

Abstract
Despite the importance of coral microbiomes for holobiont persistence, the interactions among these are not well understood. In particular, knowledge of the co‐occurrence and taxonomic importance of specific members of the microbial core, as well as patterns of specific mobile genetic elements (MGEs), is lacking. We used seawater and mucus samples collected from Mussismilia hispida colonies on two reefs located in Bahia, Brazil, to disentangle their associated bacterial communities, intertaxa correlations, and plasmid patterns. Proxies for two broad‐host‐range (BHR) plasmid groups, IncP‐1β and PromA, were screened. Both groups were significantly (up to 252 and 100%, respectively) more abundant in coral mucus than in seawater. Notably, the PromA plasmid group was detected only in coral mucus samples. The core bacteriome of M. hispida mucus was composed primarily of members of the Proteobacteria, followed by those of Firmicutes. Significant host specificity and co‐occurrences among different groups of the dominant phyla (e.g., Bacillaceae and Pseudoalteromonadaceae and the genera Pseudomonas, Bacillus, and Vibrio) were detected. These relationships were observed for both the most abundant phyla and the bacteriome core, in which most of the operational taxonomic units showed intertaxa correlations. The observed evidence of host‐specific bacteriome and co‐occurrence (and potential symbioses or niche space co‐dominance) among the most dominant members indicates a taxonomic selection of members of the stable bacterial community. In parallel, host‐specific plasmid patterns could also be, independently, related to the assembly of members of the coral microbiome.
Keywords: co‐occurrence, corals, holobiont, mobile genetic elements, plasmids
1. INTRODUCTION
Corals can harbor complex microbial ecosystems, which frequently result in the development of both specific and variable host‐associated microbial communities (reviewed in Webster & Reusch, 2017), which can benefit host fitness (Peixoto, Rosado, Leite, Rosado, & Bourne, 2017; Webster & Reusch, 2017). Despite the close relationship between corals and their associated microbiomes, which can include organisms that have effects that vary from beneficial (Damjanovic, Blackall, Webster, & van Oppen, 2017; Krediet, Ritchie, Paul, & Teplitski, 2013; Peixoto et al., 2017; Webster & Reusch, 2017) to pathogenic (Meistertzheim, Nugues, Quéré, & Galand, 2017; Sweet & Bulling, 2017; Wright et al., 2017), knowledge of these intrinsic symbiotic, or dysbiotic, that is, disrupted symbiotic relationships (Bosch & Miller, 2016; Egan & Gardiner, 2016; Petersen & Round, 2014), interactions, and associated mechanisms is sparse.
It has been proposed that important mechanisms associated with the holobiont, that is, the host and its associated microbial community (Margulis & Fester, 1991), can be regulated through microbiome shuffling (i.e., shifts in microbial abundance) and/or switching (i.e., acquisition of the microbial strains from the surrounding environment) (reviewed in Webster & Reusch, 2017). The acquired microorganisms could also be passed on from parental to offspring generations (Leite et al., 2017; Padilla‐Gamiño, Pochon, Bird, Concepcion, & Gates, 2012). This microbiome‐mediated transgenerational acclimatization (MMTA) (proposed by Webster & Reusch, 2017) could lead to the rapid adaptation (and evolution) of corals to adverse environmental conditions. This natural acclimatization could be boosted in the face of environmental stresses (Damjanovic et al., 2017; Peixoto et al., 2017), for example, through the manipulation of specific key members of the microbiome, which have recently been termed “beneficial microorganisms for corals” (BMCs) (Peixoto et al., 2017). However, several questions remain, namely who are these key beneficial players, is there a taxonomic selection of the dominant microbes, and how do they interact within the holobiont?
Knowledge of the patterns of variation and interactions within the coral microbiome is limited. Other microbial‐community studies have shown that evaluation of co‐occurrence patterns in microbiomes may offer a more comprehensive view of complex microbial communities, constituting a complementary approach to estimates of alpha and beta diversity (Barberán, Bates, Casamayor, & Fierer, 2012; Dini‐Andreote et al., 2014). Identifying microbial patterns (Andrade et al., 2012; Peixoto et al., 2011; Rachid et al., 2013; Santos, Cury, Carmo, Rosado, & Peixoto, 2010) and potential interactions among microorganisms may reveal stable populations and shared niches, indicating preferences for certain resources, and consequently, microbial groups that are more competitive for such niches, or even elucidating potential direct symbiotic relationships between these microorganisms (as suggested by Barberán et al., 2012). This approach may be especially promising in coral microbiome studies because the close relationship between the host and its microbial community reported in several studies (Ainsworth, Thurber, & Gates, 2010; Cárdenas, Rodriguez‐R, Pizarro, Cadavid, & Arévalo‐Ferro, 2012; Ceh, Keulen, & Bourne, 2013; Ceh, Raina, Soo, van Keulen, & Bourne, 2012; Kelly et al., 2014; Lema, Bourne, & Willis, 2014; Lins‐De‐barros et al., 2010, 2013; Mouchka, Hewson, & Harvell, 2010; Sharp, Ritchie, Schupp, Ritson‐Williams, & Paul, 2010; Thompson, Rivera, Closek, & Medina, 2014). We believe, in particular, that exploring the taxonomic diversity of the bacterial part of the microbiome core (the bacteriome) as well as relevant ecological rules shaping these communities could provide valuable tools to guide BMC and MMTA surveys.
Another potential key aspect of coral microbiomes that has not received much attention is horizontal gene transfer (HGT) and the presence of specific patterns to support gene exchange. HGT plays important roles in bacterial evolution and gene exchange (Bhattacharya et al., 2016; van Elsas, Turner, & Bailey, 2003; Heuer & Smalla, 2007). Conjugation, for instance, which is mediated by different classes of mobile genetic elements (MGEs), allows the acquisition of novel genes (Heuer & Smalla, 2012). Plasmids, which are the main vectors for this genetic exchange, can act in the acquisition of genes or genetic pathways (such as for antibiotic resistance, pollutant degradation, and others) (Dealtry et al., 2014; Heuer & Smalla, 2012; Izmalkova et al., 2006). This HGT could be advantageous for holobiont resilience under environmental disturbance and, therefore, constitute a key component for MMTA (Webster & Reusch, 2017). Despite their possible essential role, plasmid patterns are largely unexplored in corals.
In this study, we present a survey of proxies for two broad‐host‐range (BHR) plasmid groups, IncP‐1B and PromA, in Mussismilia hispida coral mucus and the surrounding seawater. These plasmids can efficiently transfer their genetic material to a wide range of hosts and have been widely used as proxies to evaluate the potential spread of genes in several environments (van der Auwera et al., 2009; Heuer & Smalla, 2007, 2012; Zhang, Pereira e Silva, Chaib De Mares, & Van Elsas, 2014) and as providers of bacterial HGT capacities in some soil environments (Zhang et al., 2014). We also describe the bacterial diversity in these samples, as well as the co‐occurrence patterns of the coral bacteriome. We discuss the potential impact of these results in the context of the MMTA.
2. MATERIAL AND METHODS
2.1. Ethics approval and consent to participate
Permission for sampling was obtained from the Brazilian Institute of the Environment and Renewable Natural Resources (IBAMA)/Chico Mendes Institute for Biodiversity Conservation (ICMBio), permanent permit number 16942, in accordance with the Normative Instruction No. 03/2014 of System Authorization and Information on Biodiversity (SISBIO), and from local authorities of the Municipality Environmental Agency (SMMA), Porto Seguro, Bahia, Brazil. The microbial survey permit was obtained from CNPq (National Council for Scientific and Technological Development).
2.2. Sampling procedures and total DNA extraction
Mucus samples (around 50 ml) were collected with syringes directly from the polyps of M. hispida colonies on two reefs located adjacent to a marine protected area (Parque Natural Municipal do Recife de Fora) of Porto Seguro, Bahia, Brazil, in January 2015, as described by Castro et al. (2010). Particular microhabitats, for instance the surface mucus layer (SML), function as physical and chemical barriers (Shnit‐Orland & Kushmaro, 2009) that corals can benefit from, using antimicrobial compounds and their endogenous microbiome to regulate bacterial colonization (Ritchie, 2006), as the SML closely interacts with the surrounding environment. For instance, Lee, Davy, Tang, Fan, and Kench (2015) observed shifts in the relative abundance of the genera Endozoicomonas and Vibrio during a bleaching event and suggested that these changes resulted in a decrease in coral health, as a consequence of the increased ability of potentially pathogenic bacteria to pass through the SML barrier. Based on these observations, the SML is likely a potential source of MGEs and a favorable microhabitat for HTE.
Samples were obtained at the following sites: (1) Recife Itassepocu, 2 km from the mouth of the Buranhém River (6°25.9′46.37″S, 039°01′19.42″W) (closer to the river), totaling four samples from morphologically healthy colonies (without white spots) and four samples from colonies with morphological alterations (with white spots) and (2) Recife de Fora, 9.4 km from the Buranhém River mouth (16°23′23.72″ S, 038°58′54.92″W) (more distant from the river), totaling four mucus samples from morphologically healthy colonies. Sampling was performed in quadruplicate, so that each colony constituted a replicate. Approximately 1,000 ml of surrounding seawater (10–50 cm from the colony) (four replicates at each site) was also collected and filtered through a 0.22‐μm filter, using a standard vacuum pump system (Prismatec 131B). All samples (mucus and filters) were immediately immersed in liquid nitrogen and then stored at −80°C in the laboratory. Mucus samples and seawater material scraped off the filters were homogenized, and the DNA was extracted using the PowerSoil® DNA Isolation Kit (MoBio Laboratories, Carlsbad, CA, USA) following a modification of the method described by Sunagawa, Woodley, and Medina (2010).
2.3. Bacterial diversity
The V4 variable region of the 16S rRNA gene from all samples was amplified using the primers 515F/806R (Caporaso et al., 2011), and paired‐end (2 × 250 bp) sequencing was performed at the Argonne National Laboratory in their Next Generation Sequencing Core, on an Illumina Miseq, following the manufacturer's guidelines. The QIIME software package (version 1.9.1) was used to process the raw sequence data (Caporaso et al., 2010b). In brief, sequences were trimmed using the following parameters: quality score >25, sequence length >200, maximum homopolymer length of 6, and 0 mismatched bases in the primers and barcodes.
The remaining high‐quality sequences were binned into operational taxonomic units (OTUs) at 97% sequence identity using USEARCH 6.1 (v6.1.544) followed by selection of a representative sequence for each OTU (Edgar, 2010). Chimeric sequences were also identified using USEARCH 6.1 (v6.1.544) (Edgar, 2010) and removed. A representative sequence for each phylotype was aligned against the Greengenes database (Desantis et al., 2006), using PyNAST (Caporaso et al., 2010a), with sequences classified through the Greengenes taxonomy using the RDP classifier (Wang, Garrity, Tiedje, & Cole, 2007). Before further analysis, singletons, chloroplast plastids, mitochondria, and archaeal sequences were removed from the dataset. For all OTU‐based analyses, the original OTU table was rarified to a depth of 22,900 sequences per sample to minimize the effects of sampling effort on the analysis. The QIIME package was also used to generate weighted UniFrac distance matrices (Lozupone, Hamady, & Knight, 2006) and α‐diversity metrics, including richness and diversity indices. All sequences were deposited in the NCBI Sequence Read Archive database, with the accession numbers SRR5903387–SRR5903406.
In this study, we considered “core” as the set of bacterial taxa universally present in all samples, as defined by Shade and Handelsman (2012) and Turnbaugh et al. (2007). Considering that these microbes are common across microbiomes, they could be capable of playing key roles in a given ecosystem (Shade & Handelsman, 2012; Turnbaugh et al., 2007). The mucus‐core bacteriome was identified using QIIME and determined by plotting the OTU abundance, and it was represented by OTUs shared by 100% of the samples.
Network analyses were conducted on a subset of coral mucus microbiomes (sites 1 and 2) from M. hispida, using both the 179 most abundant OTUs (obtained after filtering rare taxa, i.e., sequences <0.005%) and the core bacteriome OTUs. Significant correlations between OTUs with a minimum occurrence of 10 were determined using the following metrics: Pearson's correlation, Spearman's correlation, and Bray–Curtis dissimilarity, using the CoNet app (Faust & Raes, 2012) in Cytoscape v.3.0.2 (Shannon et al., 2003). Networks obtained by all analyses were merged by intersection, keeping only interactions that were supported by all methods. The measurements were performed with 1000 iterations. The Benjamini–Hochberg multiple test correction was applied, and clusters (highly interconnected regions) were identified using the MCODE application (Bader & Hogue, 2003).
2.4. Quantification of bacterial, plasmid and integron genes
Quantitative PCR was used to estimate the gene copy numbers per ml of 16S rRNA genes; class I integrons, which were used as a proxy for anthropogenic pollution (Gillings et al., 2015) and BHR, and IncP‐1β and PromA plasmid groups, from mucus and seawater samples (Table S1). DNA preparations from plasmids R571 and pTer331 were used as positive controls in the detection of IncP‐1β and PromA group plasmids, respectively, and DNA from plasmid R388 was used as a positive control to detect plasmids of the IncW group and class I integrons. DNA from the IncQ plasmid RSF1010 was used as a negative control for all PCRs. Quantitative PCR experiments were conducted in an ABIPrism 7300 (Applied Biosystems) detection system, following the manufacturer's recommendations. Amplification of all genes was performed in a 20 μl reaction volume, containing 10 μl of GoTaq® q‐PCR Master Mix 2× (Promega), primers, 0.02 μl T4 gene 32 protein (5 mg/ml), H2O, and 2–5 ng DNA. The temperature profile included an initial hot start for 3 min at 94°C; and PCR cycling and detection (40 cycles) for 1 min at 94°C, 45 s at the stated annealing temperature (Table S1), and 45 s at 72°C (acquiring signal at the end of this step). All samples were used in triplicate, and H2O was used as the negative control. The primers and qPCR conditions used are summarized in Table S1. The efficiency and melting curves from all reactions were determined and analyzed using the ABIPrism 7300 Detection System (Applied Biosystems). For all genes, the cut‐off value was <102.
2.5. Statistical analyses
Estimates of α‐diversity and β‐diversity were based on an evenly rarified OTU abundance matrix. Statistical differences of qPCR analyses and α‐diversity matrices (observed OTUs, phylogenetic distance—PD, and the Chao index) were determined using analysis of variance (ANOVA) followed by a Tukey post hoc test.
To analyze the difference between the profiles and compositions of the bacterial communities, we used a principal coordinates analysis—PCoA (Jolliffe, 1986), using a Bray–Curtis distance matrix with PRIMER6 (Kelly et al., 2015). To assess the variation among different samples (coral mucus and seawater), we used a permutational multivariate analysis of variance (PerMANOVA) (Kelly et al., 2015) using PRIMER6 and PERMANOVA+ (Anderson, Gorley, & Clarke, 2008). Similarity percentage (SIMPER) calculations were conducted using PRIMER6 (Kelly et al., 2015) based on Bray–Curtis dissimilarity, in order to define the OTUs primarily responsible for the differences among the groups.
3. RESULTS
3.1. Bacterial community structure
The bacterial communities detected in the coral mucus differed from those in the seawater, as evidenced using PCoA unweighted UniFrac analyses of the data (Figure 1). The replicates of each of the two biomes clustered together, whereas the biomes themselves were clearly separate. Pairwise PERMANOVA of the data confirmed that the microbial communities of the mucus and seawater were significantly different from each other (p < .001), but detected no significant effect of location (sampling sites 1 × 2, all samples together). However, the location had some influence on the microbial communities from mucus (p = .024) and seawater (p = .027). No significant differences (p > .05) were observed between the microbiome structures from healthy colonies (without white spots) and colonies with morphological alterations (with white spots), from all sites. The richness values also differed, with the highest bacterial richness observed in the seawater samples from site 2 (Table S2).
Figure 1.
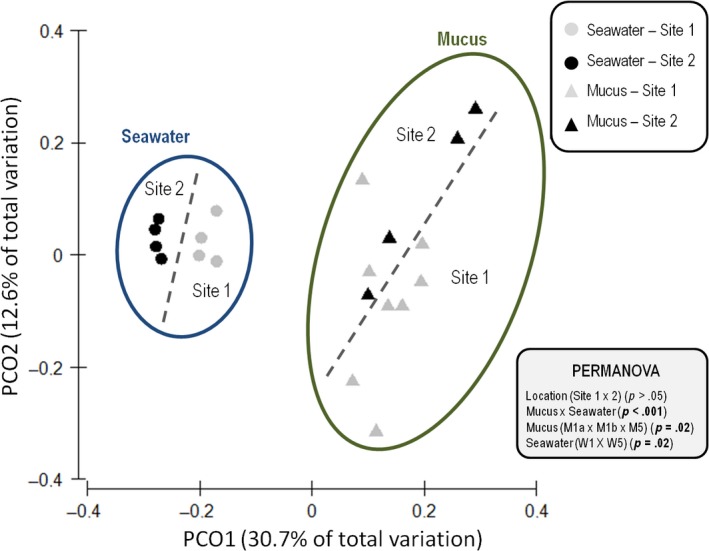
NMS (Bray–Curtis) plot of bacterial communities associated with Mussismilia hispida mucus and surrounding seawater at Recife Itassepocu (Site 1) and Recife de Fora (Site 2) based on 16S rRNA gene sequence data (n = 4). Contours and dashed lines are based on significant pairwise PERMANOVA results (p = .01). Circle represents seawater replicates, and triangle represents mucus replicates. Light gray indicates samples from site 1, and black indicates samples from site 5
Regarding the identity of the OTUs, a suite of diverse bacterial taxa was observed. Proteobacteria was the most abundant phylum associated with the coral mucus and seawater microbiomes, followed by Bacteroidetes and Firmicutes (Figure S1), and the core bacteriome was basically represented by Proteobacteria (Figure S2). The 10 most abundant OTUs represented around 58%–67% of the entire mucus bacteriome at site 1, and 38% at site 2. The top 10 OTUs consisted of members of the genera Pseudoalteromonas, Halomonas, and Rugeria, and of the families Pseudoalteromonadaceae, Rhodobacteraraceae, Desulfovibrionaceae, Desulfobulbaceae, Hyphomonadaceae, Acidaminobacteriaceae, and Flavobacteriaceae (Figure S3).
The top 23 (83.55%) OTUs responsible for the dissimilarity between M. hispida mucus and the corresponding seawater were then evaluated by SIMPER analysis (Table 1). Some microorganisms, such as members of the Pseudoalteromonadaceae, were the major OTUs that significantly constituted the general profiles observed for both bacteriomes, mucus and seawater (21.86%). However, other bacteria were specifically correlated with the mucus bacteriome, including members of the Desulfovibrionaceae (6%) and Flavobacteriaceae (3.6%), Pseudoalteromonas (2.76%), Arcobacter (2.64%), Rugeria (3.28%), and other genera of Rhodobacteraceae (2.15%).
Table 1.
SIMPER analysis results, showing top 23 operational taxonomic units (OTUs) responsible for 83.55% dissimilarity between Mussismilia hispida mucus and seawater
| OTUs | Mucus | Seawater | OTU contribution (%)b | Cumulative contribution (%) |
|---|---|---|---|---|
| Average abundancea | Average abundancea | |||
| Unclassified Pseudoalteromonadaceae | 2,203.67 | 10,112.13 | 21.86 | 21.86 |
| Unclassified Desulfovibrionaceae | 2,253.75 | 158.88 | 6.01 | 27.87 |
| Unclassified Flavobacteriaceae | 1,396.42 | 8.13 | 3.64 | 31.51 |
| Alteromonas | 45.83 | 1,417.5 | 3.58 | 35.1 |
| Ruegeria * | 1,189.92 | 372.75 | 3.28 | 38.38 |
| Vibrio | 276.67 | 1,247.75 | 2.83 | 41.21 |
| Pseudoalteromonas | 1,090.67 | 100.63 | 2.76 | 43.97 |
| Arcobacter | 1,010.17 | 2.38 | 2.64 | 46.61 |
| Unclassified Desulfobulbaceae | 847.92 | 1.25 | 2.22 | 48.82 |
| Unclassified Desulfovibrionaceae | 825.17 | 41.25 | 2.19 | 51.02 |
| Idiomarina | 76.58 | 817.13 | 2.17 | 53.18 |
| Unclassified Rhodobacteraceae | 910.33 | 276.5 | 2.15 | 55.34 |
| Unclassified Hyphomonadaceae | 804.17 | 2.75 | 2.1 | 57.44 |
| Halomonas | 560.92 | 122.13 | 1.64 | 59.08 |
| Thalassospira | 563.33 | 69.25 | 1.56 | 60.64 |
| Alcanivorax | 18.42 | 600.38 | 1.53 | 62.16 |
| Marinobacter | 10.92 | 543.63 | 1.41 | 63.57 |
| Unclassified Vibrionaceae | 613.17 | 153.75 | 1.28 | 64.85 |
| Unclassified Acidaminobacteraceae | 476.67 | 0 | 1.25 | 66.09 |
| Unclassified Flavobacteriaceae | 398.25 | 15.75 | 1.03 | 67.13 |
| Oceanicaulis | 317 | 141.38 | 0.97 | 68.1 |
| Idiomarina | 35.08 | 389 | 0.92 | 69.02 |
| Marinobacter | 0.17 | 334.25 | 0.87 | 69.9 |
| Phylum | ||||
| Proteobacteria | 63.97 | 63.97 | ||
| Firmicutes | 1.25 | 65.22 | ||
| Bacteriodetes | 4.67 | 69.89 | ||
Mean abundance of each OTU.
Contribution of each taxon to the overall dissimilarity between Mucus and seawater groups.
OTUS which are also observed in co‐occurrence analysis.
3.2. Bacterial 16S rRNA gene copy and MGE abundances
In the coral mucus samples compared to the seawater samples, a higher abundance was seen for 16S rRNA (1.7–3.1e+09 gene/ml for mucus and 5.1–5.8e+07 gene/ml for seawater; p < .01), IncP‐1β (3.9e+02–2.1e+03 gene/ml for mucus and 7.1–7.4e+00 gene/ml for seawater; p < .01), and PromA (5.1–9.7e+01 gene/ml for mucus; p < .01) plasmid groups. This represents an increase in gene copies in mucus samples of 21% for 16S rRNA genes and 252% and 100% for plasmid groups (IncP‐1 and PromA, respectively), compared to the seawater samples. PromA plasmid groups were detected only in the coral mucus and were below the detection level in the seawater bacteriomes. No significant differences (p > .05) were observed between healthy colonies and those with morphological alterations for the 16S rRNA and plasmid groups (IncP‐1 and PromA). Copies of the intl1 gene (Integron class I) were detected only in seawater, specifically from site 1, closer to the river mouth, while incW plasmid groups were not detected in any of the samples (Figure 2).
Figure 2.
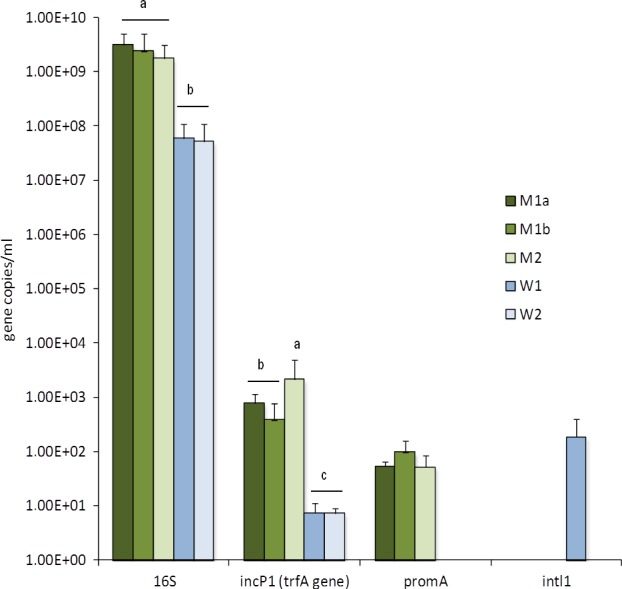
Abundances of 16S rRNA, trfA, promA and intl1 genes from Mussismilia hispida mucus and surrounding seawater at Recife Itassepocu (site 1) and Recife de Fora (site 2). Column labels are as follows: M2: M. hispida mucus at site 2, M1b: M. hispida mucus at site 1—colonies without white spots, M1a: M. hispida mucus at site 1—colonies with white spots, W1: seawater at site 1, W2: seawater at site 2
3.3. Coral core bacteriome and bacterial co‐occurrence in mucus
The core bacteriome of the M. hispida mucus (OTUs shared among all samples) was composed mostly of Proteobacteria (between 90% and 98%), followed by Firmicutes (Bacillaceae) (Table 2, Figure S2). Some of the OTUs that constituted the mucus core of M. hispida were also observed in the co‐occurrence analyses (Table 2, Figure S4), such as Pseudomonas, Bacillus, Pseudoalteromonas, Pseudoalteromonadaceae, and Rhodobacteraceae.
Table 2.
Operational taxonomic unit (OTU) table of the bacterial core community associated with Mussismilia hispida mucus at Recife Itassepocu (site 1) and Recife de Fora (site 2)
| OTU ID | Samples | ID used in this paper | Taxonomy | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M1a (i) | M1a (ii) | M1a (iii) | M1a (iv) | M1b (i) | M1b (ii) | M1b (iii) | M1b (iv) | M2 (i) | M2 (ii) | M2 (iii) | M2 (iv) | Phylum | Class | Order | Family | Genera | ||
| 540269 | 52 | 6,861 | 41 | 5 | 4 | 3 | 34 | 4 | 7 | 8 | 4 | 37 | Exiguobacteraceae_1[Link] | Firmicutes | Bacilli | Bacillales | Exiguobacteraceae | |
| NR_OTU64 | 36 | 114 | 6 | 1 | 4 | 1 | 37 | 42 | 1 | 3 | 1 | 25 | Bacillaceae[Link] | Firmicutes | Bacilli | Bacillales | Bacillaceae | |
| 1078248 | 558 | 423 | 145 | 20 | 41 | 33 | 412 | 123 | 47 | 43 | 3 | 177 | Bacillusa | Firmicutes | Bacilli | Bacillales | Bacillaceae | Bacillus |
| 1087298 | 24 | 7 | 13 | 10 | 13 | 41 | 2 | 6 | 12 | 13 | 6 | 21 | Alphaproteobacteria | Proteobacteria | Alphaproteobacteria | |||
| 309877 | 2,517 | 7 | 1 | 2 | 2 | 2 | 2 | 5 | 55 | 3,316 | 3,017 | 12,340 | Hyphomonadaceae_1 | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Hyphomonadaceae | |
| 829814 | 9,478 | 278 | 3,268 | 2,561 | 104 | 5,062 | 12 | 16 | 461 | 1,316 | 1,223 | 716 | Rhodobacteraceae_5 | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | |
| 1101488 | 2,378 | 15,366 | 131 | 491 | 12 | 9,864 | 2 | 1 | 172 | 340 | 309 | 305 | Ruegeria | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | Ruegeria |
| 2932342 | 11 | 316 | 782 | 45 | 19,637 | 12,563 | 66 | 44,095 | 3 | 2 | 1 | 57 | Desulfovibrionaceae_3[Link] | Proteobacteria | Deltaproteobacteria | Desulfovibrionales | Desulfovibrionaceae | |
| 51975 | 5 | 7 | 1 | 1 | 3 | 47 | 9 | 4 | 18,923 | 2,193 | 1,837 | 31 | Arcobacter[Link] | Proteobacteria | Epsilonproteobacteria | Campylobacterales | Campylobacteraceae | Arcobacter |
| 823476 | 258 | 146 | 10 | 35 | 8 | 102 | 193 | 65 | 56 | 16 | 23 | 220 | Alteromonas[Link] | Proteobacteria | Gammaproteobacteria | Alteromonadales | Alteromonadaceae | Alteromonas |
| 812024 | 16 | 1 | 18 | 14 | 23 | 248 | 34 | 14 | 15 | 15 | 5 | 13 | Glaciecola_2 | Proteobacteria | Gammaproteobacteria | Alteromonadales | Alteromonadaceae | Glaciecola |
| 509913 | 1,249 | 3 | 143 | 45 | 24 | 26 | 6 | 4 | 8 | 29 | 17 | 7 | Marinobacter_1[Link] | Proteobacteria | Gammaproteobacteria | Alteromonadales | Alteromonadaceae | Marinobacter |
| 956811 | 5 | 15 | 126 | 135 | 1,314 | 28 | 1 | 6 | 375 | 33 | 21 | 85 | Idiomarina_3[Link] | Proteobacteria | Gammaproteobacteria | Alteromonadales | Idiomarinaceae | Idiomarina |
| 141607 | 33 | 14 | 28 | 87 | 3 | 4 | 33 | 12 | 7 | 340 | 237 | 38 | Idiomarina_1[Link] | Proteobacteria | Gammaproteobacteria | Alteromonadales | Idiomarinaceae | Idiomarina |
| 182418 | 101 | 61 | 25 | 1 | 5 | 15 | 99 | 26 | 26 | 1 | 4 | 85 | Pseudomonasa | Proteobacteria | Gammaproteobacteria | Pseudomonadales | Pseudomonadaceae | Pseudomonas |
| 820978 | 5,005 | 8,329 | 3,379 | 341 | 3,846 | 1,047 | 15,048 | 2,042 | 1,445 | 183 | 341 | 5,630 | Pseudoalteromonadaceae_6[Link] | Proteobacteria | Gammaproteobacteria | Vibrionales | Pseudoalteromonadaceae | |
| 785565 | 386 | 519 | 238 | 23 | 312 | 78 | 443 | 154 | 100 | 14 | 37 | 367 | Pseudoalteromonadaceae_4[Link] | Proteobacteria | Gammaproteobacteria | Vibrionales | Pseudoalteromonadaceae | |
| 160928 | 144 | 162 | 136 | 19 | 126 | 31 | 422 | 48 | 18 | 3 | 5 | 142 | Pseudoalteromonadaceae_1[Link] | Proteobacteria | Gammaproteobacteria | Vibrionales | Pseudoalteromonadaceae | |
| 217623 | 15 | 22 | 19 | 2 | 70 | 19 | 78 | 225 | 24 | 1 | 2 | 21 | Pseudoalteromonadaceae_3[Link] | Proteobacteria | Gammaproteobacteria | Vibrionales | Pseudoalteromonadaceae | |
| NCUR_OTU23335 | 68 | 19 | 3 | 1 | 13 | 6 | 1 | 1 | 9 | 40 | 44 | 194 | Pseudoalteromonadaceae_7[Link] | Proteobacteria | Gammaproteobacteria | Vibrionales | Pseudoalteromonadaceae | |
| 198609 | 27 | 38 | 19 | 2 | 34 | 10 | 130 | 34 | 30 | 1 | 4 | 35 | Pseudoalteromonadaceae_2[Link] | Proteobacteria | Gammaproteobacteria | Vibrionales | Pseudoalteromonadaceae | |
| NR_OTU80 | 27 | 12 | 21 | 5 | 23 | 7 | 65 | 13 | 7 | 1 | 3 | 20 | Pseudoalteromonadaceae_8[Link] | Proteobacteria | Gammaproteobacteria | Vibrionales | Pseudoalteromonadaceae | |
| 821550 | 36 | 67 | 329 | 184 | 533 | 93 | 273 | 34 | 5 | 17 | 24 | 7 | Pseudoalteromonas_6[Link] | Proteobacteria | Gammaproteobacteria | Vibrionales | Pseudoalteromonadaceae | Pseudoalteromonas |
| 830290 | 646 | 483 | 6,481 | 8,569 | 10,253 | 1,140 | 980 | 143 | 1 | 167 | 151 | 24 | Pseudoalteromonas_7[Link] | Proteobacteria | Gammaproteobacteria | Vibrionales | Pseudoalteromonadaceae | Pseudoalteromonas |
| 939811 | 300 | 145 | 664 | 254 | 2,151 | 2,987 | 235 | 735 | 398 | 2,814 | 2,340 | 3,161 | Vibrionaceae_4[Link] | Proteobacteria | Gammaproteobacteria | Vibrionales | Vibrionaceae | |
| 837366 | 25 | 10 | 52 | 30 | 283 | 302 | 21 | 32 | 21 | 366 | 272 | 299 | Vibrionaceae_3[Link] | Proteobacteria | Gammaproteobacteria | Vibrionales | Vibrionaceae | |
| NCUR_OTU15773 | 1 | 3 | 27 | 21 | 54 | 15 | 22 | 10 | 4 | 5 | 1 | 1 | Vibrio_3 | Proteobacteria | Gammaproteobacteria | Vibrionales | Vibrionaceae | Vibrio |
| 792393 | 737 | 573 | 201 | 86 | 65 | 209 | 2,213 | 361 | 211 | 56 | 67 | 695 | Vibrio_2[Link] | Proteobacteria | Gammaproteobacteria | Vibrionales | Vibrionaceae | Vibrio |
M2, Mussismilia hispida mucus at site 2; M1b, Mussismilia hispida mucus at site 1—colonies without white spots; M1a, Mussismilia hispida mucus at site 1—colonies with white spots.
OTUS which are also observed in co‐occurrence analysis.
Members of the bacterial core that are vertically transmitted in M. hispida, as described by Leite et al., 2017.
The co‐occurrence patterns of bacterial genera revealed that for the most abundant OTUs, a total of 139 significant co‐occurrences (p = .05) were detected between 38 bacterial OTUs in a matched subset of mucus samples from sites 1 and 2 (Figure 3a). For the bacterial core, a total of 121 significant co‐occurrences (p = .05) were detected between 26 bacterial genera (Figure 4a). For both, most abundant OTUs and bacterial core, most of the co‐occurrences were positive among the OTUs, and most of the correlated members belonged to the Proteobacteria, with some Firmicutes (Bacillaceae) also found. Cluster 1, consisting of the most abundant OTUs and the core bacteriome, included core members, such as members of OTUs from the genera Pseudomonas and Bacillus (Figures 3b and 4b). The other clusters (Figures 3c,d and 4c) were composed mostly of Proteobacteria.
Figure 3.
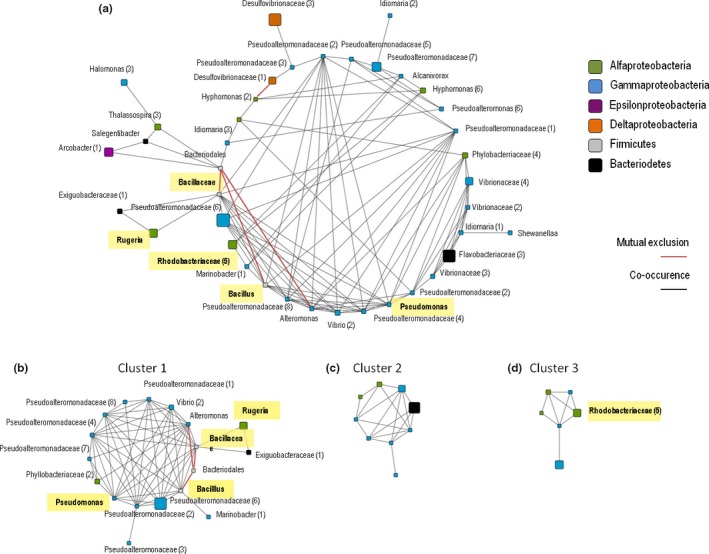
Network analysis of interactions among the more abundant bacterial operational taxonomic units (OTUs). Significant interactions among bacterial genera in Mussismilia hispida mucus from Recife Itassepocu (Site 1) and Recife de Fora (Site 2). Red lines indicate negative interactions (mutual exclusions), and black lines indicate positive interactions (co‐occurrences). The size of the nodes reflects the relative abundance of the genus in the entire data set, and the nodes are sorted and colored by phylum. (a) All significant interactions, involving 38 OTUs, sorted by class and densely interconnected regions, Cluster 1 (b), Cluster 2 (c), and Cluster 3 (d). The core bacteriome members vertically transferred (Leite et al., 2017) are highlighted in yellow
Figure 4.
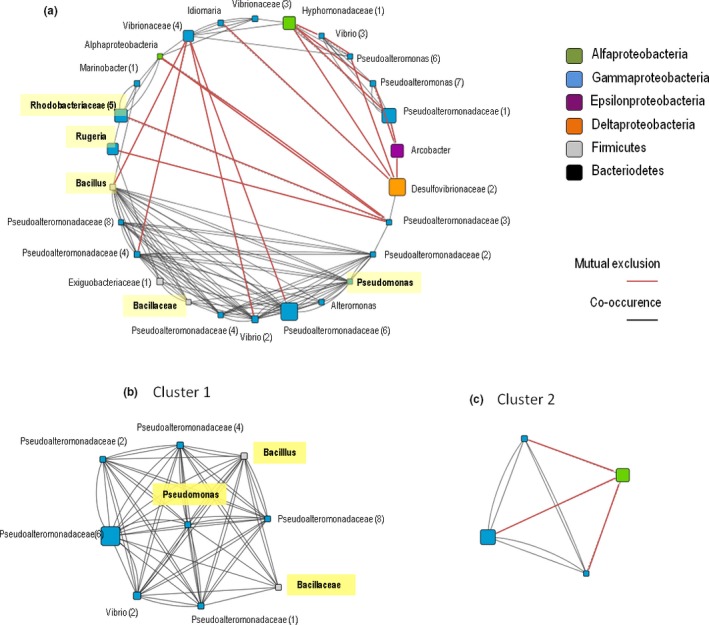
Network analysis of interactions in the core bacteriome. Significant interactions between bacterial genera in Mussismilia hispida mucus from Recife Itassepocu (Site 1) and Recife de Fora (Site 2). Red lines indicate negative interactions (mutual exclusions), and black lines indicate positive interactions (co‐occurrences). The size of the nodes reflects the relative abundance of the genus in the entire data set, and the nodes are sorted and colored by phylum. (a) All 121 significant interactions, involving 26 OTUs, sorted by class and densely interconnected regions, Cluster 1 (b) and Cluster 2 (c). The core bacteriome members vertically transferred (Leite et al., 2017) are highlighted in yellow
The co‐occurrence of taxa, considering the most dominant OTUs in the total mucus (Figure 3) and the bacterial core OTUs (Figure 4), consisted mainly of the families Bacillaceae and Pseudoalteromonadaceae and the genera Pseudomonas, Bacillus, Alteromonas, and Vibrio. Most of the inter‐relationships within the bacteriome core OTUs were also related to the same groups, except for Alteromonas (Figures 3 and 4, Table 3). OTUs related to Pseudomonas and Pseudomonadaceae, and Bacillus and Bacillaceae were the most important groups showing positive correlations (Table 3).
Table 3.
Top 10 interactions among the operational taxonomic units (OTUs) from the network analysis (Cluster 1) of the (A) most abundant OTUs and (B) core bacteriome of mucus at Recife Itassepocu (site 1) and Recife de Fora (site 2)
| OTUs | More abundant OTUs | |
|---|---|---|
| Number of positives correlations | Number of negative correlations | |
| (A) | ||
| Bacillaceae | 12 | 1 |
| Pseudomonas | 11 | — |
| Pseudoalteromonadaceae | 11 | — |
| Pseudoalteromonadaceae | 11 | — |
| Alteromonas | 11 | — |
| Pseudoalteromonadaceae | 10 | — |
| Pseudoalteromonadaceae | 10 | — |
| Bacillus | 10 | 1 |
| Vibrio | 9 | — |
| Pseudoalteromonadaceae | 9 | — |
| (B) | ||
| Pseudomonas | 16 | — |
| Pseudoalteromonadaceae | 16 | — |
| Pseudoalteromonadaceae | 15 | — |
| Bacillus | 15 | — |
| Pseudoalteromonadaceae | 15 | — |
| Vibrio | 15 | — |
| Pseudoalteromonadaceae | 14 | — |
| Pseudoalteromonadaceae | 13 | — |
| Bacillaceae | 8 | — |
The functional correlation between the abundances of Proteobacteria (OTUs) and 16S rRNA, trfA, and promA (genes/ml) in coral mucus and seawater bacteriomes generated different patterns. For mucus, no correlation (Pearson's r = .06) could be observed for the abundance of 16S rRNA copies and Proteobacteria OTUs. However, a positive correlation (Pearson's r = .54) was observed for seawater. Regarding the correlation between all tested plasmids and Proteobacteria OTUs, a positive correlation was observed (Pearson's r: trfA = 0.36 and promA = 0.62) for mucus samples, while a negative correlation was found for seawater samples (Pearson's r = −.15) (Figure 5).
Figure 5.
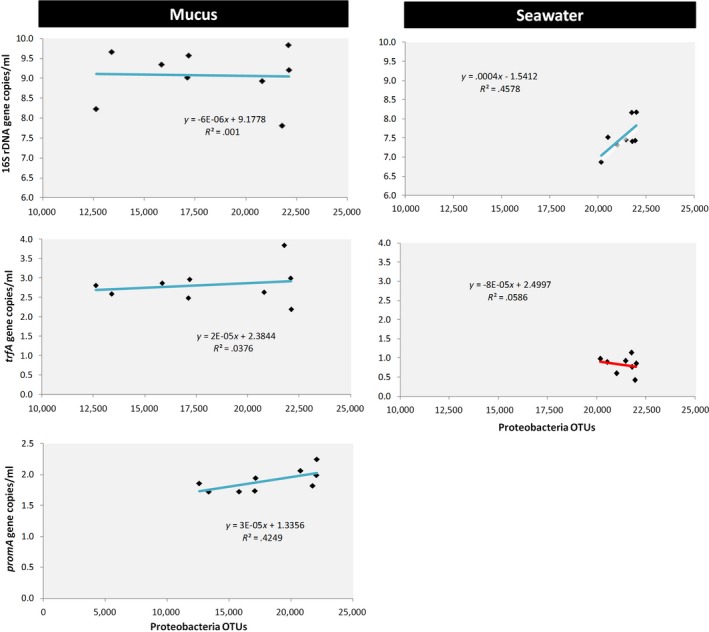
Correlation plot showing the functional correlation between Proteobacteria operational taxonomic units (OTUs) and 16S rRNA, trfA, and promA genes/ml for coral mucus and seawater bacteriome
4. DISCUSSION
Here, we describe the diversity and intercorrelations of the bacterial diversity, as well as the prevalence of MGEs associated with mucus from M. hispida and the surrounding seawater. We focused particularly on the BHR plasmid groups IncP‐1 and PromA, widely used as BHR plasmid proxies and indicated as the major providers of bacterial HGT in some soil environments (van der Auwera et al., 2009; Heuer & Smalla, 2012; Zhang et al., 2014), as well as on integron1, a good proxy for pollution (Gillings et al., 2015). The use of these plasmid groups was also based on their hypothesized importance, which is related to resistance to antibiotics and heavy metals and to the efficient mobilization among Gram‐negative bacteria.
Our main findings indicate that key groups of bacteria, that is, Proteobacteria, followed by Firmicutes, mainly represented by members of Rhodobacteraceae and the genera Pseudomonas and Bacillus associated with M. hispida, were present in the coral mucus. These dominant groups and members of the entire M. hispida microbial core have also been described as being vertically transmitted from parent to offspring in the same coral species (Leite et al., 2017). In addition, this core microbiome and some other groups in the mucus bacteriome have a positive relationship of co‐occurrence, especially the families Desulfovibrionaceae and Flavobacteriaceae, the genera Pseudoalteromonas and Arcobacter, and Rugeria and other members of Rhodobacteraceae, suggesting that these microorganisms could be selected by the holobiont. Our data also indicated that the holobiont selects (i.e., contains a higher abundance or even a specific persistence) the IncP‐1 and PromA groups of BHR plasmids, as these were significantly more abundant in coral mucus or absent in the seawater samples, respectively.
Studies on coral microbiomes have shown the importance of these organisms for host health, fitness, maintenance (Musovic, Oregaard, Kroer, & Sørensen, 2006; Peixoto et al., 2017; Santos et al., 2014, 2015; Sweet & Bulling, 2017; Webster & Reusch, 2017), and evolution (Bhattacharya et al., 2016). Microbial surveys have contributed to our understanding of how microbial communities can promote the resilience of coral reefs to environmental stress (Bhattacharya et al., 2016; Peixoto et al., 2017; Sweet & Bulling, 2017; Webster & Reusch, 2017), and have generated knowledge of the beneficial potential of the microbiome and its potential future manipulations (Damjanovic et al., 2017; Peixoto et al., 2017; Sweet & Bulling, 2017; Webster & Reusch, 2017), thereby improving the health of reef ecosystems. Knowledge of key coral microbiome microbial groups and potential intertaxa correlation patterns can improve and guide such BMC manipulations, by indicating stable, well‐adapted populations that could be involved in beneficial mechanisms and would be, at the same time, competitive, and well established in manipulative approaches.
The seawater bacteriome observed here was more diverse than the coral bacteriome, as reported in other studies (Castro et al., 2010; Garcia et al., 2013; Reis et al., 2009; Rojo, 2010; Rosenberg, Kellogg, & Rohwer, 2007). Mussismilia hispida bacterial communities from the mucus samples were composed mainly of Proteobacteria, a phylum that is widely found in M. hispida microbiomes (Castro et al., 2010; Leite et al., 2017; Lins‐De‐barros et al., 2010; Musovic et al., 2006) as well as in other species of the genus Mussismilia (Fernando et al., 2015; Garcia et al., 2013; Santos et al., 2015) and in other coral genera (Bourne & Munn, 2005; Kimes, van Nostrand, Weil, Zhou, & Morris, 2010; Kimes et al., 2013; Mouchka et al., 2010; Vega Thurber et al., 2009). Moreover, the phylum Proteobacteria is quite abundant in a range of Mussimilia microhabitats such as the mucus, tissue, and skeleton, compared with other bacterial groups (Castro et al., 2010; Fernando et al., 2015; Garcia et al., 2013; Leite et al., 2017; Lins‐De‐barros et al., 2010; Reis et al., 2009; Santos et al., 2015).
Recent studies have suggested that the coral microbial community is composed of both a stable and a variable fraction. The stable fraction is proposed to be directly involved in basic host requirements (i.e., the microbial core, which is also composed of two components, a host‐specific ubiquitous community, and a niche‐specific community). The variable fraction is proposed to vary rapidly with environmental shifts (Hernandez‐Agreda, Leggat, Bongaerts, & Ainsworth, 2016). For the maintenance of the coral holobiont, the host can acquire its symbionts directly, via parental gametes/eggs (i.e., vertical transmission (Musovic et al., 2006; Sharp, Distel, & Paul, 2011; Padilla‐Gamiño et al., 2012)) or through acquisition from the surrounding environment (i.e., horizontal transmission) (Apprill, Marlow, Martindale, & Rappé, 2009; Knowlton & Rohwer, 2003). The early acquisition and maintenance of a microbiome may ensure the establishment of key mechanisms to protect and foster the settlement and development of coral larvae (Lema et al., 2014; Sharp & Ritchie, 2012).
Leite et al. (2017) have indicated that members of the core bacteriome of M. hispida (i.e., the genera Burkholderia, Pseudomonas, Acinetobacter, Ralstonia, Inquilinus and Bacillus, and unclassified Rhodobacteraceae) were transmitted vertically to offspring, through the gametes, reinforcing the potential importance of the coral bacteriome core members. Therefore, we have focused on the core bacteriome from the M. hispida mucus samples from different sampling points. Our data have also indicated core members that have been described by Leite et al. (2017) at early life stages, such as Pseudomonas, Bacillus, and Rhodobacteraceae members, in all coral mucus samples from the two sampling sites, and showing a high level of intertaxa relationships. In addition, considering the BHR that were screened, the IncP‐1 plasmid group was the most abundant plasmid group in the coral mucus bacteriome. These plasmids have a wide distribution and are highly efficient for Gram‐negative bacteria (Popowska & Krawczyk‐Balska, 2013), but have also been reported mobilizing Gram‐positive bacteria (Musovic et al., 2006). This group of plasmids can exchange a wide range of potentially advantageous genes, such as genes for antibiotic resistance and degradation of different carbon sources (Popowska & Krawczyk‐Balska, 2013; Shintani et al., 2010; Zhang et al., 2014), which, given the abundance of “enriched” plasmids, could suggest a key role of HGT in the coral–microbiome interactions.
The second most abundant group of plasmids, PromA, proposed by van der Auwera et al. (2009), was detected only in coral samples. Previous studies have found that IncP‐1 and PromA, BHR groups of plasmids, are extremely important gene carriers in other systems, such as for soil bacterial communities (van der Auwera et al., 2009; Heuer & Smalla, 2012; Zhang et al., 2014), and are both efficient plasmids for gene exchanges between members of the Proteobacteria group (Zhang et al., 2014). We find it interesting that this group was detected only in coral mucus samples. Although there are multiple possible explanations, this could also indicate that the holobiont can indeed select and concentrate a specific diversity of MGEs.
When considering the network analyses from the total mucus samples, that is, not considering only the bacteriome core, we have found a large number of related OTUs, mainly based on co‐occurrence among Proteobacteria and Firmicutes members. More specifically, separate clusters harboring core microbiome members, previously described as vertically transmitted in M. hispida (Pseudomonas, Bacillus and Rhodobacteraceae members) (Leite et al., 2017), were observed. There are multiple possible explanations for these patterns of co‐occurrence and dominance, and we discuss a few of them below. One possibility is that taxonomic relationships, at the OTU level, are indeed relevant for the coral microbiome assembly. In this case, it could suggest that something about these specific taxa (e.g., key functions), that is best, or exclusively, provided by these members, could not be replaced by other taxa or HGT. This could explain the stable taxonomic selection observed. Alternatively, or complementarily, interactions between taxa, or between the host and these taxa, maintain their presence or absence and the observed correlations. In addition, these patterns could also be merely a consequence of history, that is, successive vertical transmission of specific groups that leads to correlations.
The observed co‐occurrence and specific taxonomic persistence indicate that these taxa are potential key players in coral health, give that their presence in the offspring is ensured. This co‐occurrence and taxonomic persistence could also suggest that these members might be actively involved in the persistence of other bacterial groups through symbiotic relationships. On the other hand, these data could indicate that these coexisting and dominant groups are independently influenced by environmental factors. Thus, these groups would be selected as the most able to survive in this environment (Barberán et al., 2012), due to their potential key irreplaceable functions. In this case, they are only sharing the M. hispida mucus niche. Nevertheless, this would mean that these are the most competitive groups within this niche, which clearly indicates them as important targets for M. hispida BMC manipulative studies.
In parallel, positive correlations were observed between the coral mucus and the abundance and/or the specific presence of the screened plasmids. Although plasmid abundance is not supported by the observed stable bacterial taxonomic diversity, as, in this case, the relevant role seems to be related to the taxonomic level rather than to transferrable functions, it could be related to the variable fraction of the coral microbiome. Thus, it is possible that these “holobiont‐enriched” plasmids could be in fact associated with the noncore, non‐co‐occurring taxa. The “enriched” presence of these MGEs within the holobiont could indicate that advantageous genes could be eventually exchanged between all members of the coral bacteriome. This advantageous exchange of genes could eventually support the transient (and even the stable) members under adverse conditions, which can, in turn, contribute to the resilience of the host in the face of environmental shifts. However, Hall, Williams, Paterson, Harrison, and Brockhurst (2017) have recently suggested that conjugation can be reduced by positive selection, indicating that HGT can be inhibited by those beneficial elements. The conjugative mobilization would be more related to infections and parasitic elements. Thus, the remaining questions are as follows: To what taxa do these “enriched” plasmids belong? And is there active HGT, mediated by these plasmids, occurring?
Taken together, the high prevalence of co‐occurrence between core bacterial groups and the specific plasmid‐pattern data could suggest separate roles in the coral bacterial assembly. It is also possible that both mechanisms could be correlated, as a random consequence of the high prevalence of the dominant microbial diversity, Proteobacteria. This could, in turn, randomly select those plasmids that can be established by the abundance of this dominant group, though not being necessarily relevant for this dominance. This suggestion is supported by the correlations between Proteobacteria OTUs and plasmids in the mucus samples. On the other hand, this role could be associated with specific mechanisms, evolved to selectively permit the persistence of the dominant components of the bacteriome and associated plasmids, which could allow eventual cooperation between other (and transient) members, mediated by gene exchange. Both hypotheses are somehow driven by the holobiont and its microbial diversity.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHORS’ CONTRIBUTIONS
RSP, DCAL and ENC performed study conception and design. DCAL and ENC performed acquisition and identification of coral samples. DCAL, ENC performed acquisition of data (conducting of experiments). RSP, DCAL, JFS and JDE performed analyses and interpretation of data. RSP and DCAL drafted the manuscript. All authors critically revised the manuscript. RSP, JFS and JDE provided financial support.
DATA ACCESSIBILITY
The raw data from each sample are available at the NCBI Sequence Read Archive (SRA) under Accession Numbers SRR5903387—SRR5903406.
Supporting information
ACKNOWLEDGMENTS
We thank Jonathan A. Eisen, ORCID ID 0000‐0002‐0159‐2197, and Alexandre Rosado for their helpful comments to improve the manuscript. We also thank Coral Vivo and its sponsors (Arraial d'Ajuda Eco Parque and Petrobras) for logistical support and the use of its research base and Clovis B. Castro, National Museum, Federal University of Rio de Janeiro, for helping with acquisition of coral samples. We thank the National Council for Scientific and Technological Development (CNPq), the National Council for the Improvement of Higher Education (CAPES), and the Carlos Chagas Filho Foundation for Research Support of Rio de Janeiro State (FAPERJ) for their support, and the Secretary for the Environment of the Municipality of Porto Seguro for the collection license.
Leite DCA, Salles JF, Calderon EN, van Elsas JD, Peixoto RS. Specific plasmid patterns and high rates of bacterial co‐occurrence within the coral holobiont. Ecol Evol. 2018;8:1818–1832. https://doi.org/10.1002/ece3.3717
Funding information
This study was supported by the National Council for Scientific and Technological Development (CNPq), the National Council for the Improvement of Higher Education (CAPES), and the Carlos Chagas Filho Foundation for Research Support of Rio de Janeiro State (FAPERJ)
REFERENCES
- Ainsworth, T. D. , Thurber, R. V. , & Gates, R. D. (2010). The future of coral reefs: A microbial perspective. Trends in Ecology & Evolution, 25, 233–240. https://doi.org/10.1016/j.tree.2009.11.001 [DOI] [PubMed] [Google Scholar]
- Anderson, M. , Gorley, R. N. , & Clarke, R. K. (2008). PERMANOVA+ for primer: Guide to software and statistical methods. Plymouth, UK: Primer‐E Limited. [Google Scholar]
- Andrade, L. L. , Leite, D. C. , Ferreira, E. M. , Ferreira, L. Q. , Paula, G. R. , Maguire, M. J. , … Rosado, A. S. (2012). Microbial diversity and anaerobic hydrocarbon degradation potential in an oil‐contaminated mangrove sediment. BMC Microbiology, 12, 186 https://doi.org/10.1186/1471-2180-12-186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apprill, A. , Marlow, H. Q. , Martindale, M. Q. , & Rappé, M. S. (2009). The onset of microbial associations in the coral Pocillopora meandrina . The ISME Journal, 3, 685–699. https://doi.org/10.1038/ismej.2009.3 [DOI] [PubMed] [Google Scholar]
- van der Auwera, G. A. , Król, J. E. , Suzuki, H. , Foster, B. , van Houdt, R. , Brown, C. J. , … Top, E. M. (2009). Plasmids captured in C. metallidurans CH34: Defining the PromA family of broad‐host‐range plasmids. Antonie van Leeuwenhoek, 96, 193–204. https://doi.org/10.1007/s10482-009-9316-9 [DOI] [PubMed] [Google Scholar]
- Bader, G. D. , & Hogue, C. W. (2003). An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics, 4, 2 https://doi.org/10.1186/1471-2105-4-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberán, A. , Bates, S. T. , Casamayor, E. O. , & Fierer, N. (2012). Using network analysis to explore co‐occurrence patterns in soil microbial communities. The ISME Journal, 6, 343–351. https://doi.org/10.1038/ismej.2011.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya, D. , Agrawal, S. , Aranda, M. , Baumgarten, S. , Belcaid, M. , Drake, J. L. , … Gruber, D. F. (2016). Comparative genomics explains the evolutionary success of reef‐forming corals. eLife, 5, e13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch, Thomas C. G. , & Miller, David J. (2016). Bleaching as an obvious dysbiosis in corals In: The holobiont imperative. Vienna: Springer; 10: 978–3. [Google Scholar]
- Bourne, D. G. , & Munn, C. B. (2005). Diversity of bacteria associated with the coral Pocillopora damicornis from the Great Barrier Reef. Environmental Microbiology, 7, 1162–1174. https://doi.org/10.1111/j.1462-2920.2005.00793.x [DOI] [PubMed] [Google Scholar]
- Caporaso, J. G. , Bittinger, K. , Bushman, F. D. , Desantis, T. Z. , Andersen, G. L. , & Knight, R. (2010a). PyNAST: A flexible tool for aligning sequences to a template alignment. Bioinformatics, 26, 266–267. https://doi.org/10.1093/bioinformatics/btp636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso, J. G. , Kuczynski, J. , Stombaugh, J. , Bittinger, K. , Bushman, F. D. , Costello, E. K. , … Gordon, J. I. (2010b). QIIME allows analysis of high‐throughput community sequencing data. Nature Methods, 7, 335–336. https://doi.org/10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso, J. G. , Lauber, C. L. , Walters, W. A. , Berg‐Lyons, D. , Lozupone, C. A. , Turnbaugh, P. J. , … Knight, R. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences of the USA, 108, 4516–4522. https://doi.org/10.1073/pnas.1000080107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cárdenas, A. , Rodriguez‐R, L. M. , Pizarro, V. , Cadavid, L. F. , & Arévalo‐Ferro, C. (2012). Shifts in bacterial communities of two Caribbean reef‐building coral species affected by white plague disease. The ISME Journal, 6, 502–512. https://doi.org/10.1038/ismej.2011.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro, A. P. , Araújo Jr, S. D. , Reis, A. M. , Moura, R. L. , Francini‐Filho, R. B. , Pappas Jr, G. , … Krüger, R. H. (2010). Bacterial community associated with healthy and diseased reef coral Mussismilia hispida from eastern Brazil. Microbial Ecology, 59, 658–667. https://doi.org/10.1007/s00248-010-9646-1 [DOI] [PubMed] [Google Scholar]
- Ceh, J. , Keulen, M. , & Bourne, D. G. (2013). Intergenerational transfer of specific bacteria in corals and possible implications for offspring fitness. Microbial Ecology, 65, 227–231. https://doi.org/10.1007/s00248-012-0105-z [DOI] [PubMed] [Google Scholar]
- Ceh, J. , Raina, J.‐B. , Soo, R. M. , van Keulen, M. , & Bourne, D. G. (2012). Coral‐bacterial communities before and after a coral mass spawning event on Ningaloo Reef. PLoS ONE, 7, e36920 https://doi.org/10.1371/journal.pone.0036920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damjanovic, K. , Blackall, L. L. , Webster, N. S. , & van Oppen, M. J. (2017). The contribution of microbial biotechnology to mitigating coral reef degradation. Microbial Biotechnology, 10, 1236–1243. https://doi.org/10.1111/1751-7915.12769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dealtry, S. , Holmsgaard, P. N. , Dunon, V. , Jechalke, S. , Ding, G.‐C. , Krögerrecklenfort, E. , … Zühlke, S. (2014). Shifts in abundance and diversity of mobile genetic elements after the introduction of diverse pesticides into an on‐farm biopurification system over the course of a year. Applied and Environmental Microbiology, 80, 4012–4020. https://doi.org/10.1128/AEM.04016-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desantis, T. Z. , Hugenholtz, P. , Larsen, N. , Rojas, M. , Brodie, E. L. , Keller, K. , … Andersen, G. L. (2006). Greengenes, a chimera‐checked 16S rRNA gene database and workbench compatible with ARB. Applied and Environmental Microbiology, 72, 5069–5072. https://doi.org/10.1128/AEM.03006-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dini‐Andreote, F. , De Cássia Pereira e Silva, M. , Triadó‐Margarit, X. , Casamayor, E. O. , Van Elsas, J. D. , & Salles, J. F. (2014). Dynamics of bacterial community succession in a salt marsh chronosequence: Evidences for temporal niche partitioning. The ISME Journal, 8, 1989–2001. https://doi.org/10.1038/ismej.2014.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics, 26, 2460–2461. https://doi.org/10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- Egan, S. , & Gardiner, M. (2016). Microbial dysbiosis: Rethinking disease in marine ecosystems. Frontiers in Microbiology, 7, 991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Elsas, J. D. , Turner, S. , & Bailey, M. J. (2003). Horizontal gene transfer in the phytosphere. New Phytologist, 157, 525–537. https://doi.org/10.1046/j.1469-8137.2003.00697.x [DOI] [PubMed] [Google Scholar]
- Faust, K. , & Raes, J. (2012). Microbial interactions: From networks to models. Nature Reviews. Microbiology, 10, 538 https://doi.org/10.1038/nrmicro2832 [DOI] [PubMed] [Google Scholar]
- Fernando, S. C. , Wang, J. , Sparling, K. , Garcia, G. D. , Francini‐Filho, R. B. , de Moura, R. L. , … Thompson, J. R. (2015). Microbiota of the major South Atlantic reef building coral Mussismilia. Microbial Ecology, 69, 267–280. https://doi.org/10.1007/s00248-014-0474-6 [DOI] [PubMed] [Google Scholar]
- Garcia, G. D. , Gregoracci, G. B. , Santos, E. D. O. , Meirelles, P. M. , Silva, G. G. , Edwards, R. , … Iida, T. (2013). Metagenomic analysis of healthy and white plague‐affected Mussismilia braziliensis corals. Microbial Ecology, 65, 1076–1086. https://doi.org/10.1007/s00248-012-0161-4 [DOI] [PubMed] [Google Scholar]
- Gillings, M. R. , Gaze, W. H. , Pruden, A. , Smalla, K. , Tiedje, J. M. , & Zhu, Y.‐G. (2015). Using the class 1 integron‐integrase gene as a proxy for anthropogenic pollution. The ISME Journal, 9, 1269–1279. https://doi.org/10.1038/ismej.2014.226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, J. , Williams, D. , Paterson, S. , Harrison, E. , & Brockhurst, M. (2017). Positive selection inhibits gene mobilisation and transfer in soil bacterial communities. Nature Ecology and Evolution, 1, 1348–1353. https://doi.org/10.1038/s41559-017-0250-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez‐Agreda, A. , Leggat, W. , Bongaerts, P. , & Ainsworth, T. D. (2016). The microbial signature provides insight into the mechanistic basis of coral success across reef habitats. MBio, 7, e00560‐16 https://doi.org/10.1128/mBio.00560-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuer, H. , & Smalla, K. (2007). Horizontal gene transfer between bacteria. Environmental Biosafety Research, 6, 3–13. https://doi.org/10.1051/ebr:2007034 [DOI] [PubMed] [Google Scholar]
- Heuer, H. , & Smalla, K. (2012). Plasmids foster diversification and adaptation of bacterial populations in soil. FEMS Microbiology Reviews, 36, 1083–1104. https://doi.org/10.1111/j.1574-6976.2012.00337.x [DOI] [PubMed] [Google Scholar]
- Izmalkova, T. Y. , Mavrodi, D. V. , Sokolov, S. L. , Kosheleva, I. A. , Smalla, K. , Thomas, C. M. , & Boronin, A. M. (2006). Molecular classification of IncP‐9 naphthalene degradation plasmids. Plasmid, 56, 1–10. https://doi.org/10.1016/j.plasmid.2005.12.004 [DOI] [PubMed] [Google Scholar]
- Jolliffe, I. T. (1986). Principal component analysis. New York, NY: Springer; https://doi.org/10.1007/978-1-4757-1904-8 [Google Scholar]
- Kelly, B. J. , Gross, R. , Bittinger, K. , Sherrill‐Mix, S. , Lewis, J. D. , Collman, R. G. , … Li, H. (2015). Power and sample‐size estimation for microbiome studies using pairwise distances and PERMANOVA. Bioinformatics, 31, 2461–2468. https://doi.org/10.1093/bioinformatics/btv183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly, L. W. , Williams, G. J. , Barott, K. L. , Carlson, C. A. , Dinsdale, E. A. , Edwards, R. A. , … Rohwer, F. (2014). Local genomic adaptation of coral reef‐associated microbiomes to gradients of natural variability and anthropogenic stressors. Proceedings of the National Academy of Sciences of the USA, 111, 10227–10232. https://doi.org/10.1073/pnas.1403319111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimes, N. E. , Johnson, W. R. , Torralba, M. , Nelson, K. E. , Weil, E. , & Morris, P. J. (2013). The Montastraea faveolata microbiome: Ecological and temporal influences on a Caribbean reef‐building coral in decline. Environmental Microbiology, 15, 2082–2094. https://doi.org/10.1111/1462-2920.12130 [DOI] [PubMed] [Google Scholar]
- Kimes, N. E. , van Nostrand, J. D. , Weil, E. , Zhou, J. , & Morris, P. J. (2010). Microbial functional structure of Montastraea faveolata, an important Caribbean reef‐building coral, differs between healthy and yellow‐band diseased colonies. Environmental Microbiology, 12, 541–556. https://doi.org/10.1111/j.1462-2920.2009.02113.x [DOI] [PubMed] [Google Scholar]
- Knowlton, N. , & Rohwer, F. (2003). Multispecies microbial mutualisms on coral reefs: The host as a habitat. The American Naturalist, 162, 51–62. https://doi.org/10.1086/378684 [DOI] [PubMed] [Google Scholar]
- Krediet, C. J. , Ritchie, K. B. , Paul, V. J. , & Teplitski, M. (2013). Coral‐associated micro‐organisms and their roles in promoting coral health and thwarting diseases. Proceedings of the Royal Society B: Biological Sciences, 280, 20122328 https://doi.org/10.1098/rspb.2012.2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S. T. M. , Davy, S. K. , Tang, S.‐L. , Fan, T.‐Y. , & Kench, P. S. (2015). Successive shifts in the microbial community of the surface mucus layer and tissues of the coral Acropora muricata under thermal stress. FEMS Microbiology Ecology, 91, fiv142 https://doi.org/10.1093/femsec/fiv142 [DOI] [PubMed] [Google Scholar]
- Leite, D. C. , Leão, P. , Garrido, A. G. , Lins, U. , Santos, H. F. , Pires, D. O. , … Rosado, A. S. (2017). Broadcast spawning coral Mussismilia hispida can vertically transfer its associated bacterial core. Frontiers in Microbiology, 8, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lema, K. A. , Bourne, D. G. , & Willis, B. L. (2014). Onset and establishment of diazotrophs and other bacterial associates in the early life history stages of the coralAcropora millepora. Molecular Ecology, 23, 4682–4695. https://doi.org/10.1111/mec.12899 [DOI] [PubMed] [Google Scholar]
- Lins‐De‐barros, M. M. , Cardoso, A. M. , Silveira, C. B. , Lima, J. L. , Clementino, M. M. , Martins, O. B. , … Vieira, R. P. (2013). Microbial community compositional shifts in bleached colonies of the Brazilian reef‐building coral Siderastrea stellata . Microbial Ecology, 65, 205–213. https://doi.org/10.1007/s00248-012-0095-x [DOI] [PubMed] [Google Scholar]
- Lins‐De‐barros, M. M. , Vieira, R. P. , Cardoso, A. M. , Monteiro, V. A. , Turque, A. S. , Silveira, C. B. , … Martins, O. B. (2010). Archaea, bacteria, and algal plastids associated with the reef‐building corals Siderastrea stellata and Mussismilia hispida from Buzios, South Atlantic Ocean, Brazil. Microbial Ecology, 59, 523–532. https://doi.org/10.1007/s00248-009-9612-y [DOI] [PubMed] [Google Scholar]
- Lozupone, C. , Hamady, M. , & Knight, R. (2006). UniFrac – An online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics, 7, 371 https://doi.org/10.1186/1471-2105-7-371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulis, L. , & Fester, R. (1991). Symbiosis as a source of evolutionary innovation: Speciation and morphogenesis. Cambridge, MA: MIT Press. [PubMed] [Google Scholar]
- Meistertzheim, A.‐L. , Nugues, M. M. , Quéré, G. , & Galand, P. E. (2017). Pathobiomes differ between two diseases affecting reef building coralline algae. Frontiers in Microbiology, 8, 1686 https://doi.org/10.3389/fmicb.2017.01686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouchka, M. E. , Hewson, I. , & Harvell, C. D. (2010). Coral‐associated bacterial assemblages: Current knowledge and the potential for climate‐driven impacts. Integrative and Comparative Biology, 50, 662–674. https://doi.org/10.1093/icb/icq061 [DOI] [PubMed] [Google Scholar]
- Musovic, S. , Oregaard, G. , Kroer, N. , & Sørensen, S. J. (2006). Cultivation‐independent examination of horizontal transfer and host range of an IncP‐1 plasmid among gram‐positive and gram‐negative bacteria indigenous to the barley rhizosphere. Applied and Environmental Microbiology, 72, 6687–6692. https://doi.org/10.1128/AEM.00013-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla‐Gamiño, J. L. , Pochon, X. , Bird, C. , Concepcion, G. T. , & Gates, R. D. (2012). From parent to gamete: Vertical transmission of symbiodinium (dinophyceae) ITS2 sequence assemblages in the reef building coral Montipora capitata . PLoS ONE, 7, e38440 https://doi.org/10.1371/journal.pone.0038440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peixoto, R. , Chaer, G. M. , Carmo, F. L. , Araújo, F. V. , Paes, J. E. , Volpon, A. , … Rosado, A. S. (2011). Bacterial communities reflect the spatial variation in pollutant levels in Brazilian mangrove sediment. Antonie van Leeuwenhoek, 99, 341–354. https://doi.org/10.1007/s10482-010-9499-0 [DOI] [PubMed] [Google Scholar]
- Peixoto, R. S. , Rosado, P. M. , Leite, D. C. D. A. , Rosado, A. S. , & Bourne, D. G. (2017). Beneficial microorganisms for corals (BMC): Proposed mechanisms for coral health and resilience. Frontiers in Microbiology, 8, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, C. , & Round, J. L. (2014). Defining dysbiosis and its influence on host immunity and disease. Cellular Microbiology, 16, 1024–1033. https://doi.org/10.1111/cmi.12308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popowska, M. , & Krawczyk‐Balska, A. (2013). Broad‐host‐range IncP‐1 plasmids and their resistance potential. Frontiers in Microbiology, 4, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachid, C. T. C. C. , Santos, A. L. , Piccolo, M. C. , Balieiro, F. C. , Coutinho, H. L. C. , Peixoto, R. S. , … Rosado, A. S. (2013). Effect of sugarcane burning or green harvest methods on the Brazilian Cerrado soil bacterial community structure. PLoS ONE, 8, e59342 https://doi.org/10.1371/journal.pone.0059342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis, A. , Araújo JR, S. , Moura, R. , Francini‐Filho, R. , Pappas Jr, G. , Coelho, A. , … Thompson, F. (2009). Bacterial diversity associated with the Brazilian endemic reef coral Mussismilia braziliensis . Journal of Applied Microbiology, 106, 1378–1387. https://doi.org/10.1111/j.1365-2672.2008.04106.x [DOI] [PubMed] [Google Scholar]
- Ritchie, K. B. (2006). Regulation of microbial populations by coral surface mucus and mucus‐associated bacteria. Marine Ecology Progress Series, 322, 1–14. https://doi.org/10.3354/meps322001 [Google Scholar]
- Rojo, F. (2010). Carbon catabolite repression in Pseudomonas: Optimizing metabolic versatility and interactions with the environment. FEMS Microbiology Reviews, 34, 658–684. https://doi.org/10.1111/j.1574-6976.2010.00218.x [DOI] [PubMed] [Google Scholar]
- Rosenberg, E. , Kellogg, C. A. , & Rohwer, F. (2007). Coral microbiology. Oceanography, 20, 147–154. [Google Scholar]
- Santos, H. F. , Carmo, F. L. , Duarte, G. , Dini‐Andreote, F. , Castro, C. B. , Rosado, A. S. , … Peixoto, R. S. (2014). Climate change affects key nitrogen‐fixing bacterial populations on coral reefs. The ISME Journal, 8, 2272–2279. https://doi.org/10.1038/ismej.2014.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos, H. F. , Cury, J. C. , Carmo, F. L. , Rosado, A. S. , & Peixoto, R. S. (2010). 18S rDNA sequences from microeukaryotes reveal oil indicators in mangrove sediment. PLoS ONE, 5, e12437 https://doi.org/10.1371/journal.pone.0012437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos, H. F. , Duarte, G. A. S. , Rachid, C. T. C. C. , Chaloub, R. M. , Calderon, E. N. , Marangoni, L. F. B. , … Peixoto, R. S. (2015). Impact of oil spills on coral reefs can be reduced by bioremediation using probiotic microbiota. Scientific Reports, 5, 18268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shade, A. , & Handelsman, J. (2012). Beyond the Venn diagram: The hunt for a core microbiome. Environmental Microbiology, 14, 4–12. https://doi.org/10.1111/j.1462-2920.2011.02585.x [DOI] [PubMed] [Google Scholar]
- Shannon, P. , Markiel, A. , Ozier, O. , Baliga, N. S. , Wang, J. T. , Ramage, D. , … Ideker, T. (2003). Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Research, 13, 2498–2504. https://doi.org/10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp, K. H. , Distel, D. , & Paul, V. J. (2011). Diversity and dynamics of bacterial communities in early life stages of the Caribbean coral Porites astreoides . The ISME Journal, 6, 790–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp, K. H. , & Ritchie, K. B. (2012). Multi‐partner interactions in corals in the face of climate change. The Biological Bulletin, 223, 66–77. https://doi.org/10.1086/BBLv223n1p66 [DOI] [PubMed] [Google Scholar]
- Sharp, K. H. , Ritchie, K. B. , Schupp, P. J. , Ritson‐Williams, R. , & Paul, V. J. (2010). Bacterial acquisition in juveniles of several broadcast spawning coral species. PLoS ONE, 5, e10898 https://doi.org/10.1371/journal.pone.0010898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani, M. , Takahashi, Y. , Tokumaru, H. , Kadota, K. , Hara, H. , Miyakoshi, M. , … Nojiri, H. (2010). Response of the Pseudomonas host chromosomal transcriptome to carriage of the IncP‐7 plasmid pCAR1. Environmental Microbiology, 12, 1413–1426. [DOI] [PubMed] [Google Scholar]
- Shnit‐Orland, M. , & Kushmaro, A. (2009). Coral mucus‐associated bacteria: A possible first line of defense. FEMS Microbiology Ecology, 67, 371–380. https://doi.org/10.1111/j.1574-6941.2008.00644.x [DOI] [PubMed] [Google Scholar]
- Sunagawa, S. , Woodley, C. M. , & Medina, M. (2010). Threatened corals provide underexplored microbial habitats. PLoS ONE, 5, e9554 https://doi.org/10.1371/journal.pone.0009554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet, M. J. , & Bulling, M. T. (2017). On the importance of the microbiome and pathobiome in coral health and disease. Frontiers in Marine Science, 4, 9. [Google Scholar]
- Thompson, J. R. , Rivera, H. E. , Closek, C. J. , & Medina, M. (2014). Microbes in the coral holobiont: Partners through evolution, development, and ecological interactions. Frontiers in Cellular and Infection Microbiology, 4, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh, P. J. , Ley, R. E. , Hamady, M. , Fraser‐Liggett, C. , Knight, R. , & Gordon, J. I. (2007). The human microbiome project: Exploring the microbial part of ourselves in a changing world. Nature, 449, 804 https://doi.org/10.1038/nature06244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega Thurber, R. , Willner‐Hall, D. , Rodriguez‐Mueller, B. , Desnues, C. , Edwards, R. A. , Angly, F. , … Rohwer, F. (2009). Metagenomic analysis of stressed coral holobionts. Environmental Microbiology, 11, 2148–2163. https://doi.org/10.1111/j.1462-2920.2009.01935.x [DOI] [PubMed] [Google Scholar]
- Wang, Q. , Garrity, G. M. , Tiedje, J. M. , & Cole, J. R. (2007). Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology, 73, 5261–5267. https://doi.org/10.1128/AEM.00062-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster, N. S. , & Reusch, T. B. (2017). Microbial contributions to the persistence of coral reefs. The ISME Journal, 11, 2167–2174. https://doi.org/10.1038/ismej.2017.66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, R. M. , Kenkel, C. D. , Dunn, C. E. , Shilling, E. N. , Bay, L. K. , & Matz, M. V. (2017). Intraspecific differences in molecular stress responses and coral pathobiome contribute to mortality under bacterial challenge in Acropora millepora . Scientific Reports, 7, 2609 https://doi.org/10.1038/s41598-017-02685-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, M. , Pereira e Silva, M. D. C. , Chaib De Mares, M. , & Van Elsas, J. D. (2014). The mycosphere constitutes an arena for horizontal gene transfer with strong evolutionary implications for bacterial‐fungal interactions. FEMS Microbiology Ecology, 89, 516–526. https://doi.org/10.1111/1574-6941.12350 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data from each sample are available at the NCBI Sequence Read Archive (SRA) under Accession Numbers SRR5903387—SRR5903406.