

Abstract
Autophagy is a lysosome-dependent degradation process that is essential for maintaining cellular homeostasis. In recent years, more studies have focused on the late stages of autophagy. Our group discovered and studied the terminal step of autophagy, namely autophagic lysosome reformation (ALR). ALR is the process that regenerates functional lysosomes from autolysosomes, thus maintaining lysosome homeostasis. ALR involves clathrin-mediated membrane budding from autolysosomes, elongation of membrane tubules along microtubules with the pulling force provided by the motor protein KIF5B, proto-lysosome scission by dynamin 2, and finally maturation of proto-lysosomes to functional lysosomes. In this review, we will summarize progress in unveiling the molecular mechanisms underlying ALR and its potential pathophysiological roles.
Keywords: autophagic lysosome reformation (ALR), autophagy, in vitro reconstitution, membrane deformation, phospholipid
INTRODUCTION
Autophagy, a lysosome-dependent degradation process, is essential for maintenance of cellular homeostasis (Mizushima and Komatsu, 2011; Rubinsztein et al., 2012; Yang and Klionsky, 2010). It is an important mechanism for adaptation to stress and intracellular quality control. The Nobel Prize in Physiology or Medicine 2016 was awarded to Yoshinori Ohsumi for his discoveries of the mechanisms of autophagy. In the past decade, autophagy research has largely focused on the early stages of the process, namely omegasome formation, isolation membrane elongation and autophagosome formation. This is because most autophagy genes (ATGs) have been identified in screens that use autophagosome formation as the readout. The molecular mechanisms underlying the function of lysosomes, for example lysosome consumption and regeneration after autolysosome formation, have received less attention. In recent years, our group has discovered and studied the terminal step of autophagy, namley autophagic lysosome reformation (ALR), which restores the level of free lysosomes to maintain lysosome homeostasis. This process also bridges the final gap to complete the autophagy cycle. In this review, we will summarize recent progress in understanding the molecular mechanisms of ALR and some pioneering work related to its pathophysiological roles.
DISCOVERY OF ALR
While studying the kinetics of autophagy, we observed that the number of lysosomes significantly decreased after 4 h of starvation and recovered after 12 h in cell lines derived from multiple species (Yu et al. 2010). We thus speculated that there must be a conserved mechanism to maintain lysosome homeostasis. Using both confocal microscopy and transmission electron microscopy (TEM), we found that long tubular structures extended from autolysosomes, and free vesicle emerged from the tips of the tubules. The newly formed tubules and vesicles were positive for the lysosome membrane protein LAMP1 and negative for the autophagosome marker protein LC3. The degradation substrates were retained within the autolysosome structure. The emerging vesicles were initially pH-neutral but gradually became acidic and acquired degradative capacity. Based on these features, we named the vesicles proto-lysosomes, because they later matured into functional lysosomes. The process of tubular formation and lysosome regeneration is called autophagic lysosome reformation.
REGULATION OF ALR
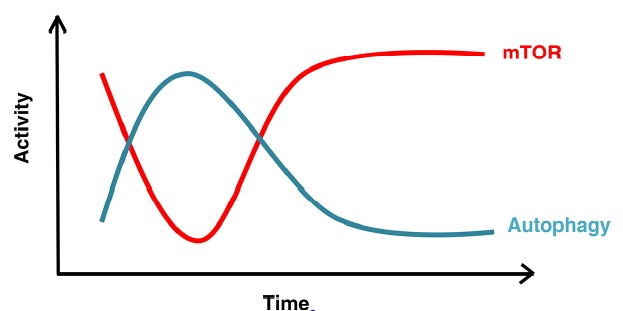
ALR is not only a lysosome biogenesis process. Regulation of ALR is tightly correlated with the regulation of autophagy, in which mTOR (mammalian target of rapamycin) is the central signaling cue. We found that the activity of mTOR oscillated in a manner that corresponded to the autophagy cycle (Yu et al., 2010). mTOR is activated in nutrient-rich conditions and is inhibited under starvation conditions when autophagy is induced. After prolonged starvation, intracellular nutrients generated by autophagy stimulate mTOR signaling and thereby provide a negative feedback signal to avoid excessive degradation and to trigger ALR (Fig. 1). Reactivation of mTOR is autophagy dependent and requires the degradation of autolysosomal products since knockdown of ATG5/7 abolished the reactivation of mTOR as judged by the level of phosphorylation of the mTOR substrate S6K. While further investigating how mTOR is reactivated upstream of ALR, we identified a role of Spinster (Spin), a putative lysosomal efflux permease and sugar transporter. Defect of Spinster in NRK cells showed accumulation of enlarged autolysosomes after prolonged starvation, which is the typical ALR-defective phenotype. This may be because defective sugar transportation activity led to impaired autolysosomal degradation capacity, resulting in the failure of mTOR reactivation (Rong et al. 2011). In addition, we found that starvation coupled autophagy with the general amino acid control (GAAC) pathway through which cells enhance amino acid uptake and synthesis (Chen et al., 2014). Starvation triggered the activation of ATF4 (activating transcription factor 4) and resulted in the upregulation of its downstream effector SLC7A5, a leucine transporter on the plasma membrane. The elevation of amino acid uptake contributed to mTOR reactivation. So far, we have identified a few effectors responsible for activation of mTOR after prolonged starvation. However, the direct substrates or effectors that trigger ALR downstream of the reactivated mTOR are still missing and require further research.
Fig. 1. Schematic illustration of the correlation between mTOR activity and autophagy during starvation.
CENTRAL PLAYERS IN ALR
To make progress in understanding the molecular basis of ALR, a screening system for the central players is needed. We combined mass spectrometry with large-scale RNAi screening by RNAi chips. We first purified the reformation tubules by density gradient centrifugation and checked the purified structures using TEM. The fractions with the most reformation tubules were subjected to mass spectrometry analysis. We carried out the screen of our primary targets identified from mass spectrometry using “SAMCell” RNAi chips, which efficiently deliver a large number of individual RNAi molecules to cells on single glass slides that are compatible with confocal microscopy observation. Accumulation of large autolysosomes positive for LC3 and LAMP1 after prolonged starvation was used as the readout in the screen.
By this method, we identified clathrin, the PI(4,5)P2-related kinase PIP5K1B, and the motor protein KIF5B as essential factors of ALR (Rong et al., 2012). Clathrin is well known for clathrin-mediated endocytosis, in which clathrin-coated vesicles are formed for cargo sorting and transportation from the plasma membrane (Kirchhausen, 2000). During ALR, clathrin is localized on the surface of autolysosomes as small buds, reminiscent of the formation of clathrin-coated buds on the surface of plasma membrane. On the surface of autolysosomes, PIP5K1B catalyzes the conversion of PI4P to PI(4,5)P2, which is the central molecule for ALR. A lack of PIP4K1B causes enlarged autolysosomes. Nanoparticles caused liver damage because PI(4)P can not be converted in nanoparticle-treated hepatocytes (Zhang et al., 2017). Adaptor protein 2 (AP2) links clathrin to PI(4,5)P2 on the autolysosomal membrane. Formation of a clathrin lattice on the autolysosome membrane may serve as a platform for subsequent recruitment of more AP2 and PI(4,5)P2 to form PI(4,5)P2-enriched microdomains. KIF5B mediated the generation of the reformation tubules at the sites of the clathrin buds by providing a pulling force along microtubules (Du et al., 2016).
After the formation of tubules from autolysosomes, scission happens at the tubule tips to generate proto-lysosomes. The large GTPase DNM2/dynamin 2, which is known to function in the scission of plasma membrane invaginations during endocytosis (Warnock et al., 1997), was shown to provide the mechanical torque for the scission event that generates proto-lysosomes (Schulze et al., 2013). Multiple phospholipids, including PI4P, PI(4,5)P2 and PI3P, play essential roles during this process. Knocking down the PIP5K1A caused extensive tubulation of autolysosomes and no proto-lysosome formation (Rong et al., 2012). Lysosomal PI4KIIIβ, a PI4K, regulates the extent of lysosome tubulation and efficient excision through in situ generation of PI4P (Sridhar et al., 2013). A lack of PI4KIIIβ causes inefficient vesicle fission, leading to formation of elongated tubular structures. In addition, PI3P generated by VPS34, which was previously shown to function during the initiation of autophagosome formation, has been shown to function in the scission step. Two specific phosphorylation sites on UVRAG are required for activation of VPS34, and disruption of these sites led to an increase in the number and length of lysosomal tubules and massive cell death (Chen and Yu, 2015; Munson et al., 2015). Although it is clear that different phospholipids function at different stages of ALR, it remains to be determined whether different phospholipids work in a coordinated manner to regulate ALR, or whether each one regulates ALR in a different way.
IN VITRO RECONSTITUTION OF ALR – GENERATION OF REFORMATION TUBULES
The art of the in vitro reconstitution system is to precisely manipulate the parameters and study the key questions at a single-molecule or single-vesicle level, which cannot usually be achieved by in vivo or intracellular studies. Our goal is to set up an in vitro system to reconstitute the KIF5B-mediated tubulation of autolysosomes. Firstly, using KIF5B, purified autolysosomes and in vitro-polymerized microtubule networks, we successfully reconstituted the generation of reformation tubules from autolysosomes along microtubule tracks (Du et al., 2016). Using this system as a basis, we were able to study in detail the role of PI(4,5)P2, an essential molecule for ALR. We showed that autolysosomes purified from PIP5K1B-deficient cells were unable to generate tubules under otherwise similar conditions, confirming that PI(4,5)P2 on autolysosomes is essential for ALR. We substituted the purified autolysosomes with man-made liposomes and found that liposomes gave a satisfactory tubulation rate with a PI(4,5)P2 concentration up to 25%. The estimated physiological concentration of PI(4,5)P2 on autolysosomes is 5%. However, in our in vitro system, we scarcely detected tubule structures when we used liposomes with 5% PI(4,5)P2. Thus, there must be a mechanism to concentrate PI(4,5)P2 at a limited site to recruit enough KIF5B molecules and provide sufficient pulling force. To address this problem, we created “stripped” autolysosomes by digesting their surface proteins using trypsin. The “stripped” autolysosomes still contained the normal lipid composition but had no surface proteins. “Stripped” autolysosomes did not generate tubules, but this phenotype was rescued by treatment with cytosol. In the presence of an anti-clathrin antibody, the “stripped” cytosol-treated autolysosomes did not give rise to tubules, which confirmed that clathrin is essential for concentrating PI(4,5)P2 into microdomains. We also showed that giant unilamellar vesicles (GUVs) containing 5% PI(4,5)P2 showed clustering of PI(4,5)P2 and KIF5B when treated with cytosol, further indicating that something in the cytosol functions to concentrate PI(4,5)P2. In the absence of cytosol, we are unable to reconstitute ALR using liposomes containing 5% PI(4,5)P2, clathrin and KIF5B; therefore, it is still not clear which proteins in the cytosol function as adaptors together with clathrin to form the PI(4,5)P2-enriched domains. Nevertheless, the in vitro reconstitution system has led us to identify new regulators of ALR.
PHYSIOLOGICAL ROLE OF ALR
Our understanding of the physiological role of ALR is at a very early stage. Several studies have shown that ALR is related to lipophagic turnover or cell death. Defective ALR resulted in accumulation of lipid droplets in hepatocytes or caused massive cell death during long-term starvation (Munson et al., 2015; Schulze et al., 2013). However, these findings are all limited to intracellular studies with no clinical evidence. Only a few studies have shed some light on the pathophysiological function of ALR in vivo. Chang et al identified that the two most common autosomal recessive hereditary spastic paraplegia gene products, spastizin and spatacsin, are pivotal for ALR. Chang et al provided intracellular evidence showing that a lack of spastizin or spatacsin caused depletion of free lysosomes and accumulation of enlarged autolysosomes, which are typical phenotypes of defective ALR. They also studied the detailed molecular mechanism underlying the function of these proteins in ALR (Chang et al., 2014). Another group reported that disruption of spatacsin in mice indeed causes hereditary spastic paraplegia-like phenotypes with loss of cortical neurons and Purkinje cells (Varga et al., 2015). It is possible that ALR defects result in a decreased number of functional lysosomes in vivo, which could be linked to inefficient autophagic clearance, accumulation of undegraded material and finally neuronal death. Another group has linked the loss of GCase activity, inhibition of autophagy and increased α-synuclein levels with ALR. Lysosome recycling by ALR was impaired in neurons so that there were not enough functional lysosomes to sustain autophagic clearance of α-synuclein. The impaired autophagy and accumulation of pathogenic α-synuclein species in the brain may be the cause of Parkinson disease (Magalhaes et al., 2016).
CONCLUSION
Since the first discovery of ALR, we have observed significant progress in understanding its regulation and molecular mechanisms (Fig. 2). We can now divide the ALR process into 4 steps: (1) clathrin-mediated budding of tubular structures from autolysosomes; (2) KIF5B-mediated tubule extension along microtubules; (3) dynamin 2-mediated scission to form proto-lysosomes; and (4) maturation of protolysosomes. Multiple phospholipids including PI(4,5)P2, PI3P and PI4P function as signals and adaptors for the formation of microstructures. Our strategy of studying ALR can be summarized as purification of ALR-associated structures, mass spectrometry analysis, large-scale RNAi screens, characterization of essential proteins, and in vitro reconstitution. This strategy can be applied to study other cellular organelles or structures. For example, we have applied the in vitro system to study the dynamic tubulation of mitochondria (a new process for forming mitochondrial networks) and lysosome motility (Su et al., 2016; Wang et al., 2015).
Fig. 2. Schematic illustration of the process of ALR involving clathrin-mediated membrane budding from autolysosomes, elongation of membrane tubules along microtubules by the motor protein KIF5B and proto-lysosome scission by dynamin 2.

Many questions still remain to be answered. Firstly, although we have some knowledge about how mTOR is reactivated, we still do not know the direct substrate of reactivated mTOR in this signaling pathway that initiates ALR. Secondly, the mechanism that sorts the lysosomal membrane and lumen proteins during ALR is largely unknown. AP4 interacts with lysosomal proteins on autolysosomes and reformation tubules, indicating that AP4 may play a role in sorting lysosome cargo. PI4KIIIβ may regulate retention of intrinsic lysosomal components since lysosomal contents were present in the reformation tubules in PI4KIIIβ knockdown cells. There should be other mechanisms to sort transmembrane proteins and luminal proteins of lysosomes without AP-binding motifs or to sort large protein complexes with a limited tendency to diffuse. Thirdly, how proto-lysosomes mature into functional lysosomes is a missing link in ALR. Fourthly, the destiny of the undigested debris retained in autolysosomes is still mysterious. We can speculate that cells may actively exocytose the debris, but further evidence is needed. Fifthly, we do not know whether ALR occurs during selective autophagy.
Last but not least, the main reason that there has not been a significant breakthrough in our knowledge of the physiological role of ALR is that most of the known ALR regulators, such as clathrin, KIF5B and kinases, are central nodes in other important biological processes. It is therefore challenging to specify their functions in ALR under pathophysiological conditions. A more specific screening of the mass spectrometry database needs to be done to identify the second layer of ALR regulation. ALR-specific mutations of genes encoding key components may also help us understand the physiological role of ALR.
ACKNOWLEDGMENTS
Research in Li Yu’s lab was supported by the Ministry of Science and Technology of the People’s Republic of China (2016YFA0500202 and 2017YFA0503404), the National Natural Science Foundation of China (31430053 and 31321003), the Natural Science Foundation of China International Cooperation and Exchange Program (31561143002), and the Independent Research of Tsinghua University (20161080135).
REFERENCES
- Chang J, Lee S, Blackstone C. Spastic paraplegia proteins spastizin and spatacsin mediate autophagic lysosome reformation. J Clin Invest. 2014;124:5249–5262. doi: 10.1172/JCI77598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Yu L. Scissors for autolysosome tubules. EMBO J. 2015;34:2217–2218. doi: 10.15252/embj.201592519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Zou Y, Mao D, Sun D, Gao G, Shi J, Liu X, Zhu C, Yang M, Ye W, et al. The general amino acid control pathway regulates mTOR and autophagy during serum/glutamine starvation. J Cell Biol. 2014;206:173–182. doi: 10.1083/jcb.201403009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W, Su QP, Chen Y, Zhu Y, Jiang D, Rong Y, Zhang S, Zhang Y, Ren H, Zhang C, et al. Kinesin 1 drives autolysosome tubulation. Dev Cell. 2016;37:326–336. doi: 10.1016/j.devcel.2016.04.014. [DOI] [PubMed] [Google Scholar]
- Kirchhausen T. Clathrin. Annu Rev Biochem. 2000;69:699–727. doi: 10.1146/annurev.biochem.69.1.699. [DOI] [PubMed] [Google Scholar]
- Magalhaes J, Gegg ME, Migdalska-Richards A, Doherty MK, Whitfield PD, Schapira AH. Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: relevance to Parkinson disease. Hum Mol Genet. 2016;25:3432–3445. doi: 10.1093/hmg/ddw185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- Munson MJ, Allen GF, Toth R, Campbell DG, Lucocq JM, Ganley IG. mTOR activates the VPS34-UVRAG complex to regulate autolysosomal tubulation and cell survival. EMBO J. 2015;34:2272–2290. doi: 10.15252/embj.201590992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong Y, McPhee CK, Deng S, Huang L, Chen L, Liu M, Tracy K, Baehrecke EH, Yu L, Lenardo MJ. Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proc Natl Acad Sci USA. 2011;108:7826–7831. doi: 10.1073/pnas.1013800108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong Y, Liu M, Ma L, Du W, Zhang H, Tian Y, Cao Z, Li Y, Ren H, Zhang C, et al. Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat Cell Biol. 2012;14:924–934. doi: 10.1038/ncb2557. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Shpilka T, Elazar Z. Mechanisms of autophagosome biogenesis. Curr Biol. 2012;22:R29–34. doi: 10.1016/j.cub.2011.11.034. [DOI] [PubMed] [Google Scholar]
- Schulze RJ, Weller SG, Schroeder B, Krueger EW, Chi S, Casey CA, McNiven MA. Lipid droplet breakdown requires dynamin 2 for vesiculation of autolysosomal tubules in hepatocytes. J Cell Biol. 2013;203:315–326. doi: 10.1083/jcb.201306140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridhar S, Patel B, Aphkhazava D, Macian F, Santambrogio L, Shields D, Cuervo AM. The lipid kinase PI4KIIIbeta preserves lysosomal identity. EMBO J. 2013;32:324–339. doi: 10.1038/emboj.2012.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su QP, Du W, Ji Q, Xue B, Jiang D, Zhu Y, Lou J, Yu L, Sun Y. Vesicle size regulates nanotube formation in the cell. Sci Rep. 2016;6:24002. doi: 10.1038/srep24002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga RE, Khundadze M, Damme M, Nietzsche S, Hoffmann B, Stauber T, Koch N, Hennings JC, Franzka P, Huebner AK, et al. In vivo evidence for lysosome depletion and impaired autophagic clearance in hereditary spastic paraplegia type SPG11. PLoS Genet. 2015;11:e1005454. doi: 10.1371/journal.pgen.1005454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Du W, Su QP, Zhu M, Feng P, Li Y, Zhou Y, Mi N, Zhu Y, Jiang D, et al. Dynamic tubulation of mitochondria drives mitochondrial network formation. Cell Res. 2015;25:1108–1120. doi: 10.1038/cr.2015.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnock DE, Baba T, Schmid SL. Ubiquitously expressed dynamin-II has a higher intrinsic GTPase activity and a greater propensity for self-assembly than neuronal dynamin-I. Mol Biol Cell. 1997;8:2553–2562. doi: 10.1091/mbc.8.12.2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–131. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942–946. doi: 10.1038/nature09076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JQ, Zhou W, Zhu SS, Lin J, Wei PF, Li FF, Jin PP, Yao H, Zhang YJ, Hu Y, et al. Persistency of enlarged autolysosomes underscores nanoparticle-induced autophagy in hepatocytes. Small. 2017;13 doi: 10.1002/smll.201602876. [DOI] [PubMed] [Google Scholar]