

Abstract
Mitochondrial quality control systems are essential for the maintenance of functional mitochondria. At the organelle level, they include mitochondrial biogenesis, fusion and fission, to compensate for mitochondrial function, and mitophagy, for degrading damaged mitochondria. Specifically, in mitophagy, the target mitochondria are recognized by the autophagosomes and delivered to the lysosome for degradation. In this review, we describe the mechanisms of mitophagy and the factors that play an important role in this process. In particular, we focus on the roles of mitophagy adapters and receptors in the recognition of damaged mitochondria by autophagosomes. In addition, we also address a functional association of mitophagy with mitochondrial dynamics through the interaction of mitophagy adaptor and receptor proteins with mitochondrial fusion and fission proteins.
Keywords: autophagy, mitochondria, mitochondrial quality control, mitophagy, selective autophagy
INTRODUCTION
Mitochondria are the main site of adenosine triphosphate (ATP) synthesis. Mitochondria are also crucial in apoptosis, necrosis, autophagy, stress regulation, production of lipids and carbohydrates, Ca2+ storage and innate immunity (Galluzzi et al., 2012). Therefore, mitochondrial dysfunction is associated with many diseases (Suomalainen and Battersby, 2017). Mitochondria undergo constant turnover and their half-life varies from tissue to tissue (Gottlieb and Stotland, 2015). Mitochondria are exposed to damages caused by intra- and extra-mitochondrial events, such as mutated mitochondrial proteins and reactive oxygen species (ROS) (Galluzzi et al., 2012). Mitochondrial quality control systems actively function, both in physiological and pathological conditions, to protect the mitochondria from stress and damage at the protein and organelle level. Damage beyond the protein level activates a bigger response that includes mitochondrial biogenesis, fusion, fission and mitochondrial degradation, also known as mitophagy. In this review, we will focus on recent data regarding the molecular mechanisms of mitochondrial quality control, especially in relation to mitophagy, and describe how these mechanisms can affect each other.
MITOCHONDRIAL QUALITY CONTROL
Mitochondria first respond to stress by attempting to maintain their structure and composition. This is achieved through the action of antioxidants, DNA repair, protein folding and degradation. Mitochondrial DNA is highly exposed to oxidative damage due to the production of ROS during oxidative phosphorylation. DNA damage is repaired by mitochondrial DNA polymerase gamma (encoded by POLG) that has base excision repair and exonuclease function. Mutation of POLG causes neurodegeneration, progressing into Leigh or Alpers-Huttenlocher syndrome (Scheibye-Knudsen et al., 2015). Antioxidants, such as superoxide dismutase, catalase, glutathione peroxidase/glutathione, peroxiredoxins and vitamin E/C, directly protect mitochondria by reducing reactive intermediates (Scheibye-Knudsen et al., 2015). In addition, mitochondrial chaperones, such as heat shock protein (Hsp)60 and Hsp70, import nuclear-encoded mitochondrial proteins (NEMP) into the mitochondrial matrix and refold misfolded proteins (Hammerling and Gustafsson, 2014). When misfolded or damaged proteins accumulate, the proteases LON and ClpP, in the matrix, and ATPase associated with diverse cellular activities (AAA), in the membrane, degrade them to maintain normal mitochondria(Cenini and Voos, 2016). Proteins in the outer mitochondrial membrane are ubiquitinated by the E3 ligase MARCH5, and targeted for degradation to the 26S proteasome (Bragoszewski et al., 2017). If the damage is beyond the capacity of these systems, a broader quality control system is activated.
At the organelle level, mitochondria quality is sustained through synthesis of new mitochondria, fusion and fission, and elimination of the damaged ones. Mitochondrial biogenesis requires the delivery of NEMP to the mitochondria and is induced by endogenous (e.g. ROS, nitrogen oxide, carbon monoxide, and H2S), and external signals (e.g. estrogen), which increase the amount of NEMP through nuclear respiration factor-1 (NRF1), peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) and estrogen receptor. Calorie restriction and exercise also stimulate mitochondrial biogenesis through changes in the ratio of oxidized/reduced nicotinamide adenine dinucleotide (NAD+/NADH) and adenosine mono-phosphate (AMP)/ATP, and increase NEMP levels via sirtuins (SIRT) and AMP-activated protein kinase (AMPK) pathways (Suliman and Piantadosi, 2016).
Compared to other organelles, mitochondria frequently change their shape under physiologic and pathologic conditions. Specifically, mitochondrial dynamics (fusion and fission) regulates mitochondrial network connectivity, which depends on the specific metabolic needs of the cell (Miettinen and Björklund, 2017; Rambold and Pearce, 2017). The processes controlling mitochondria dynamics are well understood and mediated by specific proteins. Mitochondrial outer membrane fusion is induced by MitoPLD, a member of the phospholipase D family, which converts cardiolipin into phosphatidic acid, and by homo or hetero dimerization of the guanosine triphosphate hydrolases (GTPases) mitofusin (MFN) 1 and 2. In addition to outer membrane, fusion of the mitochondrial inner membrane requires optic atrophy 1 (OPA1), anchored in the inner membrane and exposed to the intermembrane space. On the other hand, mitochondrial fission occurs when the GTPase dynamin related protein-1 (Drp1) translocates to the mitochondrial outer membrane to bind its receptors, such as FIS1, MFF, MID49 and MID51, and forms a multimeric structure around the fission site of the mitochondrion. While the precise reason is not yet clarified, mitochondrial protein 18 kDa (MTP18) gene ablation induces fusion of mitochondria and expression of cleaved OPA1 (S-OPA1) induces fission (Wai and Langer, 2016). Through mechanisms not fully understood, mitochondrial dynamics is also engaged in the repair of a part of the damaged mitochondria. If the mitochondria cannot be restored to a healthy state through these processes, they are eliminated by selective autophagy.
MITOPHAGY: ELIMINATION OF MITOCHONDRIA THROUGH SELECTIVE AUTOPHAGY
Autophagy, a lysosome-dependent degradation, is categorized into macroautophagy, microautophagy and chaperone-mediated-autophagy (CMA). Macroautophagy is characterized by the formation of an autophagosome: the phosphatidylinositol 3-kinase (PI3K) class III produces phosphatidylinositol 3-phosphate (PI3P) and induces the formation of the isolation membrane (phagophore), which progressively expands by using two-ubiquitin-like systems till the formation of a double-membrane vesicle (the autophagosome). Cytoplasmic cargos are sequestered into the autophagosome and subsequently degraded through the lysosomes. In microautophagy, cytosolic cargo is directly engulfed by lysosomes while CMA selectively recruits target proteins into the lysosomes through chaperone proteins without autophagosome formation (Nah et al., 2015).
Unlike non-selective removals of various targets by macroautophagy, targets ranging from proteins to organelles, including aggregated proteins, mitochondria, endoplasmic reticulum, peroxisomes, invaded bacteria, viruses and membranes, are often degraded by selective autophagy (Galluzzi et al., 2017). Among them, mitophagy is the selective removal of mitochondria via autophagy. Then, an important question is how mitochondria are selectively recognized and removed. A report shows that mitochondria-derived vesicles are directly degraded to lysosomes (Lemasters, 2014). Most of the studies, however, have focused on autophagosome-mediated mitophagy and on the importance of mitophagy mediators that interact with LC3. Recently, many studies have investigated the mechanisms of this process. Details on mitophagy are discussed below.
MECHANISMS OF MITOPHAGY: HOW ARE THE MITOCHONDRIA RECOGNIZED?
The recognition of target mitochondria by the autophagosome occurs through LC3 adapters, in an ubiquitin-dependent and independent pathway, and through the direct interaction of LC3 with its receptors.
Ubiquitin-mediated interaction of LC3 adapters
In this pathway, the first event is the ubiquitination of mitochondrial substrates, which are then recognized by LC3 adapters.
PINK1/Parkin pathway
PTEN-induced putative kinase 1 (PINK1, PARK6) and Parkin (PARK2) were first identified as genetic factors of Parkinson’s disease in which mitochondrial dysfunction has been suggested as one of the causes (Pickrell and Youle, 2015). Parkin, a cytosolic E3-ubiquitin ligase, and PINK, a mitochondrial serine/threonine-protein kinase, are now known to function in mitophagy (Matsuda et al., 2010; Narendra et al., 2008). Under normal conditions, PINK1 is continuously targeted to the mitochondria through a mitochondrial targeting sequence, degraded by matrix processing peptidases (MPP) and subsequently cleaved by presenilin-associated rhomboid like (PARL), a protease in the mitochondrial inner membrane. Cleaved PINK translocates to the cytosol and is degraded by the proteasome (Matsuda et al., 2010).
Depolarization of the mitochondrial membrane due to damage, reduces the cleavage of PINK1. PINK1 accumulates on the mitochondrial outer membrane through the translocase of the outer membrane (TOM). Mitochondrial pyruvate level can also affect the accumulation of PINK1 by inducing the interaction between PINK1 and TOM (Park et al., 2015). Accumulated PINK1 is autophosphorylated and activated, and phosphorylates ubiquitin on serine 65 (Ser65), which recruits Parkin. On the mitochondrial membrane, Parkin is phosphorylated and activated by PINK1, and polyubiquitinates its substrates (Nguyen et al., 2016). Specifically, the polyubiquitination of Parkin substrates, such as voltage-dependent anion channel-1 (VDAC1), MFN 1/2 and Miro 1, leads to their degradation by the proteasome (Chan et al., 2011; Tanaka et al., 2010). The degradation of MFN 1/2 induces mitochondrial fission and mitophagy (Tanaka et al., 2010), linking mitochondrial dynamics to mitophagy.
LC3 adapters: p62, OPTN, NDP52, TAX1BP1 and NBR1
Five LC3 adapters, sequestosome-1 (p62), optineurin (OPTN), nuclear domain 10 protein 52 (NDP52), TAX1 binding protein 1 (TAX1BP1) and neighbor of BRCA1 gene 1 (NBR1), are recruited to the K63-linked polyubiquitinated substrates on the mitochondria through their ubiquitin-binding domain. These adapters contain an LC3-interacting region (LIR) motif that is recognized by LC3 to recruit the tagged mitochondria to the autophagosome (Lazarou et al., 2015). Studies have shown that OPTN is the most important adapter for the recruitment of the phagophore to the mitochondria (Moore and Holzbaur, 2016). Specifically, Tank-binding kinase 1 (TBK1) is activated after recruitment by OPTN and promotes mitophagy through the phosphorylation of OPTN (Richter et al., 2016). While other adapters were also reported to be phosphorylated by TBK1, only OPTN and NDP52 were its common substrates (Heo et al., 2015; Matsumoto et al., 2015; Moore and Holzbaur, 2016; Richter et al., 2016).
Recently, a Parkin-independent role of PINK1 in mitophagy has been uncovered. In this pathway, PINK1 recruits OPTN and NDP52 on the mitochondria, which subsequently recruit autophagy initiation factors, such as ULK1, double FYVE-containing protein 1 (DFCP1) and WD repeat domain phosphoinositide-interacting protein 1 (WIPI1) (Lazarou et al., 2015). In addition, overexpressed synphilin-1, a synuclein alpha-interacting protein, interacts with PINK1 and induces its accumulation. The PINK1-synphilin1 complex recruits seven in absentia homolog-1 (SIAH-1), an E3 ubiquitin ligase, to increase mitochondrial ubiquitination (Szargel et al., 2016), indicating that E3 ligases other than Parkin target the mitochondria for degradation in different ways.
Ubiquitin-independent interaction of LC3 adapters
Damaged mitochondria can also be recognized by LC3 adapters in an ubiquitin-independent manner.
CHDH and TBC1D15
Choline dehydrogenase (CHDH) is located in the mitochondrial inner and outer membranes under normal conditions. When the mitochondrial membrane potential is disrupted, CHDH accumulates in the mitochondrial outer membrane and interacts with p62 through its Phox and Bem1 (PB1) domain, leading to the formation of the CHDH-p62-LC3 complex that mediates mitophagy (Park et al., 2014). CHDH’s role in mitophagy, shown in gene ablation experiments, is Parkin-independent. In addition, TBC1 domain family member 15 (TBC1D15), a mitochondrial Rab GTPase-activating protein (Rab-GAP), forms a complex with TBC1D17 and migrates to the mitochondrial outer membrane by interacting with mitochondrial fission 1 protein (FIS1). The TBC1D15/17 complex then interacts with LC3 (Yamano et al., 2014). These adapters sense mitochondrial damage and consequently change their subcellular location or the protein they interact with, guiding the damaged mitochondria to the autophagosome.
Interaction between LC3 and its receptors
Unlike aforementioned LC3 adapters which require additional proteins to interact with LC3, several LC3 receptors are located on the mitochondria; they directly bind to LC3 and recruit the damaged mitochondria to autophagosomes.
NIX/BNIP3L, BNIP3 and FUNDC1
Nip3-like protein X (NIX) and BCL2/Adenovirus E1B 19 kDa Interacting Protein 3 (BNIP3) contain a BH3 domain and interact with LC3 (Hanna et al., 2012; Novak et al., 2010). NIX was initially reported to function in the removal of mitochondria from red blood cells during maturation (Sandoval et al., 2008). Additionally, NIX participates in mitophagy under hypoxic conditions. NIX mRNA levels are increased by HIF1α (Sowter et al., 2001) and NIX protein is phosphory-lated at Ser81 to mediate mitophagy under hypoxia conditions (Yuan et al., 2017). NIX-mediated mitophagy is Parkin-independent and, in mice, plays a protective role in ischemic brain injury (Yuan et al., 2017). NIX also participates in the Parkin-dependent mitophagy as a substrate of Parkin that recruits NBR1 to the mitochondria (Gao et al., 2015) and knockdown of both Parkin and NIX synergistically reduces mitophagy (Yuan et al., 2017). Thus, NIX might primarily regulate basal levels of mitophagy under physiological conditions and induce mitophagy under hypoxic conditions42.
Homodimerization of BNIP3 is necessary for its interaction with LC3 and this interaction is regulated by the phosphorylation on Ser17 and Ser24 near the LIR motif (Hanna et al., 2012; Zhu et al., 2013). BNIP3 increases mitophagy by suppressing the cleavage of PINK1 (Zhang et al., 2016). Like NIX, BNIP3 levels are increased to mediate excessive mitophagy in the cortical neurons of a mouse ischemia/hypoxia model (Shi et al., 2014). As found in BNIP3−/− mice, BNIP3 deficiency significantly decreases both neuronal mitophagy and apoptosis under hypoxic conditions (Shi et al., 2014). In addition, BNIP3 deficiency also increases non-selective autophagy following ischemic/hypoxic insults (Shi et al., 2014). While NIX expression is upregulated upon BNIP3 silencing, this upregulation does not functionally compensate for the loss of BNIP3 in activating excessive mitophagy (Shi et al., 2014).
FUNDC1 is a mitochondrial outer membrane protein that mediates hypoxia-induced mitophagy in mammalian cells (Liu et al., 2012). FUNDC1 regulates Parkin-independent mitophagy by binding to LC3 under hypoxia. Specifically, the phosphorylation of FUNDC1 at Tyr18 by SRC and Ser13 by casein kinase II (CK2) suppresses its interaction with LC3 under normoxic conditions (Chen et al., 2014; Liu et al., 2012). Under hypoxia, FUNDC1 is dephosphorylated at Ser13 by phosphoglycerate mutase 5 (PGAM5) and phosphorylated at Ser17 by ULK1 (Chen et al., 2014). In addition, FUNDC1 is Lys119-ubiquitinated by MARCH5 and degraded in the initial phase of hypoxia; the remaining FUNDC1 participates in mitophagy under prolonged hypoxia (Chen et al., 2017). Inhibition of FUNDC1-mediated mitophagy by Receptor-interacting serine/threonine-protein kinase 3 (Ripk3), which participates in necroptosis- and mitochondria-mediated apoptosis, increases mitochondrial apoptosis in cardiac ischemia/reperfusion injury (Zhou et al., 2017). On the contrary, a report showed that knockdown or overexpression of FUNDC1 has insignificant effect on starvation- or hypoxia-induced mitophagy (Hirota et al., 2015), which remains to be clarified.
BCL2L13 and FKBP8
Bcl2 like 13 (BCL2L13) is the mammalian homologue of atg32, the only mitophagy receptor found in yeast (Mao et al., 2011); its expression somewhat compensates the reduced mitophagy in atg32-null yeast. In mammalian cells, BCL2L13 mediates mitophagy independently of Parkin. Like other LC3 receptors, BCL2L13 locates on the mitochondrial outer membrane and binds to LC3 via the LIR motif. Specifically, phosphorylation at Ser272 increases the binding of BCL2L13 to LC3 (Murakawa et al., 2015).
FK506-binding protein 8 (FKBP8), was identified as a LC3-interacting protein using yeast two-hybrid screening (Bhujabal et al., 2017). FKBP8 is located on the mitochondrial outer membrane and mediates mitophagy by interacting with LC3A (Bhujabal et al., 2017); the effect of FKBP8 on mitophagy is Parkin-independent. In particular, FKBP8 migrates from the mitochondria into the ER following the treatment with carbonyl cyanide m-chlorophenylhydrazone (CCCP), a chemical ionophore (Saita et al., 2013). Accordingly, the FKBP8 N412K mutant, which cannot be targeted to ER, plays a role in mitophagy only, suggesting that the subcellular localization of FKBP8 regulates mitophagy (Bhujabal et al., 2017).
PHB2 and cardiolipin
Unlike other LC3 receptors, prohibitin 2 (PHB2) is a mitochondrial inner membrane protein (Wei et al., 2017). Parkin-mediated degradation of mitochondrial outer membrane proteins results in rupture of the mitochondrial outer membrane, leading to exposure of PHB2 to LC3, and subsequent mitophagy. The function of PHB2 is crucial for the selective elimination of paternal mitochondria in C. elegans (Wei et al., 2017). However, the detailed mechanism responsible for this process remains to be elucidated.
Cardiolipin, a membrane lipid in the mitochondrial inner membrane, can also function as a LC3 receptor in mitophagy. Cardiolipin is translocated from the inner membrane to the outer membrane in the presence of external toxic signals, such as 6-hydroxydopamine (6-OHDA), rotenone and 1-methyl-4-phenylpyridinium (MPP+). Externalized cardiolipin directly interacts with the N-terminal helices of LC3 (Chu et al., 2013). This interaction seems to be LC3 subtype-specific. In vitro experiments revealed that cardiolipin directly interacts with gamma-aminobutyric acid receptor-associated protein (GABARAP), a member of the LC3 family, but does not translocate GABARAP to the mitochondria in rotenone-treated cells, implying that cardiolipin interacts with GABARAP in different autophagic processes (Anton et al., 2016). These data indicate that mitochondrial inner membrane components also participate in mitophagy.
FUNCTIONAL ASSOCIATION OF MITOCHONDRIAL DYNAMICS AND MITOPHAGY
Changes in mitochondrial shape and size are associated to the fission and fusion of the mitochondria according to the cell’s needs (Trotta and Chipuk, 2017). Since the size of the autophagosome in mammalian cells is approximately 500–1500 nm (Wang and Klionsky, 2003), it is reasonable to predict that a huge mitochondria network should be fragmented through fission prior to mitophagy. In that regard, it has been reported that either mitochondrial fission-related factors or degradation of mitochondrial fusion-related factors are required for mitophagy (Shirihai et al., 2015). Proteins with a role in mitochondrial dynamics are thus implicated in mitophagy to function together with the aforementioned LC3 adapters/receptors. That is, Drp1, a mitochondrial fission protein, interacts with over-expressed FUNDC1 and BCL2L13, and both FUNDC1 and BNIP3 require Drp1 for mitochondrial fragmentation and Parkin-mediated mitophagy (Lee et al., 2011; Wu et al., 2016). In contrast, a recent report showed that mitophagy occurs without mitochondrial fission (Yamashita and Kanki, 2017). Thus, the role of Drp1 in mitophagy needs to be further addressed.
In addition, OPA1, a mitochondrial fusion protein, may also play a role as a mitophagic factor. BNIP3 induces mitochondrial fragmentation through its interaction with OPA1 (Landes et al., 2010). Ablation of PHB2 leads to loss of OPA1 long isoform, resulting in an abnormal cristae morphogenesis and mitochondrial fragmentation (Merkwirth et al., 2008). A recent report has shown that the fusion of the inner membrane of the mitochondria requires the binding of OPA1 with cardiolipin and high concentration of cardiolipin in the inner membrane (Ban et al., 2017). Additionally, TBC1D15 mediates the autophagic encapsulation of mitochondria downstream of Parkin activation, upon interaction with FIS1 (Yamano et al., 2014). Thus, it is likely that mitophagy is functionally associated with mitochondrial dynamics, through the interaction between mitochondrial dynamics factors and LC3 adapter/receptors. The details of the interplay between mitochondrial dynamics and mitophagy remain, however, to be further assessed.
PHYSIOLOGIC ROLE OF MITOPHAGY
To understand the molecular mechanism of mitophagy, CCCP has widely been used to disrupt the membrane potential of the mitochondria and induce mitophagy, in vitro. Despite its great contribution to the elucidation of mitophagy, the conditions it creates are different from physiologic conditions. Mitophagy constantly occurs in basal conditions (Bingol et al., 2014) and is induced under specific physiological conditions. During development, NIX is responsible for the removal of mitochondria from maturing erythrocytes (Sandoval et al., 2008), and Parkin and mitochondrial E3 ubiquitin protein ligase 1 (MUL1) are required for degradation of paternal mitochondria after fertilization in mice (Rojansky et al., 2016). Mitophagy is also involved in cellular differentiation. PINK1-dependent mitophagy affects pluripotency and differentiation state in stem cells (Vazquez-Martin et al., 2016). Mitophagy appears to contribute to metabolic changes in the differentiation of retinal ganglion cells (Esteban-Martinez et al., 2017) and the transition from beige to white adipocytes (Wrighton, 2016). In addition, mitophagy plays a role in the activation of the NOD-like receptor protein 3 (NLRP3) inflammasome (Lazarou, 2015) and FUNDC1-dependent mitophagy decreases the activation of platelet following acute ischemia/reperfusion injury (Zhang et al., 2016). Together, these reports show diverse roles of mitophagy in development and cell differentiation.
MITOPHAGY AND HUMAN DISEASES
A dysfunction in the mitochondria quality control and/or mitophagy is associated with many pathological conditions: one of the most studied examples are some neurodegenerative diseases (Rodolfo et al., 2017). Parkin and PINK1 are genetic factors in Parkinson’s disease and play a protective role in neurons through mitophagy (Ashrafi et al., 2014). In Alzheimer disease progression, Parkin is involved in the removal of mitochondria (Ye et al., 2015) and its overexpression alleviates the symptoms of the disease (Martin-Maestro et al., 2016). In Huntington’s disease, mitophagy is altered. Mutant huntingtin acts as a scaffold inducing selective autophagy (Martinez-Vicente et al., 2010) or negatively affecting mitochondrial delivery to the lysosome (Wong and Holzbaur, 2014). The details of this process are unclear. Mitochondrial dysfunction is also observed in amyotrophic lateral sclerosis (ALS), though the role of autophagy in this disease is uncertain (Rodolfo et al., 2017). Mutant superoxide dismutase-1 (SOD1), involved in ALS, induces mitochondrial fragmentation and inhibits mitochondrial retrograde axonal transport (Magrane et al., 2014). In addition, mutant SOD1 binds to p62, OPTN and LC3 (Rodolfo et al., 2017). Thus, reduced targeting of ubiquitinated mitochondria to autophagosomes might contribute to ALS pathology.
Mitophagy is also associated with cancer (Drake et al., 2017). Metabolic-reprogramming in cancer cells is associated with decreased mitophagy and increased mitochondrial biogenesis (Vazquez et al., 2013) and inhibition of mitophagy reduces cell stemness in cell self-renewal (Drake et al., 2017). In relation to inflammation-induced cell death, p62-mediated elimination of damaged mitochondria attenuates inflammation by reducing NLRP3 inflammasome activation in macrophages (Zhong et al., 2016). When DNA is damaged, mitophagy is induced through the poly (ADP-ribose) polymerase-1 (PARP1)-SIRT1-BNIP3/NIX pathway to limit the generation of ROS or inflammasomes, which are harmful to the cell (Drake et al., 2017). In cardiovascular diseases, mitophagy factors are either protective or damaging. It was shown that Parkin and PGAM5 prevent cardiac damage, while FUNDC1, MFN1 and MFN2 in the absence of BNIP3 have an opposite effect. Additionally, Parkin and PINK1 have a protective role in atherosclerosis (Bravo-San Pedro et al., 2017). Likewise, the role of mitophagy in lung disease is not clear. The absence of PINK1 has a protective effect on mitophagy-dependent necroptosis in chronic obstructive pulmonary disease (COPD) (Mizumura et al., 2014), but also induces idiopathic pulmonary fibrosis (IPF). Mitophagy reduces mitochondrial ROS, which modulate senescence in human bronchial epithelial cells. Thus, the role of mitophagy in lung disease depends on the cell type (Cloonan and Choi, 2016).
In acute kidney injury, mitochondrial dysfunction precedes renal dysfunction. Mitophagy protects renal cells against damage in acute kidney injury and metabolic acidosis, a cause of chronic kidney disease. A high-calorie diet causes renal injury, reduces autophagy and mitophagy, and is associated with abnormal mitochondrial morphology (Parikh et al., 2015). Mitophagy also affects non-alcoholic fatty liver disease (NAFLD); NAFLD is relieved by the thyroid hormone (TH), which stimulates mitophagy and mitochondrial biogenesis. BNIP3, NIX, ULK1, p62 and LC3 mRNA levels are increased by TH (Sinha and Yen, 2016). Additionally, activation of AMPK contributes to NAFLD reduction through mitophagy (Smith et al., 2016). Of course, there are active ongoing researches to show the roles of mitophagy in the pathogenesis in other diseases as well.
CONCLUSION
Failure in the removal of dysfunctional mitochondria induces cellular stress and is linked to human diseases. The function of mitophagy-regulating proteins depends on the tissues and the stimuli they are exposed to. Many LC3 adaptors and receptors were originally reported to function independently of Parkin. However, some of them, such as NIX, participate in Parkin-mediated mitophagy (Gao et al., 2015). NDP52, FUNDC1, BCL2L13, FKBP8 and TBC1D15 are also Parkin substrates under certain conditions (Sarraf et al., 2013). Importantly, mitochondrial dynamics and mitophagy affect each other to maintain the quality of mitochondria. Here again, mitochondrial dynamic factors, such as Miro 1 and 2, Drp1 and FIS1 are also Parkin substrates (Sarraf et al., 2013). Therefore, future studies on mitophagy need also to consider the molecular connection among proteins involved in mitophagy, and among proteins involved in mitophagy and mitochondrial dynamics.
Fig. 1. Mitochondrial quality control.
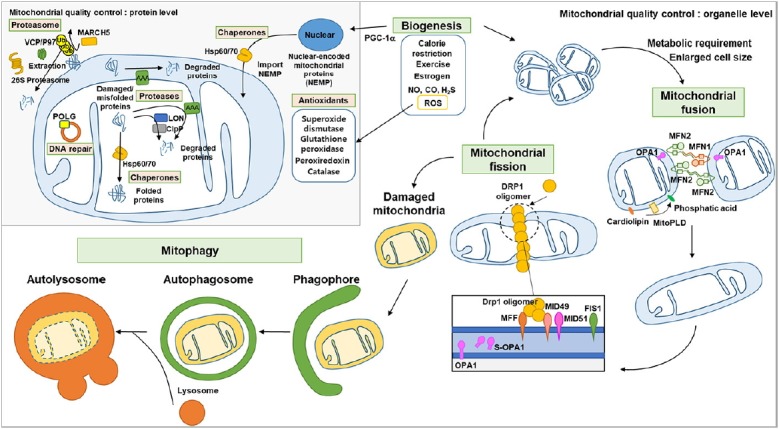
At the protein level, DNA repair proteins (mitochondrial DNA polymerase gamma) repair mitochondrial DNA; Chaperones import nuclear-encoded mitochondrial proteins into the mitochondria and correctly fold misfolded proteins in the matrix; Mitochondrial outer membrane proteins are ubiquitinated by MARCH5 and extracted by P97 to be degraded by the proteasome; Proteases in the mitochondrial matrix and inter membrane space degrade damaged proteins; Antioxidants reduce reactive oxygen species to protect mitochondria. At the organelle level, mitochondria biogenesis occurs upon specific signaling; the mitochondrial network changes mitochondrial shape through fusion and fission to accommodate metabolic requirement; Mitofusin 1/2 and OPA1 mediate fusion in the mitochondrial outer and inner membrane, respectively. MitoPLD converts cardiolipin to phosphatic acid inducing the fusion of mitochondria. When mitochondrial fission occurs, Drp1 binds to its receptors (MFF, MID49, MID51 and FIS1) to form Drp1 oligomers and tightens mitochondria to divide it. Also, increase of short form OPA1 induces mitochondrial fission. Damaged mitochondria are recognized by autophagosome, isolated and then fused with the lysosome to form an autolysosome and finally degraded.
Fig. 2. Mitochondria recognition by LC3 adaptors and receptors during mitophagy.
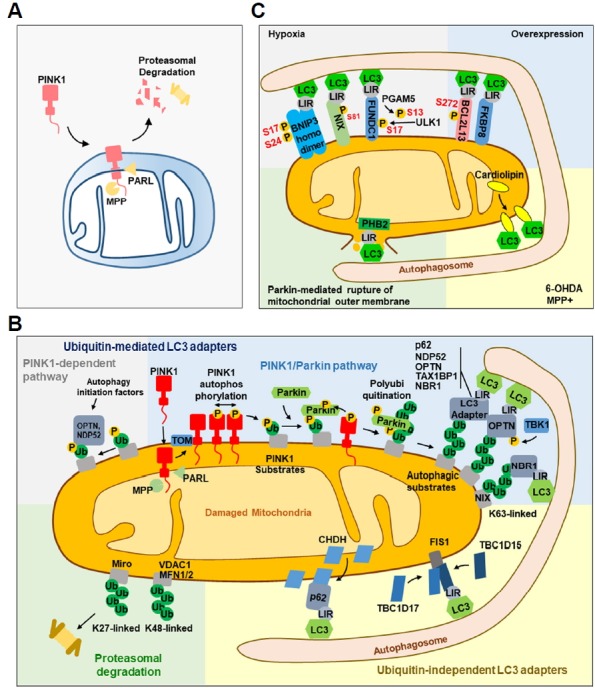
(A) In healthy mitochondria, PINK1 is targeted to the mitochondria and is cleaved by the proteases PARL and MPP in the matrix and mitochondrial inner membrane, respectively. Cleaved PINK1 is degraded in the cytosol through the proteasome. (B) In damaged mitochondria, PINK1 accumulates on the mitochondrial outer membrane and is activated through autophosphorylation. Activated PINK1 phosphorylates ubiquitin on its substrates. Phosphorylated ubiquitin-substrates interact with OPTN or NDP52 to subsequently recruit initiation factors of autophagy or Parkin, an E3 ubiquitin ligase, which is then activated by PINK1 for polyubiquitination. K63-linked polyubiquitinated substrates are recognized by five LC3 adapters (p62, NDP52, OPTN, TAX1BP1 and NBR1) to interact with LC3 on autophagosomes through the LIR motif. TBK1 phosphorylates OPTN to facilitate the recognition by LC3 and NIX is polyubiquitinated to be recognized by LC3 through NBR1. K27-linked Miro and K48-linked VDAC1 and MFN 1 and 2 are degraded by proteasome. The ubiquitin-independent LC3 adapters CHDH and TBC1D15 recognize p62 and FIS1, respectively. (C) Under hypoxic condition, homodimerized BNIP3 is phosphorylated at Ser17 and 24, and NIX is phosphory-lated at Ser81 to facilitate the interaction with LC3. FUNDC1 is dephosphorylated at Ser13 by PGAM5 and phosphorylated at Ser17 by ULK1. Phosphorylation of Ser272 in BCL2L13 enhances the interaction with LC3. PHB2 in the mitochondrial inner membrane interacts with LC3 when PHB2 LIR is exposed to the cytosol following Parkin-mediated rupture of the mitochondrial outer membrane. Cardiolipin translocates to the mitochondrial outer membrane and promotes mitophagy through direct interaction with LC3.
ACKNOWLEDGMENTS
This work was supported by a Bio & Medical Technology Development Program of the National Research Foundation (NRF-2017M3A9G7073521) and a Global Research Laboratory grant (NRF-2010-00341) funded by the Ministry of Science & ICT, Korea.
REFERENCES
- Anton Z, Landajuela A, Hervas JH, Montes LR, Hernandez-Tiedra S, Velasco G, Goni FM, Alonso A. Human Atg8-cardiolipin interactions in mitophagy: Specific properties of LC3B, GABARAPL2 and GABARAP. Autophagy. 2016;12:2386–2403. doi: 10.1080/15548627.2016.1240856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol. 2014;206:655–670. doi: 10.1083/jcb.201401070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban T, Ishihara T, Kohno H, Saita S, Ichimura A, Maenaka K, Oka T, Mihara K, Ishihara N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat Cell Biol. 2017;19:856–863. doi: 10.1038/ncb3560. [DOI] [PubMed] [Google Scholar]
- Bhujabal Z, Birgisdottir AB, Sjottem E, Brenne HB, Overvatn A, Habisov S, Kirkin V, Lamark T, Johansen T. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017;18:947–961. doi: 10.15252/embr.201643147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 2014;510:370–375. doi: 10.1038/nature13418. [DOI] [PubMed] [Google Scholar]
- Bragoszewski P, Turek M, Chacinska A. Control of mitochondrial biogenesis and function by the ubiquitin-proteasome system. Open Biol. 2017:7. doi: 10.1098/rsob.170007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo-San Pedro JM, Kroemer G, Galluzzi L. Autophagy and mitophagy in cardiovascular disease. Circ Res. 2017;120:1812–1824. doi: 10.1161/CIRCRESAHA.117.311082. [DOI] [PubMed] [Google Scholar]
- Cenini G, Voos W. Role of mitochondrial protein quality control in oxidative stress-induced neurodegenerative diseases. Curr Alzheimer Res. 2016;13:164–173. doi: 10.2174/1567205012666150921103213. [DOI] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20:1726–1737. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H, Huang L, Zhou C, Cai X, Fu C, et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell. 2014;54:362–377. doi: 10.1016/j.molcel.2014.02.034. [DOI] [PubMed] [Google Scholar]
- Chen Z, Liu L, Cheng Q, Li Y, Wu H, Zhang W, Wang Y, Sehgal SA, Siraj S, Wang X, et al. Mitochondrial E3 ligase MARCH5 regulates FUNDC1 to fine-tune hypoxic mitophagy. EMBO Rep. 2017;18:495–509. doi: 10.15252/embr.201643309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, Tyurin VA, Yanamala N, Shrivastava IH, Mohammadyani D, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15:1197. doi: 10.1038/ncb2837. U1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloonan SM, Choi AM. Mitochondria in lung disease. J Clin Invest. 2016;126:809–820. doi: 10.1172/JCI81113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake LE, Springer MZ, Poole LP, Kim CJ, Macleod KF. Expanding perspectives on the significance of mitophagy in cancer. Semin Cancer Biol. 2017;47:110–124. doi: 10.1016/j.semcancer.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban-Martinez L, Sierra-Filardi E, McGreal RS, Salazar-Roa M, Marino G, Seco E, Durand S, Enot D, Grana O, Malumbres M, et al. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J. 2017;36:1688–1706. doi: 10.15252/embj.201695916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Trojel-Hansen C, Kroemer G. Mitochondrial control of cellular life, stress, and death. Circ Res. 2012;111:1198–1207. doi: 10.1161/CIRCRESAHA.112.268946. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, Choi AM, Chu CT, Codogno P, Colombo MI, et al. Molecular definitions of autophagy and related processes. EMBO J. 2017;36:1811–1836. doi: 10.15252/embj.201796697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, Chen D, Si JM, Hu QS, Qin ZH, Fang M, Wang GH. The mitochondrial protein BNIP3L is the substrate of PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum Mol Genet. 2015;24:2528–2538. doi: 10.1093/hmg/ddv017. [DOI] [PubMed] [Google Scholar]
- Gottlieb RA, Stotland A. MitoTimer: a novel protein for monitoring mitochondrial turnover in the heart. J Mol Med (Berl) 2015;93:271–278. doi: 10.1007/s00109-014-1230-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerling BC, Gustafsson AB. Mitochondrial quality control in the myocardium: cooperation between protein degradation and mitophagy. J Mol Cell Cardiol. 2014;75:122–130. doi: 10.1016/j.yjmcc.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. 2012;287:19094–19104. doi: 10.1074/jbc.M111.322933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell. 2015;60:7–20. doi: 10.1016/j.molcel.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota Y, Yamashita S, Kurihara Y, Jin XL, Aihara M, Saigusa T, Kang DC, Kanki T. Mitophagy is primarily due to alternative autophagy and requires the MAPK1 and MAPK14 signaling pathways. Autophagy. 2015;11:332–343. doi: 10.1080/15548627.2015.1023047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landes T, Emorine LJ, Courilleau D, Rojo M, Belenguer P, Arnaune-Pelloquin L. The BH3-only Bnip3 binds to the dynamin Opa1 to promote mitochondrial fragmentation and apoptosis by distinct mechanisms. EMBO Rep. 2010;11:459–465. doi: 10.1038/embor.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M. Keeping the immune system in check: a role for mitophagy. Immunol Cell Biol. 2015;93:3–10. doi: 10.1038/icb.2014.75. [DOI] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Lee HY, Hanna RA, Gustafsson AB. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol-Heart C. 2011;301:H1924–H1931. doi: 10.1152/ajpheart.00368.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasters JJ. Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3) Redox Biol. 2014;2:749–754. doi: 10.1016/j.redox.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–185. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- Magrane J, Cortez C, Gan WB, Manfredi G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum Mol Genet. 2014;23:1413–1424. doi: 10.1093/hmg/ddt528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao K, Wang K, Zhao M, Xu T, Klionsky DJ. Two MAPK-signaling pathways are required for mitophagy in Saccharomyces cerevisiae. J Cell Biol. 2011;193:755–767. doi: 10.1083/jcb.201102092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Maestro P, Gargini R, Perry G, Avila J, Garcia-Escudero V. PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease. Hum Mol Genet. 2016;25:792–806. doi: 10.1093/hmg/ddv616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Vicente M, Talloczy Z, Wong E, Tang GM, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci. 2010;13:567. doi: 10.1038/nn.2528. U574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–221. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto G, Shimogori T, Hattori N, Nukina N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum Mol Genet. 2015;24:4429–4442. doi: 10.1093/hmg/ddv179. [DOI] [PubMed] [Google Scholar]
- Merkwirth C, Dargazanli S, Tatsuta T, Geimer S, Lower B, Wunderlich FT, von Kleist-Retzow JC, Waisman A, Westermann B, Langer T. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Gene Dev. 2008;22:476–488. doi: 10.1101/gad.460708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettinen TP, Björklund M. Mitochondrial Function and Cell Size: An Allometric Relationship. Trends Cell Biol. 2017;27:393–402. doi: 10.1016/j.tcb.2017.02.006. [DOI] [PubMed] [Google Scholar]
- Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, Glass K, Owen CA, Mahmood A, Washko GR, et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest. 2014;124:3987–4003. doi: 10.1172/JCI74985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AS, Holzbaur EL. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci USA . 2016;113:E3349–3358. doi: 10.1073/pnas.1523810113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AS, Holzbaur EL. Spatiotemporal dynamics of autophagy receptors in selective mitophagy. Autophagy. 2016;12:1956–1957. doi: 10.1080/15548627.2016.1212788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakawa T, Yamaguchi O, Hashimoto A, Hikoso S, Takeda T, Oka T, Yasui H, Ueda H, Akazawa Y, Nakayama H, et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun. 2015;6:7527. doi: 10.1038/ncomms8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nah J, Yuan J, Jung YK. Autophagy in neurodegenerative diseases: from mechanism to therapeutic approach. Mol Cells. 2015;38:381–389. doi: 10.14348/molcells.2015.0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TN, Padman BS, Lazarou M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol. 2016;26:733–744. doi: 10.1016/j.tcb.2016.05.008. [DOI] [PubMed] [Google Scholar]
- Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Lohr F, Popovic D, Occhipinti A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45–51. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh SM, Yang Y, He LY, Tang CY, Zhan M, Dong Z. Mitochondrial Function and Disturbances in the Septic Kidney. Semin Nephrol. 2015;35:108–119. doi: 10.1016/j.semnephrol.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Choi SG, Yoo SM, Nah J, Jeong E, Kim H, Jung YK. Pyruvate stimulates mitophagy via PINK1 stabilization. Cell Signal. 2015;27:1824–1830. doi: 10.1016/j.cellsig.2015.05.020. [DOI] [PubMed] [Google Scholar]
- Park S, Choi SG, Yoo SM, Son JH, Jung YK. Choline dehydrogenase interacts with SQSTM1/p62 to recruit LC3 and stimulate mitophagy. Autophagy. 2014;10:1906–1920. doi: 10.4161/auto.32177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85:257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Pearce EL. Mitochondrial dynamics at the interface of immune cell metabolism and function. Trends Immunol. 2017 doi: 10.1016/j.it.2017.08.006. pii: S1471-4906(17)30170-9. [DOI] [PubMed] [Google Scholar]
- Richter B, Sliter DA, Herhaus L, Stolz A, Wang CX, Beli P, Zaffagnini G, Wild P, Martens S, Wagner SA, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci USA. 2016;113:4039–4044. doi: 10.1073/pnas.1523926113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodolfo C, Campello S, Cecconi F. Mitophagy in neurodegenerative diseases. Neurochem Int. 2017 doi: 10.1016/j.neuint.2017.08.004. pii: S0197-0186(17)30087-6. [DOI] [PubMed] [Google Scholar]
- Rojansky R, Cha MY, Chan DC. Elimination of paternal mitochondria in mouse embryos occurs through autophagic degradation dependent on PARKIN and MUL1. Elife. 2016;5 doi: 10.7554/eLife.17896. pii: e17896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saita S, Shirane M, Nakayama KI. Selective escape of proteins from the mitochondria during mitophagy. Nat Commun. 2013;4:1410. doi: 10.1038/ncomms2400. [DOI] [PubMed] [Google Scholar]
- Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232–235. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496:372–376. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheibye-Knudsen M, Fang EF, Croteau DL, Wilson DM.3rdBohr VA. Protecting the mitochondrial powerhouse. Trends Cell Biol. 2015;25:158–170. doi: 10.1016/j.tcb.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi RY, Zhu SH, Li V, Gibson SB, Xu XS, Kong JM. BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci Ther. 2014;20:1045–1055. doi: 10.1111/cns.12325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirihai OS, Song MS, Dorn GW. How mitochondrial dynamism orchestrates mitophagy. Circ Res. 2015;116:1835–1849. doi: 10.1161/CIRCRESAHA.116.306374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha RA, Yen PM. Thyroid hormone-mediated autophagy and mitochondrial turnover in NAFLD. Cell Biosci. 2016;6 doi: 10.1186/s13578-016-0113-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BK, Marcinko K, Desjardins EM, Lally JS, Ford RJ, Steinberg GR. Treatment of nonalcoholic fatty liver disease: role of AMPK. Am J Physiol Endocrinol Metab. 2016;311:E730–E740. doi: 10.1152/ajpendo.00225.2016. [DOI] [PubMed] [Google Scholar]
- Sowter HM, Ratcliffe PJ, Watson P, Greenberg AH, Harris AL. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001;61:6669–6673. [PubMed] [Google Scholar]
- Suliman HB, Piantadosi CA. Mitochondrial quality control as a therapeutic target. Pharmacol Rev. 2016;68:20–48. doi: 10.1124/pr.115.011502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suomalainen A, Battersby BJ. Mitochondrial diseases: the contribution of organelle stress responses to pathology. Nat Rev Mol Cell Biol. 2017 doi: 10.1038/nrm.2017.66. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Szargel R, Shani V, Abd Elghani F, Mekies LN, Liani E, Rott R, Engelender S. The PINK1, synphilin-1 and SIAH-1 complex constitutes a novel mitophagy pathway. Hum Mol Genet. 2016;25:3476–3490. doi: 10.1093/hmg/ddw189. [DOI] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotta AP, Chipuk JE. Mitochondrial dynamics as regulators of cancer biology. Cell Mol Life Sci. 2017;74:1999–2017. doi: 10.1007/s00018-016-2451-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Martin A, Van den Haute C, Cufi S, Corominas-Faja B, Cuyas E, Lopez-Bonet E, Rodriguez-Gallego E, Fernandez-Arroyo S, Joven J, Baekelandt V, et al. Mitophagy-driven mitochondrial rejuvenation regulates stem cell fate. Aging (Albany NY) 2016;8:1330–1352. doi: 10.18632/aging.100976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, Clish CB, Granter SR, Widlund HR, Spiegelman BM, et al. PGC1 alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell. 2013;23:287–301. doi: 10.1016/j.ccr.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai T, Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab. 2016;27:105–117. doi: 10.1016/j.tem.2015.12.001. [DOI] [PubMed] [Google Scholar]
- Wang CW, Klionsky DJ. The molecular mechanism of autophagy. Mol Med. 2003;9:65–76. [PMC free article] [PubMed] [Google Scholar]
- Wei YJ, Chiang WC, Sumpter R, Mishra P, Levine B. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell. 2017;168:224–238. doi: 10.1016/j.cell.2016.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Holzbaur ELF. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J Neurosci. 2014;34:1293–1305. doi: 10.1523/JNEUROSCI.1870-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrighton KH. Metabolism: Mitophagy turns beige adipocytes white. Nat Rev Mol Cell Biol. 2016;17:607. doi: 10.1038/nrm.2016.125. [DOI] [PubMed] [Google Scholar]
- Wu WX, Lin CX, Wu K, Jiang L, Wang XJ, Li W, Zhuang HX, Zhang XL, Chen H, Li SP, et al. FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J. 2016;35:1368–1384. doi: 10.15252/embj.201593102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano K, Fogel AI, Wang C, van der Bliek AM, Youle RJ. Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. Elife. 2014;3:e01612. doi: 10.7554/eLife.01612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita S, Kanki T. How autophagy eats large mitochondria: Autophagosome formation coupled with mitochondrial fragmentation. Autophagy. 2017;13:980–981. doi: 10.1080/15548627.2017.1291113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Sun X, Starovoytov V, Cai Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum Mol Genet. 2015;24:2938–2951. doi: 10.1093/hmg/ddv056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Zheng Y, Zhang X, Chen Y, Wu X, Wu J, Shen Z, Jiang L, Wang L, Yang W, et al. BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy. 2017;13:1754–1766. doi: 10.1080/15548627.2017.1357792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Xue L, Li L, Tang C, Wan Z, Wang R, Tan J, Tan Y, Han H, Tian R, et al. BNIP3 Protein Suppresses PINK1 Kinase Proteolytic Cleavage to Promote Mitophagy. J Biol Chem. 2016;291:21616–21629. doi: 10.1074/jbc.M116.733410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Ren H, Xu C, Zhu C, Wu H, Liu D, Wang J, Liu L, Li W, Ma Q, et al. Hypoxic mitophagy regulates mitochondrial quality and platelet activation and determines severity of I/R heart injury. Elife. 2016;5 doi: 10.7554/eLife.21407. pii: e21407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, He F, Boassa D, Perkins G, Ali SR, et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell. 2016;164:896–910. doi: 10.1016/j.cell.2015.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Zhu P, Guo J, Hu N, Wang S, Li D, Hu S, Ren J, Cao F, Chen Y. Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol. 2017;13:498–507. doi: 10.1016/j.redox.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Massen S, Terenzio M, Lang V, Chen-Lindner S, Eils R, Novak I, Dikic I, Hamacher-Brady A, Brady NR. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J Biol Chem. 2013;288:1099–1113. doi: 10.1074/jbc.M112.399345. [DOI] [PMC free article] [PubMed] [Google Scholar]