

Abstract
Autophagy is an intracellular degradation pathway for large protein aggregates and damaged organelles. Recent studies have indicated that autophagy targets cargoes through a selective degradation pathway called selective autophagy. Peroxisomes are dynamic organelles that are crucial for health and development. Pexophagy is selective autophagy that targets peroxisomes and is essential for the maintenance of homeostasis of peroxisomes, which is necessary in the prevention of various peroxisome-related disorders. However, the mechanisms by which pexophagy is regulated and the key players that induce and modulate pexophagy are largely unknown. In this review, we focus on our current understanding of how pexophagy is induced and regulated, and the selective adaptors involved in mediating pexophagy. Furthermore, we discuss current findings on the roles of pexophagy in physiological and pathological responses, which provide insight into the clinical relevance of pexophagy regulation. Understanding how pexophagy interacts with various biological functions will provide fundamental insights into the function of pexophagy and facilitate the development of novel therapeutics against peroxisomal dysfunction-related diseases.
Keywords: pexophagy, peroxisomal disorder, peroxisomal protein, peroxisome, selective autophagy receptor
INTRODUCTION
Peroxisomes are ubiquitous organelles found in most eukaryotic cells. They are key metabolic platforms for purine catabolism, fatty acid beta-oxidation (FAO), bile acid synthesis, and ether phospholipid synthesis (Wanders et al., 2016). Various peroxisomal enzymes are involved in modulation of metabolic responses to various signals (Poirier et al., 2006). In addition, peroxisomes are important redox regulating organelles because of their dual functions in the generation and scavenging of reactive oxygen species (ROS) and reactive nitrogen species (RNS) (Du et al., 2015; Fransen, 2012). Peroxisomal biogenesis and degradation must be tightly regulated to maintain appropriate peroxisomal sizes, numbers, and functions to prevent unwanted pathological situations (Honsho et al., 2016; Waterham et al., 2016). Numerous genes and proteins, including peroxins (PEX), dynamin-related protein 1 (Drp1), and mitochondrial fission factor, have been identified in the regulation of peroxisomal biogenesis and division (Kiel et al., 2006; Mayerhofer, 2016; Schlüter et al., 2006).
The estimated half-life for peroxisomes is approximately 2 days, suggesting that biogenesis and degradation of peroxisomes are dynamic processes. Selective autophagy of cellular organelles is an important event for the maintenance of homeostasis in various internal and external stress responses (Anding and Baehrecke, 2017). Pexophagy is a catabolic progress for selective degradation of peroxisomes by autophagy, and several key regulators for pexophagy have been recently identified in yeast and mammals. Here, we present a brief introduction of peroxisomal biogenesis/degradation, and then focus on recent advances of the molecular mechanisms of pexophagy, and their relevance to physiological and pathological responses.
OVERVIEW OF PEROXISOMES: GENERAL FUNCTIONS, BIOGENESIS, AND DEGRADATION
The first description of peroxisomes, which contained hydrogen peroxide-generating oxidases, was published by the biochemist Christian de Duve in the 1960s (Bowers, 1998; Braverman et al., 2013). Peroxisomes are single, membrane-bounded and small (0.2–1 μm diameter) organelles, with several hundred found in each mammalian cell (Braverman et al., 2013). They are highly dynamic, ubiquitous, and multi-functional organelles present in most eukaryotic cells (Hiltunen et al., 2003; Hu et al., 2012; Islinger et al., 2010). They play critical roles in various biological responses, as “multipurpose organelles” that function in both catabolic and anabolic pathways, as well as in diverse responses to different tissues and single organisms (Islinger et al., 2010).
Peroxisomal functions
Peroxisomes are usually key compartments for two major functions involving FAO and generation, and detoxification of hydrogen peroxide (Deb and Nagotu, 2017; Hiltunen et al., 2003; Hu et al., 2012). Peroxisomes are important organelles for FAO, which occurs in both mitochondria and peroxisomes in animal cells and are important for the production of fatty acids and their derivatives (Deb and Nagotu, 2017). In peroxisomes, beta-oxidation of fatty acids mainly involves very long-chain fatty acids (> C22) and branched fatty acids, and usually starts with the conversion of fatty acids to their acyl-CoA derivatives in the cytosol (Deb and Nagotu, 2017; Demarquoy and Le Borgne, 2015). The fatty acyl-CoA derivatives are transported into peroxisomes via specific peroxisomal proteins, including mammalian ATP Binding Cassette Subfamily D Member 1 (ABCD1) transporter (Demarquoy and Le Borgne, 2015). FAO also contributes to the generation of hydrogen peroxide in plant peroxisomes (Hu et al., 2012), also in animal cells (Fransen et al., 2012). Thus, peroxisomes are major organelles that link redox regulation and FAO to play multiple functional roles in living cells (Deb and Nagotu, 2017).
Because peroxisomes are also involved in secondary metabolism, i.e., catabolic pathways of oleic acid, methanol, polyamines, and purines, it is inevitable that high levels of hydrogen peroxide are produced (Kumar et al., 2014). Peroxisomes produce important enzymes that generate hydrogen peroxide, including glycolate oxidase, urate oxidase, aspartate oxidase, polyamine oxidase, and acyl-CoA oxidase (Bonekamp et al., 2009; Deb and Nagotu, 2017). In addition, peroxisomes produce superoxides using the enzyme, xanthine oxidase (Bonekamp et al., 2009). To counterbalance oxidative stresses, the peroxisomes are major centers for antioxidant enzyme generation, especially catalase (Bonekamp et al., 2009). Furthermore, peroxisomes are principal sources for the production of RNS and reactive sulfur species (Bonekamp et al., 2009). Because ROS and RNS play important roles in the activation or inhibition of multiple signal transduction kinases and phosphatases, peroxisomes serve as key centers for the regulation of various biological responses through the generation or removal of redox messengers in cells (Fransen et al., 2012; Gào and Schöttker, 2017).
Biogenesis and degradation
The number, morphology, and size of peroxisomes are dynamically regulated in response to environmental and developmental cues (Heiland and Erdmann, 2005; Huybrechts et al., 2009). The biogenesis of peroxisomes is very complicated and regulated by more than 30 different PEX proteins (Kiel et al., 2006; Mayerhofer, 2016; Schlüter et al., 2006). Because peroxisomes do not have an independent system for genome or protein synthesis, they usually import all soluble proteins post-translationally from the cytosol into the peroxisomal matrix (Mayerhofer, 2016). PEX plays important, but in many cases redundant, roles in many biological processes including targeting peroxisomal membrane proteins (PMPs) to the peroxisomes, PMPs insertion and import into the matrix, control of peroxisome size, and the functional maintenance of peroxisomes (Giannopoulou et al., 2016; Kiel et al., 2006; Mayerhofer, 2016; Schlüter et al., 2006). The functions and molecular mechanisms of peroxisomal proteins are briefly described in this review (Fig. 1 and Table 1), because they are comprehensively described in a series of recent review articles (Honsho et al., 2016; Kim, 2017; Waterham et al., 2016).
Fig. 1. A model of peroxisomal biogenesis.
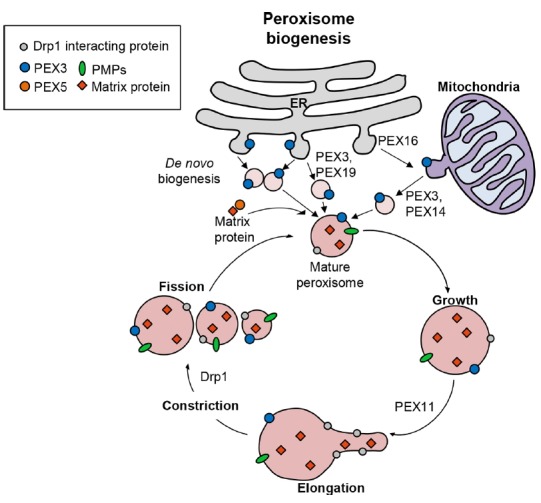
Peroxisome biogenesis is coordinated by two different pathways, de novo biogenesis and the ‘growth and division’. First, peroxisomes can be formed by peroxisome assembly and maturation of pre-peroxisomal vesicles originated from ER or mitochondria, which contain pre-peroxisomal carriers, including PEX3 and PEX16. Second, peroxisomes can proliferate the numbers by a growth and division progress from existing peroxisomes. PEX11 and Drp1 proteins mediate elongation and fission of the peroxisomes.
Table 1.
Peroxisomal proteins and their relevance with pathological aspects
| Gene | Function | Diseases | References |
|---|---|---|---|
| PEX1 | Peroxisome fusion and/or peroxisomal matrix protein import | IRD, NALD, ZSD, HS | Reuber et al., 1997; Smith et al., 2016; Tamura et al., 2001; Yik et al., 2009 |
| PEX2 | Peroxisomal matrix protein import | ZS, Mild ZSD, IRD | Shimozawa et al., 1999a |
| PEX3 | The assembly and maintenance of the peroxisomal membrane | ZS, Mild IRD | Muntau et al., 2000 |
| PEX5 | Peroxisomal matrix proteins carrying PTS1 | NALD, ZSD, RCDP5 | Barøy et al., 2015; Shimozawa et al., 1999c |
| PEX6 | Peroxisome fusion and/or peroxisomal matrix protein import | NALD, ZSD, HS | Smith et al., 2016; Yik et al., 2009; Zhang et al., 1999 |
| PEX7 | Peroxisomal matrix proteins carrying PTS2 | RCDP1, Mild RD | Braverman et al., 1997; van den Brink et al., 2003 |
| PEX10 | Peroxisomal matrix protein import | NALD, ZSD | Warren et al., 1998; Yik et al., 2009 |
| PEX11β | Peroxisome division | Mild ZSD | Ebberink et al., 2012 |
| PEX12 | Peroxisomal matrix protein import | IRD, NALD, ZSD | Chang et al., 1997; Yik et al., 2009 |
| PEX13 | Peroxisomal matrix protein import | NALD, ZSD | Al-Dirbashi et al., 2009; Liu et al., 1999; Shimozawa et al., 1999b |
| PEX14 | The initial docking site for PTS1 and PTS2 receptors of matrix proteins/an essential component of peroxisomal import machinery | ZS | Huybrechts et al., 2008 |
| PEX16 | Peroxisome division or peroxisomal membrane biogenesis | ZSD | South and Gould, 1999 |
| PEX19 | The import and/or membrane assembly of numerous peroxisomal membrane proteins. | ZS | Matsuzono et al., 1999 |
| PEX26 | Receptor export, membrane receptors for Pex1 and Pex6 | ZSD. NALD, IRD | Matsumoto et al., 2003; Yik et al., 2009 |
| ACBD5 (Atg37) | Acyl-CoA binding protein which acts as the peroxisome receptor for pexophagy | Retinal dystrophy, WMD | Ferdinandusse et al., 2017 |
ATM, Ataxia-telangiectasia mutated; HS, Heimler syndrome; IRD, Infantile refsum disease; NALD, Neonatal adrenoleukodystrophy; RCDP, Rhizomelic chondrodysplasia punctate; WMD, White matter dementia; ZS, Zellweger syndrome; ZSD, Zellweger spectrum disorders
Peroxisomal proteins are regulated at the transcriptional level by numerous gene regulators. Among them, peroxisome proliferator-activated receptor α (PPARα) is a well-characterized nuclear receptor that is essential for controlling the transcriptional activation of peroxisomal proteins, especially peroxisomal beta-oxidation (Pawlak et al., 2015). PPARα is essential in controlling the gene regulation of ACOX1 and EHHADH, the rate-limiting enzymes of peroxisomal beta-oxidation (Rakhshandehroo et al., 2009). Notably, PPARα has been reported as an autophagy regulator, even of feeding (Lee et al., 2014), and also in the activation of xenophagy against mycobacterial infection (Kim et al., 2017). PPARα function in the regulation of autophagy is mediated through the modulation of autophagy-related genes and lysosomal genes, including TFEB (Kim et al., 2017). Future studies are needed to characterize the role of PPARα in the regulation of pexophagy and other types of selective autophagy. Importantly, peroxisomes have multiple quality control mechanisms to maintain proper functioning and offer protection from damage through molecular chaperones and the activation of pexophagy, a selective autophagy for peroxisomes (Kumar et al., 2014). In this review, pexophagy, one of the mechanisms of peroxisomal quality control, will be discussed in detail.
MOLECULAR REGULATORY MECHANISMS OF PEXOPHAGY
Ubiquitination-mediated pexophagy
Recent advances in the understanding of selective autophagy have suggested that ubiquitination of membrane proteins of specific organelles is required for selective autophagy (Feng et al., 2017; Vives-Bauza et al., 2010; Yamashita et al., 2014). Consistent with this possibility, pexophagy is highly induced by the ubiquitination of PMPs (Kim et al., 2008). With the aid of these proteins, ubiquitin (Ub) exposed to cytoplasm is targeted by Ub-binding autophagy adaptors. For example, ectopic expression of a peroxisomal membrane protein, PMP34, fused with Ub in the cytoplasmic tail, dramatically enhanced pexophagy (Kim et al., 2008). In addition, pexophagy was only induced by overexpression of PEX3 tagged with Ub on the cytosolic side, but not by PEX3 tagged with Ub on the N-terminal luminal side of peroxisomes, indicating that ubiquitination of the peroxisomal protein, PEX3, is an important event for pexophagy (Yamashita et al., 2014). However, Yamashita et al. reported that expression of a PEX3 mutant, which was defective in PEX3 ubiquitination because of substitutions of all lysine and cysteine residues, still induced peroxisome ubiquitination and degradation (Yamashita et al., 2014). These differences suggested that ubiquitination of PEX3 is dispensable for pexophagy, and that unidentified peroxisomal proteins are further ubiquitinated on the peroxisomal membrane. Thus, the physiological consequences of ubiquitination of PEX3 still remain to be fully characterized.
Besides PEX3, recent studies have focused on PEX5 ubiquitination to elucidate the mechanism of pexophagy regulation under certain conditions such as oxidative stress (Zhang et al., 2015). PEX5 binds to the C-terminus of PTS1, the tripeptide peroxisomal targeting signal sequence of peroxisomal proteins, and imports the target proteins into peroxisomes (Hua and Kim, 2016). During the matrix protein import cycle, PEX5 is regulated in an ubiquitination-dependent manner. Monoubiquitination of PEX5 on the cysteine residue by PEX4 and the PEX10/PEX12 complex, which functions as the E2 and E3 enzymes, respectively, and deubiquitination of PEX5 by peroxisomal AAA ATPase, regulates its recycling to the cytosol. However, polyubiquitination of PEX5 on lysine residues results in malfunctioning of the PEX5 recycling machinery, followed by PEX5 extraction from the peroxisome membrane and targeted degradation by the Ub-proteasome system (Platta et al., 2014). Interestingly, mono-ubiquitination of the N-terminal cysteine residue of PEX5 serves as a quality control mechanism to remove the defective peroxisome protein import machinery. Nordgren et al. recently reported that the export-deficient monoubiquitinated PEX5, generated by fusion of a bulky C-terminal-enhanced green fluorescent protein, induced the elimination of peroxisomes in fibroblasts (Nordgren et al., 2015).
Taken together, previous studies have provided insight into ubiquitination-dependent pexophagy regulation (Fig. 2). However, the precise mechanisms of regulation by Ub, especially in pexophagy-associated disorders, still need to be further characterized.
Fig. 2. Pexophagy regulators.
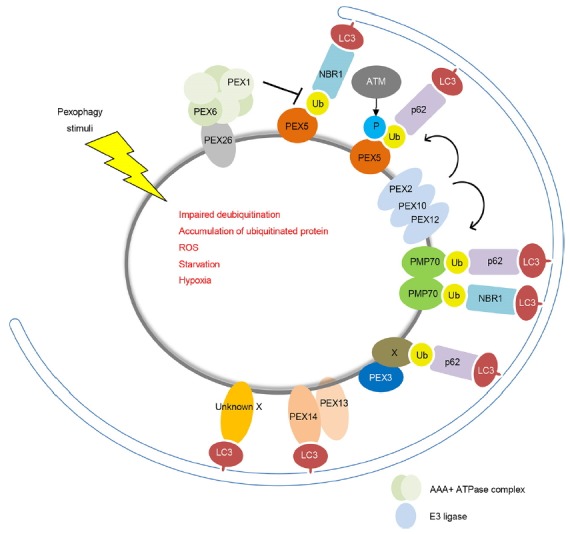
Pexophagy is triggered by both stress conditions and peroxisomal dysfunctions. Ubiquitination of PMPs, such as peroxins and PMP70, promotes pexophagy. Both NBR1 and p62 act as autophagy adaptor proteins, which interact with PMPs and sequester target peroxisome into autophagosomes. Under conditions of oxidative stress, the ataxia-telangiectasia mutation activates pexophagy by phosphorylating PEX5, leading to its ubiquitination. Pexophagy may be regulated by an unidentified protein (X).
Adaptor-mediated pexophagy
The p62 (sequestosome1/SQSTM1) protein has been identified as a mediator in the nuclear factor kappa B-signaling pathway during tumorigenesis (Duran et al., 2008), and NBR1 was originally reported as an ovarian tumor antigen monitored in ovarian cancer (Campbell et al., 1994). Recent advances in autophagy have shown that both p62 and NBR1 are involved in the degradation of protein aggregates, damaged organelles, intracellular bacteria, phagocytic membrane remnants, and in endocytic trafficking (Johansen and Lamark, 2011; Kirkin et al., 2009; Mardakheh et al., 2010). In this section, we will describe the pexophagy adaptor proteins, including NBR1 and p62 (Fig. 2).
The p62 protein plays a role as an autophagy adaptor in mammals, where it functions as an autophagy-signaling hub (Bjorkoy et al., 2005; Katsuragi et al., 2015; Komatsu et al., 2007). The p62 protein contains several conserved domains, including the phox/Bem1p (PB1) domain mediating protein interaction at the N-terminus; the ZZ-type zinc finger domain, which is a nuclear localization/export signal sequence for nucleocytoplasmic shuttling; the TNF receptor associated factor 6 (TRAF6)-binding domain; the LC3-interacting region (LIR) of the motif; a Keap1-interacting region motif in the middle part of the molecule, and a Ub-associated (UBA) domain at the C-terminus (Katsuragi et al., 2015; Vadlamudi et al., 1996). Mammalian targeting of the rapamycin (mTOR) complex 1 is a major regulator for autophagy activation, and its kinase activity is tightly regulated by cellular nutrient status (Kim and Guan, 2015). In response to amino acid stimulation, p62 interacts with TRAF6, which induces ubiquitination of mTOR at K63, leading to the translocation of mTORC1 to the lysosome and subsequent activation at the lysosome membrane surface (Linares et al., 2013). The Keap1-Nrf2 signaling pathway is one of the major cellular defense signaling pathways against oxidative stress. Under stress conditions, Nrf2 is dissociated from Keap2 and translocates to activate target genes (Liu et al., 2017). The phosphorylation of Nrf2 by mTOR increases the interaction of p62 and Keap1, which inhibits the interaction of Nrf2-Keap1. As a result, Nrf2 is stabilized and functions as a transcription factor (Jain et al., 2010).
In addition, the p62 protein has been extensively studied as an autophagic substrate. It functions as a selective autophagy adaptor and is widely used as a monitoring marker for the autophagic flux (Klionsky et al., 2016). Ubiquitination is utilized as a signal for autophagic degradation of protein aggregates and damaged organelles, and p62 is localized to Ub-positive inclusions (Kirkin et al., 2009). Recent studies have reported that p62 contributes to selective autophagy of protein aggregates (aggrepahgy), damaged mitochondria (mitophagy), damaged lysosome (lysophagy), damaged endoplasmic reticulum (ER-phagy/reticulophagy), and invasive microbes (xenophagy) (Anding and Baehrecke, 2017; Katsuragi et al., 2015; Ryter et al., 2013). The p62 protein binds to mono- and polyubiquitinated components via its UBA domain, and p62 binds to the LC3 region via its LIR motif, which results in autophagic degradation of p62, as well as binding partners (Ichimura et al., 2008; Kirkin et al., 2009). The Ub modifications and p62 binding cooperate to target substrates to autophagosomes. The depletion of p62 strongly inhibits pexophagy, which further indicates that p62 is also an important regulator of pexopahgy (Kim et al., 2008). Although p62 usually recruits ubiquitinated cargos, and has a key role in the selective autophagy process, loss of p62 results in relatively little effect on selective autophagy, (Mathew et al., 2009) suggesting the involvement of other adaptor proteins.
Exogenous expression of NBR1 induces peroxisome clustering and targeting to the lysosomes to promote pexophagy, suggesting that NBR1 is another mediator for pexophagy (Deosaran et al., 2013). The molecular structure of NBR1 indicates that it shares a similar domain composition with that of p62. NBR1 also contains PB1 at the N-terminus, and the ZZ domain in the coiled-coil, LIR in the middle part of the molecule, an amphipathic alpha helical J domain (JUBA), and a UBA domain at the C-terminus (Mardakheh et al., 2010; Zientara-Rytter and Subramani, 2016). The PB1 domain of NBR1 mediates interactions with p62 and both C-terminal domains, and the JUVA and UBA domains are involved in localization of NBR1 on peroxisomes (Deosaran et al., 2013). Mutagenesis studies on the NBR1 domain have shown that JUBA, UBA, and LIR are required to mediate pexophagy (Deosaran et al., 2013). Because p62 does not have a JUBA domain that is required for subcellular localization, p62 colocalizes only with ubiquitinated peroxisomes and increases the efficiency of NBR1-mediated pexophagy by associating with NBR1 (Deosaran et al., 2013).
Besides NBR1 and p62, other proteins, including nuclear dot protein 52 kDa (NDP52), optineurin (OPTN), and Tax1-binding protein 1 (TAX1BP1), possess similar functions as a selective autophagy adaptor in mitophagy and xenophagy (Lazarou et al., 2015; Tumbarello et al., 2015; Von Muhlinen et al., 2010, Wong and Holzbaur, 2014). However, the role of NDP52, OPTN, and TAX1BP1 in pexophagy has not been elucidated. Thus, further studies of these selective autophagy adaptors in the process of pexophagy are needed.
Other regulators of pexophagy
In mitophagy, to remove mitochondria, various cytoplasmic adaptor proteins, such as NDP52, NBR1, OPTN, p62, parkin, and TAX1BP1, work together with mitochondrial membrane proteins, including Bcl-2 interacting protein 3, Bcl-2 interacting protein 3-like, FUN14 domain containing1, prohibitin-2, and PTEN-induced putative kinase 1 (PINK1) (Martinez-Vicente, 2017). Consistent with these mechanisms of action in mitophagy, several PMPs have been proposed to contribute to pexophagy in response to adaptor proteins.
PEX13, another complex protein for PEX5-mediated protein import, is also involved in selective autophagy regulation. Lee et al. reported that both mitochondrial fragmentation and mitophagy were not induced in PEX13 deficient cells (Lee et al., 2016). In addition, ectopic expression of PEX13 mutants, associated with peroxisome biogenesis disorders (PBDs) such as Zellweger syndrome (ZS), showed an increase of mitophagy impairment in PEX13 knockdown cells (Lee et al., 2016). These data suggested that PEX13 functions in mitophagy and virophagy rather than pexophagy (Lee et al., 2016). However, we recently found that the loss of PEX13 triggered pexophagy in neuroblastoma cells (unpublished data).
PEX14 and PEX13 comprise the protein import machinery involved in the docking complex for PEX5 (Hua and Kim, 2016). Jiang et al. recently reported that PEX14 directly interacted with LC3II protein and mediated pexophagy under starvation conditions (Jiang et al., 2015). In this process, NBR1 also interacted with PEX14, which is required for the localization of LC3 in peroxisomes. Not only the peroxisomal import machinery proteins but also the peroxisomal RING complex proteins are involved in pexophagy. Sargent et al. recently reported that peroxisomal E3 Ub ligase PEX2 was a causative regulator of pexophagy (Sargent et al., 2016), and PEX2 was upregulated during starvation conditions and in rapamycin-treated cells. Moreover, the overexpression of PEX2 led to gross ubiquitination of peroxisomal proteins such as PEX5 and NBR1, which further caused the degradation of peroxisomes during amino acid starvation (Sargent et al., 2016). Similar to PEX5, 70 kDa peroxisomal membrane protein (PMP70) is also a target of PEX2 and E3 ligase during autophagy activation (Sargent et al., 2016). Thus, the role of ubiquitinated PMP70 in pexophagy needs to be further investigated.
PATHOLOGICAL ASPECTS RELATED TO PEXOPHAGY AND PEROXISOMAL PROTEINS
PBDs and pexophagy
Peroxisomal disorders are caused by impaired peroxisomal biogenesis or metabolic defects of peroxisomes. They are basically genetic disorders, but mainly heterogeneous, thus making diagnosis difficult (Wanders et al., 2017). Various organs, including the brain, eye, liver, kidney, and adrenal cortex are affected in most peroxisomal disorders (Gould and Valle, 2000). PBDs are autosomal recessive disorders that involve impaired peroxisome assembly, peroxisomal enzyme deficiencies, and disabilities. The PBDs include ZS, one of the most severe forms of PBDs, manifested by various genetic and clinical heterogeneities (Zellweger et al., 1988). ZS child patients suffer from defects in brain morphogenesis, visual defects, and sensorineural deafness (Braverman et al., 2013). Later, the related disorders of ZS were reported, involving neonatal leukodystrophy, infantile Refsum disease, and rhizomelic chondrodysplasia punctata (Heymans et al., 1983; 1985; Moser et al., 1984). It is notable that patients with ZS have mitochondrial abnormalities (Goldfischer et al., 1973), which were also found in the liver-selective PEX5 null mouse model (Dirkx et al., 2005), with abnormal interconnections between mitochondrial and peroxisomal functions.
Most PBDs in humans are related to mutations in the peroxisomal AAA ATPase complex, consisting of PEX1, PEX6, and PEX26 (Nazarko, 2017). Thus, peroxisomal AAA ATPase plays an essential role in peroxisome biogenesis by fusion of the pre-peroxisomal vesicles and regulation of ubiquitinated PEX5 recycling. Importantly, Law et al. reported that a peroxisomal AAA ATPase complex prevented pexophagy and development of PBDs (Law et al., 2017). It has been reported that depletion of either PEX1 or PEX26 resulted in the loss of peroxisomes, which could be partly rescued by the simultaneous knockdown of NRB1 (Law et al., 2017). Together, these results suggested that most PBDs do not result from the inability to form peroxisomes but, rather, are caused by impairment of the ability to prevent peroxisome degradation. Thus, the function and role of pexophagy in PBDs needs to be further clarified.
In addition, that inhibition of autophagy can improve the biochemical functions of peroxisome in several PBD-related cells. It was recently reported that treatment of a lysosomal fusion inhibitor, chloroquine, leads to a decrease in very long chain fatty acid (VLCFA) and an increase in peroxisomal β-oxidation in PBD mutant fibroblasts generated from PEX1 defect or PEX-G843D PDB patients (Law et al., 2017). More importantly, recent studies suggest that pexophagy is responsible for the cause of 65% of PBDs (Nazarko, 2017). Nazarko (2017) indicated that a number of PBDs are not due to an inability in peroxisome biogenesis, but rather due to a dysregulation of autophagy. However, the relative contribution of pexophagy in PBD is largely unknown. Future studies with additional evidence will elucidate the exact contribution of pexophagy in PBD.
Peroxisomal proteins and pexophagy-related pathological conditions
Although there is no direct evidence for dysfunctions in human disease, PEX5 is able to bind an ataxia-telangiectasia mutation (ATM) whose signaling activated ULK1 to induce pexophagy (Zhang et al., 2015). ATM kinase is a well-characterized molecule causing ataxia-telangiectasia syndrome, an autosomal recessive disorder that manifests with neurodegeneration, telangiectasia, immunodeficiency, and various cancers (Choi et al., 2016; Choy and Watters, 2017; Van Os et al., 2016). Future studies will clarify the clinical relevance of ATM function in the context of human diseases related to pexophagy.
The acyl-CoA binding domain containing protein 5 (ACBD5), which is a human ortholog of Atg37, localizes to peroxisomes and functions in pexophagy (Nazarko, 2014). A recent study has reported that a pathogenic mutation in patients with ACBD5, a peroxisomal membrane protein with a cytosolic acyl-CoA binding domain, was linked to impaired very long-chain fatty acid metabolism, resulting in retinal dystrophy and white matter diseases (Ferdinandusse et al., 2017). It has also been reported that endotoxic injury caused pexophagy to maintain peroxisomal functions during lipopolysaccharide-induced inflammation. In a mouse acute kidney injury model, inhibition of pexophagy led to impaired peroxisomes, redox imbalance, and renal damage during inflammation (Vasko et al., 2013). However, these results need to be confirmed in human inflammatory diseases. Peroxisomal proteins and their pathological aspects have been summarized (Table 1).
CONCLUSION
Although peroxisomes are important organelles for numerous biological functions such as lipid metabolism, redox balance, and bile acid synthesis, the important factors and molecular mechanisms by which pexophagy is regulated still need to be fully elucidated. Overall, the biogenesis and degradation of peroxisomes are highly dynamic and strictly regulated. Emerging evidence has suggested potential relationships between the Ub-proteasome system and pexophagy as a degradation process involving peroxisomes. In addition, accumulating studies have identified the functions of adaptors such as p62 and NBR1 in mediating pexophagy. However, these adaptors do not exclusively play a role in pexophagy, and are involved in other selective autophagic processes, including xenophagy and mitophagy. Elucidating the roles of pexophagy adaptors and peroxisomal proteins will help to enable a better understanding of the molecular mechanisms of pexophagy and its interrelationships with diverse biological processes present in peroxisomes. Understanding how pexophagy is regulated and crosstalks with numerous physiological processes, especially cellular redox system, could contribute to pexophagy-based approaches for the development of novel defense strategies against pathological conditions related to peroxisomal dysfunctions.
ACKNOWLEDGMENTS
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean Government (MSIP) (No. 2017R1A5A2015385), and supported by research fund of Chungnam National University. This project was supported under the framework of international cooperation program managed by NRF of Korea (Grant Number: 2015K2A2A6002008). This research was also supported by a grant of the Korea–UK Collaborative Alzheimer’s Disease Research Project by Ministry of Health & Welfare, Republic of Korea (HI14C1913).
REFERENCES
- Al-Dirbashi OY, Shaheen R, Al-Sayed M, Al-Dosari M, Makhseed N, Abu Safieh L, Santa T, Meyer BF, Shimozawa N, Alkuraya FS. Zellweger syndrome caused by PEX13 deficiency: report of two novel mutations. Am J Med Genet A. 2009;149A:1219–1223. doi: 10.1002/ajmg.a.32874. [DOI] [PubMed] [Google Scholar]
- Anding AL, Baehrecke EH. Cleaning House: Selective Autophagy of Organelles. Dev Cell. 2017;41:10–22. doi: 10.1016/j.devcel.2017.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar⊘y T, Koster J, Stromme P, Ebberink MS, Misceo D, Ferdinandusse S, Holmgren A, Hughes T, Merckoll E, Westvik J, et al. A novel type of rhizomelic chondrodysplasia punctata, RCDP5, is caused by loss of the PEX5 long isoform. Hum Mol Genet. 2015;24:5845–5854. doi: 10.1093/hmg/ddv305. [DOI] [PubMed] [Google Scholar]
- Bj⊘rk⊘y G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonekamp NA, Völkl A, Fahimi HD, Schrader M. Reactive oxygen species and peroxisomes: struggling for balance. Biofactors. 2009;35:346–355. doi: 10.1002/biof.48. [DOI] [PubMed] [Google Scholar]
- Bowers WE. Christian de Duve and the discovery of lysosomes and peroxisomes. Trends Cell Biol. 1998;8:330–333. doi: 10.1016/s0962-8924(98)01314-2. [DOI] [PubMed] [Google Scholar]
- Braverman N, Steel G, Obie C, Moser A, Moser H, Gould SJ, Valle D. Human PEX7 encodes the peroxisomal PTS2 receptor and is responsible for rhizomelic chondrodysplasia punctata. Nat Genet. 1997;15:369–376. doi: 10.1038/ng0497-369. [DOI] [PubMed] [Google Scholar]
- Braverman NE, D’Agostino MD, Maclean GE. Peroxisome biogenesis disorders: Biological, clinical and pathophysiological perspectives. Dev Disabil Res Rev. 2013;17:187–196. doi: 10.1002/ddrr.1113. [DOI] [PubMed] [Google Scholar]
- Campbell IG, Nicolai HM, Foulkes WD, Senger G, Stamp GW, Allan G, Boyer C, Jones K, Bast RC, Jr, Solomon E. A novel gene encoding a B-box protein within the BRCA1 region at 17q21.1. Hum Mol Genet. 1994;3:589–594. doi: 10.1093/hmg/3.4.589. [DOI] [PubMed] [Google Scholar]
- Chang CC, Lee WH, Moser H, Valle D, Gould SJ. Isolation of the human PEX12 gene, mutated in group 3 of the peroxisome biogenesis disorders. Nat Genet. 1997;15:385–388. doi: 10.1038/ng0497-385. [DOI] [PubMed] [Google Scholar]
- Choi M, Kipps T, Kurzrock R. ATM mutations in cancer: therapeutic implications. Mol Cancer Ther. 2016;15:1781–1791. doi: 10.1158/1535-7163.MCT-15-0945. [DOI] [PubMed] [Google Scholar]
- Choy KR, Watters DJ. Neurodegeneration in ataxia-telangiectasia: Multiple roles of ATM kinase in cellular homeostasis. Dev Dyn. 2017 doi: 10.1002/dvdy.24522. 2017 May 2022. 2010.1002/dvdy.24522. [DOI] [PubMed] [Google Scholar]
- Deb R, Nagotu S. Versatility of peroxisomes: An evolving concept. Tissue Cell. 2017;49:209–226. doi: 10.1016/j.tice.2017.03.002. [DOI] [PubMed] [Google Scholar]
- Demarquoy J, Le Borgne F. Crosstalk between mitochondria and peroxisomes. World J Biol Chem. 2015;26:301–309. doi: 10.4331/wjbc.v6.i4.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deosaran E, Larsen KB, Hua R, Sargent G, Wang Y, Kim S, Lamark T, Jauregui M, Law K, Lippincott-Schwartz J, et al. NBR1 acts as an autophagy receptor for peroxisomes. J Cell Sci. 2013;126:939–952. doi: 10.1242/jcs.114819. [DOI] [PubMed] [Google Scholar]
- Dirkx R, Vanhorebeek I, Martens K, Schad A, Grabenbauer M, Fahimi D, Declercq P, Van Veldhoven PP, Baes M. Absence of peroxisomes in mouse hepatocytes causes mitochondrial and ER abnormalities. Hepatology. 2005;41:868–878. doi: 10.1002/hep.20628. [DOI] [PubMed] [Google Scholar]
- Du H, Kim S, Hur YS, Lee MS, Lee SH, Cheon CI. A cytosolic thioredoxin acts as a molecular chaperone for peroxisome matrix proteins as well as antioxidant in peroxisome. Mol Cells. 2015;38:187–194. doi: 10.14348/molcells.2015.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, Moscat J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13:343–354. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Ebberink MS, Koster J, Visser G, Spronsen Fv, Stolte-Dijkstra I, Smit GP, Fock JM, Kemp S, Wanders RJ, Waterham HR. A novel defect of peroxisome division due to a homozygous non-sense mutation in the PEX11beta gene. J Med Genet. 2012;49:307–313. doi: 10.1136/jmedgenet-2012-100778. [DOI] [PubMed] [Google Scholar]
- Feng L, Zhang J, Zhu N, Ding Q, Zhang X, Yu J, Qiang W, Zhang Z, Ma Y, Huang D, et al. Ubiquitin ligase SYVN1/HRD1 facilitates degradation of the SERPINA1 Z variant/alpha-1-antitrypsin Z variant via SQSTM1/p62-dependent selective autophagy. Autophagy. 2017;13:686–702. doi: 10.1080/15548627.2017.1280207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdinandusse S, Falkenberg KD, Koster J, Mooyer PA, Jones R, van Roermund CWT, Pizzino A, Schrader M, Wanders RJA, Vanderver A, et al. ACBD5 deficiency causes a defect in peroxisomal very long-chain fatty acid metabolism. J Med Genet. 2017;54:330–337. doi: 10.1136/jmedgenet-2016-104132. [DOI] [PubMed] [Google Scholar]
- Fransen M, Nordgren M, Wang B, Apanasets O. Role of peroxisomes in ROS/RNS-metabolism: implications for human disease. Biochim Biophys Acta. 2012;1822:1363–1373. doi: 10.1016/j.bbadis.2011.12.001. [DOI] [PubMed] [Google Scholar]
- Gào X, Schöttker B. Reduction-oxidation pathways involved in cancer development: a systematic review of literature reviews. Oncotarget. 2017;8:51888–51906. doi: 10.18632/oncotarget.17128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannopoulou EA, Emmanouilidis L, Sattler M, Dodt G, Wilmanns M. Towards the molecular mechanism of the integration of peroxisomal membrane proteins. Biochim Biophys Acta. 2016;1863:863–869. doi: 10.1016/j.bbamcr.2015.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfischer S, Moore CL, Johnson AB, Spiro AJ, Valsamis MP, Wisniewski HK, Ritch RH, Norton WT, Rapin I, Gartner LM. Peroxisomal and mitochondrial defects in the cerebrohepato-renal syndrome. Science. 1973;182:62–64. doi: 10.1126/science.182.4107.62. [DOI] [PubMed] [Google Scholar]
- Gould SJ, Valle D. Peroxisome biogenesis disorders: genetics and cell biology. Trends Genet. 2000;16:340–345. doi: 10.1016/s0168-9525(00)02056-4. [DOI] [PubMed] [Google Scholar]
- Heiland I, Erdmann R. Biogenesis of peroxisomes. Topogenesis of the peroxisomal membrane and matrix proteins. FEBS J. 2005;272:2362–2372. doi: 10.1111/j.1742-4658.2005.04690.x. [DOI] [PubMed] [Google Scholar]
- Heymans HS, Schutgens RB, Tan R, van den Bosch H, Borst P. Severe plasmalogen deficiency in tissues of infants without peroxisomes (Zellweger syndrome) Nature. 1983;306:69–70. doi: 10.1038/306069a0. [DOI] [PubMed] [Google Scholar]
- Heymans HS, Oorthuys JW, Nelck G, Wanders RJ, Schutgens RB. Rhizomelic chondrodysplasia punctata: another peroxisomal disorder. N Engl J Med. 1985;313:187–188. [PubMed] [Google Scholar]
- Hiltunen JK, Mursula AM, Rottensteiner H, Wierenga RK, Kastaniotis AJ, Gurvitz A. The biochemistry of peroxisomal beta-oxidation in the yeast Saccharomyces cerevisiae. FEMS Microbiol Rev. 2003;27:35–64. doi: 10.1016/S0168-6445(03)00017-2. [DOI] [PubMed] [Google Scholar]
- Honsho M, Yamashita S, Fujiki Y. Peroxisome homeostasis: Mechanisms of division and selective degradation of peroxisomes in mammals. Biochim Biophys Acta. 2016;1863:984–991. doi: 10.1016/j.bbamcr.2015.09.032. [DOI] [PubMed] [Google Scholar]
- Hu J, Baker A, Bartel B, Linka N, Mullen RT, Reumann S, Zolman BK. Plant peroxisomes: biogenesis and function. Plant Cell. 2012;24:2279–2303. doi: 10.1105/tpc.112.096586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua R, Kim PK. Multiple paths to peroxisomes: Mechanism of peroxisome maintenance in mammals. Biochim Biophys Acta. 2016;1863:881–891. doi: 10.1016/j.bbamcr.2015.09.026. [DOI] [PubMed] [Google Scholar]
- Huybrechts SJ, Van Veldhoven PP, Hoffman I, Zeevaert R, de Vos R, Demaerel P, Brams M, Jaeken J, Fransen M, Cassiman D. Identification of a novel PEX14 mutation in Zellweger syndrome. J Med Genet. 2008;45:376–383. doi: 10.1136/jmg.2007.056697. [DOI] [PubMed] [Google Scholar]
- Huybrechts SJ, Van Veldhoven PP, Brees C, Mannaerts GP, Los GV, Fransen M. Peroxisome dynamics in cultured mammalian cells. Traffic. 2009;10:1722–1733. doi: 10.1111/j.1600-0854.2009.00970.x. [DOI] [PubMed] [Google Scholar]
- Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T, Kominami E, Yamane T, Tanaka K, Komatsu M. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem. 2008;283:22847–22857. doi: 10.1074/jbc.M802182200. [DOI] [PubMed] [Google Scholar]
- Islinger M, Cardoso MJ, Schrader M. Be different--the diversity of peroxisomes in the animal kingdom. Biochim Biophys Acta. 2010;1803:881–897. doi: 10.1016/j.bbamcr.2010.03.013. [DOI] [PubMed] [Google Scholar]
- Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Øvervatn A, McMahon M, Hayes JD, Johansen T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285:22576–22591. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Hara-Kuge S, Yamashita S, Fujiki Y. Peroxin Pex14p is the key component for coordinated autophagic degradation of mammalian peroxisomes by direct binding to LC3-II. Genes Cells. 2015;20:36–49. doi: 10.1111/gtc.12198. [DOI] [PubMed] [Google Scholar]
- Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279–296. doi: 10.4161/auto.7.3.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuragi Y, Ichimura Y, Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015;282:4672–4678. doi: 10.1111/febs.13540. [DOI] [PubMed] [Google Scholar]
- Kiel JA, Veenhuis M, van der Klei IJ. PEX genes in fungal genomes: common, rare or redundant. Traffic. 2006;7:1291–1303. doi: 10.1111/j.1600-0854.2006.00479.x. [DOI] [PubMed] [Google Scholar]
- Kim PK. Peroxisome biogenesis: a union between two organelles. Curr Biol. 2017;27:R271–R274. doi: 10.1016/j.cub.2017.02.052. [DOI] [PubMed] [Google Scholar]
- Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125:25–32. doi: 10.1172/JCI73939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci USA. 2008;105:20567–20574. doi: 10.1073/pnas.0810611105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YS, Lee HM, Kim JK, Yang CS, Kim TS, Jung M, Jin HS, Kim S, Jang J, Oh GT, et al. PPAR-α activation mediates innate host defense through induction of TFEB and lipid catabolism. J Immunol. 2017;198:3283–3295. doi: 10.4049/jimmunol.1601920. [DOI] [PubMed] [Google Scholar]
- Kirkin V, Lamark T, Sou YS, Bj⊘rk⊘y G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33:505–516. doi: 10.1016/j.molcel.2009.01.020. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- Kumar S, Kawałek A, van der Klei IJ. Peroxisomal quality control mechanisms. Curr Opin Microbiol. 2014;22:30–37. doi: 10.1016/j.mib.2014.09.009. [DOI] [PubMed] [Google Scholar]
- Law KB, Bronte-Tinkew D, Di Pietro E, Snowden A, Jones RO, Moser A, Brumell JH, Braverman N, Kim PK. The peroxisomal AAA ATPase complex prevents pexophagy and development of peroxisome biogenesis disorders. Autophagy. 2017;13:868–884. doi: 10.1080/15548627.2017.1291470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Wagner M, Xiao R, Kim KH, Feng D, Lazar MA, Moore DD. Nutrient-sensing nuclear receptors coordinate autophagy. Nature. 2014;516:112–115. doi: 10.1038/nature13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MY, Sumpter R, Jr, Zou Z, Sirasanagandla S, Wei Y, Mishra P, Rosewich H, Crane DI, Levine B. Peroxisomal protein PEX13 functions in selective autophagy. EMBO Rep. 2017;18:48–60. doi: 10.15252/embr.201642443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linares JF, Duran A, Yajima T, Pasparakis M, Moscat J, Diaz-Meco MT. K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol Cell. 2013;51:283–296. doi: 10.1016/j.molcel.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Bjorkman J, Urquhart A, Wanders RJ, Crane DI, Gould SJ. PEX13 is mutated in complementation group 13 of the peroxisome-biogenesis disorders. Am J Hum Genet. 1999;65:621–634. doi: 10.1086/302534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XF, Hao JL, Xie T, Malik TH, Lu CB, Liu C, Shu C, Lu CW, Zhou DD. Nrf2 as a target for prevention of age-related and diabetic cataracts by against oxidative stress. Aging Cell. 2017;16:934–942. doi: 10.1111/acel.12645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardakheh FK, Auciello G, Dafforn TR, Rappoport JZ, Heath JK. Nbr1 is a novel inhibitor of ligand-mediated receptor tyrosine kinase degradation. Mol Cell Biol. 2010;30:5672–5685. doi: 10.1128/MCB.00878-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Vicente M. Neuronal mitophagy in neurodegenerative diseases. Front Mol Neurosci. 2017;10:64. doi: 10.3389/fnmol.2017.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–1075. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto N, Tamura S, Furuki S, Miyata N, Moser A, Shimozawa N, Moser HW, Suzuki Y, Kondo N, Fujiki Y. Mutations in novel peroxin gene PEX26 that cause peroxisome-biogenesis disorders of complementation group 8 provide a genotype-phenotype correlation. Am J Hum Genet. 2003;73:233–246. doi: 10.1086/377004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzono Y, Kinoshita N, Tamura S, Shimozawa N, Hamasaki M, Ghaedi K, Wanders RJ, Suzuki Y, Kondo N, Fujiki Y. Human PEX19: cDNA cloning by functional complementation, mutation analysis in a patient with Zellweger syndrome, and potential role in peroxisomal membrane assembly. Proc Natl Acad Sci USA. 1999;96:2116–2121. doi: 10.1073/pnas.96.5.2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayerhofer PU. Targeting and insertion of peroxisomal membrane proteins: ER trafficking versus direct delivery to peroxisomes. Biochim Biophys Acta. 2016;1863:870–880. doi: 10.1016/j.bbamcr.2015.09.021. [DOI] [PubMed] [Google Scholar]
- Moser AE, Singh I, Brown FR, 3rd, Solish GI, Kelley RI, Benke PJ, Moser HW. The cerebrohepatorenal (Zellweger) syndrome. Increased levels and impaired degradation of very-long-chain fatty acids and their use in prenatal diagnosis. N Engl J Med. 1984;310:1141–1146. doi: 10.1056/NEJM198405033101802. [DOI] [PubMed] [Google Scholar]
- Muntau AC, Mayerhofer PU, Paton BC, Kammerer S, Roscher AA. Defective peroxisome membrane synthesis due to mutations in human PEX3 causes Zellweger syndrome, complementation group G. Am J Hum Genet. 2000;67:967–975. doi: 10.1086/303071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarko TY. Atg37 regulates the assembly of the pexophagic receptor protein complex. Autophagy. 2014;10:1348–1349. doi: 10.4161/auto.29073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarko TY. Pexophagy is responsible for 65% of cases of peroxisome biogenesis disorders. Autophagy. 2017;13:991–994. doi: 10.1080/15548627.2017.1291480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordgren M, Francisco T, Lismont C, Hennebel L, Brees C, Wang B, Van Veldhoven PP, Azevedo JE, Fransen M. Export-deficient monoubiquitinated PEX5 triggers peroxisome removal in SV40 large T antigen-transformed mouse embryonic fibroblasts. Autophagy. 2015;11:1326–1340. doi: 10.1080/15548627.2015.1061846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol. 2015;62:720–733. doi: 10.1016/j.jhep.2014.10.039. [DOI] [PubMed] [Google Scholar]
- Platta HW, Hagen S, Reidick C, Erdmann R. The peroxisomal receptor dislocation pathway: to the exportomer and beyond. Biochimie. 2014;98:16–28. doi: 10.1016/j.biochi.2013.12.009. [DOI] [PubMed] [Google Scholar]
- Poirier Y, Antonenkov VD, Glumoff T, Hiltunen JK. Peroxisomal beta-oxidation--a metabolic pathway with multiple functions. Biochim Biophys Acta. 2006;1763:1413–1426. doi: 10.1016/j.bbamcr.2006.08.034. [DOI] [PubMed] [Google Scholar]
- Rakhshandehroo M, Hooiveld G, Müller M, Kersten S. Comparative analysis of gene regulation by the transcription factor PPARalpha between mouse and human. PLoS One. 2009;4:e6796. doi: 10.1371/journal.pone.0006796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuber BE, Germain-Lee E, Collins CS, Morrell JC, Ameritunga R, Moser HW, Valle D, Gould SJ. Mutations in PEX1 are the most common cause of peroxisome biogenesis disorders. Nat Genet. 1997;17:445–448. doi: 10.1038/ng1297-445. [DOI] [PubMed] [Google Scholar]
- Ryter SW, Cloonan SM, Choi AM. Autophagy: a critical regulator of cellular metabolism and homeostasis. Mol Cells. 2013;36:7–16. doi: 10.1007/s10059-013-0140-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargent G, van Zutphen T, Shatseva T, Zhang L, Di Giovanni V, Bandsma R, Kim PK. PEX2 is the E3 ubiquitin ligase required for pexophagy during starvation. J Cell Biol. 2016;214:677–690. doi: 10.1083/jcb.201511034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlüter A, Fourcade S, Ripp R, Mandel JL, Poch O, Pujol A. The evolutionary origin of peroxisomes: an ER-peroxisome connection. Mol Biol Evol. 2006;23:838–845. doi: 10.1093/molbev/msj103. [DOI] [PubMed] [Google Scholar]
- Shimozawa N, Imamura A, Zhang Z, Suzuki Y, Orii T, Tsukamoto T, Osumi T, Fujiki Y, Wanders RJ, Besley G, et al. Defective PEX gene products correlate with the protein import, biochemical abnormalities, and phenotypic heterogeneity in peroxisome biogenesis disorders. J Med Genet. 1999a;36:779–781. doi: 10.1136/jmg.36.10.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimozawa N, Suzuki Y, Zhang Z, Imamura A, Toyama R, Mukai S, Fujiki Y, Tsukamoto T, Osumi T, Orii T, et al. Nonsense and temperature-sensitive mutations in PEX13 are the cause of complementation group H of peroxisome biogenesis disorders. Hum Mol Genet. 1999b;8:1077–1083. doi: 10.1093/hmg/8.6.1077. [DOI] [PubMed] [Google Scholar]
- Shimozawa N, Zhang Z, Suzuki Y, Imamura A, Tsukamoto T, Osumi T, Fujiki Y, Orii T, Barth PG, Wanders RJ, et al. Functional heterogeneity of C-terminal peroxisome targeting signal 1 in PEX5-defective patients. Biochem Biophys Res Commun. 1999c;262:504–508. doi: 10.1006/bbrc.1999.1232. [DOI] [PubMed] [Google Scholar]
- Smith CE, Poulter JA, Levin AV, Capasso JE, Price S, Ben-Yosef T, Sharony R, Newman WG, Shore RC, Brookes SJ, et al. Spectrum of PEX1 and PEX6 variants in Heimler syndrome. Eur J Hum Genet. 2016;24:1565–1571. doi: 10.1038/ejhg.2016.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- South ST, Gould SJ. Peroxisome synthesis in the absence of preexisting peroxisomes. J Cell Biol. 1999;144:255–266. doi: 10.1083/jcb.144.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura S, Matsumoto N, Imamura A, Shimozawa N, Suzuki Y, Kondo N, Fujiki Y. Phenotype-genotype relationships in peroxisome biogenesis disorders of PEX1-defective complementation group 1 are defined by Pex1p-Pex6p interaction. Biochem J. 2001;357:417–426. doi: 10.1042/0264-6021:3570417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbarello DA, Manna PT, Allen M, Bycroft M, Arden SD, Kendrick-Jones J, Buss F. The autophagy receptor TAX1BP1 and the molecular motor myosin VI are required for clearance of salmonella typhimurium by autophagy. PLoS Pathog. 2015;11:e1005174. doi: 10.1371/journal.ppat.1005174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadlamudi RK, Joung I, Strominger JL, Shin J. p62, a phosphotyrosine-independent ligand of the SH2 domain of p56lck, belongs to a new class of ubiquitin-binding proteins. J Biol Chem. 1996;271:20235–20237. doi: 10.1074/jbc.271.34.20235. [DOI] [PubMed] [Google Scholar]
- Van den Brink DM, Brites P, Haasjes J, Wierzbicki AS, Mitchell J, Lambert-Hamill M, de Belleroche J, Jansen GA, Waterham HR, Wanders RJ. Identification of PEX7 as the second gene involved in Refsum disease. Am J Hum Genet. 2003;72:471–477. doi: 10.1086/346093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Os NJ, Roeleveld N, Weemaes CM, Jongmans MC, Janssens GO, Taylor AM, Hoogerbrugge N, Willemsen MA. Health risks for ataxia-telangiectasia mutated heterozygotes: a systematic review, meta-analysis and evidence-based guideline. Clin Genet. 2016;90:105–117. doi: 10.1111/cge.12710. [DOI] [PubMed] [Google Scholar]
- Vasko R, Ratliff BB, Bohr S, Nadel E, Chen J, Xavier S, Chander P, Goligorsky MS. Endothelial peroxisomal dysfunction and impaired pexophagy promotes oxidative damage in lipopolysaccharide-induced acute kidney injury. Antioxid Redox Signal. 2013;19:211–230. doi: 10.1089/ars.2012.4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A. 2010;107:378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Muhlinen N, Thurston T, Ryzhakov G, Bloor S, Randow F. NDP52, a novel autophagy receptor for ubiquitin-decorated cytosolic bacteria. Autophagy. 2010;6:288–289. doi: 10.4161/auto.6.2.11118. [DOI] [PubMed] [Google Scholar]
- Wanders RJ, Waterham HR, Ferdinandusse S. Metabolic Interplay between peroxisomes and other subcellular organelles including mitochondria and the endoplasmic reticulum. Front Cell Dev Biol. 2016;3:83. doi: 10.3389/fcell.2015.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanders RJ, Klouwer FC, Ferdinandusse S, Waterham HR, Poll-Thé BT. clinical and laboratory diagnosis of peroxisomal disorders. Methods Mol Biol. 2017;1595:329–342. doi: 10.1007/978-1-4939-6937-1_30. [DOI] [PubMed] [Google Scholar]
- Warren DS, Morrell JC, Moser HW, Valle D, Gould SJ. Identification of PEX10, the gene defective in complementation group 7 of the peroxisome-biogenesis disorders. Am J Hum Genet. 1998;63:347–359. doi: 10.1086/301963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterham HR, Ferdinandusse S, Wanders RJ. Human disorders of peroxisome metabolism and biogenesis. Biochim Biophys Acta. 2016;1863:922–933. doi: 10.1016/j.bbamcr.2015.11.015. [DOI] [PubMed] [Google Scholar]
- Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci USA. 2014;111:E4439–4448. doi: 10.1073/pnas.1405752111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita S, Abe K, Tatemichi Y, Fujiki Y. The membrane peroxin PEX3 induces peroxisome-ubiquitination-linked pexophagy. Autophagy. 2014;10:1549–1564. doi: 10.4161/auto.29329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yik WY, Steinberg SJ, Moser AB, Moser HW, Hacia JG. Identification of novel mutations and sequence variation in the Zellweger syndrome spectrum of peroxisome biogenesis disorders. Hum Mutat. 2009;30:E467–480. doi: 10.1002/humu.20932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellweger H, Maertens P, Superneau D, Wertelecki W. History of the cerebrohepatorenal syndrome of Zellweger and other peroxisomal disorders. South Med J. 1988;81:357–364. doi: 10.1097/00007611-198803000-00017. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Suzuki Y, Shimozawa N, Fukuda S, Imamura A, Tsukamoto T, Osumi T, Fujiki Y, Orii T, Wanders RJ, et al. Genomic structure and identification of 11 novel mutations of the PEX6 (peroxisome assembly factor-2) gene in patients with peroxisome biogenesis disorders. Hum Mutat. 1999;13:487–496. doi: 10.1002/(SICI)1098-1004(1999)13:6<487::AID-HUMU9>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Zhang J, Tripathi DN, Jing J, Alexander A, Kim J, Powell RT, Dere R, Tait-Mulder J, Lee JH, Paull TT, et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat Cell Biol. 2015;17:1259–1269. doi: 10.1038/ncb3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zientara-Rytter K, Subramani S. Autophagic degradation of peroxisomes in mammals. Biochem Soc Trans. 2016;44:431–440. doi: 10.1042/BST20150268. [DOI] [PMC free article] [PubMed] [Google Scholar]