wild-type Sprague-Dawley rats (Charles River Laboratories) and wild-type C57BL/6NJ mice (The Jackson Laboratory) of either sex were used in these experiments. All experiments were approved by the Institutional Care and Use Committee at the University of Virginia (Charlottesville, VA), in accordance with the National Institutes of Health guidelines.
Intracerebroventricular injections.
In some instances, P0–P2 rats received an intracerebroventricular (i.c.v.) injection of an AAV9.Syn.GCaMP6s.WPRE.SV40 viral vector (Penn Vector Core, AV-1-PV2824; supplied by the GENIE Project, Janelia Research Campus, HHMI; Glascock et al., 2011). Sterile microliter calibrated glass pipettes were filled with virus diluted to ∼1 × 1013 GC/ml in 0.1% trypan blue dye (Bio-Rad). Rats were cryoanesthetized and the pipette was lowered through the skull, into the lateral ventricle. A picospritzer (Picospritzer III, Parker Hannifin) was used to deliver 3 μl of virus solution into each lateral ventricle. Animals were then returned to the dam to allow time for GCaMP6s expression.
Slice preparation.
P10–P20 animals were deeply anesthetized with pentobarbital and then transcardially perfused with an ice-cold protective recovery solution containing the following (in mm): 92 NMDG, 26 NaHCO3, 25 glucose, 20 HEPES, 10 MgSO4, 5 Na-ascorbate, 3 Na-pyruvate, 2.5 KCl, 2 thiourea, 1.25 NaH2PO4, 0.5 CaCl2, titrated to a pH of 7.3–7.4 with HCl (Ting et al., 2014). Horizontal slices (250 μm) containing the thalamus were cut in ice-cold protective recovery solution using a vibratome (VT1200, Leica Biosystems). Slices were trimmed to remove the hippocampus and cortex, and then transferred to protective recovery solution maintained at 32–34°C for 12 min. Brain slices were kept in room temperature aCSF consisting of the following (in mm): 126 NaCl, 26 NaHCO3, 10 glucose, 2.5 KCl, 2 CaCl2, 1.25 NaH2PO4, 1 MgSO4. All solutions were equilibrated with 95% O2/5% CO2.
Electrophysiology.
Intracellular recordings were performed in a submerged chamber, with slices situated on nylon netting and perfused with warm (31–33°C) oxygenated aCSF at 2.5 ml/min. All experiments were performed in the presence of kynurenic acid (3 mm) and CGP 55845 (100 nm) to block AMPA, NMDA, and GABAB receptors, and TTX (1 μm) to block sodium channel activation. Cadmium chloride (100 μm) was used to block voltage-dependent calcium channel activation in all Cl− extrusion experiments. Thalamic neurons were visualized using infrared Dodt gradient contrast illumination on a Zeiss Axio Examiner.A1 microscope (Zeiss Microscopy) and an sCMOS camera (ORCA-Flash4.0, Hamamatsu). Recording pipettes were pulled from thick-walled borosilicate capillary glass (Sutter Instruments) using a P1000 puller (Sutter Instruments) and were filled with the following (in mm): 130 KCl, 1 MgCl2, 0.07 CaCl2, 10 HEPES, 0.1 EGTA (pH-adjusted to 7.3 with KOH, osmolarity 300 mOsm). Pipettes had a 3–4 MΩ tip resistance. Gramicidin (dissolved in DMSO) was added to prefiltered internal solution to produce a final concentration of 5 μg/ml and sonicated for 30 s. For one set of experiments (see Fig. 6) a CsCl internal pipette solution, containing the following (in mm): 132 CsCl, 10 HEPES, 0.5 EGTA, 2 MgCl2, 0.16 CaCl2, and 5 QX-314 (pH-adjusted to 7.3 with KOH, osmolarity 295 mOsm) was used (Sun et al., 2012).
Figure 6.

Cs alters [Cl−]i homeostasis in RT neurons. Gramicidin perforated patch recordings of muscimol-induced currents from RT (A) and VB (B) neurons at various command potentials, using a CsCl-based internal solution. The leak current has been subtracted from the representative traces. A1, Baseline EGABA was more depolarized than in prior recordings with a KCl-based internal solution. Representative recording and current–voltage plot are shown. A2, Time series revealing that CsCl-based solutions mitigated the effect of VU0463271 application in EGABA, relative to KCl-based recordings (Fig. 5). A3, Average change in EGABA before and after 10 min of VU0463271 application. B1–B3, Similar measures examined in VB neurons, as described in A1–A3. VU0463271 application still shifted EGABA in VB neurons, although the responses were blunted. *p < 0.05, **p < 0.01.
High resistance (>0.8 GΩ) cell-attached seals were obtained through the application of negative pressure. Changes in access resistance due to gramicidin pore formation were monitored by repeatedly delivering 20 mV hyperpolarizing voltage steps. Access resistance was measured from the transient responses to the applied voltage steps using established methods (Ulrich and Huguenard, 1997). Appropriate pore formation was indicated by access resistance measurements of 50–100 MΩ, typically achieved within 10–40 min. Rapid drops in access resistance at any point during these experiments were indicative of a ruptured membrane patch and these recordings were discarded. The GABAA receptor agonist muscimol (Abcam) was dissolved in aCSF (100 μm) and applied through a patch pipette adjacent to the cell body of the recorded neuron using 10 ms pressure puffs (Picospritzer III). Membrane potentials were all corrected for the voltage drop across the series resistance.
Data were acquired in pClamp software (Molecular Devices) using a Multiclamp 700B amplifier (Molecular Devices), low-pass filtered at 2 kHz, and digitized at 10 kHz (Digidata 1440A, Molecular Devices). Data analysis was performed using custom written scripts in MATLAB (MathWorks).
Calcium imaging.
As with the electrophysiology experiments described above, acute brain slices were prepared from P10–P20 animals that received an ICV injection of AAV9.Syn.GCaMP6s.WPRE.SV40 at P0–P2. Recordings were performed at 31–33°C in aCSF containing: kynurenic acid (3 mm), CGP 55845 (100 nm), and TTX (1 μm). Illumination was provided by a DG-4 arc lamp (Sutter Instruments) using a 470 ± 22 nm bandpass excitation filter. Images were acquired with HCImage software (Hamamatsu) at 10 Hz using a 10×/NA0.2 lens. Chemical stimuli were applied with a custom built local perfusion system, which enabled controlled delivery of aCSF with added 5 μm muscimol or 10 mm KCl to the field of imaged RT neurons. Images were analyzed off-line, using custom written scripts in MATLAB to measure changes in image intensity within user defined ROIs.
Drugs and solutions.
All drugs used were applied in aCSF. VU0463271 (N-cyclopropyl-N-(4-methyl-2-thiazolyl)-2-[(6-phenyl-3-pyridazinyl)thio]acetamide; Bio-Techne) was diluted from a 10 mm stock in DMSO to a concentration of 10 μm in aCSF. A 50 U/ml stock of Chondroitinase ABC (ChABC; Sigma-Aldrich) was made in 0.1% BSA and diluted down to 0.4 U/ml in aCSF.
Histochemistry.
At P5, 10, 15, 20 and 40, animals were deeply anesthetized with pentobarbital and transcardially perfused with PBS, followed by ice-cold 4% PFA in PBS (both pH 7.4). Brains were postfixed overnight in 4% PFA at 4°C. Horizontal sections (40 μm) containing the thalamus were obtained using a vibratome (VT1000S, Leica Biosystems). Free-floating sections were washed in PBS and then treated with 0.1% sodium borohydride in PBS for 15 min to reduce autofluorescence. Sections were washed with PBS, blocked with 2% normal goat serum and Fab fragment of goat anti-mouse IgG (1:500; Jackson ImmunoResearch) in PBS for 4 h at room temperature, and then washed with PBS. Sections were incubated overnight at 4°C with combined primary antibodies for either KCC2 and parvalbumin (rabbit anti-KCC2, 1:500, EMD Millipore; mouse anti-parvalbumin, 1:2000, Sigma-Aldrich) or parvalbumin and fluorescein labeled Wisteria floribunda agglutinin (1:2000; Vector Laboratories) in PBS with 1% normal goat serum. Sections were washed in PBS and then incubated overnight at 4°C with appropriate combinations of secondary antibodies (donkey α-mouse AF488 and donkey α-rabbit Cy3 for KCC2 labeling, donkey α-mouse Cy3 for WFA labeling; all 1:200 in PBS with 1% normal goat serum, Jackson ImmunoResearch). Sections were washed in PBS and mounted with Vectashield (Vector Laboratories).
A subset of histochemical experiments were performed in 300 μm sections prepared as described for use in electrophysiological experiments. Horizontal sections were then bisected and one hemisphere was incubated at 37°C in 0.4 U/ml ChABC, whereas the other hemisphere was incubated in aCSF. These sections were then postfixed in 4% PFA for 12 h before being stained for parvalbumin and WFA as described above.
All images were obtained with a Neurolucida system (MicroBrightfield) with an Axioskop microscope driven stage and an AxioCam MRc camera (Zeiss Microscopy). All staining and imaging was performed in batches where a set of sections representing all age groups were processed simultaneously. This allowed for us to control for between experiment variability in measured labeling. Images were analyzed using custom written scripts in MATLAB, which allowed for evaluation by two independent, blinded, investigators.
Computational modeling.
This model was based on those of Sohal et al. (2003) and Jedlicka et al. (2011). Simulated networks contained 100 RT cells, each of which was modeled as a single compartment. All simulations were run using NEURON (Hines and Carnevale, 1997) at a temperature of 31°C and with a time step of 0.1 ms.
Intrinsic properties.
Each RT cell consisted of a cylinder with a length of 20 μm and a diameter of 10 μm, based on the dimensions of neurons observed during our electrophysiological experiments. All cells had the following properties: (1) a specific membrane capacitance of 1 μF/cm2, (2) an axial resistivity of 100 Ω · cm (Destexhe et al., 1996), (3) a leak current with a reversal potential of −70 mV and a conductance that was randomly selected from a uniform distribution between 45 and 55 μS/cm2 (Sohal and Huguenard, 2003), (4) Na+ and K+ currents underlying action potentials (INa, IK), (5) a low-threshold Ca2+ current (ITs), and (6) a Ca2+-dependent K current (IKCa), and (7) a GABAA receptor-mediated current comprising a Cl− current (ICl) and a nonspecific current that represents HCO3− flow (IHCO3). Kinetics and details for INa, IK, ITs, IKCa and the kinetics of intracellular Ca2+ ([Ca2+]i) were taken from Sohal et al. (2003). The following changes were implemented: (1) the K+ reversal potential was fixed at −104 mV, (2) the spike threshold was set to −47 mV (Muñoz and Fuentealba, 2012), and (3) ITs followed the constant field equation with permeability pTs = 10−4 cm/s. Properties of ICl and IHCO3 were taken from Jedlicka et al. (2011), except for the following: (1) extracellular Cl− concentration was set to 130.5 mm, (2) the conductance ratio gCl:gHCO3 was 4:1, and (3) the peak GABAA conductance (gpeak) per synapse was set to 2.5 nS, or up to 20 nS per cell.
Chloride dynamics.
Kinetics of [Cl−]i were adapted from Jedlicka et al. (2011). [Cl−]i was computed in a submembrane compartment with a depth of 0.5 μm, allowed to diffuse radially with a diffusion coefficient of 2 μm2/ms, and was actively extruded (representing the action of KCC2) via Michaelis-Menten kinetics. The dissociation constant (Kd) of Cl− extrusion was 15 mm and the maximum flux was computed by: Vmax = (Cl∞ + Kd)/τKCC2. In this equation, Cl∞ (the steady-state value of [Cl−]i) was 8 mm. A range of values (4–64 s) for τKCC2 (the extrusion time constant when [Cl−]i ≈ Cl∞) were examined. A constant Cl− leak current balanced the steady-state extrusion rate.
Network architecture.
RT cells were organized in a simple, linear array. All connections were local and topographic, such that each RT cell projected to the eight nearest RT cells (Sohal and Huguenard, 2003). An action potential was counted when the membrane voltage reached 0 mV. Following a synaptic delay of 1 ms, GABAA receptors were activated according to kinetics described by Jedlicka et al. (2011). The rise and decay time constants of GABAA currents were 0.1 and 50 ms, respectively, and the baseline GABAA reversal potential was set to −62 mV, based on our data (see Fig. 2C).
Figure 2.
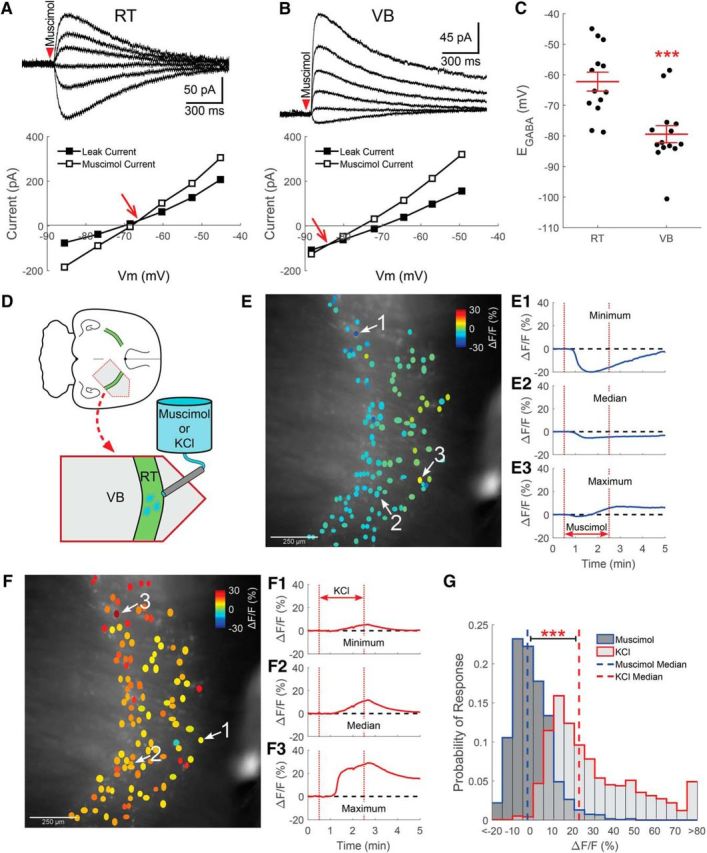
RT neurons maintain a relatively low [Cl−]i. Gramicidin perforated patch recordings of muscimol-induced (100 μm, 10 ms) currents from RT (A) and VB (B) neurons at various command potentials. The leak current has been subtracted from the representative traces. The intersection of the prestimulation leak current and the muscimol-induced current was used to determine the GABAA receptor-mediated equilibrium potential (EGABA, red arrow). C, The EGABA of RT neurons was more depolarized than in VB neurons, yet remained at levels that likely support inhibitory GABAergic signaling. D, GCaMP6s was expressed in RT neurons for calcium imaging experiments and a local perfusion system provided timed delivery of muscimol (5 μm) or elevated KCl (+10 mm) to the imaged RT nucleus. E, Two minute application of muscimol mostly decreased the fluorescence of RT neurons. ROIs drawn around GCaMP6s-expressing RT neurons are colored according to their peak change in fluorescence. Representative examples of neurons displaying the minimum (E1), median (E2), and maximum (E3) fluorescence change in a particular brain slice following muscimol application. F, Two minutes of elevated KCl produced a nearly uniform increase in the fluorescence of RT neurons. Examples of the minimum (F1), median (F2), and maximum (F3) fluorescence changes evoked by elevated KCl application. G, Histograms comparing induced responses in all cells imaged, across multiple animals, shows that the median response (dotted line) to muscimol was a slight decrease in fluorescence. In contrast, mild depolarization with elevated KCl produced a robust increase in fluorescence. Bin size: 5ΔF/F(%). ***p < 0.001.
Simulation protocol.
In each simulation, the network was allowed to initially equilibrate for 3 s. Following this period, action potential activity was elicited in three central cells (Cells 46, 51, and 56) by applying brief (100 μs, 4 nA) and repetitive current pulses. In stimulated cells pTs and gKCa (conductance of IKCa) were set to zero to better control firing behavior. We tested a range of stimulation frequencies (1–60 Hz), delivered for a total duration of 200 s. Total simulation duration was 233 s.
Experimental design and statistical analysis.
Throughout this study, measurements from neurons in the ventrobasal (VB) thalamic nuclei were used as controls for observations from RT neurons. All histochemical experiments were performed in brain slices from four rats and four mice per age group (2 male, 2 female; both hemispheres from ∼3 slices per animal were analyzed). The number of recordings for basal measures of EGABA (RT: 7 male, 6 female; VB: 7 male, 7 female) and Cl− extrusion (RT: 6 male, 5 female; VB: 7 male, 8 female) reflect the number of animals used, with recordings from RT and VB neurons at times occurring within the same subject.
When possible, drug-induced effects were compared against baseline recordings in the same neuron. Each recording of the impact of VU0463271 on basal EGABA (RT: 3 male, 2 female; VB: 1 male, 4 female), basal EGABA when using a Cs-based recording solution (RT: 4 male, 1 female; VB: 3 male, 2 female) and Cl− extrusion (RT: 2 male, 4 female; VB: 4 male, 4 female) reflects an independent subject, except where recordings from RT and VB neurons occurred within brain slices of the same animal.
The time course of ChABC experiments prohibited pretreatment and posttreatment recordings from the same neuron, so control recordings were obtained from sham-treated neurons. Each measure of the impact of ChABC treatment on basal EGABA (RT control: 3 male, 5 female; RT ChABC: 4 male, 3 female; VB control: 3 male, 7 female; VB ChABC: 4 male, 3 female), Cl− extrusion (RT control: 2 male, 2 female; RT ChABC: 1 male, 2 female; VB control: 4 male, 1 female; VB ChABC: 2 male, 2 female), and basal EGABA when combined with VU0463271 (RT control: 4 male, 4 female; RT ChABC: 2 male, 2 female) reflects the number of animals used, with recordings from RT and VB neurons at times occurring within the same subject. Control and ChABC-treated recordings were always performed in different brain slices.
In calcium imaging experiments, muscimol and elevated KCl responses were evaluated in each brain slice, with the KCl treatment being a positive control for the change in fluorescence associated with a mild depolarization. For evaluating the overall distribution of responses, the change in fluorescence of each detected RT neuron was compared (Muscimol: 1772; KCl: 1728). Photobleaching and shifting tissue, among other factors, result in a differing number of detectable cells in experiments performed in the same brain slice. The typical response to muscimol and elevated KCl was evaluated by comparing the median change in fluorescence that a treatment evoked within each subject (5 male, 2 female, average of 1.7 slices per animal). Similar comparisons were made in experiments where brain slices were preincubated in VU0463271 before calcium imaging (Muscimol+VU0463271: 1772 cells; KCl+VU0463271: 1728 cells; 3 male, 3 female, average of 1.8 slices per animal).
All statistical tests were performed in MATLAB. Unless otherwise noted, statistical tests to determine the significance of differences between groups were performed using unpaired or paired Student's t test. When used, ANOVA tests were followed by a post hoc Tukey's HSD analysis. Comparisons requiring a nonparametric factorial analysis were calculated using a two-way aligned rank transform ANOVA (Wobbrock et al., 2011). This method independently rank transforms the data to separate the contributions of the main effects and interactions. After the data has been aligned and ranked, a regular parametric ANOVA with Tukey's HSD post hoc testing can be used to analyze each individual effect. Group measures are presented as mean ± SEM. The threshold for differences to be considered significant was set at p < 0.05.
Results
The goal of our study was to resolve how intracellular Cl− is regulated in the RT nucleus, a structure proposed to serve as an important seizure choke point. We used anatomical and functional approaches to determine the relative contributions of KCC2 and impermeant anions to setting basal [Cl−]i in RT neurons. We also assessed the degree to which these two mechanisms define the activity-dependent, Cl− extrusion capacity of RT neurons. Finally, we incorporated our observations into a computational model of a local RT network to better understand how seizure propagation within this network might depend on the properties of Cl− regulation. Collectively, our experimental and computational results suggest that the weak Cl− extrusion capacity of RT neurons is sufficient to support basal inhibitory, GABAergic signaling, but that the nucleus is susceptible to an activity-dependent switch from synaptic inhibition to excitation.
KCC2 expression is low, but present in RT neurons
As a major Cl− transporter in the CNS, KCC2 is thought to critically maintain the low [Cl−]i that enables inhibitory, GABAA receptor-mediated signaling in adult neurons (Kaila et al., 2014). Interestingly, the RT nucleus is one of only a small number of regions in the adult rat brain with minimal KCC2 mRNA expression (Kanaka et al., 2001). Reduced expression of KCC2 protein is also observed in the RT nucleus of adult rats (Barthó et al., 2004) and mice (Sun et al., 2012). Anatomical assays primarily served as the basis for these previous measures.
The developmental upregulation of KCC2 in most brain regions is well established (Kaila et al., 2014). However, to our knowledge, this phenomenon has not been examined in the thalamus. Therefore, we began our study by measuring KCC2 immunoreactivity within the rodent thalamus as a function of age (P5–P40; Fig. 1). To control for experimental variability, we concurrently performed immunohistochemistry on sets of brain slices representing all age groups. As in other brain structures, thalamic KCC2 immunoreactivity increased with age in the rats we examined (F(4,146) = 2.60, p = 0.038, n = 4, two-way ANOVA; Fig. 1B). Specifically, overall KCC2 immunoreactivity was low throughout the thalamus of P5 rats (21.3 ± 1.3 A.U.), and increased thereafter (P20 rats: 47.4 ± 8.7 A.U., p = 0.049; P40 rats: 47.4 ± 6.0 A.U., p = 0.033). Whereas immunoreactivity in both the VB thalamus and RT nucleus increased with age, the effect was blunted in RT. These results suggest that KCC2 is expressed in the adult rat RT nucleus, albeit at relatively lower levels than observed in VB thalamus (F(1,146) = 8.37, p = 0.0044, n = 4, two-way ANOVA). We detected a similar pattern of KCC2 staining in P10–P40 mice (Fig. 1B).
Figure 1.

KCC2 expression is low in RT neurons throughout development. A, Immunofluorescence of parvalbumin (green) and KCC2 (red) in horizontal sections of rat thalamus at different developmental time points. All images are oriented with the anterior (A) aspect of the thalamus toward the top, the lateral (L) aspect toward the right and the internal capsule (IC) toward the top right corner of the image. Dotted lines indicate the boundaries of the RT nucleus in the enlarged images. B, KCC2 immunofluorescence increased across the ages we tested. KCC2 labeling was consistently lower in the RT nucleus, relative to VB, across the ages tested. A similar pattern of KCC2 labeling was observed in mice. C, Schematic of methodology for measuring regional variability in KCC2 intensity across segments of the RT nucleus. Intensity of KCC2 labeling was measured in an ROI, subdivided into 10 segments, that extended from the anterior to the lateral extent of the RT nucleus (A, bottom left) and values were normalized to the mean intensity across this entire span. D, KCC2 labeling was consistent throughout the span of the RT nucleus at P10, but showed significant regional variability in P40 rats and mice. *p < 0.05, **p < 0.01, ***p < 0.001.
In addition to gross, age-dependent differences in KCC2 immunoreactivity, we also observed that the distribution of KCC2 expression was not even throughout RT, particularly in adult rats and mice. To quantify KCC2 immunoreactivity, we drew a linear ROI that extended from the anterior-most edge to the lateral-most edge of the RT nucleus (Fig. 1A, bottom left). This line bisected the RT nucleus, and then automatically expanded to a width of 75 μm. This linear ROI was further subdivided into 10 equal segments that were numbered from 1 (anterior-most) to 10 (lateral-most; Fig. 1C). The mean fluorescence intensity within each segment was calculated.
We quantified KCC2 immunoreactivity in each segment along the anterior-to-lateral axis, relative to the mean intensity level across all RT segments of each slice (Fig. 1D), an approach to account for slice-to-slice staining variability. When expression was quantified in this manner in P40 rats, we observed that KCC2 levels were not uniform across segments of the RT nucleus (F(10,200) = 39.4, p < 0.001, n = 4, one-way ANOVA). This finding establishes that KCC2 immunoreactivity varies according to location within the RT nucleus. To discern specific immunoreactive differences among the segments, we compared individual segment means to the overall mean across all RT segments.
Relative to the overall RT mean, KCC2 immunoreactivity was elevated in the two anterior-most segments of the adult rat RT nucleus (Segment 1: 134 ± 5.5%, p < 0.001; Segment 2: 120 ± 3.3%, p < 0.001), as well as in the lateral-most segment (Segment 10: 116 ± 3.6%, p = 0.004). Centrally located segments, on the other hand, had much reduced KCC2 immunoreactivity (Segment 6: 82 ± 2.5%, p < 0.001; Segment 7: 79 ± 2.6%, p < 0.001; Segment 8: 81 ± 3.1%, p < 0.001).
Next, we applied the aforementioned approach to younger, P10 rats. Although KCC2 levels varied minimally along the extent of the RT nucleus in P10 rats (F(10,144) = 2.35, p = 0.013, n = 4, one-way ANOVA), no significant differences relative to the overall RT mean were observed. These results suggest that there is greater regional variability in KCC2 immunoreactivity in the RT nucleus of P40 rats than of P10 rats. We observed a broadly similar pattern of KCC2 immunoreactivity in mice (Fig. 1D).
Collectively, our results indicate that KCC2 expression is consistently lower in RT neurons than VB neurons throughout the first few postnatal weeks, in agreement with prior reports (Kanaka et al., 2001; Barthó et al., 2004; Sun et al., 2012). However, it remained unclear the extent to which detected immunoreactivity in the RT nucleus was due to KCC2 protein expression or nonspecific labeling. Therefore, we decided to use functional measurements to assess whether diminished KCC2 reduces the capacity of RT neurons to maintain a low [Cl−]i.
[Cl−]i is relatively low in RT neurons
Based on our observation that KCC2 immunoreactivity is low in RT neurons, we expected to find elevated [Cl−]i in these neurons. If true, then GABAergic signaling in RT neurons would likely be excitatory. To assess [Cl−]i in RT neurons without perturbing the intracellular Cl− gradient, we performed gramicidin perforated patch recordings in RT neurons of P10–P20 rats.
Successful patch perforation was indicated when access resistance measurements reached 50–100 MΩ. Neurons were voltage-clamped at −70 mV and then stepped to a command potential between −110 and −20 mV for 2.5 of each 15 s sweep. These 10 voltage steps were given in a randomized order to reduce experimentally induced Cl− loading. The GABAA receptor agonist muscimol (100 μm) was pressure-applied to the soma of the voltage-clamped neuron for 10 ms during each voltage step. We adjusted the pressure used for agonist application so that GABA-evoked currents fully decayed within 2 s from application.
Current–voltage (I–V) relationships were plotted for both the leak current and the muscimol-induced current, and the voltage at which these currents was equal indicated the reversal potential for the GABA-induced current (EGABA). EGABA is largely determined by ECl, with EHCO (the reversal potential for bicarbonate ions) providing a smaller contribution (Bormann et al., 1987; Staley et al., 1995). We did not isolate the relative contributions of ECl and EHCO to EGABA in our recordings, so our calculations may slightly overestimate the [Cl−]i of RT neurons (Staley et al., 1995). We corrected all measurements for the voltage drop across the series resistance.
We determined that RT neurons have an EGABA of −62 ± 3.0 mV (n = 13; Fig. 2A,C). For comparison, EGABA was also measured in VB neurons, where it was found to be significantly more hyperpolarized than in RT neurons (−79 ± 2.7 mV, t(25) = 4.15, p < 0.001, n = 14; Fig. 2B,C). Based on our recording solutions, [Cl−]i was ∼12 mm in RT neurons and ∼6 mm in VB neurons. Interestingly, although we measured a very consistent EGABA in the majority of VB neurons, this parameter was more variable among RT neurons. The RT nucleus contains a heterogeneous population of neuronal subtypes, each associated with distinct functional characteristics (Lee et al., 2007; Halassa et al., 2014; Clemente-Perez et al., 2017), and may provide a source of this variability. Our electrophysiological recordings were targeted to the central section of the RT nucleus, and likely include both parvalbumin- and somatostatin-positive RT neuron subtypes (Clemente-Perez et al., 2017).
Although gramicidin perforated patch recordings are designed to maintain physiological [Cl−]i in neurons, the low experimental yield afforded by this technique limits the ability to make observations in large populations of neurons. Therefore, we also examined the response of RT neurons to GABAA receptor stimulation using GCaMP6s-based calcium imaging. Acute brain slices, where GCaMP6s had been virally transfected into RT neurons, were prepared and a local perfusion system was used to deliver test compounds in proximity to the RT nucleus (Fig. 2D). Cell-intrinsic responses to muscimol stimulation were isolated by blocking AMPA, NMDA, and GABAB receptors, as well as voltage-gated sodium channels. A 2 min application of muscimol (5 μm) caused a decrease in the fluorescence of most, but not all, RT neurons (Fig. 2E,G). However, subsequently depolarizing the RT neurons with the addition of 10 mm KCl (12.5 mm in total) for 2 min induced a robust increase in fluorescence (Fig. 2F,G). Based on EK measurements of RT neurons (McCormick and Prince, 1986), the high-K+ test solution should depolarize neurons to ∼−45 mV. Compared across animals, 10 mm KCl produced a significantly greater median increase in fluorescence (22.3%, interquartile range 12.1–42.7%, n = 1728 cells, n = 7 animals; Fig. 2G) than the mild decrease following muscimol stimulation (−2.8%, interquartile range −7.1–4.8%, n = 1772 cells, n = 7 animals, Z = 44.86, p < 0.001, Wilcoxon rank sum test).
Because KCC2 immunoreactivity is reduced in the RT nucleus, the observation that [Cl−]i remained low, and GABAergic signaling remained inhibitory, in most RT neurons was unexpected. Considering that KCC2 immunoreactivity was low in the RT nucleus (Fig. 1), we were motivated to determine whether extracellular impermeant anions ([A]o) contribute to Cl− regulation in RT neurons (Glykys et al., 2014a).
Although elevated in RT neurons, [A]o contribute only mildly to setting EGABA
Chondroitin sulfate proteoglycans (CSPGs) are major extracellular matrix proteins that contain abundant, negatively charged, sulfate groups, thus making them a significant source of [A]o (Bandtlow and Zimmermann, 2000; Glykys et al., 2014a). The disaccharide formed by N-acetylgalactosamine and glucuronic acid is bound by the plant lectin Wisteria floribunda agglutinin (WFA), and therefore can be used to label chondroitin sulfate glycosaminoglycan elements of CSPGs (Horii-Hayashi et al., 2015). WFA staining showed that the CSPGs surrounding RT neurons increase during development from P5–P40 (Fig. 3).
Figure 3.

CSPGs are elevated around RT neurons. A, Immunofluorescence of parvalbumin (green) and labeling of CSPGs with WFA (red) in horizontal sections of rat thalamus at different developmental time points. All images are oriented with the anterior (A) aspect of the thalamus toward the top, the lateral (L) aspect toward the right. Dotted lines indicate the boundaries of the RT nucleus in the enlarged images. B, WFA staining increased across the ages we tested and was consistently elevated in the RT nucleus, relative to VB. C, Intensity of WFA labeling was measured in an ROI, subdivided into 10 segments, that extended from the anterior to the lateral extent of the RT nucleus (A, bottom left, Fig. 1C) and values were normalized to the mean intensity across this entire span. WFA labeling showed slight regional variability in both P10 and P40 rats. A similar pattern of WFA labeling was observed in mice. **p < 0.01, ***p < 0.001.
We first examined the overall levels of WFA fluorescence in the thalamus across the different ages of rats we tested. As with KCC2 immunoreactivity, WFA stain intensity increased with age (F(4,218) = 14.48, p < 0.001, n = 4, two-way ANOVA; Fig. 3B). Although overall WFA levels were initially low throughout the thalamus of P5 rats (25.0 ± 1.8 A.U.), overall WFA staining was elevated in P20 rats (49.7 ± 7.0 A.U., p = 0.0011) and further increased by P40 (72.7 ± 7.6 A.U., p < 0.001). This observation is consistent with prior findings showing that WFA staining first appears between P7–P14 in many regions of the mouse brain and further increases as the extracellular matrix matures during the next few weeks of development (Horii-Hayashi et al., 2015). Across the ages we evaluated, there was greater mean WFA staining throughout the RT nucleus (57.6 ± 3.4 A.U., n = 20), relative to mean levels in VB (39.9 ± 2.6 A.U., n = 20, F(1,218) = 16.31, p < 0.001, n = 4, two-way ANOVA). Intense WFA staining of white matter tracts is likely nonspecific as it is not eliminated by enzymatic reduction of CSPGs (Ajmo et al., 2008; Fig. 4F). An overall similar pattern of WFA staining was likewise detected in P10–P40 mice (Fig. 3).
Figure 4.

CSPGs mildly contribute to setting EGABA in RT neurons. Gramicidin perforated patch recordings of muscimol-induced (100 μm, 10 ms) currents from RT (A) and VB (B) neurons at various command potentials, following a 2 h incubation in ChABC (0.4 U/ml, 37°C). The leak current has been subtracted from the representative traces. EGABA was determined from the intersection of the prestimulation leak current and the muscimol-induced current (red arrow). C, ChABC did not alter the initial measurement of EGABA in either RT or VB neurons, relative to control neurons incubated in the absence of ChABC. D, When EGABA measurements were repeated every 2.5 min, ChABC treatment produced a small depolarizing shift in EGABA in RT neurons. E, No time-dependent shift in EGABA occurred in ChABC-treated VB neurons. F, Immunofluorescence of parvalbumin (green) and labeling of CSPGs with WFA (red) of control and ChABC-treated thalamic slices (300 μm) indicates that ChABC (0.4 U/ml, 37°C) effectively reduces CSPGs surrounding RT neurons. G, WFA labeling in the RT nucleus of ChABC-treated slices was reduced relative to control slices. H, WFA labeling in RT was first normalized to VB levels, thereby revealing a clear effect of ChABC treatment. **p < 0.01, ***p < 0.001.
As with measurements of KCC2 immunoreactivity (Fig. 1D), WFA staining was also evaluated in a linear ROI that extended from the anterior-most edge to the lateral-most edge of the RT nucleus, and was then subdivided into 10 equal segments (Fig. 3A, bottom left, Fig. 1C). We calculated the mean fluorescence intensity within each segment.
When WFA labeling in each segment along the anterior-to-lateral axis was compared with the mean intensity across all of RT, we found that WFA levels were not uniform across segments of the RT nucleus of P10 rats (F(10,202) = 9.82, p < 0.001, n = 4, one-way ANOVA; Fig. 3C). Relative to the mean WFA intensity within RT, WFA staining was increased in a central segment of RT (segment 6: 119 ± 5.5%, p = 0.0047). We also observed that WFA intensity varied along the extent of the RT nucleus in P40 rats (F(10,254) = 5.49, p < 0.001, n = 4, one-way ANOVA). There was reduced WFA staining in the anterior-most segment of the RT nucleus (segment 1: 86 ± 3.6%, p = 0.0046) relative to the mean WFA intensity within RT.
Considering that RT neurons maintain a surprisingly low [Cl−]i, despite the low expression of KCC2 in the RT nucleus, we next tested the hypothesis that elevated CSPGs surrounding RT neurons (as indicated by our WFA staining) provide a compensatory mechanism of [Cl−]i regulation. Therefore, we measured the impact of reducing [A]o surrounding RT neurons in P10–P20 rats. The enzyme ChABC digests the chondroitin sulfate polysaccharide elements of CSPGs (Yamagata et al., 1968) and releases negatively charged sulfate groups, thereby reducing [A]o (Glykys et al., 2014a). We measured changes in EGABA between neurons incubated in ChABC (0.4 U/ml, 37°C) for 2 h and control neurons incubated in the absence of ChABC.
Using perforated patch recordings, we found that the initial EGABA measurement in ChABC-treated RT neurons (−63 ± 3.2 mV, n = 7; Fig. 4A) was unchanged from control RT neurons (−60 ± 2.9 mV, t(13) = 0.66, p = 0.52, n = 8; Fig. 4C). Likewise, the initial EGABA measurement in ChABC-treated VB neurons (−70 ± 5.4 mV, n = 7; Fig. 4B) was no different from control VB neurons, (−74 ± 2.1 mV, t(15) = 0.69, p = 0.50, n = 10; Fig. 4C). Whereas the initial measurement of EGABA was not different between ChABC-treated and control RT neurons, we observed an effect of ChABC treatment when EGABA measurements were repeated at 2.5 min intervals over a 15 min period (F(1,68) = 8.74, p = 0.0043, n = 7,7, two-way ANOVA; Fig. 4D). Across all time points, ChABC-treated RT neurons underwent a greater, average shift in EGABA (Δ+3.3 ± 0.7 mV, n = 7) than control neurons (Δ+0.2 ± 0.8 mV, n = 7). The impact of ChABC treatment on the EGABA of RT neurons was particularly evident at 12.5 min (control: Δ+0.9 ± 3.4 mV, n = 7; ChABC-treated: Δ+8.1 ± 1.9 mV, n = 7). There was no effect of either ChABC treatment (F(1,82) = 1.94, p = 0.17, n = 7,9, two-way ANOVA) or time (F(5,82) = 0.57, p = 0.72, n = 7,9, two-way ANOVA) on the EGABA of VB neurons (Fig. 4E). The modest effect of ChABC treatment on EGABA motivated us to evaluate treatment efficacy in an independent manner. Toward this end, we prepared acute brain slices (300 μm) and then treated them in the same, aforementioned manner. However, rather than performing electrophysiological measurements, we performed WFA labeling protocols on control and ChABC-treated slices to localize CSPGs (Fig. 4F). We observed that mean RT WFA fluorescence was lower in ChABC-treated (34.3 ± 0.5 A.U.) versus control slices (42.5 ± 1.6 A.U., t(15) = 4.15, p = 0.0024, n = 4; Fig. 4G), an effect particularly evident when the change was measured by first normalizing RT fluorescence to adjacent, VB fluorescence levels (control: 25.3 ± 3.1%; ChABC: −1.8 ± 1.5%, t(15) = 6.19, p < 0.0001, n = 4; Fig. 4H). This observation confirms that the ChABC treatment protocol used in our perforated patch experiments was indeed associated with a significant reduction in [A]o.
Collectively, these results indicate that although elevated levels of [A]o surround RT neurons, perturbing CSPGs only affected Cl− regulation following prolonged periods of stimulation. Considering that [A]o did not exert a very large impact on the [Cl−]i of RT neurons, we next tested the hypothesis that the apparently low level of KCC2 expression in the RT nucleus nonetheless actively contributes to [Cl−]i regulation.
KCC2 is a significant regulator of [Cl−]i in RT neurons
Due to the generally low Cl− flux of resting neurons, it has been proposed that even minimal KCC2 might provide sufficient Cl− extrusion to maintain a hyperpolarizing EGABA (Blaesse et al., 2009). To determine whether the apparently reduced level of KCC2 functionally contributes to maintaining a hyperpolarized EGABA in RT neurons, the selective KCC inhibitor VU0463271 (Delpire et al., 2012) was applied during perforated patch recordings in P10–P20 rats. Although VU0463271 may act on KCCs other than KCC2, brain mRNA levels for KCC1, KCC3 and KCC4 are much lower than for KCC2 (Kaila et al., 2014). There is also no sign that KCC1, KCC3 or KCC4 localize within the thalamus (Rivera et al., 1999; Le Rouzic et al., 2006).
The addition of VU0463271 (10 μm) produced a clear shift to a more depolarized EGABA in perforated patch recordings of RT neurons (Fig. 5A). VU0463271 application produced a time-dependent shift in the EGABA of RT neurons (F(7,32) = 24.25, p < 0.001, n = 5, one-way ANOVA), which first became clear following 5 min of treatment with VU0463271 (Δ+12 ± 1.5 mV, p < 0.001; Fig. 5A2). Overall, the baseline EGABA (−58 ± 3.4 mV) became more depolarized following 10 min of VU0463271 application (−42 ± 5.4 mV, t(4) = 6.24, p = 0.0034, n = 5, paired t test; Fig. 5A3).
Figure 5.
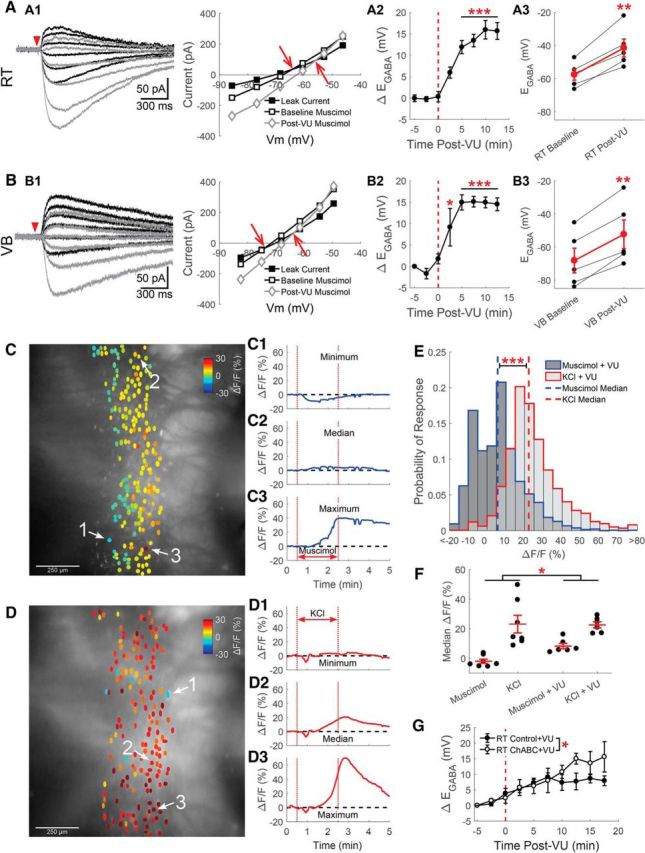
KCC2 regulates EGABA in RT. Gramicidin perforated patch recordings of muscimol-induced (100 μm, 10 ms) currents from RT (A) and VB (B) neurons at various command potentials were altered by application of the specific KCC2 antagonist VU0463271 (10 μm). A1, B1 A depolarizing shift in EGABA occurring between baseline (black traces) and after 10 min of VU0463271 application (gray traces) was reflected in a shifted intersection between the prestimulation leak current and the muscimol-induced current. The leak current has been subtracted from the representative traces. This depolarizing shift was apparent when the time course of the shift in EGABA was measured (A2, B2), and when baseline and 10 min post-VU0463271 EGABA values where compared (A3, B3). C, Calcium imaging of GCaMP6s-expressing RT neurons, preincubated in VU0463271 (10 μm) for 5 min, display a mixed response to a 2 min application of muscimol (5 μm) through a local perfusion system. ROIs drawn around GCaMP6s-expressing RT neurons are colored according to their peak change in fluorescence. Examples of the minimum (C1), median (C2), and maximum (C3) fluorescence change in a particular brain slice following muscimol application. D, Two minutes of elevated KCl still produced a nearly uniform increase in the fluorescence of RT neurons preincubated in VU0463271. Examples of the minimum (D1), median (D2), and maximum (D3) fluorescence changes evoked by elevated KCl application. E, Histograms comparing induced responses in all cells imaged, across multiple animals. Muscimol produced a slight increase in the median response. In contrast, mild depolarization with elevated KCl produced a robust increase in fluorescence. Bin size: 5ΔF/F(%). F, In a comparison of the median GCaMP6s response, per brain slice, pretreatment with VU0463271 produced a greater increase in fluorescence, across the stimuli tested. G, Gramicidin perforated patch recordings of muscimol-induced (100 μm, 10 ms) currents in RT neurons pretreated with ChABC (0.4 U/ml, 2 h at 37°C) displayed a greater depolarizing shift in EGABA following VU0463271 application (10 μm) than did control treated neurons. *p < 0.05, **p < 0.01, ***p < 0.001.
As in RT neurons, there was a time-dependent shift in the EGABA of VB neurons following VU0463271 application (F(7,32) = 14.38, p < 0.001, n = 5, one-way ANOVA; Fig. 5B). This shift in EGABA first became clear following 2.5 min of treatment with VU0463271 (Δ+9 ± 4.4 mV, p = 0.04; Fig. 5B2). Overall, VU0463271 also depolarized the EGABA of VB neurons (baseline: −69 ± 5.6 mV; post-VU0463271: −52 ± 8.7 mV, t(4) = 7.47, p = 0.0017, n = 5, paired t test; Fig. 5B3). In control experiments, no similar time-dependent shift in EGABA was observed (data not shown), comparable to the minimal change of EGABA observed in control recordings from our ChABC experiments (Fig. 4, compare D, E).
As with our initial measurements of EGABA in RT neurons, we again used calcium imaging to examine responses of larger populations of neurons, now preincubated with VU0463271 (10 μm) for 5 min before the start of recording. Preincubation was necessary because responses evoked by local muscimol perfusion attenuated following multiple rounds of application (data not shown). A 2 min application of muscimol now produced a more varied response, with an increase in fluorescence occurring in most RT neurons (Fig. 5C). In aggregate, muscimol stimulation evoked a small, but now positive, 5.5% change in median fluorescence (Interquartile range −4.9–12.4%, n = 1722 cells, n = 6 animals; Fig. 5E). As before, mildly depolarizing VU0463271-preincubated, RT neurons with the addition of 10 mm KCl for 2 min produced a robust increase in fluorescence (22.2%, interquartile range 16.0–31.1%; n = 1732 cells, n = 6 animals, Z = 35.92, p < 0.001, Wilcoxon rank sum test; Fig. 5D).
When the median calcium imaging response per animal was compared across the four conditions tested (compare Figs. 2E–G and 5C–E), larger changes in fluorescence occurred in VU0463271-treated brain slices (15.4 ± 2.5%, n = 12) than in controls (10.5 ± 4.6%, n = 14; F(1,22) = 4.76, p = 0.040, two-way aligned rank transform ANOVA; Fig. 5F). The response to a mild depolarization with KCl was very similar between control (23.1 ± 5.9%, n = 7) and VU0463271 pretreated brain slices (22.6 ± 2.1%, n = 6). Therefore, the observed treatment effect is likely due to the difference in the muscimol-induced change in fluorescence between control (−2.0 ± 1.4%, n = 7) and VU0463171 pretreated brain slices (8.2 ± 1.9%, n = 6). The impact of VU0463271 on muscimol-induced responses is consistent with the depolarizing EGABA shift we observed in VU0463271-treated RT neurons.
As we have found that either inhibiting KCC2 or reducing [A]o depolarizes the EGABA of RT neurons, we now sought to assess the interaction of these two mechanisms in regulating [Cl−]i. Similar to prior experiments, we applied VU0463271 during perforated patch recordings from P10 to P20 rats in brain slices that were either incubated in ChABC (0.4 U/ml, 37°C) for 2 h or control slices incubated in the absence of ChABC (Fig. 5G). We found that the average change in EGABA measured in ChABC-treated neurons (Δ+7.7 ± 1.2 mV), across all times of VU0463271 exposure, was larger than in control-treated neurons (Δ+5.4 ± 0.6 mV; F(1,100) = 5.66, p = 0.020, n = 4,8, two-way ANOVA). This observation suggests that VU0463271 and ChABC treatment act through distinct molecular mechanisms.
Although VU0463271 is thought to specifically antagonize the KCCs, we sought to validate the role of KCC2 in RT neurons using an additional method. Intracellular Cs+ is known to effectively reduce the ability of KCC2 to extrude Cl− (Williams and Payne, 2004). Therefore, rather than the KCl-based recording pipette solution we used in prior experiments, we now used a CsCl-based solution. Recordings from RT neurons using the aforementioned CsCl internal solution revealed a more depolarized EGABA during the 5 min of baseline recording (−54 ± 2.9 mV; Fig. 6A) than found when using a KCl pipette solution (Fig. 2C). The depolarized reversal potential observed using a Cs-based internal solution appears to result from the blockade of KCC2, because subsequent application of VU0463271 (10 μm) produced no significant, time-dependent shift in EGABA (F(7,31) = 0.62, p = 0.74, n = 5, one-way ANOVA; Fig. 6A2). Thus, VU0463271 had little overall effect on the EGABA of RT neurons (−45 ± 4.5 mV, n = 5; t(4) = 1.72, p = 0.16, paired t test; Fig. 6A3).
Similar to RT neurons, the baseline EGABA of VB neurons was less hyperpolarized when recorded with a CsCl internal solution (Fig. 6B). Nonetheless, we observed a small, time-dependent shift in EGABA in response to application of VU0463271 (F(7,31) = 16.92, p = 0.0025, n = 5, one-way ANOVA) following 7.5 min of treatment (Δ+5 ± 1.1 mV; p = 0.011; Fig. 6B2). Overall, the baseline EGABA (−69 ± 1.4 mV) became more depolarized following 10 min of VU0463271 application (−65 ± 1.0 mV; t(4) = 4.74, p = 0.009, n = 5, paired t test; Fig. 6B3). The ability to still detect VU0463271 sensitivity may reflect a greater resiliency of VB neurons to the impact of a Cs-based internal solution on KCC2 function.
Together, these findings suggest that the KCC2 expressed in the RT nucleus still contributes significantly to maintaining a relatively low baseline [Cl−]i, which in turn supports inhibitory responses to GABAergic signaling in RT neurons. However, it remains possible that reduced KCC2 expression diminishes the capacity of RT neurons to maintain a low [Cl−]i during intense periods of GABAergic signaling.
RT neurons have a diminished ability to recover from Cl− loading
Although RT neurons express sufficient levels of KCC2 to maintain a low basal [Cl−]i, it remains unclear how relatively limited KCC2 expression impacts the responses of RT neurons to more intense periods of GABAergic signaling. In the cortical undercut model, a reduction in KCC2 expression is not accompanied by a shift in basal EGABA, although Cl− accumulates faster in these neurons during stimulation (Jin et al., 2005). Therefore, we tested the capacity of RT neurons to resist Cl− loading during repetitive GABAergic signaling, and the rate at which Cl− extrusion returns EGABA to basal levels once stimulation has ceased.
Cl− loading and recovery was measured using perforated patch recordings where the membrane potential of the neuron was held at −30 mV for 500 ms, and then ramped from −100 to −10 mV over a duration of 500 ms (Fig. 7A). This voltage-clamp protocol was repeated once per second for 100 s. Following a 10 s period of baseline recording, Cl− loading was induced with ten 20 ms puffs of 100 μm muscimol, occurring once every 3 s (Fig. 7A). Muscimol was applied while the neuron was voltage-clamped at −30 mV to promote Cl− entry. EGABA was determined by finding the voltage at which the baseline and Cl− loading current traces intersected, following correction for the voltage drop across the series resistance (Fig. 7B, a vs b). Changes in EGABA associated with Cl− recovery, also determined according to the intersection with baseline current traces (Fig. 7B, a vs c), were measured for 62 s following the end of Cl− loading. For each neuron, the time constants for Cl− loading (τload) and recovery (τrec) were determined by fitting the measured EGABA values during the rising and falling phases, respectively, with single-exponential functions (Fig. 7C,D).
Figure 7.

Recovery from Cl− loading is limited in RT neurons. A, Schematics of protocol for Cl− loading experiments. Neurons were voltage-clamped at −30 mV for 500 ms and then ramped from −100 to −10 mV over a duration of 500 ms (left). This protocol lasted one second and was repeated 100 times per cell (right). After 10 s of baseline recording, Cl− was loaded by applying 10 puffs of muscimol (20 ms, 100 μm), once every 3 s, while the neuron was held at −30 mV. B, Gramicidin perforated patch recordings of RT and VB neurons during measurement of Cl− loading and recovery. EGABA was measured by finding the point in the voltage ramp where the membrane current responses from before (a) and during (b) muscimol application intersected (marked by dotted line). Cl− recovery was measured by tracking the change in EGABA following the last application of muscimol (a vs c). Single-exponential functions were fit to the shifting EGABA occurring in RT (C) and VB (D) neurons to determine time constants for Cl− loading and recovery. E, The change in EGABA (ΔEGABA) was measured between the start of Cl− loading and the final reading during Cl− recovery (C, inset). This measurement indicates that chloride loading occurs rapidly in RT neurons. F, The basal Cl− recovery rate (τrec) was slower in RT than in VB neurons. The τrec in RT neurons was unaffected by a 10 min application of VU0463271 (G, 10 μm), but became slower in VB neurons (H). I, A 2 h incubation in ChABC (0.4 U/ml, 37°C) did not alter the τrec of either RT or VB neurons. *p < 0.05, **p < 0.01, ***p < 0.001.
Our experiments revealed a clear difference in τload between RT and VB neurons, and indicated that RT neurons are particularly susceptible to Cl− loading. In general, VB neurons displayed Cl− loading profiles similar to those previously described (Jin et al., 2005), where the initial EGABA measurement at the start of loading was comparable to the final EGABA at the end of recovery (ΔEGABA: −2.0 ± 2.0 mV, n = 16; Fig. 7D,E). Figure 7, C and D (insets), illustrates how this measurement was calculated. In contrast to VB neurons, RT neurons displayed a flat Cl− loading profile and the EGABA measured at the end of recovery was substantially below the initial measurement (ΔEGABA: −17.3 ± 3.3 mV, n = 11, t(25) = 3.81, p < 0.001; Fig. 7C,E). This near-instantaneous τload is likely due to the initial application of muscimol producing a rapid shift in the EGABA of RT neurons, which then only returns to basal levels following a prolonged recovery; our experimental paradigm promotes large Cl− influxes by applying muscimol while neurons are highly depolarized (Fig. 7A). Whereas we measured a τload of 8.5 ± 0.6 s in VB neurons (data not shown), such measurements were obscured in RT neurons by the rapid dynamics of Cl− loading.
Consistent with the limited Cl− extrusion capacity of RT neurons revealed by our Cl− loading measurements, the τrec for RT neurons (30.3 ± 3.7 s, n = 11) was significantly slower than in VB neurons (18.3 ± 2.3 s, n = 15, t(24) = 2.89, p = 0.008; Fig. 7F). As with our earlier measurements of basal EGABA, we next investigated whether limited KCC2 nonetheless contributes to Cl− loading and recovery in RT neurons. Unlike in our basal EGABA measurements, application of the KCC2 antagonist VU0463271 did not produce a shift in the τrec of RT neurons between baseline (25.5 ± 3.3 s) and 10 min post-VU0463271 (10 μm) measurements (27.2 ± 3.1 s, n = 6, t(5) = 0.77, p = 0.47, paired t test; Fig. 7G). In contrast, we observed that the τrec for VB neurons became slower following 10 min of VU0463271 treatment (16.4 ± 3.6 s vs 28.2 ± 4.0 s; n = 8, t(7) = 2.44, p = 0.045, paired t test; Fig. 7H).
We also assessed the impact of reducing [A]o on the speed of recovery from Cl− loading. The τrec for RT neurons incubated in ChABC (0.4 U/ml, 37°C) for 2 h (32.8 ± 4.4 s, n = 3) was unchanged from control RT neurons that were incubated in the absence of ChABC (31.1 ± 6.6 s, n = 4, t(5) = 0.19, p = 0.86; Fig. 7I). We also detected no impact of ChABC incubation on the τrec of VB neurons (20.4 ± 3.0 s, n = 4) compared with control VB neurons (23.1 ± 1.6 s, n = 5, t(7) = 0.86, p = 0.42; Fig. 7I).
Collectively, these extrusion experiments indicate that RT neurons have a reduced capacity to resist activity-dependent shifts in [Cl−]i. While VU0463271 was effective in slowing the rate of Cl− extrusion in VB neurons, we observed no VU0463271 effect on Cl− extrusion in RT neurons. The lack of VU0463271 sensitivity on Cl− extrusion in RT may result from low KCC2 expression; that is, KCC2 is likely overwhelmed during substantial chloride loading, thereby mitigating the effects of pharmacological blockade. Due to their lack of efficient Cl− extrusion mechanisms, RT neurons may be more susceptible to undergoing a shift from inhibitory to excitatory GABAergic signaling during periods of repeated stimulation, such as during a seizure.
Low Cl− extrusion may promote the spread of activity between RT neurons
Although RT neurons maintain a basal [Cl−]i that appears to be sufficiently low to support inhibitory GABAergic signaling, reduced levels of KCC2 compromise rapid recovery from Cl− loading in these neurons. Previous computational models have examined activity-dependent Cl−accumulation in neurons (Jedlicka et al., 2011), and how even mildly reduced KCC2-mediated Cl− extrusion disrupts neural coding (Doyon et al., 2016). We expanded upon this prior work by modeling the impact of reduced Cl− extrusion on signaling within a network of RT neurons. We developed this model primarily to understand how the RT seizure choke point behaves during low and moderate activity bouts.
Based on prior models used to study seizure activity within the RT nucleus (Sohal and Huguenard, 2003), we designed a linear network of 100 RT neurons, in which each cell projected GABAergic synapses to the eight nearest neurons (Fig. 8A). To approximate excitatory input to this network, we delivered repetitive, simulated depolarizing current injections to three centrally located neurons in the array (Cells 46, 51, and 56) to evoke action potentials (Fig. 8B,C1; e.g., Cell 51). In the example shown here, the three stimulated RT neurons generated action potentials at 30 Hz, and all RT neurons extruded Cl− with a τrec of 32 s, a value comparable to our experimental results. Evoked action potentials immediately produced GABAergic currents in neurons postsynaptic to the stimulated cell (Fig. 8C1; e.g., Cell 53, inset). Although each individual postsynaptic potential only generated a transient increase in [Cl−]i, the slow decay kinetics of IPSCs in RT neurons (Huntsman and Huguenard, 2006), coupled with slow Cl− extrusion mechanisms in RT neurons, resulted in a fairly steady, activity-dependent, elevation in [Cl−]i. Once [Cl−]i became sufficiently elevated, postsynaptic potentials became suprathreshold and evoked action potentials (Fig. 8C2; e.g., Cell 53). Thus, as glutamatergic signaling was not present in our model, excitatory GABAergic signaling was sufficient to propagate activity within this simplified network.
Figure 8.
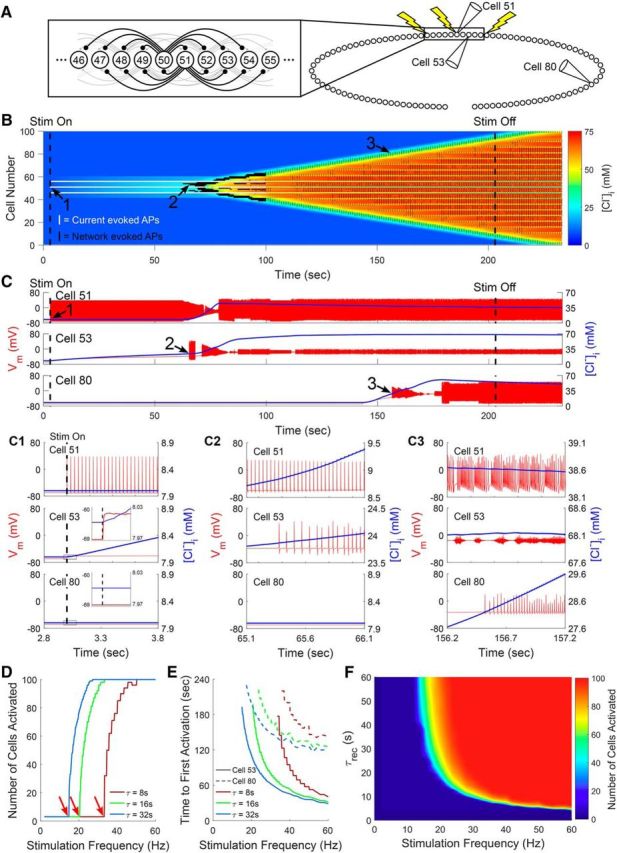
Computational modeling of [Cl−]i dynamics in a network of RT neurons. A, Network composed of a linear array of 100 RT neurons, each projecting GABAergic synapses to the eight nearest neurons. Current injections to Cells 46, 51, and 56 simulated excitatory inputs to this network. B, Graphical representation of evolving [Cl−]i within all model RT neurons over time. Action potential activity generated by individual model RT neurons is overlaid. White vertical lines indicate action potentials in neurons directly receiving current injection, whereas black vertical lines indicate synaptically evoked action potentials. In a network wherein Cells 46, 51, and 56 received 30 Hz stimulation and all cells were characterized by a Cl− recovery rate (τrec) of 32 s, [Cl−]i initially increased only in the neurons receiving direct GABAergic projections from the stimulated cells. Over time, however, activity propagated throughout the entire network. C, Example Vm (red) and [Cl−]i (blue) traces from Cells 51, 53, and 80 from B. Cell 51 received direct, current stimulation. Cell 53 received monosynaptic input from a stimulated cell. Cell 80 was more distant from the site of stimulation. Expanded insets show the shifts in Vm and [Cl−]I at various time points during the propagation of activity within the RT network (C1–C3). When [Cl−]i became sufficiently elevated for GABAergic signaling to evoke action potentials in these neurons (C2), the rise in [Cl−]i began to spread throughout the network, along with an increase in the number of cells firing action potentials (C3). D, Higher-frequency stimulation increased the number of activated neurons (arrows indicate the inflection point where activation first occurs) and (E) decreased the time required to first evoke action potentials (Cell 53, solid lines; Cell 80, dashed lines). F, Both slower τrec and more frequent stimulation were correlated with an increased likelihood of activity spreading within the RT nucleus.
Next, we examined how varying the τrec within our model altered the spread of activation. When the τrec was equal to 32 s, 30 Hz stimulation was sufficient to produce activation throughout the network, including neurons more distant from the sites of direct stimulation (Fig. 8C3; e.g., Cell 80). When we accelerated τrec, higher-frequency stimulation was required to recruit activity within our network (Fig. 8D), as KCC2 more effectively offset Cl− accumulation between postsynaptic potentials. In aggregate, we observed a rapid, τrec-dependent, inflection point between stimulation frequencies unable to recruit network activity and stimulation frequencies that triggered activity throughout the entire RT network (Fig. 8D). Increasing the stimulation frequency also effectively reduced the latency for distant neurons to begin firing action potentials (Fig. 8E). We examined the interplay between τrec and stimulation frequency on global RT network activity across a range of parameters (Fig. 8F). Across this range of parameters, only a narrow band separated conditions that activated zero neurons and all neurons in our network, suggesting a tenuous boundary between normal activity and hyperactivity. Finally, we also observed that slightly increasing the initial [Cl−]i of the neurons in our model, comparable to the shift we observed following the reduction of [A]o with ChABC, slightly promoted the spread of activity throughout our network (data not shown).
Our model demonstrates that the limited Cl− extrusion capability of RT neurons, largely dictated by expression of KCC2, has a considerable impact on the response of this nucleus to excitatory drive. Specifically, the model predicts that RT neurons with a τrec of 32 s, similar to what we measured experimentally, are more susceptible to an activity-dependent shift toward GABA-mediated excitation than neurons with a faster τrec. This propensity for undergoing a shift from inhibitory to excitatory GABAergic signaling allows weaker stimuli to produce activity that spreads throughout the RT nucleus.
Discussion
We demonstrate that RT neurons maintain a low [Cl−]i despite showing reduced immunoreactivity for the Cl− transporter KCC2. The limited KCC2 in RT neurons nonetheless sustains a hyperpolarized EGABA, but restricts Cl− extrusion in response to prolonged GABAergic signaling. RT neurons are therefore susceptible to an activity-dependent shift from inhibitory to excitatory GABAergic signaling. Elevated impermeant anions surrounding RT neurons contribute modestly to EGABA. Overall, these findings support the importance of Cl− transporters and impermeant anions in regulating [Cl−]i in the thalamus and enabling inhibitory GABAergic signaling among RT neurons.
The RT nucleus is a gatekeeper of signaling between cortical and subcortical structures (Jones, 1975; Pinault, 2004; McAlonan et al., 2008). Inhibition among RT neurons is hypothesized to prevent seizures and suppress extraneous sensory input. Regarding the former function, intra-RT inhibition is proposed to establish a seizure choke point that constrains the widespread thalamic synchrony underlying generalized seizures (Sohal and Huguenard, 2003; Paz and Huguenard, 2015; Makinson et al., 2017). Regarding the latter function, inhibitory RT signaling is proposed to focus attention by filtering extraneous sensory stimuli (Crick, 1984; McAlonan et al., 2008; Halassa et al., 2014; Wimmer et al., 2015; Wells et al., 2016). Comprising this focusing mechanism produces sensory gating deficits. Indeed, hallucinations and delusions associated with schizophrenia are hypothesized to result from RT neuron dysfunction (Krause et al., 2003; Egerton et al., 2005; Ferrarelli and Tononi, 2011; Ahrens et al., 2015). If seizure regulation and attention gating requires inhibition within the RT nucleus, then we show that they do so under tenuous circumstances.
GABAergic signaling inhibits RT neurons
Utilizing electrophysiological and calcium imaging techniques, we find that RT neuron activity is, in aggregate, inhibited by GABA. Our measured EGABA for RT neurons (−62 ± 3.0 mV) is substantially more hyperpolarized than RT action potential threshold (Dreyfus et al., 2010; Muñoz and Fuentealba, 2012; ∼−47 mV). As in vivo recordings reveal that RT neurons rest at −62 mV (Bazhenov et al., 1999), our results indicate that GABAA receptor activation clamps RT neurons at their resting membrane potential. Indeed, our calcium imaging reveals that most RT neurons respond to muscimol with minimal change in fluorescence (median ΔF/F = −2.8%, interquartile range −7.1–4.8%; Fig. 2G), suggesting that GABAA receptor activation does not alter resting membrane potential. Even if RT neurons rest at −69 mV, as reported in vitro (Gentet and Ulrich, 2003), then GABAA receptor activation would nonetheless promote shunting inhibition (Edwards, 1990; Staley and Mody, 1992; Blaesse et al., 2009).
It remains possible that a subthreshold membrane depolarization resulting from GABAA-receptor activation evokes action potentials in RT neurons. Modest GABA-induced depolarizations may recruit low voltage-activated, T-type Ca2+ channels that further depolarize RT neurons toward firing threshold (Sun et al., 2012). If true, then a widespread increase in muscimol-induced GCaMP fluorescence would likely result. However, muscimol only intensified fluorescence in a few RT neurons, possibly reflecting cells initially resting at relatively hyperpolarized membrane potentials with more available T-type channels (Perez-Reyes, 2003; Dreyfus et al., 2010). Alternatively, brighter cells may have corresponded to the small subpopulation of RT neurons with a more depolarized basal EGABA, reflecting the heterogeneity of distinct subpopulations known to exist within RT (Lee et al., 2007; Clemente-Perez et al., 2017).
KCC2 is an important regulator of [Cl−]i in RT neurons
Reduced KCC2 mRNA (Kanaka et al., 2001) and protein (Barthó et al., 2004; Sun et al., 2012) in RT neurons of adult rodents predict that GABAergic signaling is excitatory. However, anatomical experiments are unable to evaluate KCC2 function. Recording EGABA with the selective KCC antagonist VU0463271 reveals that even limited KCC2 in RT neurons contributes to Cl− homeostasis. Although VU0463271 has minimal off-target interactions, it does inhibit α1B adrenergic receptors (Delpire et al., 2012; Sivakumaran et al., 2015). As adrenergic signaling may alter KCC2 surface stability and transporter efficacy (Mahadevan and Woodin, 2016), VU0463271 may alter [Cl−]i via an indirect, albeit still KCC2-dependent mechanism. Whereas VU0463271 analogues antagonize other KCCs beyond KCC2 (Delpire and Weaver, 2016), the thalamus appears to only express KCC2 (Rivera et al., 1999; Le Rouzic et al., 2006).
To validate VU0463271, we performed a subset of perforated patch recordings with a Cs-based pipette solution. Although this approach prevents anionic contamination of recorded neurons, cations still diffuse through gramicidin pores and alter intracellular ionic concentrations (Myers and Haydon, 1972). Whereas K+ and Cs+ ions are both substrates for Cl− transport by KCC2, Cs+ translocation occurs at a significantly slower rate, restricting Cl− extrusion and effectively inhibiting KCC2 (Williams and Payne, 2004). The occlusion of a VU0463271 effect by Cs+ indicates that both compounds impact EGABA by blocking the action of KCC2. Cs-based recording solutions have previously yielded noticeably higher measurements of [Cl−]i in RT neurons (Sun et al., 2012).
[A]o only mildly impact [Cl−]i regulation in RT neurons
Our anatomical staining shows that CSPGs, a major source of [A]o, are elevated in the RT nucleus by the second postnatal week, consistent with past investigations (Vitellaro-Zuccarello et al., 2001; Gáti et al., 2010). Regional and cell-type-specific heterogeneity exists in CSPG localization (Matthews et al., 2002), and WFA preferentially labels CSPGs surrounding parvalbumin-positive interneurons (Gáti et al., 2010). Heightened WFA labeling surrounding RT neurons matches the nearly homogenous population of parvalbumin-positive interneurons in this nucleus. In contrast, the limited WFA labeling of VB neurons is attributed to a sparse localization of CSPGs around the preterminal compartments of axons (Gáti et al., 2010).
Whereas ChABC treatment increases [Cl−]i in hippocampal neurons (Glykys et al., 2014a), we observed no change in the basal EGABA of RT or VB neurons following a 2 h incubation in ChABC. However, the EGABA of ChABC-treated RT neurons became more depolarized following repeated measurement. This observation is consistent with the hypothesis that [A]o can define a set point for EGABA, but that other mechanisms likely provide the Cl− flux required for the neuron to equilibrate to this new level (Delpire and Staley, 2014). Our repeated measurements of EGABA involved repetitive GABAA receptor activation, which may have provided a sufficient Cl− flux to unmask the impact of ChABC treatment. Nevertheless, whereas Glykys et al. (2014a) found ChABC treatment of hippocampal neurons produced a 16 mm increase in [Cl−]i, the greatest change in EGABA we observed in RT neurons was only equivalent to a 4 mm increase in [Cl−]i. Utilizing a more intense ChABC treatment or a higher throughput technique to measure changes in [Cl−]i, such as Clomeleon imaging (Dzhala et al., 2012; Wimmer et al., 2015), may permit the unmasking of a greater contribution of [A]o to Cl− homeostasis in thalamic neurons.
Slow Cl− extrusion enhances spreading activation among RT neurons
While the steady-state EGABA value measured in RT neurons supports inhibition, low KCC2 expression significantly slows Cl− extrusion in RT neurons. Repeated GABAA receptor activation produces rapid [Cl−]i accumulation and an associated depolarizing GABAergic response (Staley et al., 1995; Kuner and Augustine, 2000; Jedlicka et al., 2011). Having both weaker initial GABAergic inhibition and a diminished capacity to recover from activity-dependent Cl− influx, RT neurons appear predisposed to shift toward excitatory GABAergic signaling when repeatedly stimulated. Indeed, our computational model suggests that slow Cl− extrusion leaves RT neurons susceptible to undergoing an inhibitory-to-excitatory shift after receiving a train of GABAergic inputs. Slower RT neuron Cl− extrusion rates also correlate with a reduced threshold to evoke excitatory signaling and accelerate the spread of excitatory GABAergic signaling throughout the network. We hypothesize that this recapitulates the characteristic sudden onset of absence seizures (Lüttjohann and van Luijtelaar, 2015), occurring once the inhibitory choke point formed by the RT nucleus is overwhelmed.
Years of anatomical evidence reveal the presence of GABAergic synapses onto RT neurons (Ahlsén and Lindström, 1982; Yen et al., 1985; Ohara, 1988; Cox et al., 1996), and electrophysiological studies suggest that RT neurons are functionally connected via GABAergic synapses (Zhang and Jones, 2004; Deleuze and Huguenard, 2006; Makinson et al., 2017). However, the existence of intra-RT connectivity remains debated (Landisman et al., 2002; Cruikshank et al., 2010; Hou et al., 2016). Nevertheless, the substantia nigra pars reticulata (Paré et al., 1990), globus pallidus (Nauta, 1979), and basal forebrain (Asanuma and Porter, 1990) also provide GABAergic input to RT neurons. These inputs will likewise inhibit RT neuron activity, due to the low [Cl−]i we have measured. Regardless of the source, GABAA receptor-mediated signaling onto RT neurons modulates rhythmic thalamic oscillations. Both local application of the GABAA antagonist bicuculline (Sanchez-Vives and McCormick, 1997) and targeted knockdown of the β3 GABAA subunit (Huntsman et al., 1999) selectively block GABAergic signaling in the RT nucleus, without impacting other thalamic neurons. In both cases, reducing GABAA receptor-mediated inputs to RT neurons promotes absence seizure-like hypersynchronous oscillations.
Conclusions
In sum, the findings of this study demonstrate that GABAergic inhibition is tenuous within the RT nucleus. Weak chloride extrusion mechanisms render RT neurons susceptible to an activity-dependent switch to GABAergic excitation. These findings have important implications for RT's proposed role as a seizure choke point for generalized epilepsies.
Footnotes
This work was supported by NIH/NIGMS T32 training Grant T32GM008328-24 and NIH Grant R01NS099586-01. We thank Drs. Vivek Jayaraman, Douglas S. Kim, Loren L. Looger, and Karel Svoboda from the GENIE Project, Janelia Research Campus, HHMI for permission to use the GCaMP6s construct; Drs. Tarek Deeb and Paul Davies for guidance regarding perforated patch recordings; and Kathryn Salvati and Dr. Ruth Stornetta for invaluable guidance for our anatomical experiments.
The authors declare no competing financial interests.
References
- Ahlsén G, Lindström S (1982) Mutal inhibition between perigeniculate neurones. Brain Res 236:482–486. 10.1016/0006-8993(82)90731-4 [DOI] [PubMed] [Google Scholar]
- Ahrens S, Jaramillo S, Yu K, Ghosh S, Hwang GR, Paik R, Lai C, He M, Huang ZJ, Li B (2015) ErbB4 regulation of a thalamic reticular nucleus circuit for sensory selection. Nat Neurosci 18:104–111. 10.1038/nn.3897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajmo JM, Eakin AK, Hamel MG, Gottschall PE (2008) Discordant localization of WFA reactivity and brevican/ADAMTS-derived fragment in rodent brain. BMC Neurosci 9:14. 10.1186/1471-2202-9-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asanuma C, Porter LL (1990) Light and electron microscopic evidence for a GABAergic projection from the caudal basal forebrain to the thalamic reticular nucleus in rats. J Comp Neurol 302:159–172. 10.1002/cne.903020112 [DOI] [PubMed] [Google Scholar]
- Bandtlow CE, Zimmermann DR (2000) Proteoglycans in the developing brain: new conceptual insights for old proteins. Physiol Rev 80:1267–1290. 10.1152/physrev.2000.80.4.1267 [DOI] [PubMed] [Google Scholar]
- Barthó P, Payne JA, Freund TF, Acsády L (2004) Differential distribution of the KCl cotransporter KCC2 in thalamic relay and reticular nuclei. Eur J Neurosci 20:965–975. 10.1111/j.1460-9568.2004.03562.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ (1999) Self-sustained rhythmic activity in the thalamic reticular nucleus mediated by depolarizing GABA A receptor potentials. Nat Neurosci 2:168–174. 10.1038/5729 [DOI] [PubMed] [Google Scholar]
- Blaesse P, Airaksinen MS, Rivera C, Kaila K (2009) Cation-chloride cotransporters and neuronal function. Neuron 61:820–838. 10.1016/j.neuron.2009.03.003 [DOI] [PubMed] [Google Scholar]
- Bormann J, Hamill OP, Sakmann B (1987) Mechanism of anion permeation through channels gated by glycine and gamma-aminobutyric acid in mouse cultured spinal neurones. J Physiol 385:243–286. 10.1113/jphysiol.1987.sp016493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherubini E, Rovira C, Gaiarsa JL, Corradetti R, Ben Ari Y (1990) GABA mediated excitation in immature rat CA3 hippocampal neurons. Int J Dev Neurosci 8:481–490. 10.1016/0736-5748(90)90080-L [DOI] [PubMed] [Google Scholar]
- Clemente-Perez A, Makinson SR, Higashikubo B, Brovarney S, Cho FS, Urry A, Holden SS, Wimer M, Dávid C, Fenno LE, Acsády L, Deisseroth K, Paz JT (2017) Distinct thalamic reticular cell types differentially modulate normal and pathological cortical rhythms. Cell Rep 19:2130–2142. 10.1016/j.celrep.2017.05.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CL, Huguenard JR, Prince DA (1996) Heterogeneous axonal arborizations of rat thalamic reticular neurons in the ventrobasal nucleus. J Comp Neurol 366:416–430. 10.1002/(SICI)1096-9861(19960311)366:3%3C416::AID-CNE4%3E3.0.CO;2-7 [DOI] [PubMed] [Google Scholar]
- Crick F. (1984) Function of the thalamic reticular complex: the searchlight hypothesis. Proc Natl Acad Sci U SA 81:4586–4590. 10.1073/pnas.81.14.4586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruikshank SJ, Urabe H, Nurmikko AV, Connors BW (2010) Pathway-specific feedforward circuits between thalamus and neocortex revealed by selective optical stimulation of axons. Neuron 65:230–245. 10.1016/j.neuron.2009.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleuze C, Huguenard JR (2006) Distinct electrical and chemical connectivity maps in the thalamic reticular nucleus: potential roles in synchronization and sensation. J Neurosci 26:8633–8645. 10.1523/JNEUROSCI.2333-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLorey TM, Handforth A, Anagnostaras SG, Homanics GE, Minassian BA, Asatourian A, Fanselow MS, Delgado-Escueta A, Ellison GD, Olsen RW (1998) Mice lacking the β3 subunit of the GABAA receptor have the epilepsy phenotype and many of the behavioral characteristics of Angelman syndrome. J Neurosci 18:8505–8514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpire E, Staley KJ (2014) Novel determinants of the neuronal Cl− concentration. J Physiol 592:4099–4114. 10.1113/jphysiol.2014.275529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpire E, Weaver CD (2016) Challenges of finding novel drugs targeting the K-Cl cotransporter. ACS Chem Neurosci 7:1624–1627. 10.1021/acschemneuro.6b00366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpire E, Baranczak A, Waterson AG, Kim K, Kett N, Morrison RD, Daniels JS, Weaver CD, Lindsley CW (2012) Further optimization of the K-Cl cotransporter KCC2 antagonist ML077: development of a highly selective and more potent in vitro probe. Bioorg Med Chem Lett 22:4532–4535. 10.1016/j.bmcl.2012.05.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Destexhe A, Bal T, McCormick DA, Sejnowski TJ (1996) Ionic mechanisms underlying synchronized oscillations and propagating waves in a model of ferret thalamic slices. J Neurophysiol 76:2049–2070. [DOI] [PubMed] [Google Scholar]
- Donnan FG. (1911) Theorie der membran gleichgewichte und membran potentiale bei vorhandernsein von nicht dialysierenden elektrolyten. Zeitschrift für Elektrochimie und Angewandlte Phys Chemie 17:572–581. [Google Scholar]
- Doyon N, Prescott SA, De Koninck Y (2016) Mild KCC2 hypofunction causes inconspicuous chloride dysregulation that degrades neural coding. Front Cell Neurosci 9:516. 10.3389/fncel.2015.00516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon N, Vinay L, Prescott SA, De Koninck Y (2016) Chloride regulation: a dynamic equilibrium crucial for synaptic inhibition. Neuron 89:1157–1172. 10.1016/j.neuron.2016.02.030 [DOI] [PubMed] [Google Scholar]
- Dreyfus FM, Tscherter A, Errington AC, Renger JJ, Shin HS, Uebele VN, Crunelli V, Lambert RC, Leresche N (2010) Selective T-type calcium channel block in thalamic neurons reveals channel redundancy and physiological impact of ITwindow. J Neurosci 30:99–109. 10.1523/JNEUROSCI.4305-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhala V, Valeeva G, Glykys J, Khazipov R, Staley K (2012) Traumatic alterations in GABA signaling disrupt hippocampal network activity in the developing brain. J Neurosci 32:4017–4031. 10.1523/JNEUROSCI.5139-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DH. (1990) Mechanisms of depolarizing inhibition at the crayfish giant motor synapse: I. Electrophysiology. J Neurophysiol 64:541–550. [DOI] [PubMed] [Google Scholar]
- Egerton A, Reid L, McKerchar CE, Morris BJ, Pratt JA (2005) Impairment in perceptual attentional set-shifting following PCP administration: a rodent model of set-shifting deficits in schizophrenia. Psychopharmacology 179:77–84. 10.1007/s00213-004-2109-y [DOI] [PubMed] [Google Scholar]
- Ferrarelli F, Tononi G (2011) The thalamic reticular nucleus and schizophrenia. Schizophr Bull 37:306–315. 10.1093/schbul/sbq142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gáti G, Morawski M, Lendvai D, Jäger C, Négyessy L, Arendt T, Alpár A (2010) Distribution and classification of aggrecan-based extracellular matrix in the thalamus of the rat. J Neurosci Res 88:3257–3266. 10.1002/jnr.22493 [DOI] [PubMed] [Google Scholar]
- Gentet LJ, Ulrich D (2003) Strong, reliable and precise synaptic connections between thalamic relay cells and neurones of the nucleus reticularis in juvenile rats. J Physiol 546:801–811. 10.1113/jphysiol.2002.032730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glascock JJ, Osman EY, Coady TH, Rose FF, Shababi M, Lorson CL (2011) Delivery of therapeutic agents through intracerebroventricular (ICV) and intravenous (IV) injection in mice. J Vis Exp 56:2968. 10.3791/2968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glykys J, Dzhala V, Egawa K, Balena T, Saponjian Y, Kuchibhotla KV, Bacskai BJ, Kahle KT, Zeuthen T, Staley KJ (2014a) Local impermeant anions establish the neuronal chloride concentration. Science 343:670–675. 10.1126/science.1245423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glykys J, Dzhala VI, Egawa K, Balena T, Saponjian Y, Kuchibhotla KV, Bacskai BJ, Kahle KT, Zeuthen T, Staley KJ (2014b) Response to comments on “Local impermeant anions establish the neuronal chloride concentration.” Science 345:1130. 10.1126/science.1253146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Chen Z, Wimmer RD, Brunetti PM, Zhao S, Zikopoulos B, Wang F, Brown EN, Wilson MA (2014) State-dependent architecture of thalamic reticular subnetworks. Cell 158:808–821. 10.1016/j.cell.2014.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines ML, Carnevale NT (1997) The NEURON simulation environment. Neural Comput 9:1179–1209. 10.1162/neco.1997.9.6.1179 [DOI] [PubMed] [Google Scholar]
- Homanics GE, DeLorey TM, Firestone LL, Quinlan JJ, Handforth A, Harrison NL, Krasowski MD, Rick CE, Korpi ER, Mäkelä R, Brilliant MH, Hagiwara N, Ferguson C, Snyder K, Olsen RW (1997) Mice devoid of γ-aminobutyrate type A receptor β3 subunit have epilepsy, cleft palate, and hypersensitive behavior. Proc Natl Acad Sci U S A 94:4143–4148. 10.1073/pnas.94.8.4143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horii-Hayashi N, Sasagawa T, Matsunaga W, Nishi M (2015) Development and structural variety of the chondroitin sulfate proteoglycans-contained extracellular matrix in the mouse brain. Neural Plast 2015:256389. 10.1155/2015/256389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou G, Smith AG, Zhang ZW (2016) Lack of intrinsic GABAergic connections in the thalamic reticular nucleus of the mouse. J Neurosci 36:7246–7252. 10.1523/JNEUROSCI.0607-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntsman MM, Huguenard JR (2006) Fast IPSCs in rat thalamic reticular nucleus require the GABAA receptor beta1 subunit. J Physiol 572:459–475. 10.1113/jphysiol.2006.106617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntsman MM, Porcello DM, Homanics GE, DeLorey TM, Huguenard JR (1999) Reciprocal inhibitory connections and network synchrony in the mammalian thalamus. Science 283:541–543. 10.1126/science.283.5401.541 [DOI] [PubMed] [Google Scholar]
- Jedlicka P, Deller T, Gutkin BS, Backus KH (2011) Activity-dependent intracellular chloride accumulation and diffusion controls GABAA receptor-mediated synaptic transmission. Hippocampus 21:885–898. 10.1002/hipo.20804 [DOI] [PubMed] [Google Scholar]
- Jin X, Huguenard JR, Prince DA (2005) Impaired Cl− extrusion in layer V pyramidal neurons of chronically injured epileptogenic neocortex. J Neurophysiol 93:2117–2126. 10.1152/jn.00728.2004 [DOI] [PubMed] [Google Scholar]
- Jones EG. (1975) Some aspects of the organization of the thalamic reticular complex. J Comp Neurol 162:285–308. [DOI] [PubMed] [Google Scholar]
- Jones EG. (2007) The thalamus, Ed 2 Cambridge; New York: Cambridge UP. [Google Scholar]
- Kaila K, Price TJ, Payne JA, Puskarjov M, Voipio J (2014) Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat Rev Neurosci 15:637–654. 10.1038/nrn3819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaka C, Ohno K, Okabe A, Kuriyama K, Itoh T, Fukuda A, Sato K (2001) The differential expression patterns of messenger RNAs encoding K-Cl cotransporters (KCC1,2) and Na-K-2Cl cotransporter (NKCC1) in the rat nervous system. Neuroscience 104:933–946. 10.1016/S0306-4522(01)00149-X [DOI] [PubMed] [Google Scholar]
- Krause M, Hoffmann WE, Hajós M (2003) Auditory sensory gating in hippocampus and reticular thalamic neurons in anesthetized rats. Biol Psychiatry 53:244–253. 10.1016/S0006-3223(02)01463-4 [DOI] [PubMed] [Google Scholar]
- Kuner T, Augustine GJ (2000) A genetically encoded ratiometric indicator for chloride: capturing chloride transients in cultured hippocampal neurons. Neuron 27:447–459. 10.1016/S0896-6273(00)00056-8 [DOI] [PubMed] [Google Scholar]
- Landisman CE, Long MA, Beierlein M, Deans MR, Paul DL, Connors BW (2002) Electrical synapses in the thalamic reticular nucleus. J Neurosci 22:1002–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Govindaiah G, Cox CL (2007) Heterogeneity of firing properties among rat thalamic reticular nucleus neurons. J Physiol 582:195–208. 10.1113/jphysiol.2007.134254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Rouzic P, Ivanov TR, Stanley PJ, Baudoin FM, Chan F, Pinteaux E, Brown PD, Luckman SM (2006) KCC3 and KCC4 expression in rat adult forebrain. Brain Res 1110:39–45. 10.1016/j.brainres.2006.06.055 [DOI] [PubMed] [Google Scholar]
- Luhmann HJ, Kirischuk S, Kilb W (2014) Comment on “Local impermeant anions establish the neuronal chloride concentration.” Science 345:1130. 10.1126/science.1255337 [DOI] [PubMed] [Google Scholar]
- Lüttjohann A, van Luijtelaar G (2015) Dynamics of networks during absence seizure's on- and offset in rodents and man. Front Physiol 6:16. 10.3389/fphys.2015.00016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan V, Woodin MA (2016) Regulation of neuronal chloride homeostasis by neuromodulators. J Physiol 594:2593–2605. 10.1113/JP271593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinson CD, Tanaka BS, Sorokin JM, Wong JC, Christian CA, Goldin AL, Escayg A, Huguenard JR (2017) Regulation of thalamic and cortical network synchrony by Scn8a. Neuron 93:1165–1179.e6. 10.1016/j.neuron.2017.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews RT, Kelly GM, Zerillo CA, Gray G, Tiemeyer M, Hockfield S (2002) Aggrecan glycoforms contribute to the molecular heterogeneity of perineuronal nets. J Neurosci 22:7536–7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlonan K, Cavanaugh J, Wurtz RH (2008) Guarding the gateway to cortex with attention in visual thalamus. Nature 456:391–394. 10.1038/nature07382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Prince DA (1986) Acetylcholine induces burst firing in thalamic reticular neurones by activating a potassium conductance. Nature 319:402–405. 10.1038/319402a0 [DOI] [PubMed] [Google Scholar]
- Muñoz F, Fuentealba P (2012) Dynamics of action potential initiation in the GABAergic thalamic reticular nucleus in vivo. PLoS One 7:e30154. 10.1371/journal.pone.0030154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers VB, Haydon DA (1972) Ion transfer across lipid membranes in the presence of gramicidin A: II. The ion selectivity. Biochim Biophys Acta 274:313–322. 10.1016/0005-2736(72)90179-4 [DOI] [PubMed] [Google Scholar]
- Nauta HJ. (1979) Projections of the pallidal complex: an autoradiographic study in the cat. Neuroscience 4:1853–1873. 10.1016/0306-4522(79)90060-5 [DOI] [PubMed] [Google Scholar]
- Ohara PT. (1988) Synaptic organization of the thalamic reticular nucleus. J Electron Microsc Tech 10:283–292. 10.1002/jemt.1060100306 [DOI] [PubMed] [Google Scholar]
- Paré D, Hazrati LN, Parent A, Steriade M (1990) Substantia nigra pars reticulata projects to the reticular thalamic nucleus of the cat: a morphological and electrophysiological study. Brain Res 535:139–146. 10.1016/0006-8993(90)91832-2 [DOI] [PubMed] [Google Scholar]
- Payne JA, Stevenson TJ, Donaldson LF (1996) Molecular characterization of a putative K-Cl cotransporter in rat brain: a neuronal-specific isoform. J Biol Chem 271:16245–16252. 10.1074/jbc.271.27.16245 [DOI] [PubMed] [Google Scholar]
- Paz JT, Huguenard JR (2015) Microcircuits and their interactions in epilepsy: is the focus out of focus? Nat Neurosci 18:351–359. 10.1038/nn.3950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Reyes E. (2003) Molecular physiology of low-voltage-activated T-type calcium channels. Physiol Rev 83:117–161. 10.1152/physrev.00018.2002 [DOI] [PubMed] [Google Scholar]
- Pinault D. (2004) The thalamic reticular nucleus: structure, function and concept. Brain Res Brain Res Rev 46:1–31. 10.1016/j.brainresrev.2004.04.008 [DOI] [PubMed] [Google Scholar]
- Pinault D, Smith Y, Deschênes M (1997) Dendrodendritic and axoaxonic synapses in the thalamic reticular nucleus of the adult rat. J Neurosci 17:3215–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K (1999) The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397:251–255. 10.1038/16697 [DOI] [PubMed] [Google Scholar]
- Rohrbough J, Spitzer NC (1996) Regulation of intracellular Cl− levels by Na+-dependent Cl− cotransport distinguishes depolarizing from hyperpolarizing GABAA receptor-mediated responses in spinal neurons. J Neurosci 16:82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Vives MV, McCormick DA (1997) Functional properties of perigeniculate inhibition of dorsal lateral geniculate nucleus thalamocortical neurons in vitro. J Neurosci 17:8880–8893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivakumaran S, Cardarelli RA, Maguire J, Kelley MR, Silayeva L, Morrow DH, Mukherjee J, Moore YE, Mather RJ, Duggan ME, Brandon NJ, Dunlop J, Zicha S, Moss SJ, Deeb TZ (2015) Selective inhibition of KCC2 leads to hyperexcitability and epileptiform discharges in hippocampal slices and in vivo. J Neurosci 35:8291–8296. 10.1523/JNEUROSCI.5205-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal VS, Huguenard JR (2003) Inhibitory interconnections control burst pattern and emergent network synchrony in reticular thalamus. J Neurosci 23:8978–8988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley K, Smith R (2001) A new form of feedback at the GABAA receptor. Nat Neurosci 4:674–676. 10.1038/89439 [DOI] [PubMed] [Google Scholar]
- Staley KJ, Mody I (1992) Shunting of excitatory input to dentate gyrus granule cells by a depolarizing GABAA receptor-mediated postsynaptic conductance. J Neurophysiol 68:197–212. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Soldo BL, Proctor WR (1995) Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science 269:977–981. 10.1126/science.7638623 [DOI] [PubMed] [Google Scholar]
- Sun YG, Wu CS, Renger JJ, Uebele VN, Lu HC, Beierlein M (2012) GABAergic synaptic transmission triggers action potentials in thalamic reticular nucleus neurons. J Neurosci 32:7782–7790. 10.1523/JNEUROSCI.0839-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JT, Daigle TL, Chen Q, Feng G (2014) Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. In: Patch-clamp methods and protocols, Ed 2 (Martina M, Taverna S, eds), pp 221–242. Methods in Molecular Biology New York: Springer New York. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich D, Huguenard JR (1997) Nucleus-specific chloride homeostasis in rat thalamus. J Neurosci 17:2348–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitellaro-Zuccarello L, Meroni A, Amadeo A, De Biasi S (2001) Chondroitin sulfate proteoglycans in the rat thalamus: expression during postnatal development and correlation with calcium-binding proteins in adults. Cell Tissue Res 306:15–26. 10.1007/s004410100425 [DOI] [PubMed] [Google Scholar]
- Voipio J, Boron WF, Jones SW, Hopfer U, Payne JA, Kaila K (2014) Comment on “Local impermeant anions establish the neuronal chloride concentration.” Science 345:1130. 10.1126/science.1252978 [DOI] [PubMed] [Google Scholar]
- Wells MF, Wimmer RD, Schmitt LI, Feng G, Halassa MM (2016) Thalamic reticular impairment underlies attention deficit in Ptchd1(Y/−) mice. Nature 532:58–63. 10.1038/nature17427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JR, Payne JA (2004) Cation transport by the neuronal K+-Cl− cotransporter KCC2: thermodynamics and kinetics of alternate transport modes. Am J Physiol Cell Physiol 287:C919–C931. 10.1152/ajpcell.00005.2004 [DOI] [PubMed] [Google Scholar]
- Wimmer RD, Schmitt LI, Davidson TJ, Nakajima M, Deisseroth K, Halassa MM (2015) Thalamic control of sensory selection in divided attention. Nature 526:705–709. 10.1038/nature15398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wobbrock JO, Findlater L, Gergle D, Higgins JJ (2011) The aligned rank transform for nonparametric factorial analyses using only ANOVA procedures. In: Proceedings of the ACM Conference on Human Factors in Computing Systems (CHI ‘11) Vancouver, British Columbia (May 7–12, 2011), pp 143–146. New York: ACM. [Google Scholar]
- Wong CGT, Bottiglieri T, Snead OC (2003) GABA, gamma-hydroxybutyric acid, and neurological disease. Ann Neurol 54:S3–S12. 10.1002/ana.10696 [DOI] [PubMed] [Google Scholar]
- Yamagata T, Saito H, Habuchi O, Suzuki S (1968) Purification and properties and chondrosulfatases of bacterial chondroitinases. J Biol Chem 243:1523–1535. [PubMed] [Google Scholar]
- Yen CT, Conley M, Hendry SH, Jones EG (1985) The morphology of physiologically identified GABAergic neurons in the somatic sensory part of the thalamic reticular nucleus in the cat. J Neurosci 5:2254–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Jones EG (2004) Corticothalamic inhibition in the thalamic reticular nucleus. J Neurophysiol 91:759–766. 10.1152/jn.00624.2003 [DOI] [PubMed] [Google Scholar]