

Abstract
Thirty-two gypsogenin derivatives were synthesized and screened for their cytotoxic activities. Their structures were established using IR, 1H NMR, 13C NMR, and LC-MS spectroscopic data. In MTT assays nearly all the compounds displayed good cytotoxicity in the low μM range for several human tumour cell lines (A549, LOVO, SKOV3 and HepG2). Low IC50 values were obtained especially for the carboxamides 7a–7j, for an oxime derivative 3 and a (2,4-dinitrophenyl)hydrazono derivative 4. In particular, the IC50 values of compounds 4 (IC50 = 2.97 ± 1.13 µΜ) and 7 g (IC50 = 3.59 ± 2.04 µΜ) against LOVO cells were found to be much lower than those of the other derivatives and parent compound. These compounds were submitted to an extensive biological testing and proved compounds 4 and 7 g to act mainly by an arrest of the tumour cells in the S phase of the cell cycle. In addition, compounds 4 and 7 g triggered the apoptotic pathway in cancer cells, showing high apoptosis ratios.
Keywords: gypsogenin derivatives, cytotoxic activity, apoptosis, cell cycle arrest
1. Introduction
Cancer is one of the most challenging problems in medicine. One of the several treatments is chemotherapy, and many chemotherapeutics have been developed so far. Many of them were gained from secondary natural products, for example vinca-alkaloids (from poisonous evergreen, Catharanthus roseus), taxeles (diterpenes first derived from the Pacific yew tree, Taxus brevifolia) and camptothecins (lactone alkaloids from Camptotheca acuminata). Moreover, it was reported that a regular consumption of fruits and herbs helps reduce carcinogenic risk [1,2].
Gypsophila oldhamiana, known as ‘xia cao’ in China, belongs to the Caryophyllaceae family [3]. The roots of Gypsophila species are an especially rich source of triterpenoid saponins [4,5]. Some triterpenoid saponins from Gypsophila have shown a variety of biological activities including anticarcinogenic [6], immunostimulatory [7], cytotoxicity [8–10] and α-glucosidase inhibition activities [11]. Gypsogenin (3-hydroxy-23-oxoolean-12-en-28-oic acid), a natural pentacyclic triterpenoid, has four active sites such as C-3 hydroxyl, ring-C double bond, C-23 aldehyde group and C-28 carboxylic acid, which are amenable for a wide range of chemical transformations. The structure of gypsogenin is shown in figure 1. This valuable compound has been detected and isolated from Gypsophila oldhamiana. Many biological activities have been credited to gypsogenin, such as inhibitory activity [12,13], cytotoxicity [14,15], antimicrobial and antiproliferative activities [16]. Of special interest are its cytotoxicity and its antiproliferative properties. In previous study, The C-28 carboxylic acid and C-23 aldehyde group were used to prepare nitrile and different types of esters. These compounds are the first pentacyclic triterpenoids described as a potent AChE-selective inhibitor [17]. The C-23 aldehyde group of gypsogenin was treated with hydroxylamine hydrochloride to provide oxime. This compound triggered the apoptotic pathway in cancer cells, showing high apoptosis ratios [16]. Thus, there is strong evidence that gypsogenin has anti-cancer activity.
Figure 1.

The structure of gypsogenin.
Oleanolic acid (3-hydroxy-12-en-28-oic acid), a pentacyclic triterpenoic acid, has three active sites such as C-3 hydroxyl, ring-C double bond and C-28 carboxylic acid. The structure of gypsogenin is very similar to the structure of oleanolic acid. The C-3 hydroxyl and C-28 carboxylic acid of oleanolic acid were used to prepare acetoxy and different types of amide and esters [18,19]. In particular, the different 3-O-acetyl oleanolic acid derived amides have been prepared and screened for their cytotoxic activity [20]. The results reveal that most of the carboxamides displayed good cytotoxicity in the low micromolar range for several human tumour cell lines. These compounds were submitted to an extensive biological testing and proved some compounds to act mainly by an arrest of the tumour cells in the S phase of the cell cycle. In addition, some compounds triggered the apoptotic pathway in cancer cells, showing high apoptosis ratios. These findings make oleanolic acid a promising lead compound for developing new cytotoxic/antitumour active compounds. In previous study, we have never found any references about the cytotoxicity of gypsogenin derived amides. Thus, it is valuable to investigate gypsogenin derived amides.
Gypsogenin aglycone is found at high concentrations in Gypsophila oldhamiana [21]; therefore, it can be obtained with ease [22]. In this study, the C-23 aldehyde group has been used to prepare hydrazone and oxime. The different types of esters and amide were designed, and synthesized at C-28 carboxylic acid. In addition, they were evaluated for their cytotoxic activities against four different human cancer cell cultures. More investigations about the mechanism of cell death induced by these gypsogenin derivatives were performed.
2. Results and discussion
2.1. Chemistry
Thirty-two gypsogenin derivatives were synthesized by a series of reactions as outlined in schemes 1–3. All compounds were obtained in different yields. The gypsogenin (1) was obtained by the hydrolysis of the gypsogenin saponin mixtures. For compound 1, the signals for H-3, H-12 and H-23 could be seen at δ 3.95 (1H, dd, J = 11.41, 5.15 Hz), 5.29 (1H, s) and 9.50 (1H, s), respectively. These signals were also evident by their 13C NMR spectra showing C-3, C-12 and C-23 at δ 71.52, 122.17 and 207.20, respectively. Gypsogenin was acetylated to afford compound 2 in 95.7% yields. In the 1H NMR spectrum for compound 2, the proton signal of H-3 at δ 5.23 (dd, J = 11.44, 5.16 Hz) was observed instead of at δ 3.95. Compounds 1 and 2 were mixed with hydroxylamine hydrochloride in pyridine at 105°C to provide compounds 3 and 5 in 85.1% and 96.5% yields, respectively. The structures of compounds 3 and 5 were confirmed by their respective 13C NMR spectra, which showed the characteristic C-23 carbon signals at δ 159.46 and 157.15 respectively. Compounds 1 and 2 were treated with 2,4-dinitrophenylhydrazine in acetic acid at room temperature to obtain compounds 4 and 6 in 88.9% and 86.5% yields, respectively. The 13C NMR spectra of compounds 4 and 6 showed the characteristic carbon signals of C-23 at δ 163.13 and 158.58 respectively.
Scheme 2.

Reagents and conditions: (d) CH2Cl2, oxalyl chloride, secondary amine or primary alcohol, room temperature.
Scheme 1.

Reagents and conditions: (a) 2,4-dinitrophenylhydrazine, acetic acid, room temperature; (b) hydroxylamine hydrochloride (NH2OH·HCl), pyridine, 105°C; (c) acetic anhydride, pyridine, room temperature.
Scheme 3.

Reagents and conditions: (e) ethyl alcohol absolute, 2.0 mol l−1 NaOH, 1.0 mol l−1, room temperature.
Consecutive reactions of the intermediate compounds 2 and 6 with oxalyl chloride and secondary amine in CH2Cl2 at room temperature gave the corresponding amides 7a–7g and 9a–9g in 82–93% and 85–93% yields respectively. Their 13C NMR spectra showed the characteristic carbon signals at δ 174.45–175.82 attributed to C-28. Under similar conditions, compounds 2 and 6 were coupled with oxalyl chloride and primary alcohols in CH2Cl2 at room temperature to obtain the corresponding esters 7 h–7j and 9h–9j in 89–94% and 89–92% yields, respectively. For compounds 7h–7j and 9h–9j, these carbon signals for C-28 could be seen at δ 177.42–178.22 respectively.
Compounds 7a–7g were coupled with NaOH in C2H5OH at room temperature to afford the corresponding amides 8a–8g in 82–89% yields. In the 1H NMR spectra for compounds 8a–8g, the proton signal of H-3 at δ 5.24–5.26 was converted to δ 4.06–4.10. The thirty-two compounds' spectral data are presented in the electronic supplementary material.
2.2. Biology
2.2.1. Cytotoxic activity
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay is a mitochondrial function assay that is based on the ability of viable cells to reduce the MTT to insoluble formazan crystals by mitochondrial dehydrogenase [23]. The anti-cancer activities of the synthesized compounds were studied by an MTT assay using the following human cancer cell lines in cell culture [24]: LOVO (a colon cancer cell line), SKOV3 (an ovary cancer cell line), A549 (a lung cancer cell line) and HepG2 (a liver cancer cell line). Cisplatin along with gypsogenin were taken as reference standards in this study. The IC50 values were determined for each compound and cell line. The results of tests are shown in table 1. It is evident that the synthesized compounds showed selectivity in exhibiting significant anti-cancer activity against the four cancer cell lines. Among the tested compounds, 3, 4 and 7a–7g were found to be more toxic than others of this series. The structure–activity relationship studies revealed that the introduction of an amide in the 28 position of the target compounds 7a–7g enhanced the anti-cancer activity. Similarly, the introduction of oxime and 2,4-dinitrophenylhydrazone in the 23 position of the target compounds 3 (IC50 = 12.35 ± 1.34 µΜ for LOVO cells) and 4 (IC50 = 2.97 ± 1.13 µΜ for LOVO cells) also led to an increase in anti-cancer activity. Our results revealed the superior anti-cancer activity of 4 and 7g (IC50=3.59 ± 2.04 µΜ for LOVO cells) compared to other compounds of the same series.
Table 1.
Cytotoxic activity of investigated compounds against human cancer cell lines.
| IC50 (μM) |
IC50 (μM) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| compound | A549 | LOVO | SKOV3 | HepG2 | compound | A549 | LOVO | SKOV3 | HepG2 |
| 1 | 19.60 ± 4.50 | 15.90 ± 1.87 | 20.67 ± 3.77 | 22.18 ± 2.62 | 8b | 28.83 ± 6.78 | 26.83 ± 2.44 | >30 | >30 |
| 2 | 30.86 ± 3.26 | 14.36 ± 2.21 | 21.20 ± 2.13 | 24.71 ± 3.15 | 8c | 27.77 ± 5.14 | >30 | >30 | >30 |
| 3 | 17.70 ± 2.49 | 12.35 ± 1.34 | 18.51 ± 1.18 | 19.15 ± 2.21 | 8d | 25.70 ± 2.68 | >30 | >30 | >30 |
| 4 | 3.10 ± 1.14 | 2.97 ± 1.13 | 10.04 ± 1.38 | 9.71 ± 2.06 | 8e | 15.77 ± 2.37 | 12.36 ± 2.35 | 25.63 ± 1.14 | 27.88 ± 2.42 |
| 5 | 28.23 ± 2.04 | 12.42 ± 1.03 | >30 | >30 | 8f | 15.03 ± 3.48 | 11.14 ± 1.87 | 24.59 ± 2.72 | 26.91 ± 2.74 |
| 6 | 26.50 ± 1.77 | 5.31 ± 1.26 | >30 | >30 | 8g | 14.16 ± 8.70 | 10.88 ± 1.32 | 27.38 ± 3.49 | 28.17 ± 2.88 |
| 7a | 17.08 ± 2.32 | 13.49 ± 2.86 | 16.23 ± 2.35 | 19.14 ± 2.53 | 9a | >30 | 25.98 ± 1.65 | >30 | >30 |
| 7b | 15.57 ± 2.17 | 10.44 ± 2.73 | 15.74 ± 1.44 | 17.50 ± 2.48 | 9b | >30 | 24.36 ± 1.48 | >30 | >30 |
| 7c | 7.32 ± 1.28 | 7.59 ± 1.63 | 10.10 ± 2.19 | 12.33 ± 2.63 | 9c | >30 | 24.06 ± 1.17 | >30 | >30 |
| 7d | 7.04 ± 1.54 | 6.18 ± 1.46 | 9.88 ± 1.54 | 11.68 ± 1.33 | 9d | >30 | 24.65 ± 2.05 | >30 | >30 |
| 7e | 11.05 ± 1.87 | 5.24 ± 1.69 | 17.34 ± 1.24 | 16.58 ± 1.84 | 9e | >30 | 28.54 ± 2.37 | >30 | >30 |
| 7f | 10.55 ± 2.10 | 4.96 ± 1.64 | 15.56 ± 1.68 | 13.14 ± 1.61 | 9f | >30 | 25.36 ± 2.43 | >30 | >30 |
| 7g | 9.24 ± 1.53 | 3.59 ± 2.04 | 13.16 ± 2.96 | 12.55 ± 1.41 | 9g | >30 | 24.55 ± 1.42 | >30 | >30 |
| 7h | 8.36 ± 3.97 | 22.37 ± 3.08 | 24.77 ± 2.78 | >30 | 9h | >30 | >30 | >30 | >30 |
| 7i | 8.04 ± 3.65 | 28.93 ± 2.89 | >30 | 29.61 ± 2.44 | 9i | >30 | >30 | >30 | >30 |
| 7j | 9.47 ± 2.81 | 26.42 ± 3.25 | 28.74 ± 3.24 | >30 | 9j | >30 | >30 | >30 | >30 |
| 8a | >30 | >30 | >30 | >30 | cisplatin | 0.48 ± 0.21 | 0.32 ± 0.17 | 1.55 ± 0.43 | 0.96 ± 0.51 |
2.2.2. Morphological observation by acridine orange and ethidium bromide staining
The morphological abnormalities induced by compounds 4 and 7g in LOVO cells were studied under fluorescence microscopy using the acridine orange (AO)/ethidium bromide (EB) staining technique [25]. AO permeates the intact cell membrane and stains the nuclei green, while EB can stain the cells that have lost their membrane integrity and tinge the nucleus red. Intact cells therefore exhibit homogeneous green nuclei, whereas apoptotic cells show condensed or fragmented chromatin. Early apoptotic cells have green nuclei. Late apoptotic or necrotic cells have orange to red nuclei. It can be interpreted from figures 2 and 4 that the control cells showed the normal healthy morphology with intact nuclear architecture and appeared green in colour. Fluorescence microscopic images of LOVO cells treated with compounds 4 and 7g have clearly demonstrated morphological changes which are the characteristic features of apoptotic cells such as cell shrinkage, membrane blebbing and apoptotic body formation. Compared with spontaneous apoptosis observed in control cells (figure 2), LOVO cells treated for 48 h with 5 µM of compound 4 showed a marked increase in percentage of early apoptotic/necrotic cells (figure 2). Cells treated for 48 h with 5 µM of compound 7g showed a significant increase in percentage of late apoptotic/ necrotic cells (figure 4). Apoptotic rates of LOVO cells treated with and without compounds 4 and 7g for 48 h are shown in figure 3 and table 2.
Figure 2.

(a) Morphological changes in LOVO cells treated with and without compound 4 for 48 h: (i) LOVO control cells; (ii) LOVO cells treated with 5 µM for 48 h followed by morphological observation using AO/EB cell staining method. (b) Effect of LOVO on cell cycle progression of colon cancer cells: (i) LOVO control cells; (ii,iii) LOVO cells treated with 5 and 10 µM for 48 h followed by analysis of cell cycle distribution using propidium iodide cell staining method. Cell population in each cell cycle phase was numerically depicted. Data represent one of three independent experiments.
Figure 4.
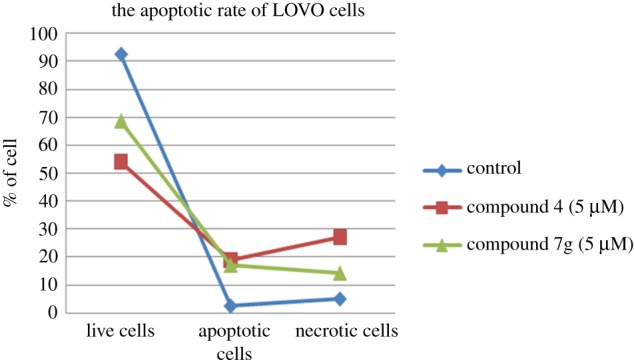
Apoptotic rate of cells treated with and without compounds 4 (5 µM) and 7 g (5 µM) for 48 h.
Figure 3.

(a) Morphological changes in LOVO cells treated with compound 7 g for 48 h: (i) LOVO control cells; (ii) LOVO cells treated with 5 µM for 48 h followed by morphological observation using AO/EB cell staining method. (b) Effect of LOVO on cell cycle progression of colon cancer cells: (i,ii) LOVO cells treated with 5 and 10 µM for 48 h followed by analysis of cell cycle distribution using propidium iodide cell staining method. Cell population in each cell cycle phase was numerically depicted. Data represent one of three independent experiments.
Table 2.
Apoptotic rate of cells treated with and without compounds 4 (5 µM) and 7 g (5 µM) for 48 h.
| live cells | apoptotic cells | necrotic cells | |
|---|---|---|---|
| control | 92.5 | 2.5 | 5.0 |
| compound 4 (5 µM) | 54.1 | 18.9 | 27.0 |
| compound 7 g (5 µM) | 68.6 | 17.1 | 14.3 |
2.2.3. Cell cycle analysis by propidium iodide staining
To further investigate the differential growth inhibition mechanism mediated by amide and 2,4-dinitrophenylhydrazone compounds on cell cycle dynamics, the effects of compounds 4 and 7g on cell cycle progression were determined by propidium iodide (PI) staining method [26]. LOVO cells treated with compounds 4 and 7g for 48 h resulted in a marked accumulation of cells in S-phase and a slight reduction in G0/G1 phase as shown. Treatment with the compound 4 at 2.5 µM and 5.0 µM in LOVO cells displayed rise in S-phase population from 11.9% (control) to 15.5% and 21.9% respectively in a dose-dependent manner. For compound 7g, the ratio of cells in the S phase increased from 11.9% (control) to 13.0% in cells treated with 2.5 µM and 27.4% in cells treated with 5.0 µM. The results of these experiments showed a dose-dependent S-phase arrest in cells treated with compound 4 and compound 7 g. These results indicated that the compounds 4 and 7g inhibited the growth of the cancer cells by inhibiting the cell cycle. Especially, the effect of compound 4 is better than compound 7g. The results are shown in figures 2 and 4.
3. Conclusion
In summary, we synthesized thirty-two gypsogenin derivatives in good yield. These compounds were found to be promising lead compounds against the majority of the studied cancer cell lines, exhibiting IC50 values much lower than that of the parent compound. The synthesized compounds showed selectivity in exhibiting significant anti-cancer activity against the human cancer cell lines. The structure–activity relationship studies revealed that the introduction of amide in the 28 position and 2,4-dinitrophenylhydrazone in the 23 position of target compounds enhanced the anti-cancer activity. Among all of these compounds, compounds 4 and 7g could be considered as possible anti-cancer agents as they were shown to affect the cell cycle, causing cell cycle arrest. In addition, compounds 4 and 7g triggered the apoptotic pathway in cancer cells, showing strong activity in promoting apoptosis.
4. Experimental
4.1. General
Melting points of all compounds were recorded on an Optimelt-100 automatic melting point apparatus and are uncorrected. IR (KBr) spectra were recorded with a Thermo Nicolet Nexus 670 FT-IR. The NMR spectra were obtained with a Bruker Avance III 600 spectrometer (1H: 600 MHz, 13C: 150 MHz) with tetramethylsilane as internal standard. Chemical shifts are given in values of ppm and coupling constants in hertz. LC/MS was recorded with an Agilent 1200 capillary spectrometer. Thin-layer chromatography (TLC) was performed on precoated silica gel plates (Qingdao Marine Chemical Industry Factory, Qingdao, China). Column chromatography was carried out using silica gel (200–300 mesh, Qingdao Marine Chemical Industry Factory, Qingdao, China).
4.2. General method for the preparation of gypsogenin (1)
The air-dried roots (20 kg) of Gypsophila oldhamiana were extracted with H2O under refluxing three times at room temperature. The water extract was evaporated in vacuo to obtain a yellow mixture (1000 ml). This mixture was hydrolysed with 10% HCl for 72 h before being neutralized with NaOH and extracted with ethyl acetate. The ethyl acetate phase was concentrated to a yellow residue (5.9 kg). Flash chromatography (silica gel, hexane–ethyl acetate, 3 : 1) followed by washing of the crude product (hexane–ethyl acetate, 10 : 1) gave pure gypsogenin (1.3 g) as a white solid.
4.2.1. (3β)-Hydroxy-23-oxoolean-12-en-28-oic acid (gypsogenin (1))
White solid, mp: 273.1–274.2°C, LC/MS (ESI-MS): m/z = 493.10 (M + 23) (positive ion mode); 1H NMR (C5D5N, 600 MHz) δ: 9.50 (1H, s, H-23), 5.29 (1H,br s, 12-H), 3.95 (1H, t, J = 7.8 Hz, H-3), 3.19 (1H, dd, J = 14.44, 4.63 Hz, H-18), 1.72 (1H, s, H-24), 1.24 (3H, s, H-27), 1.07 (3H, s, H-30), 0.94 (3H, s, H-26), 0.90 (3H, s, H-29), 0.80 (3H, s, H-25); 13C NMR (C5D5N, 150 MHz) δ: 207.20 (C-23), 180.10 (C-28), 144.83 (C-13), 122.17 (C-12), 71.52 (C-3), 56.23 (C-4), 47.91 (C-9), 47.61 (C-5), 46.56 (C-17), 46.38 (C-19), 42.14 (C-14), 41.94 (C-18), 40.00 (C-8),38.38 (C-1), 36.15 (C-10), 34.13 (C-21), 33.20 (C-29), 33.47 (C-22), 33.09 (C-7), 30.89 (C-20), 28.19 (C-15), 26.70 (C-2), 26.07 (C-27), 23.79 (C-16), 23.68 (C-30), 23.57 (C-11), 20.97 (C-6), 17.28 (C-26), 15.60 (C-25), 9.60 (C-24).
4.3. General procedure for the synthesis of compound 2
Acetic anhydride (15 ml) and dimethylaminopyridine (16.25 mg, 1.33 × 10−4 mol) were added to gypsogenin (1) (625 mg, 1.33 × 10−3 mol) in pyridine (5 ml). The mixture was stirred at room temperature for 24 h. The product was detected with TLC (chloroform–methanol, 100 : 1). After completion of the reaction, the mixture was extracted with ethyl acetate (3 × 20 ml). The organic layer was dried over anhydrous sodium sulfate and evaporated to dryness. The residue was purified by a silica gel column chromatography using chloroform–acetone (100 : 1) to afford the products.
4.3.1. (3β)-Acetyloxy-23-oxoolean-12-en-28-oic acid (2)
White solid, yield: 95.7%, mp: 164.6–166.5°C, IR (KBr) νmax: 3424, 2947, 1735, 1696, 1463, 1372, 1239, 1031, 1009, 728, 684, 634, 601, 514, 415 cm−1; LC/MS (ESI-MS): m/z = 535.20 (M + 23) (positive ion mode); 1H NMR (C5D5N, 600 MHz) δ: 9.52 (1H, s, H-23) 5.48 (1H, br s, 12-H), 5.23 (1H, dd, J = 11.44, 5.16 Hz, H-3), 3.30 (1H, dd, J = 14.44, 4.63 Hz, H-18), 1.94 (3H, s, CH3COO-), 1.28 (1H, s, H-24), 1.18 (3H, s, H-27), 1.02 (3H, s, H-30), 0.97 (3H, s, H-26), 0.96 (3 H, s, H-29), 0.85 (3H, s, H-25); 13C NMR (C5D5N, 150 MHz) δ: 204.74 (C-23), 180.18 (C-28), 170.18 (CH3COO-),144.89 (C-13), 122.10 (C-12), 73.51 (C-3), 54.65 (C-4), 47.77(C-9), 47.75 (C-5), 46.62 (C-17), 46.43 (C-19), 42.20 (C-14), 41.97 (C-18), 39.98 (C-8), 37.71 (C-1), 35.98 (C-10), 34.20 (C-21), 33.28 (C-29), 33.15 (C-22), 32.36 (C-7), 30.97 (C-20), 28.24 (C-15), 26.27 (C-27), 23.76 (C-30), 23.66 (C-11), 23.61 (C-16), 22.79 (C-2), 20.82 (CH3COO-),20.74 (C-6), 17.28 (C-26), 15.44 (C-25),9.75 (C-24).
4.4. General procedure for the synthesis of compounds 3 and 5
Gypsogenin (1) (50 mg, 1.06 × 10−4 mol) and compound 2 (60 mg, 1.17 × 10−4 mol) were mixed with hydroxylamine hydrochloride (11.05 mg and 12.19 mg, respectively) and dissolved in pyridine (5 ml). The mixture was stirred at 105°C for 4 h. After stirring, water (10 ml) was added to this mixture, and the mixture was extracted with CHCl3 (3 × 15 ml). The combined organic layer was dried over anhydrous sodium sulfate and evaporated to dryness. The crude material was purified by silica gel chromatography using chloroform–methanol (100 : 4) to give compounds 3 and 5, respectively.
4.4.1. (3β)-Hydroxy-23-(hydroxyimino) olean-12-en-28-oic acid (3)
White solid, yield: 85.1%, mp: 268.0–270.0°C, IR (KBr) νmax: 3939, 3398, 3200, 2941, 1724, 1694, 1458, 1388, 1261, 1180, 956, 816, 734, 591, 476 cm−1. LC/MS (ESI-MS): m/z = 508.30 (M + 23) (positive ion mode); 1H NMR(C5D5N, 600 MHz) δ: 7.76(s, 1H, -CHNOH), 5.51(br s, 1H, H-12), 3.88(dd, J = 11.40, 4.2 Hz, 1H, H-3), 3.32(dd, J = 13.80, 4.20 Hz, 1H, H-18), 1.47(s, 1H, H-24), 1.29(s, 3H, H-27), 1.02(s, 3H, H-30), 1.01(s, 3H, H-26), 0.97(s, 3H, H-29), 0.95(s, 3H, H-25); 13C NMR (C5D5N, 150 MHz) δ: 180.15 (C-28), 159.46 (C-23), 144.88 (C-13), 122.40 (C-12), 75.20 (C-3), 51.96 (C-4), 48.25 (C-9), 47.12 (C-5), 46.66 (C-17), 46.46 (C-19), 42.22 (C-14), 42.04 (C-18), 40.04 (C-8),38.70 (C-1), 36.89 (C-10), 34.22 (C-21), 33.29 (C-20), 33.19 (C-22), 32.87 (C-7), 30.98 (C-29), 28.31 (C-15), 27.15 (C-2), 26.20 (C-27), 23.82 (C-11), 23.78 (C-30), 23.68 (C-16), 20.25 (C-6), 17.42 (C-26), 15.97 (C-25), 12.32 (C-24).
4.4.2. (3β)-Acetyloxy-23-(hydroxyimino) olean-12-en-28-oic acid (5)
White solid, yield: 96.5%, mp: 251.2–252.8°C, IR (KBr) νmax: 3860, 3731, 3260, 2940, 2630, 1731, 1688, 1463, 1370, 1263, 1178, 1035, 1008, 942, 810, 684, 646, 608, 558 cm−1; LC/MS (ESI-MS): m/z = 528.50 (M + 1) (positive ion mode); 1H NMR (C5D5N, 600 MHz) δ: 7.63 (s, 1H, -CHNOH), 5.47 (br s, 1H, H-12), 5.16 (dd, J = 11.45, 5.17 Hz, 1H, H-3), 3.30 (dd, J = 12.00, 6.00 Hz, 1H, H-18), 1.95 (s, 3H, CH3COO-), 1.35 (s, 1H, H-24), 1.27 (s, 3H, H-27), 1.02 (s, 3H, H-30), 0.97 (s, 3H, H-26), 0.97 (s, 3H, H-29), 0.89 (s, 3H, H-25); 13C NMR (C5D5N, 150 MHz) δ: 180.14 (C-28), 170.18 (CH3COO-), 157.15 (C-23), 144.91 (C-13), 122.21 (C-12), 77.04 (C-3), 52.38 (C-4), 46.63 (C-5), 46.41(C-17), 45.42 (C-19), 42.19 (C-14), 41.98 (C-18), 39.97 (C-8), 37.97 (C-1), 36.76 (C-10), 34.22 (C-21), 33.28 (C-20), 33.19 (C-22), 32.65 (C-7), 30.98 (C-29), 47.99 (C-9), 30.01 (C-2), 28.27 (C-15), 26.21 (C-27), 23.78 (C-30), 23.65 (C-11), 23.29 (C-16), 21.10 (CH3COO-), 19.96 (C-6), 17.33 (C-26), 15.80 (C-25), 12.71 (C-24).
4.5. General procedure for the synthesis of compounds 4 and 6
Gypsogenin (50 mg, 1.06 × 10−4 mol) and compound 2 (60 mg, 1.17 × 10−4 mol) were mixed with 2,4-dinitrophenylhydrazine (31.5 mg and 34.8 mg, respectively) and dissolved in acetic acid (5 ml). The mixture was stirred at room temperature for 4 h. After stirring, the water (10 ml) was added to this mixture, and the mixture was extracted with CHCl3 (3 × 15 ml). The combined organic layer was dried over anhydrous sodium sulfate and evaporated to dryness. The crude material was purified by silica gel chromatography using chloroform–methanol (100 : 2) to afford compounds 4 and 6, respectively.
4.5.1. (3β)-Hydroxy-23-[(2,4-dinitrophenyl)hydrazono]olean-12-en-28-oic acid (4)
Yellow solid, yield: 88.9%, mp: 206–207.1°C, IR(KBr) νmax: 3452, 3302, 3101, 2941, 2860, 2853, 1697, 1618, 1589, 1519, 1463, 1425, 1386, 1332, 1277, 1216, 1139, 1075, 958, 920, 832, 712, 646, 580, 526, 454 cm−1; LC/MS (ESI-MS): m/z = 673.10 (M + 23) (positive ion mode); 1H NMR(C5D5N, 600 MHz) δ: 11.54 (s, 1H, -CHNNH-), 9.07 (d, 2.4 Hz, 1H, PhH-3), 8.33 (dd, 9.6, 2.4 Hz, 1H, PhH-5), 8.06 (d, 9.6 Hz, 1H, PhH-6), 7.90 (s, 1H, -CHNNH-), 5.52 (br s, 1H, 12-H), 3.96 (dd, J = 11.40, 5.40 Hz, 1H, H-3), 3.32 (dd, J = 13.50, 4.50 Hz, 1H, H-18), 1.51 (s, 1H, H-24), 1.27 (s, 3H, H-27), 1.03 (s, 3H, H-30), 1.03 (s, 3H, H-26),0.98 (s, 3H, H-29),0.98 (s, 3H, H-25); 13C NMR (C5D5N, 150 MHz) δ: 180.19 (C-28), 163.13 (C-23), 145.55 (Ph-1), 144. 84 (C-13), 137.44 (Ph-4), 129.79 (Ph-5), 129.06 (Ph-2), 123.84 (Ph-6), 122.34 (C-12), 116.95 (Ph-3), 74.99 (C-3), 51.50 (C-4), 49.29 (C-9), 48.32 (C-5), 46.66 (C-17), 46.49 (C-19), 42.20 (C-14), 42.02 (C-18), 40.05 (C-8), 38.64 (C-1), 36.74 (C-10), 36.74 (C-21), 34.22 (C-22), 33.30 (C-29), 33.16 (C-20), 32.75 (C-7), 30.98 (C-15), 28.31 (C-2), 27.15 (C-11), 26.17 (C-27), 23.81 (C-16), 23.77 (C-30), 20.59 (C-6), 17.42 (C-26), 15.94 (C-25), 11.77 (C-24).
4.5.2. (3β)-Acetyloxy-23-[(2,4-dinitrophenyl)hydrazono]olean-12-en-28-oic acid (6)
Yellow solid, yield: 86.5%, mp: 230.2–231.3°C, IR (KBr) νmax: 3295, 3103, 2930, 2861, 2853, 1697, 1618, 1591, 1520, 1460, 1428, 1370, 1333, 1270, 1241, 1071, 1026, 945, 920, 832, 739, 613, 515, 449 cm−1; LC/MS (ESI-MS): m/z = 692.1 (M − 1) (negative ion mode); 1H NMR(CDCl3, 600 MHz) δ: 11.00 (s, 1H, -CHNNH-), 9.12 (d, 2.4 Hz, 1H, PhH-3), 8.30 (dd, 9.6, 2.4 Hz, 1H, PhH-5), 7.91 (d, 9.6 Hz, 1H, PhH-6), 7.25 (s, 1H, -CHNNH-), 5.29 (br s, 1H, 12-H), 4.92 (dd, J = 11.70, 4.20 Hz, 1H, H-3), 2.83 (dd, J = 14.10, 4.50 Hz, 1H, H-18), 1.96 (s, 3H, CH3COO-), 1.23 (s, 1H, H-24), 1.16 (s, 3H, H-27), 1.04 (s, 3H, H-30), 0.93 (s, 3H, H-26), 0.91 (s, 3H, H-29), 0.77 (s, 3H, H-25); 13C NMR (CDCl3, 150 MHz) δ: 183.73 (C-28), 170.54 (CH3COO-), 158.58 (C-23), 145.12 (Ph-1), 143.63 (C-13), 137.84 (Ph-4), 129.97 (Ph-5), 128.99 (Ph-2), 123.53 (Ph-6), 122.17 (C-12), 116.44 (Ph-3), 76.00 (C-3), 52.06 (C-4), 47.80 (C-9), 47.26 (C-5), 46.52 (C-17), 45.78 (C-19), 41.65 (C-14), 40.98 (C-18), 39.60 (C-8), 37.84 (C-1), 36.55 (C-10), 33.75 (C-22), 33.05 (C-20), 32.41 (C-7), 32.22 (C-21), 30.68 (C-29), 29.70 (C-2), 27.63 (C-15), 25.94 (C-30), 23.56 (C-27), 22.95 (C-11), 22.78 (C-16), 21.21 (CH3 COO-), 20.11 (C-6), 17.04 (C-26), 15.80 (C-25), 11.98 (C-24).
4.6. General procedure for the synthesis of compounds 7a–7j
Oxalyl chloride (2.0 × 10−1 ml, 2.36 × 10−3 mol) was added to compound 2 (40 mg, 7.8 × 10−5 mol) in CH2Cl2 (3 ml). The mixture was allowed to stir for 12 h at room temperature. After completion, the reaction mixture was neutralized with Et3N and evaporated to dryness. To a stirred solution of the mixture in dry CH2Cl2 (3 ml) was added piperidine (4.0 × 10−2 ml, 4.04 × 10−4 mol). The stirring was continued for 8 h at room temperature. After CH2Cl2 evaporation, water (5 ml) was added to this mixture, and the mixture was extracted with ethyl acetate (3 × 5 ml). The combined organic layer was dried over anhydrous sodium sulfate and evaporated to dryness. The crude material was purified by silica gel chromatography using chloroform–methanol (100 : 2) to afford compound 7a. Compounds 7b–7j were prepared as 7a.
4.6.1. N-Piperidy-(3β)-acetyloxy-23-oxoolean-12-en-28-amide (7a)
White solid, yield: 85%, mp: 117.2–118.1°C, IR (KBr) νmax: 3435, 3358, 2937, 2854, 1735, 1630, 1465, 1446, 1371, 1240, 1028, 1002, 800, 739, 679, 641, 602, 526, 471 cm−1; LC/MS (ESI-MS): m/z = 580.50 (M + 1) (positive ion mode); 1H NMR (C5D5N, 600 MHz) δ: 9.53 (s, 1H, H-23), 5.44 (br s, 1H, 12-H), 5.24 (dd, J = 11.10, 5.70 Hz, 1H, H-3), 3.59 (m, 4H, -N(CH2)2-(CH2)3), 3.42 (dd, J = 14.70, 3.90 Hz, 1H, H-18), 1.94 (s, 3H, CH3COO-), 1.50 (m, 4H, -N(CH2)2-(CH2)2-CH2), 1.31 (m, 2H, -N(CH2)4-CH2), 1.27 (s, 1H, H-24), 1.22 (s, 3H, H-27), 0.98 (s, 3H, H-30), 0.97 (s, 3H, H-26), 0.92 (s, 3H, H-29),0.91 (s, 3H, H-25); 13C NMR (C5D5N, 150 MHz) δ: 204.79 (C-23), 174.45 (C-28), 170.19 (CH3COO-), 145.61 (C-13), 121.29 (C-12), 73.54 (C-3), 54.69 (C-4), 47.89 (C-9), 47.86 (C-5), 47.54 (-N(CH2)2-(CH2)3), 46.82 (C-17), 46.65 (C-19), 44.18 (C-14), 42.28 (C-18), 39.84 (C-8), 37.75 (C-1), 36.05 (C-10), 36.05 (C-21), 34.30 (C-22), 33.20 (C-20), 32.59 (C-7), 30.58 (C-29), 29.98 (C-2), 28.32 (C-15), 26.51 (-N(CH2)2-(CH2)2-CH2), 26.13 (C-11), 25.05 (-N(CH 2)4-CH2), 24.19 (C-16), 23.64 (C-30), 22.81 (C-27), 20.83 (CH3COO-), 20.83 (C-6), 17.20 (C-26), 15.57 (C-25), 9.78 (C-24).
4.6.2. N-Morpholino-(3β)-acetyloxy-23-oxoolean-12-en-28-amide (7b)
White solid, yield: 86.7%, mp: 129.8–135.1°C, IR(KBr) νmax: 3430, 2948, 2853, 2695, 1734, 1635, 1457, 1298, 1238, 1183, 1118, 1025, 975, 893, 849, 745, 690, 657, 526, 460 cm−1; LC/MS (ESI-MS): m/z = 582.50 (M + 1) (positive ion mode); 1H NMR (C5D5N, 600 MHz) δ: 9.54 (s, 1H, H-23), 5.44 (br s, 1H, 12-H), 5.25 (dd, J = 11.10, 5.70 Hz, 1H, H-3), 3.73–3.80 (m, 4H, -N(CH2)2-(CH2)2O), 3.69–3.72 (m, 4H, -N(CH2)2-(CH2)2O), 3.39 (dd, J = 15.00, 5.40 Hz, 1H, H-18), 1.95 (s, 3H, CH3COO-), 1.26 (s, 1H, H-24), 1.22 (s, 3H, H-27), 0.98 (s, 3H, H-30), 0.97 (s, 3H, H-26), 0.91 (s, 3H, H-29), 0.89 (s, 3H, H-25); 13C NMR (C5D5N, 150 MHz) δ: 204.84 (C-23), 175.03 (C-28), 170.23 (CH3COO-), 145.34 (C-13), 121.51 (C-12), 73.56 (C-3), 67.17 (-N(CH2)2-(CH2)2O), 54.69 (C-4), 47.86 (C-9), 47.57 (C-5), 47.57 (-N(CH2)2-(CH2)2O), 46.63 (C-17), 46.39 (C-19), 44.08 (C-14), 42.28 (C-18), 39.84 (C-8), 37.75 (C-1), 36.04 (C-10), 36.04 (C-21), 34.17 (C-22), 33.17 (C-20), 32.51 (C-7), 30.56 (C-29),30.04 (C-2), 28.16 (C-15), 26.16 (C-11), 24.14 (C-16), 23.63 (C-30), 22.80 (C-27), 20.84 (CH3COO-) ,20.82 (C-6), 17.11 (C-26), 15.57 (C-25), 9.78 (C-24).
4.6.3. N-(1-Methyl-piperazinyl)-(3β)-acetyloxy-23-oxoolean-12-en-28-amide (7c)
White solid, yield: 92.4%, mp: 129.8–131.3°C, IR(KBr) νmax: 3413, 2945, 2854, 2794, 2690, 1732, 1630, 1460, 1405, 1293, 1252, 1141, 1031, 1001, 893, 821, 772, 745, 690, 657, 531, 471 cm−1; LC/MS (ESI-MS): m/z = 595.5 (M + 1) (positive ion mode); 1H NMR (C5D5N, 600 MHz) δ: 9.54 (s, 1H, H-23), 5.44 (br s, 1H, 12-H), 5.25 (dd, J = 12.00, 6.00 Hz, 1H, H-3), 3.76–3.83 (m, 4H,-N(CH 2)2(CH2)2NCH3), 3.39 (dd, J = 12.00, 4.20 Hz, 1H, H-18), 2.38–2.44 (m, 4H, -N(CH2)2 (CH2)2 NCH3), 2.24 (s, 3H,-N(CH2)4NCH3), 1.95 (s, 3H, CH3COO-), 1.26 (s, 1H, H-24), 1.22 (s, 3H, H-27), 0.98 (s, 3H, H-30), 0.97 (s, 3H, H-26), 0.90 (s, 3H, H-29), 0.90 (s, 3H, H-25); 13C NMR (C5D5N, 150 MHz) δ: 204.78 (C-23), 174.73 (C-28), 170.18 (CH3COO-), 145.44 (C-13), 121.38 (C-12), 73.49 (C-3), 55.45 (-N(CH2)2(CH2)2NCH3), 54.70 (C-4), 47.84 (C-9), 47.78 (C-5), 47.51 (C-17), 46.67 (C-19), 45.89 (-N(CH2)4NCH3), 45. 54 (-N (CH2)2(CH2)2NCH3), 42.22 (C-14), 42.22 (C-18), 39.81 (C-8), 37.69 (C-1), 36.01 (C-10), 36.01 (C-21), 34.22 (C-22), 33.20 (C-20), 32.50 (C-7), 30.57 (C-29), 30.03 (C-2), 28.21 (C-15), 26.14 (C-11), 24.13 (C-16), 23.62 (C-30), 22.78 (C-27), 20.85 (CH3COO-), 20.80 (C-6), 17.15 (C-26), 15.55 (C-25), 9.77 (C-24).
4.6.4. N-(1-Ethyl-piperazinyl)-(3β)-acetyloxy-23-oxoolean-12-en-28-amide (7d)
White solid, yield: 91.7%, mp: 126.2–128.1°C, IR (KBr) νmax: 3534, 3424, 3194, 2943, 2849, 2805, 2695, 1732, 1630, 1455, 1407, 1378, 1251, 1141, 1163, 1023, 938, 904, 810, 767, 745, 684, 635, 602, 526, 460 cm−1; LC/MS (ESI-MS): m/z = 609.5 (M + 1) (positive ion mode); 1H NMR (C5D5N, 600 MHz) δ: 9.53 (s, 1H, H-23), 5.44 (brs, 1H, 12-H), 5.25 (dd, J = 11.10, 5.70 Hz, 1H, H-3), 3.44 (dd, J= 15.60, 4.20 Hz, 1H, H-18), 3.74–3.81 (m, 4H, -N(CH2)2(CH2)2 NC2 H5), 2.37–2.44 (m, 4H, -N(CH2)2(CH2)2NC2H5), 2.32 (q, 2H, -N (CH2)4 NCH2CH3), 1.94 (s, 3H, CH3COO-), 1.27 (s, 1H, H-24), 1.22 (s, 3H, H-27), 1.04 (t, 3H, -N(CH2)4NCH2CH3), 0.99 (s, 3H, H-30), 0.97 (s, 3H, H-26), 0.92 (s, 3H, H-29), 0.91 (s, 3H, H-25); 13C NMR (C5D5N, 150 MHz) δ: 204.75 (C-23), 174.68 (C-28), 170.15 (CH3C OO-), 145.50 (C-13), 121. 41 (C-12), 73.54 (C-3), 54.69 (-N(CH 2)2(CH2)2NC2H5), 53.41 (-N(CH2)2(CH2)2NC2H5), 52.38 (C-4), 47.90 (C-9), 47.86 (C-5), 47.55 (C-17), 46.72 (C-19), 45.89 (-N(CH2)4 NCH2CH3), 42.12 (C-14), 42.28 (C-18), 39.87 (C-8), 37.75 (C-1), 36.06 (C-10), 36.06 (C-21), 34.26 (C-22), 33.21 (C-20), 32.57 (C-7), 30.59 (C-29), 30.13 (C-2), 28.27 (C-15), 26.18 (C-11), 24.17 (C-16), 23.65 (C-30), 22.82 (C-27), 20.83 (CH3COO-), 20.83 (C-6), 17.22 (C-26), 15.57 (C-25), 12.30 (-N(C H2)4 NCH2CH3), 9.78 (C-24).
4.6.5. N,N-Dimethyl-(3β)-acetyloxy-23-oxoolean-12-en-28-amide (7e)
White solid, yield: 82.1%, mp: 184.1–186.4°C, IR (KBr) νmax: 3446, 2947, 2865, 2695, 1735, 1629, 1465, 1370, 1239, 1141, 1029, 975, 904, 816, 684, 641, 564, 526 cm−1; LC/MS (ESI-MS): m/z = 540.40 (M + 1) (positive ion mode); 1H NMR (C5D5N, 600 MHz) δ: 9.54 (s, 1H, H-23), 5.42 (br s, 1H, 12-H), 5.25 (dd, J = 11.1,5.7 Hz, 1H, H-3), 3.43 (dd, J = 13.5, 3.9 Hz, 1H, H-18), 3.01 (s, 6H,-N(CH3)2), 1.95 (s, 3H, CH3COO-), 1.25 (s, 1H, H-24), 1.21 (s, 3H, H-27), 0.97 (s, 3H, H-30), 0.96 (s, 3H, H-26), 0.88 (s, 3H, H-29), 0.84 (s, 3H, H-25); 13C NMR (C5D5N, 150 MHz) δ: 204.74 (C-23), 175.82 (C-28), 170.16 (CH3COO-), 145.51 (C-13), 121.47 (C-12), 73.54 (C-3), 54.70 (C-4), 47.88 (C-9), 47.86 (C-5), 47.57 (C-17), 46.58 (C-19), 43.84 (C-14), 42.31 (C-18), 39.76 (C-8), 38.61 (-N(CH3)2), 37.72 (C-1), 36.04 (C-10), 36.04 (C-21), 34.13 (C-22), 33.25 (C-20), 32.42 (C-7), 30.65 (C-29), 29.89 (C-2), 27.99 (C-15), 26.32 (C-11), 24.19 (C-16), 23.63 (C-30), 22.81 (C-27), 20.83 (CH3COO-), 20.79 (C-6), 17.03 (C-26), 15.51 (C-25), 9.78 (C-24).
4.6.6. N,N-Diethyl-(3β)-acetyloxy-23-oxoolean-12-en-28-amide (7f)
White solid, yield: 85.5%, mp: 175.8–176.1°C, IR (KBr) νmax: 2939, 2869, 2695, 1732, 1624, 1466, 1406, 1378, 1253, 1139, 1031, 936, 893, 739, 635, 608, 558, 526, 471 cm−1; LC/MS (ESI-MS): m/z = 568.6 (M + 1) (positive ion mode); 1H NMR (C5D5N, 600 MHz) δ: 9.55 (s, 1H, H-23), 5.46 (br s, 1H, 12-H), 5.26 (dd, J = 10.8, 6.0 Hz, 1H, H-3), 3.34–3.44 (m, 4H, -N(CH2-CH3)2), 3.43 (dd, J = 14.45, 4.62 Hz, 1H, H-18), 1.95 (s, 3H, CH3COO-), 1.27 (s, 1H, H-24), 1.22 (s, 3H, H-27), 1.15 (t, 6H, -N(CH2CH3)2), 0.97 (s, 3H, H-30), 0.96 (s, 3H, H-26), 0.92 (s, 3H, H-29), 0.89 (s, 3H, H-25); 13C NMR (C5D5 N,150 MHz) δ: 204.81 (C-23), 174.75 (C-28), 170.20 (CH3COO-), 145.61 (C-13), 121.22 (C-12), 73.50 (C-3), 54.72 (C-4), 47.83 (C-9), 47.81 (C-5), 47.65 (C-17), 47.06 (C-19), 44.13 (-N(CH2CH3)2), 42.25 (C-14), 42.36 (C-18), 39.92 (C-8), 37.74 (C-1), 36.03 (C-10), 36.03 (C-21), 34.41 (C-22), 33.18 (C-20), 32.64 (C-7), 30.56 (C-29), 30.03 (C-2), 28.41 (C-15), 25.95 (C-11), 24.11 (C-16), 23.66 (C-30), 22.79 (C-27), 20.85 (CH3COO-), 20.83 (C-6), 17.46 (C-26), 15.57 (C-25), 13.61 (-N(CH2CH3)2), 9.79 (C-24).
4.6.7. N,N-(n-Dibutyl)-(3β)-acetyloxy-23-oxoolean-12-en-28-amide (7g)
White solid, yield: 82.4%, mp: 169.7–171.1°C, IR (KBr) νmax: 3424, 2947, 1735, 1696, 1463, 1372, 1239, 1031, 1009, 728, 684, 634, 601, 514, 415 cm−1; LC/MS (ESI-MS): m/z = 624.6 (M + 1) (positive ion mode); 1H NMR (C5D5N, 600 MHz) δ: 9.52 (s, 1H, H-23), 5.46 (br s, 1H, 12-H), 5.24 (dd, J = 11.4, 5.7 Hz, 1H, H-3), 3.43 (dd, J = 14.44, 4.63 Hz, 1H, H-18), 3.35 (br s, 4H, -N(CH2-C3H7)2), 1.94 (s, 3H, CH3COO-), 1.31–1.36 (m, 8H, -N(CH2C2H4CH3)2), 1.29 (s, 1H, H-24), 1.20 (s, 3H, H-27), 0.98 (s, 3H, H-30), 0.97 (s, 3H, H-26), 0.96 (s, 3H, H-29), 0.95 (t, 6H, -N(C3H6CH3)2), 0.90 (s, 3H, H-25); 13C NMR (C5D5N, 150 MHz) δ: 204.73 (C-23), 174.98 (C-28), 170.14 (CH3COO-), 145.68 (C-13), 121.29 (C-12), 73.54 (C-3), 54.69 (C-4), 47.90 (C-9), 48.42, 44.19 (-N(CH2C3H7)2), 47.87 (C-5), 47.87 (C-17), 47.18 (C-19), 44.19 (C-14), 42.44 (C-18), 40.00 (C-8), 37.78 (C-1), 36.07 (C-10), 36.07 (C-21), 34.42 (C-22), 33.16 (C-20), 32.69 (C-7), 30.58 (C-29), 30.01 (C-2), 29.94, 26.03 (-N(CH2CH2C2H5)2), 28.55 (C-15), 26.03 (C-11), 24.23 (C-16), 23.69 (C-30), 22.82 (C-27), 20.82 (CH3COO-), 20.71 (C-6), 19.59, 14.20 (-N(C2 H4CH2CH3)2), 17.52 (C-26), 15.58 (C-25), 14.20 (-N(C3H6CH3)2), 9.79 (C-24).
4.6.8. Methyl (3β)-acetyloxy-23-oxoolean-12-en-28-oate (7h)
White solid, yield: 93.5%, mp: 160.5–162.2°C, IR (KBr) νmax: 3424, 2950, 2879, 2950, 1720, 1446, 1378, 1244, 1165, 1032, 969, 882, 810, 745, 635, 564, 465 cm−1; LC/MS (ESI-MS): m/z = 549.3 (M + 23) (positive ion mode); 1H NMR (C5D5N, 600 MHz) δ: 9.52 (s, 1H, H-23), 5.39 (br s, 1H, 12-H), 5.24 (dd, J = 11.40, 5.40 Hz, 1H, H-3), 3.72 (s, 3H, -OCH3); 3.11 (dd, J = 14.10, 4.50 Hz, 1H, H-18), 1.94 (s, 3H, CH3COO-),1.22 (s, 1H, H-24), 1.21 (s, 3H, H-27), 0.95 (3H, s, H-30), 0.94 (s, 3H, H-26), 0.89 (s, 3H, H-29), 0.80 (s, 3H, H-25); 13C NMR (C5D5N, 150 MHz) δ: 204.68 (C-23), 177.94 (C-28), 170.15 (CH3COO-), 144.22 (C-13), 122.44 (C-12), 73.50 (C-3), 54.66 (C-4), 51.64 (-OCH3), 47.74 (C-9), 47.68 (C-5), 46.90 (C-17), 46.04 (C-19), 42. 01 (C-14), 41.79 (C-18), 39.90 (C-8), 37.72 (C-1), 35.98 (C-10), 35.98 (C-21), 33.94 (C-22), 33.14 (C-20), 32.75 (C-7), 32.22 (C-2), 30.84 (C-29), 28.01 (C-15), 26.13 (C-30), 23.62 (C-27), 23.34 (C-11), 22.81 (C-16), 20.83 (CH3COO-), 20.73 (C-6), 17.04 (C-26), 15.48 (C-25), 9.79 (C-24).
4.6.9. Ethyl (3β)-acetyloxy-23-oxoolean-12-en-28-oate (7i)
White solid, yield: 91.6%, mp: 163.5–165.3°C, IR(KBr) νmax: 3430, 2977, 2940, 2876, 2739, 2677, 2604, 2494, 2356, 1731, 1654, 1475, 1444, 1397, 1243, 1172, 1073, 1036, 854, 810, 454 cm−1; LC/MS (ESI-MS): m/z = 563.2 (M + 23) (positive ion mode); 1H NMR (C5D5N, 600 MHz) δ: 9.21 (s, 1H, H-23), 5.23 (br s, 1H, 12-H), 4.91 (dd, J = 11.40, 4.80 Hz, 1H, H-3), 4.01 (q, 2H,-OCH2CH3), 2.80 (dd, J = 14.44, 4.63 Hz, 1H, H-18), 1.89 (s, 3H, CH3COO-), 1.15 (t, 3H,-OCH2 CH3), 1.08 (s, 1H, H-24), 1.01 (s, 3H, H-27), 0.91 (s, 3H, H-30), 0.86 (s, 3H, H-26), 0.83 (s, 3H, H-29), 0.68 (s, 3H, H-25); 13C NMR (C5D5N, 150 MHz) δ: 204.55 (C-23), 177.63 (C-28), 170.32 (CH3COO-), 143.95 (C-13), 121.80 (C-12), 73.38 (C-3), 60.10 (-OCH2CH3), 54.34 (C-4), 47.84 (C-9), 47.58 (C-5), 46.45 (C-17), 45.88 (C-19), 41.77 (C-14), 41.31 (C-18), 39.68 (C-8), 37.82 (C-1), 35.83 (C-10), 33.88 (C-21), 33.11 (C-22), 32.35 (C-20), 32.07 (C-7), 30.71 (C-29), 29.71 (C-15), 27.60 (C-2), 25.84 (C-11), 23.62 (C-16), 23.36 (C-30), 22.50 (C-27), 21.02 (CH3COO-), 20.46 (C-6), 16.97 (C-26), 15.88 (C-25), 14.28 (-OCH2CH3), 9.49 (C-24).
4.6.10. n-Dibutyl (3β)-acetyloxy-23-oxoolean-12-en-28-oate (7j)
White solid, yield: 89.6%, mp: 164.4–166.7°C, IR (KBr) νmax: 3424, 2958, 2926, 2843, 2794, 2679, 1742, 1715, 1468, 1369, 1232, 1205, 1161, 1178, 1117, 1172, 1079, 1024, 969, 936, 816, 756, 619, 553, 465 cm−1; LC/MS (ESI-MS): m/z = 569.3 (M + 1) (positive ion mode); 1H NMR (C5D5N, 600 MHz) δ: 9.52 (s, 1H, H-23), 5.42 (br s, 1H, 12-H), 5.24 (dd, J = 11.40, 5.40 Hz, 1H, H-3), 4.19 (q, 2H, -OCH 2C3H7), 3.14 (dd, J = 13.80, 4.80 Hz, 1H, H-18), 1.94 (s, 3H, CH3C OO-), 1.90(dd, 2H, -OCH2CH2C2H5), 1.39 (q, 2H, -OC2H4CH2CH3), 1.24 (s, 1H, H-24), 1.21 (s, 3H, H-27), 0.98 (s, 3H, H-30), 0.95 (s, 3H, H-26), 0.90 (t, 3H, -OC3H6CH3), 0.90 (s, 3H, H-29), 0.87 (s, 3H, H-25); 13C NMR (C5D5N,150 MHz) δ: 204.67 (C-23), 177.42 (C-28), 170.15 (CH3COO-), 144.25 (C-13), 122.46 (C-12), 73.50 (C-3), 64.16 (-OCH2C3H7), 54.66 (C-4), 47.74 (C-9), 47.68 (C-5), 46.90 (C-17), 46.09 (C-19), 42.10 (C-14), 41.84 (C-18), 40.00 (C-8), 37.75 (C-1), 35.99 (C-10), 34.01 (C-22), 33.16 (C-20), 32.91 (C-7), 32.36 (C-2), 31.03 (C-21), 30.88 (C-29), 30.02 (-O CH2CH2C2H5), 27.98 (C-15), 26.08 (C-30), 23.66 (C-27), 23.32 (C-11), 22.81 (C-16), 20.83 (CH3COO-), 20.74 (C-6), 19.55 (-OC2H4 CH2CH3), 17.29 (C-26), 15.49 (C-25), 14.28 (-OC3H6CH3), 9.79 (C-24).
4.7. General procedure for the synthesis of compounds 8a–8j
NaOH (1.0 ml, 2.0 mol l−1) was added to compound 7a (60 mg, 1.04 × 10−4 mol) in C2H5OH (6 ml). The mixture was allowed to stir for 6 h at room temperature. After completion, the reaction mixture was neutralized with HCl (1.0 mol l−1) and organic layer was dried over anhydrous sodium sulfate and evaporated to dryness. The crude material was purified by silica gel chromatography using chloroform–methanol (100 : 3) to obtain compound 8a. Compounds 8b–8j were prepared as 8a.
4.7.1. N-Piperidyl-(3β)-hydroxy-23-oxoolean-12-en-28-amide (8a)
White solid, yield: 86.5%, mp: 220.2–223.4°C, IR (KBr) νmax: 3423, 3178, 2932, 2851, 2673, 1728, 1600, 1479, 1421, 1364, 1243, 1134, 1052, 997, 767, 673, 635, 580, 526, 438 cm−1; LC/MS (ESI-MS): m/z = 538.4 (M + 1) (positive ion mode); 1H NMR (C5D5N, 600 MHz) δ: 9.66 (s, 1H, H-23), 5.43 (br s, 1H, 12-H), 4.06 (dd, J = 11.10, 5.70 Hz, 1H, H-3), 3.59 (m, 4H, -N(CH2)2-(CH2)3), 3.45 (dd, J= 15.60, 3.90 Hz, 1H, H-18), 1.50 (m, 4H, -N(CH2)2-(CH2)2-CH2), 1.40 (s, 1H, H-24), 1.31 (m, 2H, -N(CH2)4-CH2), 1.27 (s, 3H, H-27), 0.98 (s, 3H, H-30), 0.95 (s, 3H, H-26), 0.95 (s, 3H, H-29), 0.89 (s, 3H, H-25); 13C NMR (C5D5N, 150 MHz) δ: 207.43 (C-23), 174.49 (C-28), 145.64 (C-13), 121.51 (C-12), 71.73 (C-3), 56.39 (C-4), 48.15 (C-9), 47.84 (C-5), 47.59 (-N(CH2)2-(CH2)3), 46.87 (C-17), 46.69 (C-19), 44.27 (C-14), 42.33 (C-18), 39.95 (C-8), 38.54 (C-1), 36.27 (C-10), 36.27 (C-21), 34.34 (C-22), 33.24 (C-20), 32.82 (C-7), 30.63 (C-2), 30.04 (C-29), 28.39 (C-15), 27.13 (C-11), 26.55 (-N(CH2)2-(CH2)2-C H2), 26.16 (-N(CH2)4-CH2), 25.09 (C-16), 24.22 (C-30), 23.79 (C-27), 21.16 (C-6), 17.34 (C-26), 15.84 (C-25), 9.76 (C-24).
4.7.2. N-Morpholino-(3β)-hydroxy-23-oxoolean-12-en-28-amide (8b)
White solid, yield: 84.3%, mp: 223.8–225.1°C, IR (KBr) νmax: 3456, 2946, 2852, 2668, 1729, 1605, 1456, 1417, 1262, 1183, 1114, 1025, 1003, 893, 849, 734, 679, 597, 526 cm−1; LC/MS (ESI-MS): m/z = 540.5 (M + 1) (positive ion mode); 1H NMR (C5D5N, 600 MHz) δ: 9.66 (s, 1H, H-23), 5.46 (br s, 1H, 12-H), 4.10 (dd, J = 9.30, 6.90 Hz, 1H, H-3), 3.72–3.80 (m, 4H, -N(CH2)2(CH2)2O), 3.67–3.71 (m, 4H, -N(CH2)2(CH2)2O), 3.42 (dd, J = 14.40, 4.80 Hz, 1H, H-18), 1.40 (s, 1H, H-24), 1.27 (s, 3H, H-27), 0.98 (s, 3H, H-30), 0.96 (s, 3H, H-26), 0.96 (s, 3H, H-29), 0.92 (s, 3H, H-25); 13C NMR (C5D5N, 150 MHz) δ: 207.36 (C-23), 174.95 (C-28), 145.34 (C-13), 121.70 (C-12), 71.70 (C-3), 67.18 (-N(CH2)2(CH2)2O), 56.35 (C-4), 48.10 (C-9), 47.82 (C-5), 47.58 (-N(CH2)2(CH2)2O), 46.66 (C-17), 46.40 (C-19), 44.13 (C-14), 42.30 (C-18), 39.93 (C-8), 38.51 (C-1), 36.23 (C-10), 36.23 (C-21), 34.19 (C-22), 33.18 (C-20), 32.71 (C-7), 30.58 (C-29), 30.08 (C-2), 28.21 (C-15), 27.11 (C-11), 26.16 (C-16), 24.15 (C-30), 23.75 (C-27), 21.13 (C-6), 17.21 (C-26), 15.81 (C-25), 9.73 (C-24).
4.7.3. N-(1-Methyl-piperazinyl)-(3β)-hydroxy-23-oxoolean-12-en-28-amide (8c)
White solid, yield: 86.7%, mp: 118.3–189.7°C, IR (KBr) νmax: 3431, 2941, 2859, 2793, 2678, 1731, 1632, 1459, 1417, 1293, 1254, 1141, 1052, 1002, 810, 777, 738, 684, 645, 596, 470 cm−1; LC/MS (ESI-MS): m/z = 553.4 (M + 1) (positive ion mode); 1H NMR (C5D5N, 600 MHz) δ: 9.54 (1H, s, H-23), 5.46 (1H, br s,12-H), 4.10 (1H, dd, J = 12.00, 6.00 Hz, H-3), 3.81–3.88 (4H, m, -N(CH2)2(CH2)2NCH3), 3.43 (1H, dd, J = 14.40, 4.20 Hz, H-18), 2.47–2.53 (4H, m, -N(CH 2)2(CH2)2 N CH3), 2.31 (3H, s, -N(CH2)4NCH3), 1.40 (1H, s, H-24), 1.26 (3H, s, H-27), 0.98 (3H, s, H-30), 0.96 (3H, s, H-26), 0.96 (3H, s, H-29), 0.92 (3H, s, H-25); 13C NMR (C5D5N, 150 MHz) δ: 207.40 (C-23), 174.86 (C-28), 145.41 (C-13), 121.63 (C-12), 71.71 (C-3), 56.35 (C-4), 55.31 (-N(CH2)2(CH2)2NCH3), 48.10 (C-9), 47.80 (C-5), 47.59 (C-17), 46.72 (C-19), 45.64 (-N(CH2)4N-CH3), 45.35 (-N(CH 2)2(CH2)2NCH3), 44.15 (C-14), 42.27 (C-18), 39.93 (C-8), 38.50 (C-1), 36.22 (C-10), 36.22 (C-22), 34.24 (C-21), 33.19 (C-20), 32.71 (C-7), 30.57 (C-29), 30.10 (C-2), 28.28 (C-15), 27.09 (C-11), 26.15 (C-16), 24.15 (C-30), 23.76 (C-27), 21.12 (C-6), 17.26 (C-26), 15.81 (C-25), 9.72 (C-24).
4.7.4. N-(1-Ethyl-piperazinyl)-(3β)-hydroxy-23-oxoolean-12-en-28-amide (8d)
White solid, yield: 87.2%, mp: 124.7–126.1°C, IR (KBr) νmax: 3442, 2943, 2848, 2815, 2695, 1729, 1604, 1451, 1385, 1353, 1265, 1226, 1089, 1050, 1007, 925, 766, 689, 634, 602, 596, 536 cm−1; LC/MS (ESI-MS): m/z = 567.5 (M + 1) (positive ion mode); 1H NMR (C5D5N, 600 MHz) δ: 9.65(1H, s, H-23), 5.47 (1H, br s, 12-H), 4.10 (1H, t, J = 8.00 Hz, H-3), 3.75–3.82 (4H,m,-N(CH2)2(CH2)2NC2H5), 3.44 (1H, d, J = 13.8 Hz, H-18), 2.40–2.46 (4H, m, -N(CH2)2(CH2)2NC2H5), 2.33 (2H, q, -N(CH2)4NCH2CH3), 1.39 (1H, s, H-24), 1.27 (3H, s, H-27), 1.05 (3H, t, -N(CH2)4NCH2CH3), 0.99 (3H, s, H-30), 0.97 (3H, s, H-26), 0.96 (3H, s, H-29), 0.94 (3H, s, H-25); 13C NMR (C5D5N, 150 MHz) δ: 207.38 (C-23), 174.74 (C-28), 145.47 (C-13), 121.59 (C-12), 71.70 (C-3), 56.34 (C-4), 53.39 (-N(CH2)2(CH2)2N C2H5), 52.38 (-N(CH2)2(CH2)2 NC2H5), 48.11 (C-9), 47.79 (C-5), 47.57 (C-17), 46.74 (C-19), 45.79 (-N(CH2)4NCH2 CH3), 44.16 (C-14), 42.28 (C-18), 39.94 (C-8), 38.50 (C -1), 36.22 (C-10), 36.22 (C-21), 34.25 (C-22), 33.21 (C-20), 32.74 (C-7), 30.58 (C-29), 30.13 (C-2), 28.30 (C-15), 27.07 (C-11), 26.18 (C-16), 24.18 (C-30), 22.82 (C-27), 21.12 (C-6), 17.30 (C-26), 15.80 (C-25), 12.28 (-N(CH 2)4 NCH2CH3), 9.71 (C-24).
4.7.5. N,N-Dimethyl-(3β)-hydroxy-23-oxoolean-12-en-28-amide (8e)
White solid, yield: 88.3%, mp: 121.4–123.3°C, IR (KBr) νmax: 3388, 2923, 2852, 2723, 1720, 1605, 1468, 1381, 1265, 1149, 1090, 1052, 975, 904, 823, 739, 690, 613, 569, 531, 465 cm−1; LC/MS (ESI-MS): m/z = 498.5 (M + 1) (positive ion mode); 1H NMR (C5D5N, 600 MHz) δ: 9.65 (s, 1H, H-23), 5.44 (br s, 1H, 12-H), 4.09 (dd, J = 10.2, 6.0 Hz, 1H, H-3), 3.47 (dd, J= 15.3, 3.9 Hz, 1H, H-18), 3.00 (s, 6H, -N(CH3)2), 1.39 (s, 1H, H-24), 1.25 (s, 3H, H-27), 0.97 (s, 3H, H-30), 0.95 (s, 3H, H-26), 0.93 (s, 3H, H-29), 0.87 (s, 3H, H-25); 13C NMR (C5D5N, 150 MHz): δ: 207.36 (C-23), 175.84 (C-28), 145.50 (C-13), 121.64 (C-12), 71.69 (C-3), 56.36 (C-4), 48.10 (C-9), 47.80 (C-5), 47.59 (C-17), 46.60 (C-19), 43.88 (C-14), 42.32 (C-18), 39.84 (C-8), 38.61 (-N(CH3)2), 38.48 (C-1), 36.21 (C-10), 34.13 (C-21), 33.25 (C-20), 32.60 (C-7), 30.65 (C-29), 30.02 (C-22), 29.91 (C-2), 28.02 (C-15), 27.10 (C-11), 26.32 (C-16), 24.20 (C-30), 23.74 (C-27), 21.20 (C-6), 17.11 (C-26), 15.75 (C-25), 9.71 (C-24).
4.7.6. N,N-Diethyl-(3β)-hydroxy-23-oxoolean-12-en-28-amide (8f)
White solid, yield: 87.6%, mp: 121.7–123.5°C, IR (KBr) νmax: 3459, 2926, 2854, 2679, 1732, 1607, 1466, 1416, 1382, 1260, 1205, 1139, 1019, 969, 827, 794, 690, 630, 564, 526, 460 cm−1; LC/MS (ESI-MS): m/z = 526.6 (M + 1) (positive ion mode); 1H NMR (C5D5N, 600 MHz) δ: 9.65 (s, 1H, H-23), 5.47 (br s, 1H, 12-H), 4.09 (dd, J= 10.2, 5.7 Hz, 1H, H-3), 3.43 (dd, J = 14.45, 4.62 Hz, 1H, H-18), 3.31–3.44 (m, 4H, -N(CH2CH3)2), 1.39 (s, 1H, H-24), 1.27 (s, 3H, H-27), 1.15 (t, 6H, -N(CH2CH3)2), 0.95 (s, 3H, H-30), 0.96 (s, 3H, H-26), 0.96 (s, 3H, H-29), 0.94 (s, 3H, H-25); 13C NMR (C5D5N, 150 MHz) δ: 207.34 (C-23), 174.77 (C-28), 145.60 (C-13), 121.42 (C-12), 71.69 (C-3), 56.35 (C-4), 48.09 (C-9), 47.82 (C-5), 47.71 (C-17), 47.12 (C-19), 44.18 (C-14), 42.43 (-N(CH2CH3)2), 42.40 (C-18), 40.04 (C-8), 38.54 (C-1), 36.23 (C-10), 36.23 (C-21), 34.44 (C-22), 33.17 (C-20), 32.86 (C-7), 30.56 (C-29), 30.02 (C-2), 28.48 (C-15), 27.11 (C-11), 25.97 (C-16), 24.14 (C-30), 24.14 (C-27), 21.14 (C-6), 17.56 (C-26), 15.83 (C-25), 13.61 (-N(CH2CH3)2), 9.73 (C-24).
4.7.7. N,N-(n-Dibutyl)-(3β)-hydroxy-23-oxoolean-12-en-28-amide (8g)
White solid, yield: 82.0%, mp: 124.3–125.7°C, IR (KBr) νmax: 3419, 2955, 2926, 1736, 1623, 1465, 1409, 1377, 1249, 1200, 1139, 1106, 734, 663, 593, 526, 476 cm−1; LC/MS (ESI-MS): m/z = 582.5 (M + 1) (positive ion mode); 1H NMR (C5D5N, 600 MHz) δ: 9.65 (s, 1H, H-23), 5.49 (br s, 1H, 12-H), 4.10 (dd, J = 11.4, 5.7 Hz, 1H, H-3), 3.46 (dd, J = 14.46, 4.62 Hz, 1H, H-18), 3.40 (br s, 4H, -N(CH2-C3H 7)2), 1.39 (s, 1H, H-24), 1.31–1.36 (m, 8H, -N(CH2C2H4CH3)2), 1.30 (s, 3H, H-27), 1.00 (s, 3H, H-30), 0.98 (s, 3H, H-26), 0.96 (s, 3H, H-29), 0.95 (s, 3H, H-25), 0.95 (t, 6H, -N(C3H6CH3)2); 13C NMR (C5D5N, 150 MHz) δ: 207.35 (C-23), 174.99 (C-28), 145.68 (C-13), 121.47 (C-12), 71.70 (C-3), 56.35 (C-4), 48.43 (-N(CH2C3H7)2), 48.11 (C-9), 47.92 (C-5), 47.81 (C-17), 47.21 (C-19), 44.3 (C-14), 42.46 (C-18), 40.08 (C-8), 38.54 (C-1), 36.24 (C-10), 36.24 (C-21), 34.43 (C-22), 33.16 (C-20), 32.88 (C-7), 30.58 (C-29), 30.01 (C-2), 28.59 (C-15), 27.11 (C-11), 26.03 (-N(CH2CH2C2H5)2), 24.23 (C-16), 23.80 (C-30), 23.01 (-N(C2H4CH2CH3)2), 21.13 (C-27), 20.71 (C-6), 14.20 (-N(C3H6CH3)2), 17.61(C-26), 15.82(C-25), 9.73 (C-24).
4.8. General procedure for the synthesis of compounds 9a–9j
Oxalyl chloride (2.0 × 10−1 ml, 2.36 × 10−3 mol) was added to compound 6 (46 mg, 7.8 × 10−5 mol) in CH2Cl2 (3 ml). The mixture was allowed to stir for 12 h at room temperature. After completion, the reaction mixture was neutralized with Et3N and evaporated to dryness. To a stirred solution of the mixture in dry CH2Cl2 (3 ml) was added piperidine (4.0 × 10−2 ml, 4.04 × 10−4 mol). The stirring was continued for 8 h at room temperature. After CH2Cl2 evaporation, water (5 ml) was added to this mixture, and the mixture was extracted with ethyl acetate (3 × 5 ml). The combined organic layer was dried over anhydrous sodium sulfate and evaporated to dryness. The crude material was purified by silica gel chromatography using petroleum ether–ethyl acetate (1 : 1) to afford compound 9a. Compounds 9b–9j were prepared as 9a.
4.8.1. N-Piperidyl-(3β)-acetyloxy-23-[(2,4-dinitrophenyl) hydrazono]olean-12-en-28-amide (9a)
Yellow solid, yield: 92.4%, mp: 117.2–118.1°C, IR(KBr) νmax: 3441, 3283, 3099, 2928, 2854, 2036, 1726, 1618, 1590, 1520, 1465, 1423, 1332, 1308, 1244, 1136, 1030, 951, 914, 825, 741, 599, 530, 441 cm−1; LC/MS (ESI-MS): m/z = 760.6 (M + 1) (positive ion mode); 1H NMR (CDCl3, 600 MHz) δ: 10.99 (s, 1H, -CHNNH-), 9.11 (d, 1H, 2.4 Hz,PhH-3), 8.30 (dd, 9.6, 2.4 Hz, 1H, PhH-5), 7.90 (d, 9.6 Hz, 1H, PhH-6), 7.25 (s, 1H, -CHNNH-), 5.28 (brs, 1H, 12-H), 4.90 (dd, J = 11.40, 4.80 Hz, 1H, H-3), 3.52–3.57 (m, 4H, -N(CH 2)2(CH2)3), 3.10 (dd, J = 14.10, 4.50 Hz, 1H, H-18), 1.94 (s, 3H, CH3 COO-), 1.46–1.51 (m, 4H, -N(CH2)2(CH2)2 CH2), 1.30–1.36 (m, 2H, -N(CH2)4CH2), 1.23 (s, 1H, H-24), 1.16 (s, 3H, H-27), 1.04 (s, 3H, H-30), 0.93 (s, 3H, H-26), 0.90 (s, 3H, H-29), 0.78 (s, 3H, H-25); 13C NMR (CDCl3, 150 MHz) δ: 174.64 (C-28), 170.52 (CH3COO-), 158.78 (C-23), 145.15 (Ph-1), 145.14 (C-13), 137.79 (Ph-4), 129.99 (Ph-5), 128.93 (Ph-2), 123.50 (Ph-6), 120.81 (C-12), 116.44 (Ph-3), 76.06 (C-3), 58.45, 53.44 (-N(CH2)2(CH2)), 52.20 (C-4), 48.02 (C-17), 47.34 (C-9), 47.30 (C-5), 46.56 (C-19), 43.66 (C-14), 41.92 (C-18), 39.44 (C-8), 37.86 (C-1), 36.59 (C-10), 36.59 (C-21), 34.09 (C-22), 33.09 (C-20), 32.49 (C-7), 30.40 (C-29), 29.70 (C-2), 28.01 (C-15), 26.16 (C-30), 25.91, 24.10 (-N(CH2)2 (CH2)2CH2), 24.82 (C-11), 23.35 (C-16), 22.96 (C-27), 21.21 (CH3COO-), 20.19 (C-6), 18.45 (-N(CH2)4CH2), 16.94 (C-26), 15.84 (C-25), 11.99 (C-24).
4.8.2. N-Morpholino-(3β)-acetyloxy-23-[(2,4-dinitrophenyl) hydrazono]olean-12-en-28-amide (9b)
Yellow solid, yield: 85%; mp: 114.2–116.1°C, IR(KBr) νmax: 3293, 3112, 2924, 2853, 1727, 1619, 1591, 1521, 1456, 1425, 1378, 1332, 1246, 1120, 1026, 1004, 946, 915, 832, 734, 613, 520 cm−1; LC/MS (ESI-MS): m/z = 762.6 (M + 1) (positive ion mode); 1H NMR (CDCl3, 600 MHz) δ: 10.99 (s, 1H,-CHNNH-), 9.12 (d,3.0 Hz, 1H, PhH-3), 8.30 (dd, 9.6, 2.4 Hz, 1H, PhH-5), 7.90 (d, 9.6 Hz, 1H, PhH-6), 7.23 (s, 1H, -CHNNH-), 5.29 (br s, 1H, 12-H), 4.90 (dd, J = 11.40, 4.80 Hz, 1H, H-3), 3.59–3.69 (m, 8H, -N(CH2)4O), 3.09 (dd, J = 15.00, 4.20 Hz, 1H, H-18), 1.94 (s, 3H, CH3COO-), 1.23 (s, 1H, H-24), 1.17 (s, 3H, H-27), 1.03 (s, 3H, H-30), 0.94 (s, 3H, H-26), 0.91 (s, 3H, H-29), 0.76 (s, 3H, H-25); 13C NMR (CDCl3, 150 MHz) δ: 175.10 (C-28), 170.52 (CH3COO-), 158.67 (C-23), 145.14 (Ph-1), 144.77 (C-13), 137.82 (Ph-4), 130.00 (Ph-5), 128.95 (Ph-2), 123.50 (Ph-6), 121.13 (C-12), 116.44 (Ph-3), 76.02 (C-3), 66.95 (-N(CH 2)2(CH2)2O), 52.20 (C-4), 47.98 (C-9), 47.98 (C-5), 47.40 (-N(CH 2)2(CH2)2O), 47.29 (C-17), 46.35 (C-19), 43.57 (C-14), 41.92 (C-18), 39.46 (C-8), 37.86 (C-1), 36.58 (C-10), 33.96 (C-22), 33.04 (C-20), 32.45 (C-7), 31.93 (C-21), 30.39 (C-29), 27.86 (C-2), 25.95 (C-15), 24.04 (C-11), 23.34 (C-30), 22.95 (C-27), 22.73 (C-16), 21.21 (CH3COO-), 20.16 (C-6), 16.90 (C-26), 15.83 (C-25), 11.99 (C-24).
4.8.3. N-(1-Methyl-piperazinyl)-(3β)-acetyloxy-23-[(2,4-dinitrophenyl) hydrazono] olean-12-en-28-amide (9c)
Yellow solid, yield: 85%, mp: 115.3–117.1°C, IR (KBr) νmax : 3446, 3298, 3112, 2927, 2853, 2778, 1732, 1618, 1591, 1519, 1458, 1425, 1371, 1332, 1308, 1243, 1139, 1027, 915, 832, 739, 602, 539, 493 cm−1; LC/MS (ESI-MS): m/z = 775.8 (M + 1) (positive ion mode); 1H NMR (CDCl3, 600 MHz) δ: 11.00 (s, 1H, CHNNH-), 9.12 (d, 2.4 Hz, 1H, PhH-3), 8.30 (dd, 9.6, 2.4 Hz, 1H, PhH-5), 7.90 (d, 9.6 Hz, 1H, PhH-6), 7.24 (s, 1H, -CHNNH-), 5.28 (br s, 1H, 12-H), 4.90 (dd, J = 11.40, 4.20 Hz, 1H, H-3), 3.61–3.75 (m, 4H, -N(CH2)2 (CH2)2NCH3), 3.08 (dd, J = 14.40, 4.20 Hz, 1H, H-18), 2.38–2.46 (m, 4H, -N(CH2)2(CH2)2NCH3), 2.32 (s, 3H, -N(CH2)4NCH3), 1.95 (s, 3H, CH3COO-), 1.23 (s, 1H, H-24), 1.16 (s, 3H, H-27), 1.03 (s, 3H, H-30), 0.93 (s, 3H, H-26), 0.90 (s, 3H, H-29), 0.76 (s, 3H, H-25); 13C NMR (CDCl3, 150 MHz) δ: 174.97 (C-28), 170.53 (CH3COO-), 158.72 (C-23), 145.15 (Ph-1), 144.86 (C-13), 137.80 (Ph-4), 130.00 (Ph-5), 128.93 (Ph-2), 123.50 (Ph-6), 122.05 (C-12), 116.44 (Ph-3), 76.06 (C-3), 55.09 (-N(CH2)2(CH2)2NCH3), 52.18 (C-4), 47.99 (-N (CH2)2(CH2)2NCH3), 47.39 (C-9), 47.29 (C-5), 46.39 (C-17), 45.81 (C-19), 45.04 (-N(CH2)4NCH3), 43.58 (C-14), 41.90 (C-18), 39.45 (C-8), 37.86 (C-1), 36.58 (C-10), 34.00 (C-22), 33.05 (C-20), 32.45 (C-7), 30.39 (C-29), 29.93 (C-21), 29.70 (C-2), 27.90 (C-15), 22.94 (C-11), 24.05 (C-16), 23.34 (C-30), 22.95 (C-27), 21.21 (CH3COO-), 20.18 (C-6), 16.92 (C-26), 15.84 (C-25),11.99 (C-24).
4.8.4. N-(1-Ethyl-piperazinyl)-(3β)-acetyloxy-23-[(2,4-dinitrophenyl) hydrazono] olean-12-en-28-amide (9d)
Yellow solid, yield: 85%, mp: 124.4–126.5°C, IR (KBr) νmax 3446, 3293, 3101, 2923, 2852, 1732, 1618, 1590, 1519, 1463, 1376, 1332, 1308, 1240, 1068, 1024, 991, 920, 832, 739, 635, 536, 460 cm−1; LC/MS (ESI-MS): m/z = 789.8 (M + 1) (positive ion mode); 1H NMR (CDCl3, 600 MHz) δ: 11.00 (s, 1H, -CHNNH-), 9.11 (d, 2.4 Hz, 1H, PhH-3), 8.30 (dd, 9.6, 2.4 Hz, 1H, PhH-5),7.90 (d, 9.6 Hz, 1H, PhH-6), 7.24 (s, 1H, -CHNNH-), 5.28 (br s, 1H, 12-H), 4.90 (dd, J = 11.70, 4.50 Hz, 1H, H-3), 3.64–3.74 (m, 4H, -N(CH2)2(CH2)2NC2 H5), 3.09 (dd, J = 14.40, 4.20 Hz, 1H, H-18), 2.40–2.48 (m, 4H, -N (CH2)2 (CH2)2NC2H5), 2.45 (q, 2H, -N(CH2)4NCH2CH3), 1.94 (s, 3H, CH3COO-), 1.23 (s, 1H, H-24), 1.16 (s, 3H, H-27), 1.11 (t, 3H, -N(CH2)4 NCH2CH3), 1.03 (s, 3H, H-30), 0.93 (s, 3H, H-26), 0.92 (s, 3H, H-29), 0.76 (s, 3H, H-25); 13C NMR (CDCl3, 150 MHz) δ: 174.90 (C-28), 170.51 (CH3COO-), 158.72 (C-23), 145.14 (Ph-1), 144.89 (C-13), 137.80 (Ph-4), 129.99 (Ph-5), 128.93 (Ph-2), 123.49 (Ph-6), 122.03 (C-12), 116.44 (Ph-3), 76.06 (C-3), 52.90 (-N(CH2)2 (CH2)2NC2 H5), 52.30 (-N(CH2)2(CH2)2NC2H5), 52.17 (C-4), 47.99 (C-9), 47.38 (C-5), 47.29 (C-17), 46.39 (C-19), 45.08 (-N(C H2)4 NCH2CH3), 43.57 (C-14), 41.90 (C-18), 39.45 (C-8), 37.86 (C-1), 36.58 (C-10), 34.00 (C-22), 33.06 (C-20), 32.45 (C-7), 31.93 (C-21), 30.39 (C-29), 29.36 (C-2), 27.90 (C-15), 24.16 (C-11), 23.34 (C-16), 22.95 (C-30), 22.69 (C-27), 21.20 (CH3COO-), 20.18 (C-6), 16.94 (C-26), 15.83 (C-25), 14.13 (-N(CH2)4NCH2CH3), 11.98 (C-24).
4.8.5. N,N-Dimethyl-(3β)-acetyloxy-23-[(2,4-dinitrophenyl) hydrazono]olean-12-en-28-amide (9e)
Yellow solid, yield: 85%, mp: 121.5–123.2°C, IR (KBr) νmax: 3446, 3298, 3101, 2928, 2854, 1732, 1618, 1590, 1519, 1464, 1428, 1371, 1332, 1308, 1242, 1139, 1079, 1028, 958, 926, 827, 734, 641, 526, 432 cm−1; LC/MS (ESI-MS): m/z = 720.4 (M + 1) (positive ion mode); 1H NMR (CDCl3, 600 MHz) δ: 10.99 (s, 1H, -CHNNH-), 9.12 (d, 3.0 Hz, 1H, PhH-3), 8.30 (dd, 9.3, 2.7 Hz, 1H, PhH-5), 7.90 (d, 9.6 Hz, 1H, PhH-6), 7.24 (s, 1H, -CHNNH-), 5.28 (br s, 1H, 12-H), 4.90 (dd, J = 11.70, 4.50 Hz, 1H, H-3), 3.15 (m, 1H, H-18), 3.01 (s, 6H, -N(CH3)2), 1.94 (s, 3H, CH3COO-),1.23 (s, 1H, H-24), 1.16 (s, 3H, H-27), 1.03 (s, 3H, H-30), 0.94 (s, 3H, H-26), 0.90 (s, 3H, H-29), 0.75 (s, 3H, H-25); 13C NMR (CDCl3, 150 MHz) δ: 176.21 (C-28), 170.53 (CH3COO-), 158.76 (C-23), 145.15 (Ph-1), 144.99 (C-13), 137.80 (Ph-4), 130.00 (Ph-5), 128.94 (Ph-2), 123.50 (Ph-6), 121.08 (C-12), 116.45 (Ph-3), 76.05 (C-3), 52.20 (C-4), 48.01 (C-9), 47.42 (C-5), 47.30 (C-17), 46.31 (C-19), 43.33 (C-14), 41.96 (C-18), 39.39 (C-8), 38.72 (-N(CH3)2), 37.84 (C-1), 36.58 (C-10), 33.93 (C-22), 33.13 (C-20), 32.37 (C-7), 30.48 (C-29), 29.70 (C-2), 29.63 (C-21), 27.69 (C-15), 26.10 (C-11), 24.09 (C-16), 23.35 (C-30), 22.96 (C-27), 21.21 (CH3COO-), 20.18 (C-6), 16.84 (C-26), 15.80 (C-25), 11.97 (C-24).
4.8.6. N,N-Diethyl-(3β)-acetyloxy-23-[(2,4-dinitrophenyl) hydrazono]olean-12-en-28-amide (9f)
Yellow solid, yield: 85%, mp: 119.7–121.2°C, IR (KBr) νmax: 3479, 3292, 3101, 2949, 2849, 1726, 1619, 1520, 1468, 1423, 1332, 1307, 1246, 1137, 1069, 1029, 963, 908, 837, 744, 601, 530, 448 cm−1; LC/MS (ESI-MS): m/z = 748.6 (M + 1) (positive ion mode); 1H NMR (CDCl3, 600 MHz) δ: 10.99 (s, 1H, -CHNNH-), 9.11 (d, 3.0 Hz, 1H, PhH-3), 8.30 (dd, 9.6, 2.4 Hz, 1H, PhH-5), 7.90 (d, 9.6 Hz, 1H, PhH-6), 7.24 (s, 1H, -CHNNH-), 5.28 (br s, 1H, 12-H), 4.90 (dd, J = 11.40, 4.20 Hz, 1H, H-3), 3.26–3.33 (m, 4H, -N(CH2CH3)2), 3.09 (dd, J = 14.40, 4.2 Hz, 1H, H-18), 1.94 (s, 3H, CH3COO-), 1.23 (s, 1H, H-24), 1.17 (s, 3H, H-27), 1.11–1.14 (m, 6H, -N(CH2CH3)2), 1.04 (s, 3H, H-30), 0.93 (s, 3H, H-26), 0.90 (s, 3H, H-29), 0.80 (s, 3H, H-25); 13C NMR (CDCl3, 150 MHz) δ: 174.85 (C-28), 170.53 (CH3COO-), 158.78 (C-23), 145.15 (Ph-1), 145.11 (C-13), 137.79 (Ph-4), 129.99 (Ph-5), 128.92 (Ph-2), 123.50 (Ph-6), 120.77 (C-12), 116.44 (Ph-3), 76.06 (C-3), 52.20 (C-4), 48.00 (C-9), 47.48 (C-5), 47.30 (C-17), 46.80 (C-19), 43.67 (C-14), 42.14 (-N(CH2CH3)2), 42.01 (C-18), 39.54 (C-8), 37.89 (C-1), 36.59 (C-10), 36.59 (C-21), 34.24 (C-22), 33.05 (C-20), 32.57 (C-7), 30.37 (C-29), 29.70 (C-2), 28.12 (C-15), 25.77 (C-11), 24.07 (C-16), 23.35 (C-30), 22.52 (C-27), 21.21 (CH3 COO-), 20.19 (C-6), 17.16 (C-26), 15.88 (C-25), 13.31 (-N(CH2CH 3)2), 11.99 (C-24).
4.8.7. N,N-(n-Dibutyl)-(3β)-acetyloxy-23-[(2,4-dinitrophenyl) hydrazono]olean-12-en-28-amide (9g)
Yellow solid, yield: 85%, mp: 112.6–113.9°C, IR (KBr) νmax: 3293, 3112, 2954, 2871, 1733, 1619, 1592, 1520, 1465, 1426, 1332, 1308, 1241, 1139, 1068, 1030, 958, 920, 832, 745, 641, 536, 443 cm−1; LC/MS (ESI-MS): m/z = 803.0 (M − 1) (negative ion mode); 1H NMR (CDCl3, 600 MHz) δ: 10.99 (s, 1H, -CHNNH-),9.12 (d, 3.0 Hz, 1H, PhH-3), 8.30 (dd, 9.6, 2.4 Hz, 1H, PhH-5), 7.90 (d, 9.0 Hz, 1H, PhH-6), 7.24 (s, 1H, -CHNNH-), 5.28 (br s, 1H, 12-H), 4.90 (dd, J = 11.40, 4.80 Hz, 1H, H-3), 3.07–3.17 (m, 4H, -N(CH2C3H7)2), 3.08 (d, J = 13.8 Hz, 1H, H-18), 1.94 (s, 3H, CH3COO-), 1.48–1.51 (m, 4H, -N (CH2CH2C2H5)2), 1.27–1.29 (m, 4H, -N(C2H4CH2CH3)2), 1.23 (s, 1H, H-24), 1.16 (s, 3H, H-27), 1.04 (s, 3H, H-30), 0.93 (s, 3H, H-26), 0.92 (t, 6H, -N(C3H6CH3)2), 0.90 (s, 3H, H-29), 0.79 (s, 3H, H-25); 13C NMR (C5D5N, 150 MHz) δ: 174.98 (C-28), 170.52 (CH3COO-), 158.79 (C-23), 145.16 (Ph-1), 145.16 (C-13), 137.79 (Ph-4), 129.99 (Ph-5), 128.93 (Ph-2), 123.50 (Ph-6), 120.77 (C-12), 116.44 (Ph-3), 76.09 (C-3), 52.18 (C-4), 48.01 (C-9), 48.01 (C-5), 47.60 (C-17), 47.30 (-N(CH2C3H7)2), 46.89 (C-19), 43.67 (C-14), 42.03 (C-18), 39.54 (C-8), 37.89 (C-1), 36.60 (C-10), 36.60 (C-21), 34.25 (C-22), 33.03 (C-20), 32.56 (C-7), 30.38 (C-29), 29.70 (C-2), 28.18 (C-15), 25.76 (C-11), 24.10 (C-16), 23.34 (C-30), 22.97 (C-27), 21.21 (CH3 COO-), 20.42 (-N(CH2CH2C2H5)2), 20.19 (C-6), 18.45 (-N(C2H4C H2CH3)2), 17.16 (C-26), 15.89 (C-25), 13.98 (-N(C3H6CH3)2), 12.00 (C-24).
4.8.8. Methyl (3β)-acetyloxy-23-[(2,4-dinitrophenyl)hydrazono] olean-12-en-28-oate (9h)
Yellow solid, yield: 85%, mp: 134.4–136.1°C, IR (KBr) νmax: 3287, 3106, 2936, 2860, 2853, 1736, 1616, 1583, 1523, 1518, 1463, 1435, 1336, 1232, 1079, 1030, 1029, 969, 920, 832, 739, 613, 515, 449 cm−1; LC/MS (ESI-MS): m/z = 705.7 (M − 1) (negative ion mode); 1H NMR (CDCl3, 600 MHz) δ: 10.99 (s, 1H, -CHNNH-), 9.12 (d, 2.4 Hz, 1H, PhH-3), 8.30 (dd, 9.6, 2.4 Hz, 1H, PhH-5), 7.90 (d, 9.6 Hz, 1H, PhH-6), 7.24 (s, 1H, -CHNNH-), 5.30 (br s, 1H, 12-H), 4.90 (dd, J = 11.70, 4.50 Hz, 1H, H-3), 3.62 (q, 3H, -OCH3), 2.87 (dd, J = 14.10, 4.50 Hz, 1H, H-18), 1.95 (s, 3H, CH3COO-), 1.24 (s, 1H, H-24), 1.16 (s, 3H, H-27), 1.04 (s, 3H, H-30), 0.93 (s, 3H, H-26), 0.90 (s, 3H, H-29), 0.75 (s, 3H, H-25); 13C NMR (CDCl3,150 MHz) δ: 178.22 (C-28), 170.53 (CH3COO-), 158.62 (C-23), 145.15 (Ph-1), 143.89 (C-13), 137.82 (Ph-4), 130.00 (Ph-5), 128.96 (Ph-2), 123.51 (Ph-6), 121.91 (C-12), 116.44 (Ph-3), 76.04 (C-3), 52.10 (C-4), 51.56 (-OCH3), 47.83 (C-9), 47.27 (C-5), 46.69 (C-17), 45.84 (C-19), 41.70 (C-14), 41.30 (C-18), 39.61 (C-8), 37.89 (C-1), 36.52 (C-10), 33.82 (C-22), 33.10 (C-20), 32.31 (C-7), 32.25 (C-21), 30.70 (C-29), 29.70 (C-2), 27.68 (C-15), 25.93 (C-30), 22.63 (C-27), 23.37 (C-11), 23.00 (C-16), 21.21 (CH3COO-), 20.15 (C-6), 16.85 (C-26), 15.80 (C-25), 12.01 (C-24).
4.8.9. Ethyl(3β)-acetyloxy-23-[(2,4-dinitrophenyl)hydrazono] olean-12-en-28-oate (9i)
Yellow solid, yield: 85%, mp: 135.5–136.8°C, IR (KBr) νmax: 3291, 2977, 2924, 2853, 1739, 1719, 1621, 1591, 1518, 1464, 1420, 1369, 1338, 1250, 1135, 1029, 958, 920, 838, 745, 602, 536 cm−1; LC/MS (ESI-MS): m/z = 720.2 (M − 1) (negative ion mode); 1H NMR (CDCl3, 600 MHz) δ: 10.99 (s, 1H, -CHNNH-), 9.12 (d, 2.4 Hz, 1H, PhH-3), 8.30 (dd, 9.3, 2.7 Hz, 1H, PhH-5), 7.90 (d, 9.6 Hz, 1H, PhH-6), 7.23 (s, 1H, -CHNNH-), 5.30 (br s, 1H, 12-H), 4.90 (dd, J = 11.70, 4.50 Hz, 1H, H-3), 4.08 (q, 2H, -OCH2CH3), 2.88 (dd, J = 14.44, 4.63 Hz, 1H, H-18), 1.94 (s, 3H, CH3COO-), 1.23 (s, 1H, H-24), 1.22 (t, 3H, -OCH2CH3), 1.16 (s, 3H, H-27), 1.04 (s, 3H, H-30), 0.93 (s, 3H, H-26), 0.90 (s, 3H, H-29), 0.77 (s, 3H, H-25); 13C NMR (CDCl3, 150 MHz) δ: 177.64 (C-28), 170.52 (CH3COO-), 158.60 (C-23), 145.13 (Ph-1), 143.90 (C-13), 137.82 (Ph-4), 129.99 (Ph-5), 128.95 (Ph-2), 123.50 (Ph-6), 121.83 (C-12), 116.42 (Ph-3), 76.03 (C-3), 60.10 (-OCH2 CH3), 52.10 (C-4), 47.82 (C-9), 47.27 (C-5), 46.46 (C-17), 45.88 (C-19), 41.76 (C-14), 41.30 (C-18), 39.67 (C-8), 37.90 (C-1), 36.51 (C-10), 36.51 (C-21), 33.87 (C-22), 33.10 (C-20), 32.34 (C-7), 30.71 (C-29), 29.66 (C-2), 27.63 (C-15), 25.84 (C-11), 23.61 (C-16), 23.38 (C-30), 22.93 (C-27), 21.21 (CH3COO-), 20.14 (C-6), 16.98 (C-26), 15.81 (C-25), 14.27 (-OCH2CH3), 12.01 (C-24).
4.8.10. n-Butyl(3β)-acetyloxy-23-[(2,4-dinitrophenyl) hydrazono]olean-12-en-28-oate (9j)
Yellow solid, yield: 85%, mp: 137.6–139.3°C; IR (KBr) νmax: 3298, 3106, 2951, 2872, 1728, 1618, 1591, 1519, 1463, 1426, 1366, 1333, 1240, 1139, 1071, 1029, 969, 909, 827, 739, 646, 536, 454 cm−1; LC/MS (ESI-MS): m/z = 747.8 (M − 1) (negative ion mode); 1H NMR (CDCl3, 600 MHz) δ: 10.99 (s, 1H, -CHNNH-), 9.12 (d, 2.4 Hz, 1H, PhH-3), 8.30 (dd, 9.6, 2.4 Hz, 1H, PhH-5), 7.90 (d, 9.6 Hz, 1H, PhH-6), 7.23 (s, 1H, -CHNNH-), 5.30 (br s, 1H, 12-H), 4.90 (dd, J = 11.40, 4.80 Hz, 1H, H-3), 4.01 (t, 2H, -OCH2C3H7), 2.88 (dd, J = 13.80, 4.20 Hz, 1H, H-18), 1.95 (s, 3H, CH3COO-), 1.92 (q, 2H, -O CH2CH2C2H5), 1.38 (q, 2H, -OC2H4CH2CH3), 1.23 (s, 1H, H-24), 1.16 (s, 3H, H-27), 1.04 (s, 3H, H-30), 0.93 (s, 3H, H-26), 0.93 (t, 3H, -OC3H6CH3), 0.90 (s, 3H, H-29), 0.76 (s, 3H, H-25); 13C NMR (CDCl3, 150 MHz) δ: 177.70 (C-28), 170.53 (CH3COO-), 158.62 (C-23), 145.15 (Ph-1), 143.93 (C-13), 137.82 (Ph-4), 130.00 (Ph-5), 128.96 (Ph-2), 123.51 (Ph-6), 121.89 (C-12), 116.44 (Ph-3), 76.04 (C-3), 64.00 (-OCH2C3H7), 52.09 (C-4), 47.82 (C-9), 47.28 (C-5), 46.63 (C-17), 45.87 (C-19), 41.77 (C-14), 41.34 (C-18), 39.66 (C-8), 37. 91 (C-1), 36.52 (C-10), 33.87 (C-22), 33.11 (C-20), 32.40 (C-7), 32.35 (C-21), 30.72 (C-29), 30.69 (-OCH2CH2C2H5), 29.71 (C-2), 27.63 (C-15), 25.87 (C-30), 23.61 (C-27), 23.39 (C-11), 27.97 (C-16), 21.21 (CH3COO-), 20.16 (C-6), 19.25 (-OC2H4CH2CH3), 17.01 (C-26), 15.81 (C-25), 13.73 (-OC3H6CH3), 12.01 (C-24).
4.9. Biology
A549, LOVO, SKOV3 and HepG2 cell lines were purchased from American Type Culture Collection (ATCC). Trypsin, EDTA and fetal bovine serum (FBS) were purchased from Sigma Chemicals Co. (St. Louis, MO). AO and EB were purchased from KeyGEN BioTECH Co. (Nanjing, China). PI, RNase A, RPMI-1640 Hyclone, phosphate buffered solution (PBS), MTT and 6- and 96-well flat bottom tissue culture plates were purchased from Beyotime Biotechnology Co. (Shanghai, China).
4.9.1. Cytotoxicity screening using MTT assay
MTT assay is a standard colorimetric assay for measuring cellular proliferation, in which MTT is taken up by living cells and reduced by a mitochondrial dehydrogenase enzyme to a purple formazan product that is impermeable to the cell membrane. Solubilization with solvents like dimethylsulfoxide (DMSO) leads to liberation of product and amount of purple formazan product is directly related to the cell viability. Cells were seeded in 96-well plates at a density of 1.0 × 105 cells ml−1 and incubated with increasing concentrations of agents (corresponding to 100, 10, 1, 0.1, and 0.01 µM of the compounds or controls) for 48 h at 37°C in RPMI-1640 with 10% FBS medium. Then the above media were replaced with 90 µl of fresh serum free media and 10 µl of MTT reagent (5 mg ml−1) and plates were incubated at 37°C for 4 h. Then the above media were replaced with 150 ml of DMSO and incubated at 37°C for 20 min. The absorbance at 570 nm was recorded using Thermo Scientific SkanIt software. The IC50 values were analysed using IBM SPSS 22.0 software.
4.9.2. Morphological observation
Cellular morphological effects of compounds 4 and 7g were determined by the AO/EB double-staining method [24]. Cells were seeded in 6-well plates at a concentration of 1 × 106 cells ml−1 for 24 h at 37°C in RPMI-1640 with 10% FBS medium. After incubation, cells were treated with different concentrations of compounds 4 and 7 g for 48 h. Then the cells were collected and washed two times by PBS. Cells were seeded in 48-well plates at a concentration of 4 × 106 cells ml−1. 10 µl of fluorescent dyes containing AO and EB added into each well in equal volumes (10 mg ml−1) respectively and within 10 min the cells were visualized under an inverted fluorescence microscope (Nikon ECLIPSE Ti, Japan) with a blue filter at 200× magnification [27]. At least 200 cells were randomly counted in various fields. The tests were repeated three times. The percentage of apoptotic cells was calculated by the following formula: apoptotic rate (%) = (numbers of early apoptotic cells + numbers of late apoptotic cells)/(numbers of all the cells counted).
4.9.3. Cell cycle analysis
To determine the effect of compounds 4 and 7g on the cell cycle, cells were seeded in 6-well plates at a concentration of 1 × 105 cells ml−1 for 24 h at 37°C in RPMI-1640 with 10% FBS medium. After incubation, cells were treated with different concentrations of compounds 4 and 7 g for 48 h. Then the cells were collected, washed and fixed in 70% ethanol at 4°C. After 12 h, fixed cells were pelleted and stained with cell cycle analysis reagent as per the manufacturer instructions for 30 min at 37°C in the dark. The data were recorded by a COULTER® EPICS® XL™ flow cytometer (Beckman Coulter, USA). The results were analysed by Expo 32 ADC software.
Data accessibility
Further details are available in the electronic supplementary material. This includes further experimental data and various NMR, UV and MS spectra.
Authors' contributions
Jingyong Sun, F.W. and Y.M. contributed to the conception of the study. Jie Sun contributed significantly to analysis and manuscript preparation. H.Z. and L.S. performed the data analyses and wrote the manuscript. Y.L. helped perform the analysis with constructive discussions.
Competing interests
We declare we have no competing interests.
Funding
The authors for correspondence are grateful to support from the Project of Shandong Province Higher Educational Science and Technology Program (J15LM07) and the Innovation Project of Shandong Academy of Medical Sciences.
Supplementary Material
Acknowledgements
The authors are grateful for support from the Innovation Project of Shandong Academy of Medical Sciences. We are also grateful to two anonymous reviewers and editors, who provided comments that substantially improved the manuscript.
References
- 1.Calza S, Ferraroni M, La Vecchia C, Franceschi S, Decarli A. 2001. Low-risk diet for colorectal cancer in Italy. Eur. J. Cancer Prev. 10, 515–521. (doi:10.1097/00008469-200112000-00006) [DOI] [PubMed] [Google Scholar]
- 2.Levi F, Pasche C, La Vecchia C, Lucchini F, Franceschi S. 1999. Food groups and colorectal cancer risk. Br. J. Cancer 79, 1283–1287. (doi:10.1038/sj.bjc.6690206) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiangsu New Medical College. 1977. The Chinese medicine dictionary, pp. 2170 Shanghai, China: Shanghai People's Publishing House. [Google Scholar]
- 4.Hostettmann SK, Marston A. 1995. Saponins. In Chemistry and pharmacology of natural products (eds Phillipon JD, Ayres DC, Baxter H), pp. 326–327. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 5.Yotova M, Krasteva I, Nikolov S. 2012. Triterpenoid saponins from genus Gypsophila L. (Caryophyllaceae). In Saponins: properties, applications and health benefits (eds Koh R, Tay I), pp. 99–122. New York, NY: Nova Science Publishers. [Google Scholar]
- 6.Sung MK, Kendall CW, Rao AV. 1995. Effect of soybean saponins and gypsophila saponin on morphology of colon carcinoma cells in culture. Food Chem. Toxicol. 33, 357–366. (doi:10.1016/0278-6915(95)00007-O) [DOI] [PubMed] [Google Scholar]
- 7.Marciani DJ. 1999. Triterpene saponin analogs having adjuvant and immunostimulatory activity. US Patent no. 5977081.
- 8.Voutquenne-Nazabadioko L, Gevrenova R, Borie N, Harakat D, Sayagh C, Weng A, Thakur M, Zaharieva M, Henry M. 2013. Triterpenoid saponins from the roots of Gypsophila trichotoma Wender. Phytochemistry 90, 114–127. (doi:10.1016/j.phytochem.2013.03.001) [DOI] [PubMed] [Google Scholar]
- 9.Arslan I, Celik A, Chol JH. 2012. A cytotoxic triterpenoid saponin from underground parts of Gypsophila pilulifera Boiss. & Heldr. Fitoterapia 83, 699–703. (doi:10.1016/j.fitote.2012.02.005) [DOI] [PubMed] [Google Scholar]
- 10.Arslan I, Celik A, Melzig MF. 2013. Nebulosides A-B, novel triterpene saponins from under-ground parts of Gypsophila arrostii Guss. var. nebulosa. Bioorg. Med. Chem. 21, 1279–1283. (doi:10.1016/j.bmc.2012.12.036) [DOI] [PubMed] [Google Scholar]
- 11.Yao S, Ma L, Luo J, Wang J, Kong L. 2010. New triterpenoid saponins from the roots of Gypsophila paniculata L. Helv. Chim. Acta 93, 361–374. (doi:10.1002/hlca.200900194) [Google Scholar]
- 12.Luo J, Ma L, Kong L. 2008. New triterpenoid saponins with strong α-glucosidase inhibitory activity from the roots of Gypsophila oldhamiana. Bioorg. Med. Chem. 16, 2912–2920. (doi:10.1016/j.bmc.2007.12.053) [DOI] [PubMed] [Google Scholar]
- 13.Gevrenova R, Stancheva T, Voynikov Y, Laurain-Mattar D, Henry M. 2010. Root in vitro cultures of six Gypsophila species and their saponin contents. Enzyme Microb. Technol. 47, 97–104. (doi:10.1016/j.enzmictec.2010.05.007) [Google Scholar]
- 14.Bai H, Zhong Y, Xie Y, Wang Y, Liu L, Zhou L, Mu Y, Zuo C. 2007. A major triterpenoid saponin from Gypsophila oldhamiana. Chem. Biodivers. 4, 955–960. (doi:10.1002/cbdv.200790085) [DOI] [PubMed] [Google Scholar]
- 15.Yotova M, Krasteva I, Jenett-Siems K, Zdraveva P, Nikolov S. 2012. Triterpenoids in Gypsophila trichotoma Wend. Phytochem. Lett. 5, 752–755. (doi:10.1016/j.phytol.2012.08.006) [Google Scholar]
- 16.Emirdağ-Öztürk S, Karayıldırım T, Çapcı-Karagöz A, Alankuş-Çalışkan Ö, Özmen A, Poyrazoğlu-Çoban E. 2014. Synthesis, antimicrobial and cytotoxic activities, and structure-activity relationships of gypsogenin derivatives against human cancer cells. Eur. J. Med. Chem. 82, 565–573. (doi:10.1016/j.ejmech.2014.05.084) [DOI] [PubMed] [Google Scholar]
- 17.Heller L, Schwarz S, Weber BA, Csuk R. 2014. Gypsogenin derivatives: an unexpected class of inhibitors of cholinesterases. Arch. Pharm. Chem. Life Sci. 347, 707–716. (doi:10.1002/ardp.201400103) [DOI] [PubMed] [Google Scholar]
- 18.Cheng K, Liu J, Sun H, Xie J. 2010. Synthesis of oleanolic acid dimmers as inhibitors of glycogen phosphorylase. Chem. Biodivers. 7, 690–697. (doi:10.1002/cbdv.200900086) [DOI] [PubMed] [Google Scholar]
- 19.Mallavadhani UV, Mahapatra A, Pattnaik B, Nagireddy V, Suri N, Saxena AK. 2012. Synthesis and anti-cancer activity of some novel C-17 analogs of ursolic and oleanolic acids. Med. Chem. Res. 22, 1263–1269. (doi:10.1007/s00044-012-0106-y) [Google Scholar]
- 20.Heller L, Knorrscheidt A, Flemming F, Wiemann J, Sommerwerk S, Pavel IZ, Al-Harrasi A, Csuk R. 2016. Synthesis and proapoptotic activity of oleanolic acid derived amides. Bioorg. Chem. 68, 137–151. (doi:10.1016/j.bioorg.2016.08.004) [DOI] [PubMed] [Google Scholar]
- 21.Böttger S, Melzig MF. 2011. Triterpenoid saponins of the Caryophyllaceae and Illecebraceae family. Phytochem. Lett. 4, 59–68. (doi:10.1016/j.phytol.2010.08.003) [Google Scholar]
- 22.Kim YC, Higuchi R, Komori T. 1992. Thermal degradation of glycosides, VII. Application of hydrothermolysis to the studies on the constituents of the Merck saponin. Liebigs Ann. Chem. 9, 941–946. (doi:10.1002/jlac.1992199201155) [Google Scholar]
- 23.Van AA, Beelen RH, Ossenkoppele GJ, Broekhoven MG, Langenhuijsen MM. 1994. A tetrazolium-based colorimetric MTT assay to quantitate human monocyte mediated cytotoxicity against leukemic cells from cell lines and patients with acute myeloid leukemia. J. Immunol. Methods 174, 311–320. (doi:10.1016/0022-1759(94)90034-5) [DOI] [PubMed] [Google Scholar]
- 24.Naidu V, Mahesh BU, Giddam AK, Babu KRD, Ding J, Babu KS, Ramesh B, Pragada RR, Gopalakrishnakone P. 2013. Apoptogenic activity of ethyl acetate extract of leaves of Memecylon edule on human gastric carcinoma cells via mitochondrial dependent pathway. Asian Pac. J. Trop. Med. 6, 337–345. (doi:10.1016/S1995-7645(13)60036-X) [DOI] [PubMed] [Google Scholar]
- 25.Ribble D, Goldstein NB, Norris DA, Shellman YG. 2005. A simple technique for quantifying apoptosis in 96-well plates. BMC Biotechnol. 5, 5–12. (doi:10.1186/1472-6750-5-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee EJ, Oh SY, Sung MK. 2012. Luteolin exerts anti-tumor activity through the suppression of epidermal growth factor receptor-mediated pathway in MDA-MB-231 ER-negative breast cancer cells. Food Chem. Toxicol. 50, 4136–4143. (doi:10.1016/j.fct.2012.08.025) [DOI] [PubMed] [Google Scholar]
- 27.Mallavadhani UV, Vanga NR, Jeengar MK, Naidu VG. 2014. Synthesis of novel ring-A fused hybrids of oleanolic acid with capabilities to arrest cell cycle and induce apoptosis in breast cancer cells. Eur. J. Med. Chem. 74, 398–404. (doi:10.1016/j.ejmech.2013.12.040) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Further details are available in the electronic supplementary material. This includes further experimental data and various NMR, UV and MS spectra.