

Abstract
Key points
K+ channels are important in intestinal epithelium as they ensure the ionic homeostasis and electrical potential of epithelial cells during anion and fluid secretion.
Intestinal epithelium cAMP‐activated anion secretion depends on the activity of the (also cAMP dependent) KCNQ1–KCNE3 K+ channel, but the secretory process survives after genetic inactivation of the K+ channel in the mouse.
Here we use double mutant mice to investigate which alternative K+ channels come into action to compensate for the absence of KCNQ1–KCNE3 K+ channels.
Our data establish that whilst Ca2+‐activated KCa3.1 channels are not involved, K2P two‐pore domain TASK‐2 K+ channels are major players providing an alternative conductance to sustain the intestinal secretory process.
Work with double mutant mice lacking both TASK‐2 and KCNQ1–KCNE3 channels nevertheless points to yet‐unidentified K+ channels that contribute to the robustness of the cAMP‐activated anion secretion process.
Abstract
Anion and fluid secretion across the intestinal epithelium, a process altered in cystic fibrosis and secretory diarrhoea, is mediated by cAMP‐activated CFTR Cl− channels and requires the simultaneous activity of basolateral K+ channels to maintain cellular ionic homeostasis and membrane potential. This function is fulfilled by the cAMP‐activated K+ channel formed by the association of pore‐forming KCNQ1 with its obligatory KCNE3 β‐subunit. Studies using mice show sizeable cAMP‐activated intestinal anion secretion in the absence of either KCNQ1 or KCNE3 suggesting that an alternative K+ conductance must compensate for the loss of KCNQ1–KCNE3 activity. We used double mutant mouse and pharmacological approaches to identify such a conductance. Ca2+‐dependent anion secretion can also be supported by Ca2+‐dependent KCa3.1 channels after independent CFTR activation, but cAMP‐dependent anion secretion is not further decreased in the combined absence of KCa3.1 and KCNQ1–KCNE3 K+ channel activity. We show that the K2P K+ channel TASK‐2 is expressed in the epithelium of the small and large intestine. Tetrapentylammonium, a TASK‐2 inhibitor, abolishes anion secretory current remaining in the absence of KCNQ1–KCNE3 activity. A double mutant mouse lacking both KCNQ1–KCNE3 and TASK‐2 showed a much reduced cAMP‐mediated anion secretion compared to that observed in the single KCNQ1–KCNE3 deficient mouse. We conclude that KCNQ1–KCNE3 and TASK‐2 play major roles in the intestinal anion and fluid secretory phenotype. The persistence of an, admittedly reduced, secretory activity in the absence of these two conductances suggests that further additional K+ channel(s) as yet unidentified contribute to the robustness of the intestinal anion secretory process.
Keywords: epithelial transport, fluid secretion, K+ channel
Key points
K+ channels are important in intestinal epithelium as they ensure the ionic homeostasis and electrical potential of epithelial cells during anion and fluid secretion.
Intestinal epithelium cAMP‐activated anion secretion depends on the activity of the (also cAMP dependent) KCNQ1–KCNE3 K+ channel, but the secretory process survives after genetic inactivation of the K+ channel in the mouse.
Here we use double mutant mice to investigate which alternative K+ channels come into action to compensate for the absence of KCNQ1–KCNE3 K+ channels.
Our data establish that whilst Ca2+‐activated KCa3.1 channels are not involved, K2P two‐pore domain TASK‐2 K+ channels are major players providing an alternative conductance to sustain the intestinal secretory process.
Work with double mutant mice lacking both TASK‐2 and KCNQ1–KCNE3 channels nevertheless points to yet‐unidentified K+ channels that contribute to the robustness of the cAMP‐activated anion secretion process.
Introduction
The secretion of fluid across the small and large intestine is secondary to electrogenic anion transport. The disruption of intestinal secretion in pathophysiological scenarios such as secretory diarrhoea and cystic fibrosis bear witness to the importance of the fine regulation of this process (reviewed by Barrett & Keely, 2000; Frizzell & Hanrahan, 2012). Secretion occurs when CFTR Cl− channels become activated by phosphorylation thus allowing for the efflux of Cl− accumulated intracellularly above electrochemical equilibrium across apical membranes into the intestinal lumen. Influx across basolateral membrane through NKCC1 Na+–K+–2Cl− cotransport accumulates Cl− harnessing the Na+ gradient maintained by the basolateral membrane Na+–K+ pump. Cl− secretion across intestinal epithelium is stimulated by an increase in cAMP (or cGMP) that promotes phosphorylation of CFTR. A K+ conductance of the basolateral membranes of intestinal epithelial cells is an essential player in the process of transepithelial Cl− secretion by virtue of its role in recycling of K+ ions taken up through the action of the Na+–K+ pump and the Na+–K+–2Cl− cotransporter, and in the maintenance of the electrochemical driving force for apical Cl− exit through their hyperpolarizing effect on the enterocytes. Additionally, but not further discussed here, the colon is able to secrete K+ using apically located K+ channels (Halm & Frizzell, 1986; Field, 2003).
A convincing body of work has demonstrated that the function of cAMP‐activated basolateral K+ channels is essential for intestinal Cl− secretion, a process that is abolished using the cAMP‐activated K+ channel inhibitor chromanol 293B (C293B) (Lohrmann et al. 1995; Diener et al. 1996; Warth et al. 1996; Greger et al. 1997; MacVinish et al. 1998). The identification of KCNE3 as a regulatory subunit of the KCNQ1 K+ channel revealed that a KCNE3–KCNQ1 complex yields a functional conductance remarkably suited to represent the basolateral K+‐selective counterpart of CFTR Cl− channels (Schroeder et al. 2000): KCNQ1–KCNE3‐mediated, cAMP‐activated K+ current is virtually time‐ and voltage‐independent and abolished by low micromolar concentration of C293B. The mRNAs for KCNQ1 and KCNE3 are found in the crypt cells of small and large intestine where the proteins localize to the basolateral membranes (Preston et al. 2010). Formal proof of the involvement of KCNQ1–KCNE3 in cAMP‐activated anion secretion in the intestine came from work using knockout (KO) mice invalidated for the genes encoding either KCNQ1 (Vallon et al. 2005) or KCNE3 (Preston et al. 2010), which are markedly affected in their intestinal anion secretory function.
A puzzling observation in these studies was that despite the marked decreases in cAMP‐activated anion secretion in the colon of both the Kcnq1 and Kcne3 KO mice, a sizeable remnant activity persisted that could be as high as 40% of that in WT epithelia (Vallon et al. 2005; Preston et al. 2010). This suggests that an alternative K+ conductance might compensate for the loss of KCNQ1–KCNE3 activity, a concept that had also been advanced on the basis of a pharmacological study of colon anion secretion (Liao et al. 2005). We have attempted to identify such a conductance using double mutant mouse and pharmacological approaches. Ca2+‐dependent anion secretion can also be supported by activation of Ca2+‐dependent KCa3.1 channels provided CFTR channels are independently activated (Warth et al. 1999) and this type of secretion is absent in the KCa3.1‐encoding gene Kcnn4 null mouse (Flores et al. 2007; Matos et al. 2007). Double mutant mice lacking both KCNQ1–KCNE3 and KCa3.1 were not different from single KCNQ1–KCNE3 KOs in their cAMP‐dependent anion secretion. Pharmacological and genetic ablation experiments, on the other hand, strongly suggest a major role for the K2P K+ channel TASK‐2 (Cid et al. 2013). Surprisingly, however, double mutant mice lacking both KCNQ1–KCNE3 and TASK‐2 showed a persistent, albeit small, cAMP‐mediated anion secretion. We conclude that KCNQ1–KCNE3 and TASK‐2 are important actors in the intestinal anion and fluid secretory phenotype, which nevertheless persists in spite of the knockouts of K+ channel genes predicted to be crucial to the physiological process. This contrasts with the single ablation of CFTR that obliterates function altogether. The K+ channel(s) functioning in the absence of those recognized as supporting intestinal anion secretion remain to be identified and the mechanism by which they are put into action represents a future challenging subject of research.
Methods
Ethical approval
All animal procedures were reviewed and approved by the local Institutional Animal Care and Use Committee. The animal facility at the Centro de Estudios Científicos (CECs) is accredited by AAALAC and it adheres to the guidelines in the Guide for the Care and Use of Laboratory Animals (the Guide, NRC 2011). Mice were bred at the Animal Facility of the CECs from founders purchased at The Jackson Laboratory (Bar Harbor, ME, USA), unless otherwise stated. Animals had access to food and water ad libitum. The authors of the present investigation understand the ethical principles under which The Journal operates and confirm that the work complies with The Journal’s animal ethics checklist.
Generation of Tg(Kcne3‐D90N) transgenic mice
Transgenic mice were generated at the Animal Facility of the CECs. A mammalian expression construct with the villin promoter was used to insert the hemagglutinin (HA)‐tagged human KCNE3‐D90N coding sequence, downstream of the villin promoter. The transgene cassette was sequenced, released from the parental vector by digestion with SalI, and injected into zygotes from (C57BL/6J × CBA) F0 hybrid mice. Transgenic animals were back‐crossed to C57BL/6 WT mice for at least 10 generations to generate the Tg(Kcne3‐D90N) mice on the C57BL/6 strain background.
PCR genotyping was performed using the following primers: P1 (5′‐GGT CGA GGC TAA AGA AGA G‐3′), and P2 (5′‐CTA CGC TTG TCC ACT TTG C‐3′).
Kcne3, Kcnn4 and Kcnk5 null and Cftr‐ΔF508 mutant mice
Kcne3 KO animals have been generated by deletion of exon 4 that contains the whole coding region of the Kcne3 gene (Preston et al. 2010). Kcnn4 KO mice were those generated by Begenisich et al. (2004). The Kcnk5 KO mouse was generated using a trapping vector encoding a β‐galactosidase (Taniwaki et al. 2005) and lacks expression of K2P channel TASK‐2. The Cftrtm1Eur mouse (van Doorninck et al. 1995) carries the mutant ΔF508 of the CFTR protein.
Only male mice were used throughout, except for the Cftrtm1Eur mice for which three males and three females were used.
Tissue isolation and Ussing chamber experiments
Mice (C57BL/6J) were killed by cervical dislocation carried out by trained personnel at approved premises. Colon was excised, rinsed with warm 0.9% NaCl and cut open lengthwise through the mesenteric border. A partially stripped mucosal sheet, from distal colon, was obtained by scraping the mucosa surface with a glass microscope slide. The sheet obtained was mounted on a tissue‐holding slider (aperture 0.1 cm2) and put as a dividing membrane in a modified Ussing chamber (Physiologic Instruments Inc., San Diego, CA, USA). The bath solution contained (in mm): 120 NaCl, 25 NaHCO3, 3.3 KH2PO4, 0.8 K2HPO4, 1.2 MgCl2, 1.2 CaCl2 and 10 d‐glucose, and was continuously gassed with 5% CO2 and 95% O2.
The transepithelial potential difference (referred to the serosal compartment) was measured continuously using a VCC MC2 amplifier (Physiologic Instruments Inc.). The current was clamped at zero and 1 s pulses of 10 μA were given at 0.2 s intervals. The voltage pulses were generated with Acquire & Analyse v. 2.3 software through a DI‐720 data acquisition system (DataQ Instruments, Akron, OH, USA). The difference in current and voltage was used to calculate the tissue resistance and equivalent short‐circuit currents according to Ohm's law.
Apical 10 μm amiloride (Sigma‐Aldrich, St Louis, MO, USA) was added to block Na+ current through the epithelial sodium channels (ENaC). Increasing intracellular cAMP was achieved with 1 μm forskolin (Merck, Darmstadt, Germany) and isobutylmethylxanthine μm 100 (IBMX, Sigma‐Aldrich). K+ channel inhibitors used were 10 μm chromanol 293B (Tocris Bioscience, Bristol, UK) and 100 μm tetrapentylammonium (TPeA; Sigma‐Aldrich). Carbachol 100 μm (Sigma‐Aldrich) was used as muscarinic cholinergic agonist.
Intestinal fluid secretion measured in vivo
Mice of 2–3 months of age and body weight between 20 and 30 g were used. Animals were anaesthetized by exposure to isoflurane (Baxter Healthcare, Guayama, PR, USA) using the RC2 rodent circuit controller (Vetequipment, Pleasanton, CA, USA). The experiment was conducted at a separate, ad hoc laboratory accessible to the trained personnel involved only. Animals were placed in a 1‐litre induction chamber under 1000 ml min−1 flow of air containing 2.5% isoflurane, until recumbent. Animals were then kept under anaesthesia with 2% isoflurane at a constant flow rate of 500 ml min−1 using a mask. Body temperature was maintained by placing the mouse on a heating pad at ∼38°C. Prior to the operation mice were hydrated by subcutaneous injection of 17 ml kg−1 0.9% NaCl at 35°C. The small intestine was externalized from the abdominal cavity using anatomical tweezers after making a small abdominal incision that exposed the caecum. Great care was taken not to break any blood vessels in the process. Two ileal loops (∼20 mm) proximal to the caecum were isolated. The closed loops were injected with 100 μl of phosphate‐buffered saline (PBS) or PBS containing cholera toxin (0.5 μg per loop) (Gentaur, Kampenhout, Belgium). The wound was sutured, and mice were kept under isoflurane anaesthesia (2% isoflurane and air flow rate 500 ml min−1).
Animals were constantly monitored (respiratory frequency, temperature, skin colour and depth of anaesthesia judged by tail pinching) at 30 min intervals. After 5 h postoperatory, mice were killed without recovery from anaesthesia by cervical dislocation, and intestinal loops were removed, weighed and their length measured. Fluid secretion was calculated as the weight difference of loops before and after draining all fluid and corrected for tissue length.
Immunofluorescence
Distal colon paraffin‐embedded sections (4 μm) from 8‐week‐old mice were incubated with rat anti‐HA (1:300; cat. no. 11867423001, Sigma‐Aldrich) or rabbit anti‐KCNQ1 (1:200; cat. no. AB9888, Millipore) primary antibodies overnight at 4°C. The sections were then incubated with goat anti‐rat and goat anti‐rabbit IgG coupled to Alexa Fluor 488 (cat. no. A‐11006, Thermo Fisher Scientific, Waltham, MA, USA) or 568 (cat. no. A‐11011, Thermo Fisher Scientific), respectively, for 1h at room temperature. Analysis was performed using confocal microscopy (Olympus FV1000).
β‐Galactosidase activity
The expression of TASK‐2 channel was observed indirectly through the detection of β‐galactosidase activity. Intestine sections were fixed with 10% formol for 1 h at 4°C. Cryopreserved tissues were cut into 5 μm sections, rinsed with PBS and incubated overnight at 37°C in X‐Gal staining solution (PBS pH 7.4, containing 1 mg ml−1 X‐Gal, 2.5 mm K3Fe(CN)6, 4 mm K4Fe(CN)6, 1.2 mm MgCl2, 0.01% sodium deoxycholate and 0.02% Igepal). Sections were washed with PBS and then counterstained with eosin. Digital images were collected using an Olympus CX31 microscope and recorded with a digital microscope camera (Mshot MD90).
Statistical analysis
Data are presented as means with standard deviation (SD) and significance of differences was assessed by Student t test or ANOVA as indicated in the figure legends.
Results and discussion
Generation of a transgenic mouse expressing KCNE3‐D90N mutant in the intestinal epithelium
In order to confirm the role of KCNQ1–KCNE3 channels in intestinal epithelium (Preston et al. 2010) we wanted to generate a mouse in which the channel was inactivated in a tissue‐specific manner. To this end we have made a transgenic mouse expressing the mutated KCNE3‐D90N auxiliary subunit known to exert a dominant‐negative effect on the KCNQ1–KCNE3 channel complex (Abbott & Goldstein, 2002) under the control of intestinal tissue‐specific villin promoter. Villin is a protein in intestinal epithelial cells and the regulatory region of its gene efficiently targets heterologous constructs to immature and differentiated epithelial cells of the small and large intestinal epithelium (Pinto et al. 1999; Sandoval et al. 2011). Figure 1 A shows the transgene used to generate the Tg(Kcne3‐D90N) transgenic animals. In addition to being under the control of a villin promoter the construct was designed to carry an HA epitope to facilitate its localization by immunofluorescence. Figure 1 B identifies the transgene expressed in four out of nine mice originating from microinjected zygotes, which were used to generate the lines analysed here. The immunolocalization of the KCNE3‐D90N protein was then compared with that of the conducting subunit KCNQ1. As shown in Fig. 1 C, KCNQ1 was, as expected (Dedek & Waldegger, 2001; Liao et al. 2005; Preston et al. 2010), found at the basolateral membranes in the crypts of colonic epithelium from both WT and KCNE3‐D90N‐expressing animals. Immunoreactivity for KCNE3‐D90N, on the other hand, was only observed in the transgenic tissue where it was basolateral at the base of the crypts but appeared also in the cytoplasm of cells at or near the surface. These data reveal that expression of KCNE3‐D90N did not alter the general distribution of the KCNQ1 subunit, but we cannot discern whether the levels of expression are equivalent in the two genotypes.
Figure 1. Generation and characterization of transgenic mice expressing KCNE3‐D90N mutant in intestinal epithelium.
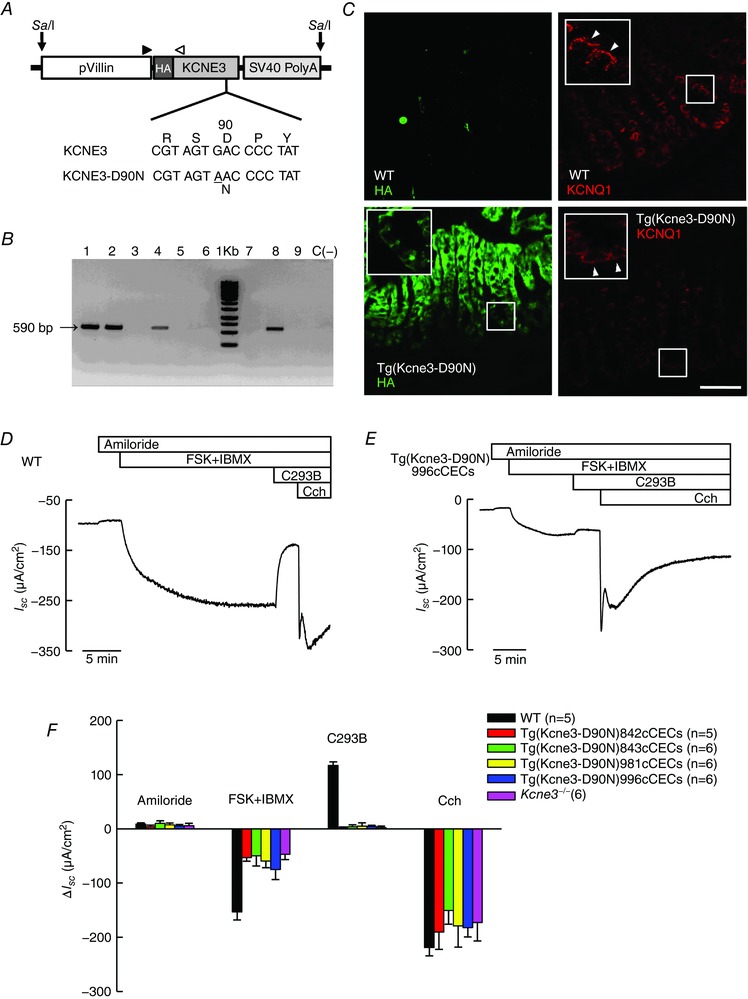
A, scheme of the pVillin KCNE3‐D90N‐HA transgene used for the generation of tissue‐specific transgenic mice expressing hemagglutinin (HA)‐tagged KCNE3‐D90N protein under intestinal epithelium‐specific promoter villin; SV40(A), simian virus 40 polyadenylation signal. SalI, restriction site used for transgene (9600 bp) linearization. The arrowheads indicate the primer binding sites. Bottom, sequence of KCNE3, showing the G>A mutation results in KCNE3‐D90N protein. B, a 560 bp PCR fragment is detectable with the P1–P2 primer pair in transgenic mice. Lanes labelled 1, 2, 4 and 8 correspond to different KCNE3‐D90N transgenic lines (later named as in panel F) and lanes 3, 5, 6, 7 and 9 to mice that did not incorporate the transgene; negative control C (−) is DNA from a WT biopsy. C, tissue distribution of KCNE3‐D90N and KCNQ1. KCNE3‐D90N was detected using immunofluorescence against HA (green) in mouse distal colon. Top panels show WT control sections; bottom, transgenic tissues from the Tg(Kcne3‐D90N)842cCECs line. Right, staining for KCNQ1 (red). Insets show higher magnifications of regions shown in frames with arrowheads pointing to basolateral aspect of enterocytes. Bar represents 40 μm. D and E, traces showing recordings of short circuit currents (I SC) as function of time obtained using distal colon from WT (D) or Tg(Kcne3‐D90N) (E) mice mounted in Ussing chambers. Additions, during the times shown in the upper bars, were 10 μm amiloride, 100 μm isobutylmethylxanthine (IBMX) plus 1 μm forskolin (FSK), 10 μm chromanol 293B (C293B) and 100 μm carbachol (Cch). All additions were made to the serosal side of the epithelium save for amiloride, which was added apically. Tissue resistances were, respectively, 78 and 71 Ω cm2 at the beginning and end of the experiment shown in D. The respective numbers in E were 91 and 67 Ω cm2. F, bar graph summarizing the mean change in short circuit‐current (ΔI SC) after the different additions in D and E. Each colour corresponds to a different Tg(Kcne3‐D90N) transgenic line. Data for the Kcne3 −/− mouse are also shown. Results are means ± SD of the number of animals given in parentheses. FSK+IBMX and C293B treatments in transgenic and Kcne3 −/− tissues are all different from those in WT judged by Bonferroni t test with P < 0.001.
Effect of intestinal inactivation of KCNQ1–KCNE3 K+ channels in intestinal Cl− secretion
Direct evidence for a role of the KCNE3/KCNQ1 channel in intestinal secretion has come from functional studies of mice null for the KCNQ1 or KCNE3 subunits (Vallon et al. 2005; Preston et al. 2010). We confirm those results here using the intestinal epithelium‐specific expression of the dominant‐negative KCNE3 mutant D90N. As seen in Fig. 1 D, increasing cellular cAMP using forskolin and isobutylmethylxanthine (FSK+IBMX) evoked a sustained transepithelial anion secretory current across the colon of WT mice. This was markedly diminished in tissues from the Tg(Kcne3‐D90N) transgenic mouse (Fig. 1 E) that behaved virtually identically to the total KO, the Kcne3 −/− mouse (Preston et al. 2010). Average responses to agonists and inhibitors in colon from WT, Tg(Kcne3‐D90N) and Kcne3 −/− mice are shown in Fig. 1 F, those for the transgenic animal as results from the four lines generated, which are identified by different numbers. The colon of the Tg(Kcne3‐D90N) transgenic mouse had a remnant cAMP‐activated current that was about 40% of that in WT epithelium. The current was very markedly inhibited by basolaterally added C293B in WT but not in the KCNE3‐D90N‐expressing or Kcne3 −/− tissues, which were not affected by the blocker. The anion secretory current activated with the Ca2+ agonist carbachol was not different in WT and transgenic or KO tissues. This anion secretory current is known to reflect the activation of Ca2+‐dependent KCa3.1 channels that provide the driving force for apical Cl− efflux through previously activated CFTR channels (Warth et al. 1999) and is absent in Kcnn4 null mice (Flores et al. 2007; Matos et al. 2007). The small component of the current attributable to amiloride‐sensitive Na+ absorption was not different between the genotypes. Table 1 shows the same results as Fig. 1 F but before ΔI SC calculation. There is a tendency for smaller basal or amiloride‐insensitive current in the models with inactivated KCNE3 but otherwise these raw data give the same message as those in Fig. 1 F. The results are very similar to those obtained by inactivation of the KCNE3/KCNQ1 channels through ablation of either the conducting KCNQ1 subunit (Vallon et al. 2005) or the KCNE3 β‐subunit essential for function at physiological potentials (Preston et al. 2010).
Table 1.
Absolute I SC values in colon from WT and different genotypes with inactivated KCNE3 expression
| WT (5) | 842c (5) | 843c (6) | 982c (6) | 996c (6) | Kcne3 −/− (6) | |
|---|---|---|---|---|---|---|
| Basal | −53 ± 18.2 | −19 ± 7.9 | −29 ± 13.0 | −33 ± 13.8 | −51 ± 24.7 | −36 ± 8.2 |
| Amiloride | −45 ± 19.9 | −16 ± 9.3 | −19 ± 16.6 | −26 ± 16.4 | −45 ± 24.7 | −32 ± 9.5 |
| FSK+IBMX | −198 ± 25.4 | −69 ± 3.8 | −69 ± 33.9 | −86 ± 27.8 | −120 ± 59.6 | −77 ± 13.4 |
| C293B | −81 ± 15.2 | −67 ± 4.7 | −65 ± 36.2 | −81 ± 31.3 | −116 ± 59.8 | −75 ± 14.7 |
| Carbachol | −299 ± 40.3 | −257 ± 32.4 | −216 ± 30.0 | −260 ± 63.7 | −298 ± 91.2 | −240 ± 46.5 |
Values are given as means ± SD of the number of mice indicated in parentheses. The genotype of the Kcne3‐D90N transgenic mice is abbreviated to the line number only. The data are from the individual values used to calculate the changes in I SC under different treatments in Fig. 1 F.
Effect of simultaneous inactivation of KCNQ1–KCNE3 and KCa3.1 K+ channels on intestinal Cl− secretion
The result shown above indicates that an alternative K+ conductance, insensitive to C293B, must support CFTR‐mediated Cl− efflux in the absence of KCNQ1–KCNE3 activity. As already suggested in previous KCNE3 inactivation work (Preston et al. 2010) an obvious candidate to take such a place is the KCa3.1 K+ channel encoded by the Kcnn4 gene, which has been demonstrated to be involved in Ca2+‐activated intestinal secretion (Flores et al. 2007; Matos et al. 2007). To test this hypothesis we generated double mutant mice null for Kcnn4 and Kcne3, and Kcnn4‐deficient mice additionally transgenic for the mutant KCNE3‐D90N subunit. The intestinal secretory function in these animals was tested and the results are shown in Fig. 2. The upper panels of Fig. 2 show current traces across colonic epithelium indicating that, like in tissues from the single intestinal transgenic KCNE3‐D90N‐expressing or the Kcne3 KO mice, the double mutant deletants with Kcnn4 respond to increases in cAMP with sizeable anion secretory currents. The Ca2+‐activated anion secretory currents normally elicited by carbachol are absent from these animals, which only show a transient current consistent with K+ secretion (Flores et al. 2007). The bottom panel shows a summary of the results and a comparison with those of the single Tg(Kcne3‐D90N) transgenic and the Kcne3 KO mice. There were no significant differences between any of the genotypes with the exception of the response to the muscarinic agonist addition that yielded the expected large anion secretory response in the animals with normal expression of KCa3.1 K+ channel encoded by the Kcnn4 gene and that was absent in those deficient for this gene. From these results we can discard KCa3.1 as being responsible for the cAMP‐activated anion secretory current in the absence of KCNQ1–KCNE3 activity.
Figure 2. Effect of additional inactivation of the Ca2+‐dependent K+ channel KCa3.1 on anion secretion in the colon of Kcne3 null animals.
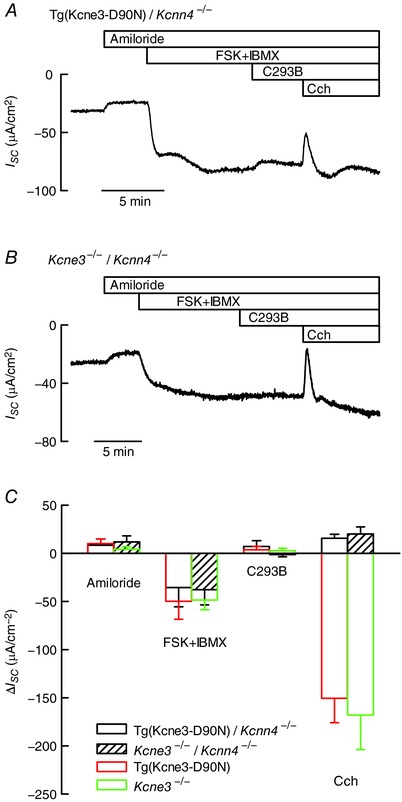
The double mutants were obtained by crossing single mutants expressing a dominant‐negative KCNE3‐D90N subunit or Kcne3 −/− mice with KCa3.1 knockouts (Kcnn4 −/−). A and B, traces of short circuit currents (I SC) as function of time obtained using mouse distal colon of Tg(KCNE3‐D90N)/Kcnn4 −/− or Kcne3 −/−/Kcnn4 −/− homozygous double mutants mounted in Ussing chambers. Details of compound additions are as in Fig. 1. Tissue resistances were, respectively, 68 and 60 Ω cm2 at the beginning and end of the experiment shown in A. The respective values in B were 78 and 67 Ω cm2. C, summary of the results of experiments in A and B. Data are means ± SD (number of animals: 5 for Tg(KCNE3‐D90N)/Kcnn4 −/− and 6 for Kcne3 −/−/Kcnn4 −/−). The results for single Tg(Kcne3‐D90N) transgenic and Kcne3 −/− shown in Fig. 1 F are reproduced for comparison. The transgenic line used in these experiments was Tg(Kcne3‐D90N)843cCECs. There was no difference between the results obtained using single or double mutants, except for the complete absence of carbachol‐induced anion secretion in the tissues of animals lacking KCa3.1.
Tetrapentylammonium inhibits intestinal Cl− secretion remaining in the absence of KCNE3–KCNQ1 activity
A variety of K+ channels are inhibited by intracellular quaternary ammonium (QA) ions and these inhibitors have become useful tools to probe their gating processes (Armstrong, 1971; Holmgren et al. 1997). To explore whether the conductance supporting Cl− secretion in the absence of KCNE3–KCNQ1 activity corresponds to a K+ channel, we have tested its sensitivity to the membrane‐permeant QA tetrapentylammonium (TPeA). Figure 3 A illustrates an experiment on colonic epithelium of a KCNE3‐D90N transgenic mouse showing that cAMP‐activated anion secretory current was not inhibited by basolateral addition of C293B or additionally increasing apical TPeA from 3 to 100 μm. The current was abolished, however, by basolaterally added 100 μm TPeA, which nevertheless did not impede KCa3.1 activation, as shown by the secretory current induced after carbachol addition. We do not think that the sidedness of TPeA action is an indication of the membrane at which the action is taking place. TPeA is quite hydrophobic and readily permeates the plasma membrane of cells such as HEK‐293. The apical, unlike the basolateral, membrane of colon has been reported to be rather impermeable (Endeward & Gros, 2005) and this might explain the observed sidedness. Figure 3 B shows that a component of the secretory current refractory to inhibition by C293B was also inhibited by TPeA, bringing current levels almost to zero. Figure 3 C corroborates the finding with colonic epithelium of a transgenic mouse expressing the mutant KCNE3‐D90N and shows that TPeA inhibition is graded, whilst Fig. 3 D shows that C293B‐indifferent colonic anion secretory current is also inhibited by TPeA in the Kcne3 KO model. The half‐maximal inhibition occurred at 14.8 μm TPeA and the dose–response curve appeared cooperative with a Hill coefficient (n H) of 1.9 (Fig. 3 E). Use of a Cftrtm1Eur mouse that expresses the CFTR‐ΔF508 mutant and is deficient in anion secretion (Fig. 3 F) suggests that the effect of TPeA is not due to activation of an unidentified conductance, e.g. mediating cation absorption, obliterating the anion secretion signal. Instead the results are consistent with TPeA exerting its action by affecting a K+ channel separate from KCNE3–KCNQ1 and KCa3.1.
Figure 3. Inhibiting cAMP‐dependent intestinal anion secretion by pharmacological K+ channel blockade.
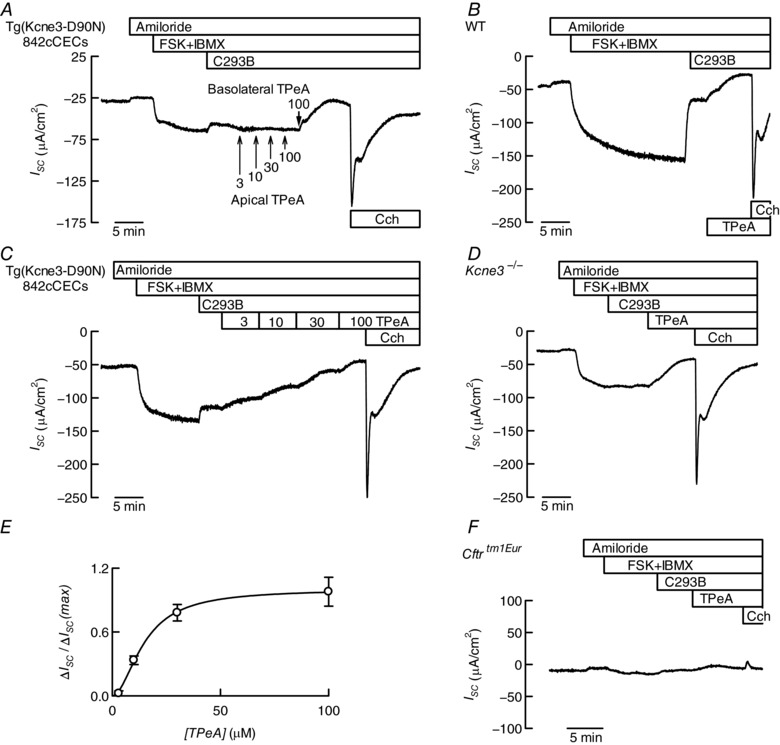
A–D and F, Ussing chamber traces of short circuit currents (I SC) as function of time obtained using distal colon of transgenic mice expressing the KCNE3‐D90N mutant subunit (A and C), a WT mouse (B), a Kcne3 −/− (D) and a Cftrtm1Eur mouse expressing the CFTR‐ΔF508 mutation (F). Details of compound additions are as in Fig. 1 except for the use of tetrapentylammonium (TPeA) added, unless otherwise indicated, basolaterally at 100 μm. Tissue resistances at the beginning and end of the experiment were 82 and 68 Ω cm2 in A, 98 and 70 Ω cm2 in B, 66 and 58 Ω cm2 in D, and 82 and 82 Ω cm2 in F. E, concentration dependence of the effect of basolateral TPeA on the cAMP‐activated anion secretion (normalized) measured using distal colon from Tg(Kcne3‐D90N)842cCECs mice expressing the KCNE3‐D90N subunit (means ± SD, n = 4).
Intestinal expression of K2P TASK‐2 K+ channels and possible role in intestinal Cl− secretion
Intracellularly applied quaternary ammonium compounds with long alkyl chains, such as TPeA, are high affinity inhibitors of K2P channels (Piechotta et al. 2011) including TASK‐2 (Niemeyer et al. 2017). RT‐PCR experiments performed by Reyes et al. at the time of the discovery of TASK‐2 (Reyes et al. 1998) have shown the presence of the messenger for this channel both in the colon and small intestine of the mouse. Sizeable amounts of TASK‐2 mRNA have also been found in human small intestinal tissue (Medhurst et al. 2001). To confirm these observations we have now used a TASK‐2 deficient mutant, the Kcnk5 KO mouse obtained using a trapping vector encoding β‐galactosidase (Taniwaki et al. 2005). The activity of the enzyme revealed by cytochemistry, which is expressed under the control of the Kcnk5 promoter, becomes a proxy for endogenous sites of TASK‐2 expression (Cid et al. 2013). Cytochemical staining revealed β‐galactosidase expression on the epithelium of the ileum (Fig. 4 A and E) and the colon (Fig. 4 C and G) of Kcnk5 −/− mice that was absent in the tissue from WT animals (Fig. 4 B, F, D and H, respectively). Some activity is observed the length of the crypts but maximal staining is at the upper third of the crypt and the surface epithelium in the case of the colon. A similar epithelial expression of β‐galactosidase was seen in Kcnk5 −/− small intestine with highest expression at the villi but significant expression also in the crypts. These results suggest that TASK‐2 is expressed over the epithelium. The higher expression in the villi or surface epithelium has to be interpreted with caution as we ignore the half‐lives of the TASK‐2 and β‐galactosidase proteins in these rapidly renewing tissues. The evidence that TASK‐2 is present in the intestinal epithelium suggests it might contribute to the driving force and K+ recycling necessary to efficient Cl− secretion.
Figure 4. Expression of TASK‐2 in the intestinal epithelium of the colon revealed by β‐galactosidase activity in Kcnk5 −/− mice.
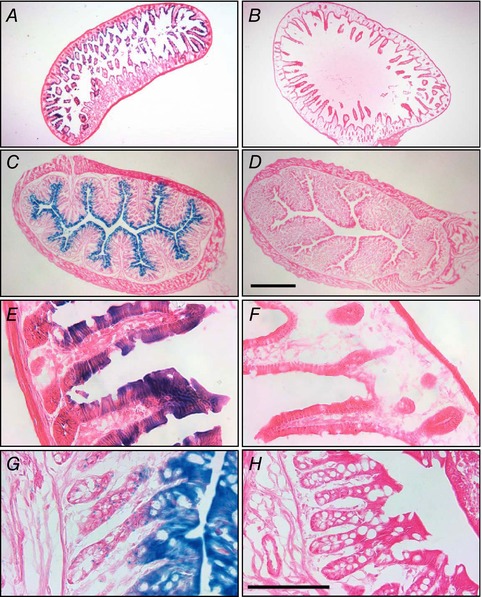
Ileum (A, B, E and F) or colon (C, D, G and H) sections from Kcnk5 −/− (A, C, E and G) and WT (B, D, F and H) mice were stained for X‐gal (blue) thus revealing β‐galactosidase activity used here as a proxy for endogenous Kcnk5 gene expression. Bars: 500 μm for the four upper panels and 100 μm for the lower panels.
Anion secretion across the colonic epithelium of TASK‐2 deficient mice
Figure 5 A illustrates an experiment on colonic epithelium from a Kcnk5 KO mouse lacking the TASK‐2 channel that shows a robust cAMP‐activated anion secretory current. As expected, the current was markedly inhibited by basolateral addition of C293B but additional basolateral TPeA at 100 μm was without effect. KCa3.1 activation, as evidenced by the secretory current induced by carbachol in the continued presence of both inhibitors, occurred normally. The summary in Fig. 5 C compares these results with those obtained with WT mouse tissues. The cAMP‐activated anion secretory current in TASK‐2 KO colon (black empty column) was not different from that in across WT epithelium (red empty column). Inhibition by C293B was significantly smaller in WT, but that by TPeA roughly made up the difference. Significantly, there was no effect of TPeA addition in Kcnk5 KO colon. Carbachol‐induced Ca2+‐dependent anion secretion was not significantly different between WT and the mouse lacking TASK‐2 channel. The simplest interpretation of this result is that a TPeA‐inhibitable TASK‐2 K+ channel is present and active in mouse colon and is capable of contributing to part of the driving force for anion secretion through cAMP‐activated CFTR Cl− channels.
Figure 5. Anion secretion in the colon of mice deficient in K2P K+ channel TASK‐2 or in double mutants lacking expression of both TASK‐2 and KCNQ1–KCNE3 K+ channels.
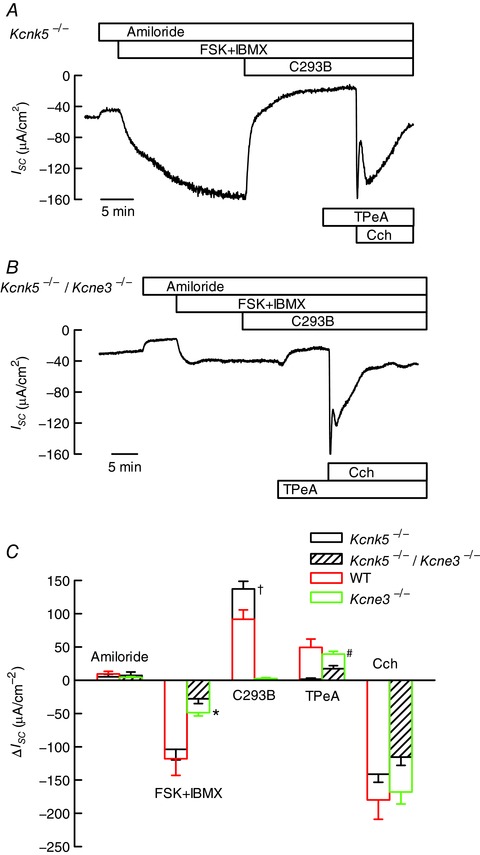
The double mutants were obtained by crossing single mutant mice. A and B, traces of short circuit currents (I SC) as function of time obtained using mouse distal colon of Kcnk5 −/− or Kcnk5 −/−/Kcne3 −/− homozygous double mutants mounted in Ussing chambers. Details of compound additions are as in Figs 1 and 3. Tissue resistances were, respectively, 81 and 66 Ω cm2 at the beginning and end of the experiment shown in A. The respective numbers in B were 102 and 71 Ω cm2. C, summary of the results of experiments in A and B. Data are means ± SD (number of animals: 6 for Kcnk5 −/− and 6 for Kcnk5 −/−/Kcne3 −/−). The results for WT and single Kcne3 −/− from Fig. 1 are reproduced for comparison. Results for double Kcnk5 −/−/Kcne3 −/− and single Kcne3 −/− mutants were different when tested by t test as indicated: * P < 0.01; † and #, P < 0.001.
From the result presented above, it would be reasonable to assume that TASK‐2 is responsible for the persistence of cAMP‐activated anion secretion in the absence of activity of the cAMP‐activated KCNQ1–KCNE3 K+ channel. If this were the case the simultaneous ablation of KCNE3 and TASK‐2 should lead to a phenotype with complete absence of cAMP‐activated anion secretion akin to that seen in the mice expressing the mutant CFTR‐ΔF508. To test this prediction we generated double Kcne3 −/−/Kcnk5 −/− mice. The double mutant mice, which had no obvious general phenotypic alteration, were then assayed for their colonic anion secretion in Ussing chambers. The result of a typical experiment is illustrated in Fig. 5 B, which shows that a small cAMP‐activated current consistent with anion secretion was present in these Kcne3 −/−/Kcnk5 −/− mice. This current was not affected by basolateral C293B but, surprisingly, addition of basolateral TPeA did have an effect in diminishing partially the current activated by cAMP increase. A robust carbachol‐induced secretory current was present in this Kcne3 −/−/Kcnk5 −/− double mutant tissues. Figure 5 C shows a summary of these results (hatched bars), which are compared with results obtained with Kcne3 −/− single mutant mouse tissues (green bars). The response to cAMP increase (FSK+IBMX) was significantly decreased in the Kcne3 −/−/Kcnk5 −/− double mutant compared to that in the single Kcne3 −/− mouse (P = 0.00704, by t test). As observed with the Kcne3 −/− mutants, tissues from the Kcne3 −/−/Kcnk5 −/− mouse cAMP‐activated currents were indifferent to C293B but not to TPeA, which abolished cAMP‐dependent currents in mice lacking both KCNE3 and TASK‐2 channels. This result is surprising in view of the lack of effect of TPeA added after C293B on the single TASK‐2−/− mutant. One possible explanation is that in the absence of KCNQ1–KCNE3 and TASK‐2 activity, a third K+ channel, only partially inhibited by TPeA, becomes active and is able to support a small but still measurable cAMP‐dependent anion secretion.
The absolute value of I SC after amiloride inhibition in the WT colon was (mean ± SD (n animals)) −55 ± 21 μA cm−2 (n = 19). This must correspond to basal anion secretion in the unstimulated tissue as it is absent in the Cftrtm1Eur mouse, in which the corresponding figure is 2.0 ± 9.9 μA cm−2 (n = 6). In tissues from the Kcne3 −/− and Kcnk5 −/− animals the respective figures were −32 ± 9.5 (n = 6) and −48 ± 19 μA cm−2 (n = 5), not significantly different from those in WT suggesting that neither KCNQ1–KCNE3 nor TASK‐2 is essential for this basal anion secretion. The result obtained with the double Kcne3 −/−/Kcnk5 −/− mutant was −11 ± 11.3 μA cm−2 (n = 5), significantly different from WT but not from the Cftrtm1Eur (ANOVA, P < 0.05) suggesting that either KCNQ1–KCNE3 or TASK‐2 must be present to support the residual CFTR‐mediated secretion in the absence of stimulation.
Cholera toxin‐induced small intestinal fluid secretion
The inactivation of KCNQ1–KCNE3 by genetic silencing of either Kcne3 or Kcnq1 genes leads to partial inhibition of Cl− secretion measured in the small intestine in vitro (Vallon et al. 2005; Preston et al. 2010). Cholera toxin‐stimulated fluid secretion measured in vivo, however, did not differ between the ilea of WT and Kcne3 −/− mice (Preston et al. 2010). The conditions of those experiments, involving rather long secretion times and strong stimulus with cholera toxin, lend themselves to the demonstration that a small remaining K+ conductance is capable of sustaining the same accumulated fluid secretion as that seen in WT animals. We have measured toxin‐stimulated fluid secretion in vivo using the same protocol in the different animal models used above with the results shown in Fig. 6. Net cholera toxin‐induced fluid accumulation in the lumen of ileal loops (third bar in each case) was not different in Kcne3 −/− animals when compared with WT intestine, confirming previously published results (Preston et al. 2010). The same was true for single Kcnn4 −/− and Kcnk5 −/− mutants, and for the double deficient Kcne3 −/−/Kcnn4 −/− and Kcne3 −/−/Kcnk5 −/− animals. These results point to a high resilience of the cAMP‐activated intestinal fluid secretory process to K+ channel inactivation and suggest that regulated expression of channels providing alternative conductance to K+ must be taking place to ensure this mutational tolerance. A caveat in the interpretation of these results is that in order to achieve significant fluid secretion we had to use long incubations and might have missed differences between the genotypes that we might have detected had we been able to measure at shorter, initial velocity times. Alternative explanations include the fact that cholera toxin action on small intestinal fluid secretion can be attributed in part to inhibition of electroneutral NaCl absorption through a decrease in activity of Na+/H+ exchanger NHE3 (Zachos et al. 2005). This antiabsorptive effect, well documented in the small intestine, would not be detectable in transepithelial current measurements. Additionally, vasodilatation with increased hydraulic conductivity (Lucas, 2010) and changes in motility (Fung et al. 2010) have been proposed to contribute to cholera toxin‐induced fluid secretion. Again, these might play a role in the fluid secretion measured in vivo but are absent in the in vitro measurements.
Figure 6. Cholera toxin‐induced small intestinal fluid secretion measured in vivo .
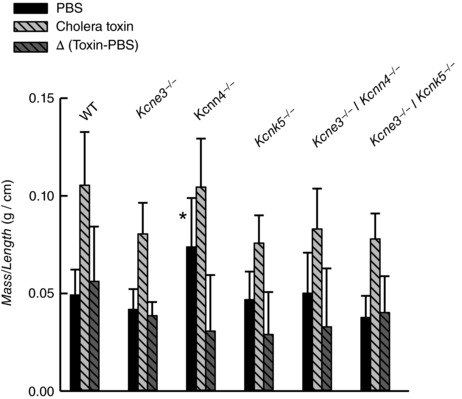
Mass over length relations of ileal loops 5 h after luminal injection of PBS or PBS + cholera toxin (0.5 μg per loop) in wild‐type (WT) and various single and double knockout mouse models. The difference between the measurements taken without the toxin was subtracted from the toxin‐containing number to give the net secretion stimulated by cholera toxin (Δ (Toxin‐PBS)). Data are means ± SD. The numbers of experiments were as follows: WT, n = 10; Kcne3 −/−, n = 10; Kcnn4 −/−, n = 9; Kcnk5 −/−, n = 4; Kcnn4 −/−/Kcne3 −/−, n = 11; Kcnk5 −/−/Kcne3 −/−, n = 10. There was no difference between measurements on mutant mice and that obtained with WT except for measurement with PBS alone identified with *(P = 0.002 by ANOVA).
Implications of the present results
The activity of K+ channels is an essential component of the cAMP‐activated intestinal anion and, consequently, fluid secretion process. K+ channels serve to recycle K+ ions that enter secretory epithelial cells through the action of the Na+–K+ pump and the NKCC1 Na+–K+–2Cl− cotransporter, and to counteract the depolarization caused by activation of the CFTR Cl− channel. Abundant research documents the presence of cAMP‐activated K+ channels at the basolateral membrane of intestinal epithelial cells (Lohrmann & Greger, 1995; Lohrmann et al. 1995; see also review by Heitzmann & Warth, 2008). In the intestine, KCNQ1 and KCNE3 mRNAs colocalize in crypt cells. This localization and the pharmacology, voltage dependence and stimulation by cAMP of KCNQ1–KCNE3 currents indicate that these proteins assemble to form the potassium channel that is important for cAMP‐stimulated intestinal chloride secretion and that it is therefore involved in secretory diarrhoea and cystic fibrosis (Schroeder et al. 2000).
Direct corroboration of the importance of KCNQ1–KCNE3 in intestinal secretion has emerged more recently from the use of mice made genetically deficient in the pore‐bearing KCNQ1 subunit (Vallon et al. 2005) or the KCNE3 regulatory subunit essential for physiological function of the complex (Preston et al. 2010). Surprisingly, genetic inactivation of KCNQ1–KCNE3 in mice did not abolish anion secretion in either small or large intestine measured in vitro and, in the case of the Kcne3 KO animal, it did not affect cholera toxin‐dependent intestinal fluid secretion. This observation, coupled to the evidence of the participation of the KCa3.1 K+ channel in CFTR‐mediated Ca2+‐dependent intestinal anion secretion (Flores et al. 2007; Matos et al. 2007) prompted Preston et al. (2010) to postulate a role for this channel to partially or even fully compensate for the loss of KCNQ1–KCNE3 activity in maintaining cAMP‐mediated Cl− and fluid secretion, perhaps under some sort of cross‐talk between cAMP and intracellular Ca2+ signalling pathways as in other epithelia (Ahuja et al. 2014). As we show here, however, the simultaneous inactivation of KCNQ1–KCNE3 and KCa3.1 K+ channels fails to affect cAMP‐dependent intestinal anion or fluid secretion suggesting the participation of a third K+ channel in this process.
Pharmacological experiments, gene expression and the use of a TASK‐2 deficient mouse model all converge to provide evidence that K2P K+ channel TASK‐2 is expressed in the intestinal epithelium where it contributes to the process of anion secretion initiated by cAMP. This novel function comes to join important roles of TASK‐2 in cell volume regulation (Niemeyer et al. 2001; Barriere et al. 2003), renal proximal tubule bicarbonate reabsorption (Warth et al. 2004), sensing of CO2 in the retrotrapezoid nucleus (Gestreau et al. 2010; Wang et al. 2013), and K+ recycling in cochlear outer sulcus cells (Cazals et al. 2015). TASK‐2 is gated by intra‐ and extracellular pH, by membrane phosphatidylinositol and directly by the Gβ subunit of G proteins (Añazco et al. 2013; Cid et al. 2013; López‐Cayuqueo et al. 2015; Niemeyer et al. 2017). We do not know if any of these regulatory pathways is involved in the function TASK‐2 plays in the intestinal epithelium. Although our results point clearly to a function of TASK‐2 as an alternative, or additional, K+ conductance compensating the absence of KCNQ1–KCNE3 to support anion transport, a small electrogenic anion secretion is still present in double TASK‐2 and KCNQ1–KCNE3 inactivated mutants, which allows for fluid secretion under the strong stimulation of cholera toxin. This leads to the conclusion that a further K+ conductance emerges under these conditions. This conductance, whose molecular identity we ignore, also shows some sensitivity to TPeA, which is a generally promiscuous inhibitor of K+ channels. Interestingly, however, TPeA did not inhibit Ca2+‐activated secretion suggesting the emerging conductance in this case did not correspond to KCa3.1 activation through cross‐talk with the cAMP‐signalling pathway.
The complete absence of intestinal secretion after inactivation of the cAMP‐dependent Cl− channel CFTR (Grubb & Boucher, 1999; Greger, 2000) is in contrast with the resistance of this process to inactivation of the K+ channels purported to be essential to support Cl− flux. Perhaps an explanation for this contrast is the high abundance of K+ channels in mammals. There are at least 88 different human proteins of the K+ channel superfamily pore domains that can be divided into five clades (Brohawn et al. 2012). TASK‐2 is one among 15 members of the K2P clade, KCNQ1 belongs into a clade made up of 12 α‐subunits whilst KCa3.1 is one of eight KCa channels. CFTR, on the other hand, is alone in being a Cl− channel in a family of ABC transporters (Gadsby et al. 2006).
The persistent intestinal anion secretion after genetic inactivation of K+ channels also appears at odds with its complete pharmacological inhibition by a combination of C293B and TPeA in the WT or by C293B alone in the Kcnk5 KO mouse colon. Given that both KCNQ1–KCNE3 and TASK‐2 are druggable, our result suggests a pharmacological approach aimed at modulating secretion, for instance in a scenario of secretory diarrhoea. Resistance to double genetic inactivation, on the other hand, is possibly an intestinal epithelial example of what has been termed mutational robustness or tolerance, a mechanism by which the phenotype of a given organism remains unaltered in the face of mutation (Fares, 2015; Payne & Wagner, 2015).
Both KCNQ1–KCNE3 and KCa3.1 have been shown to be present in colonic crypt enterocytes from intestinal human biopsies (Al‐Hazza et al. 2012, 2016) and their function in anion secretion has been demonstrated in cultured epithelial cells derived from colon carcinomas (Dharmsathaphorn & Pandol, 1986; Warth et al. 1999; Alzamora et al. 2011). In addition, the expression of TASK‐2 messenger has been reported in human intestine (Reyes et al. 1998) but there is no evidence for its possible function in ion transport. The interplay between these different K+ channels in supporting anion secretion awaits future studies.
We have uncovered an important role for TASK‐2 in the process of intestinal secretion that we propose can buffer changes in the expression of KCNQ1–KCNE3. But the picture of robustness of this physiological process is yet more complex as a new conductance emerges in the simultaneous absence of both KCNQ1–KCNE3 and TASK‐2. Identifying the gene(s) coding for what looks like a network of K+ channels supporting intestinal secretion is important although beyond the scope of the present work. The mechanism by which the physiological function is preserved by putting into action different gene products, perhaps by transcriptional regulation, is also a challenging and fascinating question for future research, as recently suggested in a review (Noble, 2011) advocating an integrative approach in physiology, in which ‘the activity of the proteins and RNAs formed from each DNA template is analysed in networks of interactions’.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
The experiments were performed at the Centro de Estudios Centíficos (CECs) biological laboratory. L.P.C., T.J.J. and F.V.S. conceived and designed the work. F.J.‐K., S.V., J.B. and M.O., performed the experiments and together with L.P.C., T.J.J. and F.V.S. analysed and interpreted the data. F.V.S., L.P.C. and T.J.J. drafted the work that was additionally revised critically by F.J.‐K., S.V., J.B. and M.O. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was funded by the FONDECYT‐Chile grants 1120743 and 1160043. The Centro de Estudios Científicos (CECs) is funded by the Base Financing Programme of Conicyt.
Acknowledgements
We are grateful to the following colleagues for providing genetically modified mice: Dr James E. Melvin from the NIH National Institute of Dental and Craniofacial Research, USA, for the Kcnn4 KO, Drs Masatake Araki and Kimi Araki from Kumamoto University, Japan for the Kcnk5 KO, and Dr Bob J. Scholte from Erasmus University, The Netherlands for the Cftrtm1Eur mutants. Juan Manuel Baamonde and the staff of CECs animal facility are gratefully thanked for their untiring help.
Linked articles This article is highlighted by a Perspective by O'Grady. To read this Perspective, visit https://doi.org/10.1113/JP275567.
Edited by: Peying Fong & Kim Barrett
This is an Editor's Choice article from the 1 February 2018 issue.
References
- Abbott GW & Goldstein SA (2002). Disease‐associated mutations in KCNE potassium channel subunits (MiRPs) reveal promiscuous disruption of multiple currents and conservation of mechanism. FASEB J 16, 390–400. [DOI] [PubMed] [Google Scholar]
- Ahuja M, Jha A, Maleth J, Park S & Muallem S (2014). cAMP and Ca2+ signaling in secretory epithelia: crosstalk and synergism. Cell Calcium 55, 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Hazza A, Linley J, Aziz Q, Hunter M & Sandle G (2016). Upregulation of basolateral small conductance potassium channels (KCNQ1/KCNE3) in ulcerative colitis. Biochem Biophys Res Commun 470, 473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Hazza A, Linley JE, Aziz Q, Maclennan KA, Hunter M & Sandle GI (2012). Potential role of reduced basolateral potassium (IKCa3.1) channel expression in the pathogenesis of diarrhoea in ulcerative colitis. J Pathol 226, 463–470. [DOI] [PubMed] [Google Scholar]
- Alzamora R, O'Mahony F, Ko WH, Yip TW, Carter D, Irnaten M & Harvey BJ (2011). Berberine reduces cAMP‐induced chloride secretion in T84 human colonic carcinoma cells through inhibition of basolateral KCNQ1 channels. Front Physiol 2, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Añazco C, Peña‐Münzenmayer G, Araya C, Cid LP, Sepúlveda FV & Niemeyer MI (2013). G protein modulation of K2P potassium channel TASK‐2: a role of basic residues in the C‐terminus domain. Pflugers Arch 465, 1715–1726. [DOI] [PubMed] [Google Scholar]
- Armstrong CM ( 1971). Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. J Gen Physiol 58, 413–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett KE & Keely SJ (2000). Chloride secretion by the intestinal epithelium: molecular basis and regulatory aspects. Annu Rev Physiol 62, 535–572. [DOI] [PubMed] [Google Scholar]
- Barriere H, Belfodil R, Rubera I, Tauc M, Lesage F, Poujeol C, Guy N, Barhanin J & Poujeol P (2003). Role of TASK2 potassium channels regarding volume regulation in primary cultures of mouse proximal tubules. J Gen Physiol 122, 177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begenisich T, Nakamoto T, Ovitt CE, Nehrke K, Brugnara C, Alper SL & Melvin JE (2004). Physiological roles of the intermediate conductance, Ca2+‐activated potassium channel Kcnn4. J Biol Chem 279, 47681–47687. [DOI] [PubMed] [Google Scholar]
- Brohawn SG, del Mármol J & MacKinnon R (2012). Crystal structure of the human K2P TRAAK, a lipid‐ and mechano‐sensitive K+ ion channel. Science 335, 436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazals Y, Bevengut M, Zanella S, Brocard F, Barhanin J & Gestreau C (2015). KCNK5 channels mostly expressed in cochlear outer sulcus cells are indispensable for hearing. Nat Commun 6, 8780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cid LP, Roa‐Rojas HA, Niemeyer MI, González W, Araki M, Araki K & Sepúlveda FV (2013). TASK‐2: a K2P K+ channel with complex regulation and diverse physiological functions. Front Physiol 4, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedek K & Waldegger S (2001). Colocalization of KCNQ1/KCNE channel subunits in the mouse gastrointestinal tract. Pflugers Arch 442, 896–902. [DOI] [PubMed] [Google Scholar]
- Dharmsathaphorn K & Pandol SJ (1986). Mechanism of chloride secretion induced by carbachol in a colonic epithelial cell line. J Clin Invest 77, 348–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diener M, Hug F, Strabel D & Scharrer E (1996). Cyclic AMP‐dependent regulation of K+ transport in the rat distal colon. Br J Pharmacol 118, 1477–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endeward V & Gros G (2005). Low carbon dioxide permeability of the apical epithelial membrane of guinea‐pig colon. J Physiol 567, 253–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fares MA ( 2015). The origins of mutational robustness. Trends Genet 31, 373–381. [DOI] [PubMed] [Google Scholar]
- Field M ( 2003). Intestinal ion transport and the pathophysiology of diarrhea. J Clin Invest 111, 931–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores CA, Melvin JE, Figueroa CD & Sepúlveda FV (2007). Abolition of Ca2+‐mediated intestinal anion secretion and increased stool dehydration in mice after inactivation of the intermediate conductance Ca2+‐dependent K+ channel Kcnn4. J Physiol 583, 705–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frizzell RA & Hanrahan JW (2012). Physiology of epithelial chloride and fluid secretion. Cold Spring Harb Perspect Med 2, a009563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung C, Ellis M & Bornstein JC (2010). Luminal cholera toxin alters motility in isolated guinea‐pig jejunum via a pathway independent of 5‐HT3 receptors. Front Neurosci 4, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadsby DC, Vergani P & Csanady L (2006). The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature 440, 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gestreau C, Heitzmann D, Thomas J, Dubreuil V, Bandulik S, Reichold M, Bendahhou S, Pierson P, Sterner C, Peyronnet‐Roux J, Benfriha C, Tegtmeier I, Ehnes H, Georgieff M, Lesage F, Brunet JF, Goridis C, Warth R & Barhanin J (2010). Task2 potassium channels set central respiratory CO2 and O2 sensitivity. Proc Natl Acad Sci USA 107, 2325–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greger R ( 2000). Role of CFTR in the colon. Annu Rev Physiol 62, 467–491. [DOI] [PubMed] [Google Scholar]
- Greger R, Bleich M, Riedemann N, Van Driessche W, Ecke D & Warth R (1997). The role of K+ channels in colonic Cl− secretion. Comp Biochem Physiol A 118, 271–275. [DOI] [PubMed] [Google Scholar]
- Grubb BR & Boucher RC (1999). Pathophysiology of gene‐targeted mouse models for cystic fibrosis. Physiol Rev 79, S193–S214. [DOI] [PubMed] [Google Scholar]
- Halm DR & Frizzell RA (1986). Active K transport across rabbit distal colon: relation to Na absorption and Cl secretion. Am J Physiol Cell Physiol 251, C252–C267. [DOI] [PubMed] [Google Scholar]
- Heitzmann D & Warth R (2008). Physiology and pathophysiology of potassium channels in gastrointestinal epithelia. Physiol Rev 88, 1119–1182. [DOI] [PubMed] [Google Scholar]
- Holmgren M, Smith PL & Yellen G (1997). Trapping of organic blockers by closing of voltage‐dependent K+ channels: evidence for a trap door mechanism of activation gating. J Gen Physiol 109, 527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao T, Wang L, Halm ST, Lu L, Fyffe RE & Halm DR (2005). K+ channel KVLQT1 located in the basolateral membrane of distal colonic epithelium is not essential for activating Cl− secretion. Am J Physiol Cell Physiol 289, C564–C575. [DOI] [PubMed] [Google Scholar]
- Lohrmann E, Burhoff I, Nitschke RB, Lang HJ, Mania D, Englert HC, Hropot M, Warth R, Rohm W, Bleich M & Greger R (1995). A new class of inhibitors of cAMP‐mediated Cl− secretion in rabbit colon, acting by the reduction of cAMP‐activated K+ conductance. Pflugers Arch 429, 517–530. [DOI] [PubMed] [Google Scholar]
- Lohrmann E & Greger R (1995). The effect of secretagogues on ion conductances of in vitro perfused, isolated rabbit colonic crypts. Pflugers Arch 429, 494–502. [DOI] [PubMed] [Google Scholar]
- López‐Cayuqueo KI, Peña‐Münzenmayer G, Niemeyer MI, Sepúlveda FV & Cid LP (2015). TASK‐2 K2P K+ channel: thoughts about gating and its fitness to physiological function. Pflugers Arch 467, 1043–1053. [DOI] [PubMed] [Google Scholar]
- Lucas ML ( 2010). Diarrhoeal disease through enterocyte secretion: a doctrine untroubled by proof. Exp Physiol 95, 479–484. [DOI] [PubMed] [Google Scholar]
- MacVinish LJ, Hickman ME, Mufti DA, Durrington HJ & Cuthbert AW (1998). Importance of basolateral K+ conductance in maintaining Cl− secretion in murine nasal and colonic epithelia. J Physiol 510, 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos JE, Sausbier M, Beranek G, Sausbier U, Ruth P & Leipziger J (2007). Role of cholinergic‐activated KCa1.1 (BK), KCa3.1 (SK4) and KV7.1 (KCNQ1) channels in mouse colonic Cl− secretion. Acta Physiol (Oxf) 189, 251–258. [DOI] [PubMed] [Google Scholar]
- Medhurst AD, Rennie G, Chapman CG, Meadows H, Duckworth MD, Kelsell RE, Gloger II & Pangalos MN (2001). Distribution analysis of human two pore domain potassium channels in tissues of the central nervous system and periphery. Brain Res Mol Brain Res 86, 101–114. [DOI] [PubMed] [Google Scholar]
- Niemeyer MI, Cid LP, Barros LF & Sepúlveda FV (2001). Modulation of the two‐pore domain acid‐sensitive K+ channel TASK‐2 (KCNK5) by changes in cell volume. J Biol Chem 276, 43166–43174. [DOI] [PubMed] [Google Scholar]
- Niemeyer MI, Cid LP, Paulais M, Teulon J & Sepúlveda FV (2017). Phosphatidylinositol (4,5)‐bisphosphate dynamically regulates the K2P background K+ channel TASK‐2. Sci Rep 7, 45407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble D (2011). Neo‐Darwinism, the modern synthesis and selfish genes: are they of use in physiology? J Physiol 589, 1007–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne JL & Wagner A (2015). Mechanisms of mutational robustness in transcriptional regulation. Front Genet 6, 322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piechotta PL, Rapedius M, Stansfeld PJ, Bollepalli MK, Erhlich G, Andres‐Enguix I, Fritzenschaft H, Decher N, Sansom MS, Tucker SJ & Baukrowitz T (2011). The pore structure and gating mechanism of K2P channels. EMBO J 30, 3607–3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Robine S, Jaisser F, El Marjou FE & Louvard D (1999). Regulatory sequences of the mouse villin gene that efficiently drive transgenic expression in immature and differentiated epithelial cells of small and large intestines. J Biol Chem 274, 6476–6482. [DOI] [PubMed] [Google Scholar]
- Preston P, Wartosch L, Gunzel D, Fromm M, Kongsuphol P, Ousingsawat J, Kunzelmann K, Barhanin J, Warth R & Jentsch TJ (2010). Disruption of the K+ channel beta‐subunit KCNE3 reveals an important role in intestinal and tracheal Cl− transport. J Biol Chem 285, 7165–7175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes R, Duprat F, Lesage F, Fink M, Salinas M, Farman N & Lazdunski M (1998). Cloning and expression of a novel pH‐sensitive two pore domain K+ channel from human kidney. J Biol Chem 273, 30863–30869. [DOI] [PubMed] [Google Scholar]
- Sandoval M, Burgos J, Sepúlveda FV & Cid LP (2011). Extracellular pH in restricted domains as a gating signal for ion channels involved in transepithelial transport. Biol Pharm Bull 34, 803–809. [DOI] [PubMed] [Google Scholar]
- Schroeder BC, Waldegger S, Fehr S, Bleich M, Warth R, Greger R & Jentsch TJ (2000). A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature 403, 196–199. [DOI] [PubMed] [Google Scholar]
- Taniwaki T, Haruna K, Nakamura H, Sekimoto T, Oike Y, Imaizumi T, Saito F, Muta M, Soejima Y, Utoh A, Nakagata N, Araki M, Yamamura K & Araki K (2005). Characterization of an exchangeable gene trap using pU‐17 carrying a stop codon‐beta geo cassette. Dev Growth Differ 47, 163–172. [DOI] [PubMed] [Google Scholar]
- Vallon V, Grahammer F, Volkl H, Sandu CD, Richter K, Rexhepaj R, Gerlach U, Rong Q, Pfeifer K & Lang F (2005). KCNQ1‐dependent transport in renal and gastrointestinal epithelia. Proc Natl Acad Sci USA 102, 17864–17869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Doorninck JH, French PJ, Verbeek E, Peters RH, Morreau H, Bijman J & Scholte BJ (1995). A mouse model for the cystic fibrosis delta F508 mutation. EMBO J 14, 4403–4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Benamer N, Zanella S, Kumar NN, Shi Y, Bevengut M, Penton D, Guyenet PG, Lesage F, Gestreau C, Barhanin J & Bayliss DA (2013). TASK‐2 channels contribute to pH sensitivity of retrotrapezoid nucleus chemoreceptor neurons. J Neurosci 33, 16033–16044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warth R, Barrière H, Meneton P, Bloch M, Thomas J, Tauc M, Heitzmann D, Romeo E, Verrey F, Mengual R, Guy N, Bendahhou S, Lesage F, Poujeol P & Barhanin J (2004). Proximal renal tubular acidosis in TASK2 K+ channel‐deficient mice reveals a mechanism for stabilizing bicarbonate transport. Proc Natl Acad Sci USA 101, 8215–8220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warth R, Hamm K, Bleich M, Kunzelmann K, von Hahn T, Schreiber R, Ullrich E, Mengel M, Trautmann N, Kindle P, Schwab A & Greger R (1999). Molecular and functional characterization of the small Ca2+‐regulated K+ channel (rSK4) of colonic crypts. Pflugers Arch 438, 437–444. [DOI] [PubMed] [Google Scholar]
- Warth R, Riedemann N, Bleich M, Van Driessche W, Busch AE & Greger R (1996). The cAMP‐regulated and 293B‐inhibited K+ conductance of rat colonic crypt base cells. Pflugers Arch 432, 81–88. [DOI] [PubMed] [Google Scholar]
- Zachos NC, Tse M & Donowitz M (2005). Molecular physiology of intestinal Na+/H+ exchange. Annu Rev Physiol 67, 411–443. [DOI] [PubMed] [Google Scholar]