

Abstract

Huntington disease (HD) is an inherited neurodegenerative disease caused by the expansion beyond a critical threshold of a polyglutamine (polyQ) tract near the N-terminus of the huntingtin (htt) protein. Expanded polyQ promotes the formation of a variety of oligomeric and fibrillar aggregates of htt that accumulate into the hallmark proteinaceous inclusion bodies associated with HD. htt is also highly associated with numerous cellular and subcellular membranes that contain a variety of lipids. As lipid homeostasis and metabolism abnormalities are observed in HD patients, we investigated how varying both the sphingomyelin (SM) and ganglioside (GM1) contents modifies the interactions between htt and lipid membranes. SM composition is altered in HD, and GM1 has been shown to have protective effects in animal models of HD. A combination of Langmuir trough monolayer techniques, vesicle permeability and binding assays, and in situ atomic force microscopy (AFM) were used to directly monitor the interaction of a model, synthetic htt peptide and a full-length htt-exon1 recombinant protein with model membranes comprised of total brain lipid extract (TBLE) and varying amounts of exogenously added SM or GM1. The addition of either SM or GM1 decreased htt insertion into the lipid monolayers. However, TBLE vesicles with an increased SM content were more susceptible to htt-induced permeabilization, whereas GM1 had no effect on permeablization. Pure TBLE bilayers and TBLE bilayers enriched with GM1 developed regions of roughened, granular morphologies upon exposure to htt-exon1, but plateau-like domains with a smoother appearance formed in bilayers enriched with SM. Oligomeric aggregates were observed on all bilayer systems regardless of induced morphology. Collectively, these observations suggest that the lipid composition and its subsequent effects on membrane material properties strongly influence htt binding and aggregation on lipid membranes.
Introduction
The expansion of a polyglutamine (polyQ) tract near the N-terminus of the huntingtin (htt) protein is the primary cause of Huntington’s disease (HD), a fatal neurodegenerative disorder.1 Expanded polyQ domains in htt are directly correlated with a propensity to aggregate into a variety of proteinacious structures ranging from small oligomers to fibrils.2−7 There is also a strong correlation of the age of onset and disease severity with the length of the polyQ domain, with ∼35 repeat glutamines being the critical threshold required for the disease.8−10 The exact mass of htt depends on the size of the polyQ domain, but full-length htt is approximately 350 kDa and 3144 amino acids in size, based on a polyQ domain of 23. The polyQ domain is contained within the 1st exon of htt and begins at the 18th residue. htt undergoes a variety of cleavages to produce a variety of N-terminal fragments comparable with exon1,11,12 and numerous lines of evidence support the notion that N-terminal fragments are directly involved in HD pathogenesis.2,12−14
Several functions have been attributed to the htt protein,15 and many of these functions are directly related to interactions between htt and lipid membrane interfaces.16−20 htt localizes to specific subcellular surfaces21 and functions in the transport of lipid vesicles (endocytic, synaptic, and lysosomal), especially along microtubules.17−20 htt is also essential for the normal development of several perinuclear membrane organelles, including mitochondria and endoplasmic reticulum (ER).22,23 Furthermore, htt localizes with brain membrane fractions24 and intimately interacts with a variety of specific membrane lipid components,25−29 including anionic phosphatidylinositol phosphates, consistent with an electrostatic-based mechanism for membrane association.26−29 With regard to htt-exon1, the first 17 amino acids of htt (Nt17) directly preceding the polyQ tract functions as a lipid-binding domain. Nt17 lipid-binding is facilitated by the formation of highly conserved amphipathic α-helix,21,30−32 and post-translational modifications of Nt17 can alter the affinity of htt for lipid membranes.33
Beyond these normal functions of htt, there is increasing evidence that lipid interactions may play a role in the toxic gain of functions associated with the expansion of polyQ in htt, as membrane-related changes (including mutant htt membrane association and induced disruption as well as altered membrane composition) are observed in HD.27,29,34−36 Whereas mutant htt is primarily associated with the formation of microscopic inclusion bodies in the cytoplasm and nucleus,13 about one-half of the endogenous htt partitions with the membranes after subcellular fractionation of neuronlike clonal striatal cells.25 Furthermore, htt associates with a variety of membranous organelles, including mitochondria, ER, tubulovesicles, endosomes, lysosomes, and synaptic vesicles.37−39 Lipids are even incorporated within htt aggregates observed in mouse models, and the surface of htt inclusion bodies contain membranous structural elements.24,40 A variety of amino-terminal mutant htt fragments directly bind the lipid membranes, aggregate and alter the mechanical properties of the membrane, and ultimately cause membrane leakage in vitro.41−46 Perinuclear inclusions of htt have been linked to disruption of the nuclear envelope in HD mouse models,36 and expanded polyQ can embed into ER, resulting in membrane distortion.47
Protein/lipid membrane interactions have been shown to potentially play a role in a number of amyloid-related diseases.43 Membrane composition may directly influence htt aggregation and related toxicity. In this regard, there are a variety of lipid homeostasis and metabolism abnormalities associated with HD. Specifically, sphingomyelin (SM) fatty acid composition in cerebral white matter is altered in both juvenile and adult onset HD with an increase in short-chain fatty acids; this finding has been attributed to disturbed myelination of nerves.48,49 Metabolic profiling of presymptomatic HD in sheep found a marked decrease in plasma sphingolipids, which may reflect a disease signature,50 whereas an untargeted metabolomics approach in the STHdH cell line model of HD identified altered sphingolipid metabolism.51 Furthermore, reduced glycosphingolipid concentration is observed in erythrocytes, striatum, and caudate in both HD patients and animal models.52,53 Glycosphingolipid species play a number of roles in maintaining neuronal viability, and in particular, GM1 has neuroprotective properties upon exposure to a variety of toxic entities ranging from glutamate to amyloids.54,55 Reduced levels of gangliosides may be related to the observed reduced synthesis of GM1 in fibroblasts from HD patients as well as in cell and animal models of HD.56 The expression of glycosyltransferases is altered in HD, resulting in disrupted glycolipid and ganglioside metabolic pathways in the caudate.53 GM1 deficiencies have been observed in the YAC128, R6/1, and R6/2 transgenic mouse model of HD as well as in caudate samples from human HD patients.53,56,57 Interestingly, the GM1 content in the plasma membrane of HD cells correlated with a cell’s susceptibility to apoptosis.56 Collectively, these observations have led to a number of studies exploring the use of GM1 as a potential therapeutic agent. For example, addition of GM1 to the striatal cell line models of HD resulted in a protective effect against htt toxicity, restoring cell survival to wild-type levels.56 The additional GM1 activates AKT (Ser/Thr-specific protein kinase), resulting in enhanced phosphorylation of mutant htt,56 and phosphorylation is known to reduce mutant-htt accumulation and induce toxicity.58−60 The intraventricular infusion of GM1 in a symptomatic transgene HD mouse model induced phosphorylation of residues 13 and 16 within Nt17 and attenuated htt toxicity, restoring normal motor function.61 Beyond simply adding GM1, mitochondrial modulators, alpha-lipoic acid and acetyle-l-carnitine, were able to enhance glycosphingolipid levels in an experimental HD cell model.62 Although the primary importance of htt aggregation in HD has long been appreciated, the impact of specific lipid components to modulating htt aggregation and its direct interaction with membrane interfaces is poorly understood.
Here, we characterize the interaction of N-terminal htt fragments with total brain lipid extract (TBLE) lipid membranes that contain varying exogenous amounts of sphingolipids, SM or GM1. The use of a variety of membrane models [lipids monolayers at the air/buffer interface, large unilamellar vesicles (LUVs), and supported lipid bilayers] and biophysical techniques [in situ atomic force microscopy (AFM), Langmuir trough techniques, and vesicle association/permeability assays] enabled us to test a range of SM and GM1 concentrations, which can amplify any weak htt–sphingolipid interaction to determine how varying the SM and GM1 concentrations affects the interactions of htt with lipid membrane interfaces.
Results and Discussion
Addition of SM or GM1 Modifies the Insertion of htt into Lipid Membranes
To understand how the sphingolipids, SM and GM1, with a common sphingosine backbone but varying headgroups (SM—phosphocholine; GM1—complex oligosaccharide) alter the interactions between htt and lipid membranes, monomeric htt peptide (Nt17-Q35-P10-KK) insertion into TBLE monolayers with varying amounts of exogenously added SM or GM1 was measured using a Langmuir trough (Figure 1). Upon injection to the subphase, the peptide molecules partition between the bulk and air/buffer interface because of the presence of the amphipathic N-terminal lipid-binding domain, Nt17, until an equilibrium spreading pressure is reached (Π = 17.3 mN/m at 900 nM peptide42). Pure TBLE monolayers and those doped with 10 or 20 wt % exogenous SM or 5 or 10 wt % GM1 were spread at the air/buffer interface and compressed to a physiologically relevant surface pressure of 30 mN/m.63,64 The amounts of exogenous SM and GM1 to be added were determined from preliminary screening experiments that suggested the SM-impacted htt/lipid interactions up to an addition of 20%, whereas the impact of GM1 saturated after the 10% increase. The exogenous amounts of SM and GM1 added to the TBLE system are not physiological. Though TBLE with 10% GM1 or 30% SM will not be found within a cell membrane, these limits were chosen to provide a clear experimental system to determine how these lipid components interacted with htt. The monolayers were held at this pressure as freshly prepared monomeric peptide was injected into the subphase, and the average area per molecule at the interface, an indication of peptide insertion, was monitored until equilibrium was reached. Then, the pressure was lowered 4 mN/m, and the process was repeated. The addition of either SM or GM1 at any concentration decreased the extent of htt peptide insertion into the monolayer at each surface pressure (Figure 1). These decreases were statistically significant (p < 0.05) compared to control using a t-test, except for addition of GM1 at surface pressures greater than 26 mN/m and addition of SM at a surface pressure of 26 mN/m. The reduced insertion scales with the SM content; pure TBLE had a 28% total increase in the area, whereas adding 10 wt % exogenous SM resulted in a 23% area increase and 20 wt % SM had an 18% increase. However, the reduction in insertion associated with increased GM1 concentration was similar for both 5 and 10 wt % used here at ∼20% total increase in the area. Of note, regardless of the amount of exogenous SM or GM1, Nt17-Q35-P10-KK inserted into the lipid monolayers at surface pressures higher than the previously measured equilibrium spreading pressure, indicative of a driving force for the peptide to interact with the TBLE membrane. This experiment measures htt peptide association with and insertion into the lipid monolayer, as determined by the headgroup chemistry and the lipid packing density dictated by the phase state of the layer.
Figure 1.
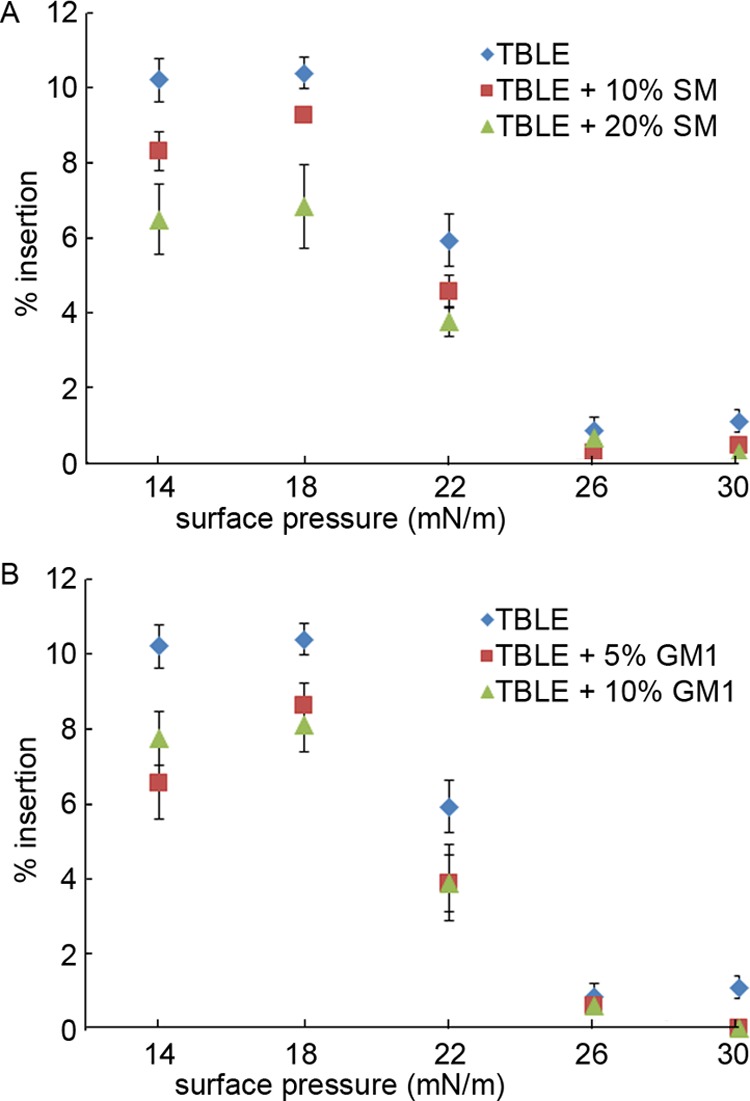
Insertion of Nt17-Q35-P10-KK into lipid monolayers of varying (A) TBLE/SM and (B) TBLE/GM1 ratios at the air/buffer interface, expressed as % insertion at each surface pressure. The error bars represent the standard deviation (n = 3 separate experiments).
To ascertain how the addition of exogenous SM or GM1 affects TBLE membranes in decreasing the amount of htt peptide insertion, individual surface pressure versus mean molecular area isotherms were measured for the pure components and a series of binary mixtures of TBLE with SM or GM1 (Figure 2A,B). As pure TBLE and SM had similar isotherms, an increase in the SM content did not dramatically affect the TBLE monolayer isotherm. In terms of lipid mixtures, the lipid raft hypothesis proposes that naturally occurring lipids such as sphingolipids, glycosphingolipids, and cholesterol specifically aggregate in the plane of the membrane driven primarily by hydrogen bonding between the lipid headgroups and alignment of saturated hydrocarbon chains.65 The SM brain extract used here is a heterogonous mixture consisting of SM molecules with a sphingosine backbone bound to one of a variety of amide-linked fatty acids, of which the prominent ones are stearic (C18:0, 50%) and lignoceric (C24:1, 21%) acids. Adding this mixture of SM lipids to TBLE has a complex effect. Because of the hydrogen-bonding capabilities in the sphingosine backbone, neighboring headgroups can be tightly bound and locally ordered.66 The SM molecules containing saturated hydrocarbon fatty acid chains contribute to this close packing, but the lignoceric acid has a cis-double bond in the chain that promotes disorder and fluidity, resulting in a film with a packing density (on average) similar to that of TBLE. The decreased amount of htt insertion, as measured by an increase in the monolayer area at a constant surface pressure in Figure 1, that scales with the SM content suggests that the solidifying effect of the increased hydrogen-bonding network in the lipid headgroup region may prevent the physical insertion of Nt17.
Figure 2.
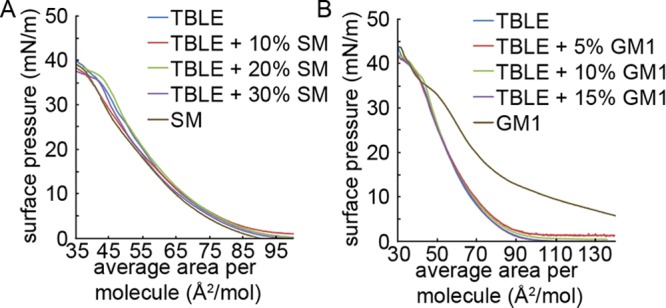
Monolayer compression isotherms of (A) pure TBLE, pure SM, and binary mixtures of TBLE and SM and (B) pure TBLE, pure GM1, and binary mixtures of TBLE and GM1, measured at 30 °C. Average area per molecule was calculated assuming an average TBLE molecular weight of 850 g/mol.
By contrast, the TBLE/GM1 isotherms clearly demonstrate TBLE monolayer condensation upon addition of GM1, relative to ideal mixing (Figure 2B) where the inclusion of negatively charged bulky oligosaccharide headgroups did not appreciably change the area per molecule at a specific pressure. Comparison of the mean molecular area of the binary mixtures to that of a theoretical mean area consistent with ideal mixing of the TBLE with GM1 suggests that exogenous GM1 causes condensation or stiffening of the monolayer. This is in agreement with both the lipid raft hypothesis, where ordered regions of the membrane are enriched in gangliosides, and the GM1-induced lateral condensation of lipids seen in binary DPPC/GM1 membranes.67 The GM1 condensation effect can be explained by the electrostatic interactions between the headgroups, alignment of dipole moments within their headgroups, and the tendency of gangliosides to have cooperative interactions qualitatively attributed to hydrogen bond formation between adjacent sugar residues.68,69 Our interpretation is that the small condensation effect seen at low GM1 concentrations dramatically decreases the amount of htt peptide insertion that leads to membrane lateral expansion.
SM and GM1 Alter the Susceptibility of Vesicles to htt-Induced Permeability
Although lipid monolayers are a convenient model for the outer leaflet of a cell membrane, it should be noted that in addition to in-plane interactions between monolayer components, cross-leaflet interactions between the lipids in bilayer membranes also affect lipid packing, and these effects are not captured in a monolayer model,70 prompting the need for lipid bilayer assays to complement the measurements. As we have shown previously, Nt17-Q35-P10-KK peptides permeablize TBLE vesicles, allowing content leakage.42,45 To determine if altering the SM or GM1 content of TBLE bilayers changed the membrane susceptibility to permeabilization by the htt peptide, calcein dye leakage assays with LUVs were performed. Disruption of the LUV membrane by external agents via mechanisms ranging from short-lived pore formation to complete vesicle lysis results in calcein release, dilution, and increased fluorescence emission from the unquenched dye, allowing permeabilization to be correlated with the fluorescence intensity. Solutions of predominately monomeric Nt17-Q35-P10-KK were mixed with vesicles comprised of TBLE containing varying amounts of exogenously added SM or GM1, and maximum fluorescence signal was measured (Figure 3). Measurements were taken after 3 min of exposure to the peptide, as both our previous studies on related systems42,45 and our current work indicate that the peptide interacts with membranes to induce the maximal amount of leakage, and hence the fluorescence, within this timeframe. Nt17-Q35-P10-KK induced a significant calcein release from LUVs comprised of pure TBLE, that is, approximately 30% increase in relative fluorescence at the highest peptide concentrations compared to 0.7% arising from control volumes of trifluoroacetic acid (TFA)/phosphate-buffered saline (PBS) buffer (Figure 3). As the SM content of the LUVs was increased, the extent of calcein dye leakage induced by exposure to Nt17-Q35-P10-KK drastically increased, with maximum leakage at the highest SM levels (Figure 3A). Just 5% addition by mass of SM to TBLE LUVs significantly increased permeabilization at all peptide concentrations, except at a peptide concentration of 0.484 μM (based on t-tests, p ≤ 0.02), and the addition of 30 wt % SM approximately doubled the permeabilization. All increases in calcein dye leakage associated with SM content greater than 5% were statistically significant (based on t-tests, p ≤ 0.02). Increased susceptibility of SM containing LUVs to Nt17-Q35-P10-KK-induced permeabilization was unexpected based on the decreased amount of peptide insertion into TBLE monolayers enriched in SM, as measured on a Langmuir trough. This suggests that SM increases the efficiency of lipid-bound htt to induce membrane permeabilization, such that a lower concentration of membrane-inserted htt resulted in greater content leakage. This may be attributed to the disruption of the sphingosine H-bonding network, which could increase leakage for comparable amounts of peptide insertion. By contrast, decreased insertion of htt into membranes containing exogenous GM1, as measured by Langmuir isotherms, did not correlate with a significant change in membrane permeabilization of LUVs based on t-tests (Figure 3B).
Figure 3.
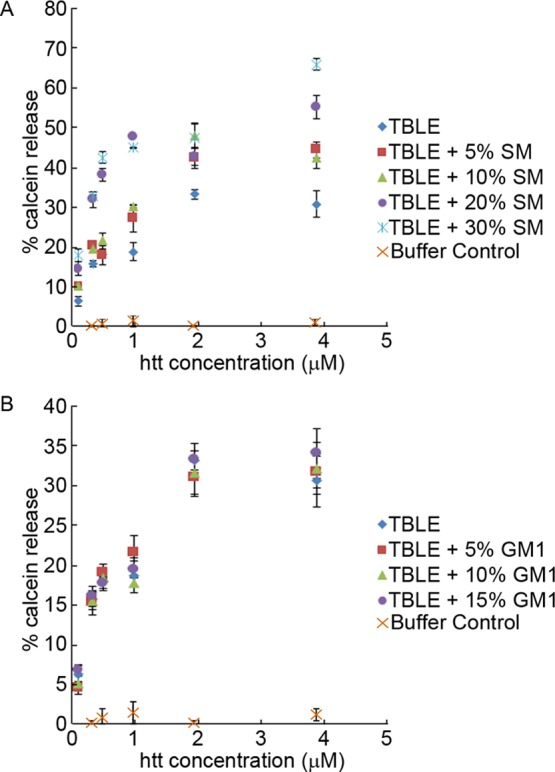
Calcein leakage, measured as relative fluorescence, from LUVs composed of different (A) TBLE/SM ratios or (B) TBLE/GM1 ratios, exposed to Nt17-Q35-P10-KK (or buffer acting as a control) as a function of the peptide concentration. The error bars indicate one standard deviation (n = 3 separate experiments).
Enriching TBLE with SM or GM1 Alters the Extent of htt-Exon1 Binding to Lipid Vesicles, as Determined by a TBLE/PDA Binding Assay
To verify that enhancing the lipid content of TBLE with SM or GM1 would modulate the interaction of a larger fragment of htt, full-length htt-exon1, with lipids, we performed a TBLE/PDA vesicle lipid-binding assay (Figure 4). This assay uses mixed vesicles comprised of phospholipids and polymerized PDA, which appear blue because of absorbance λmax ≈ 640 nm. Upon interaction with an exogenous particle or macromolecule, the PDA side-chain groups in the vesicle can become disordered, leading to an irreversible distortion of the side chains that strains the polymer backbone. This disruption leads to a change in the effective conjugated length of the PDA bonds, resulting in a rapid, visible color change to red.71,72 The colorimetric response can be used to quantify peptide/membrane interactions and interfacial membrane processes such as protein aggregation that would cause structural perturbations of the lipid interface. PDA-based vesicles have been used to study a variety of protein/lipid interactions, including those involving amyloid proteins.73−75 The TBLE/PDA vesicles were systematically prepared with different ratios of lipid components. These included pure TBLE, TBLE enriched with 10, 20, or 30% SM by mass, and TBLE enriched with 5, 10, or 15% GM1 by mass. These different vesicle preparations were exposed to freshly prepared htt-exon1(51Q) at a concentration of 20 μM, and % CR was measured for 16 h (Figure 4). Typically, an initial steady increase in % CR is observed upon exposure of TBLE/PDA vesicles to htt-exon1(51Q), and the % CR eventually reaches a quasi-steady state. As the exogenous SM content was enriched from 0 to 10%, the interaction of htt-exon1(51Q) with the vesicles was significantly reduced (p ≤ 0.02 at each time point, based on a t-test). However, an increase in the SM content resulted in enhanced htt/lipid interaction (p ≤ 0.02 at each time point based on a t-test). The interactions between htt and vesicles enriched with 20 or 30% SM were indistinguishable. The enhanced % CR associated with the addition of SM at 20% or higher is consistent with the increased leakage measured by the calcein dye assay, suggesting that the PDA assay detects an induced change in the membrane associated with disruption. In comparison with the calcein dye leakage assay, the reduced % CR associated with the addition of 10% SM suggests that SM enhances the susceptibility of membranes to htt-induced damage. Though less htt peptide is physically inserted, leading to reduced lateral expansion in the SM-doped TBLE membrane, the associated htt disrupts and permeabilizes the membrane more effectively. A small 5% increase in GM1 to the vesicles modestly decreased the htt/lipid interaction at early time points. Increasing the GM1 content by 10 or 15% dramatically inhibited the interaction of htt-exon1(51Q) with the lipid vesicles (p ≤ 0.02 at each time point based on a t-test), which was consistent with the previous measurements of decreased peptide insertion into the lipid monolayer.
Figure 4.
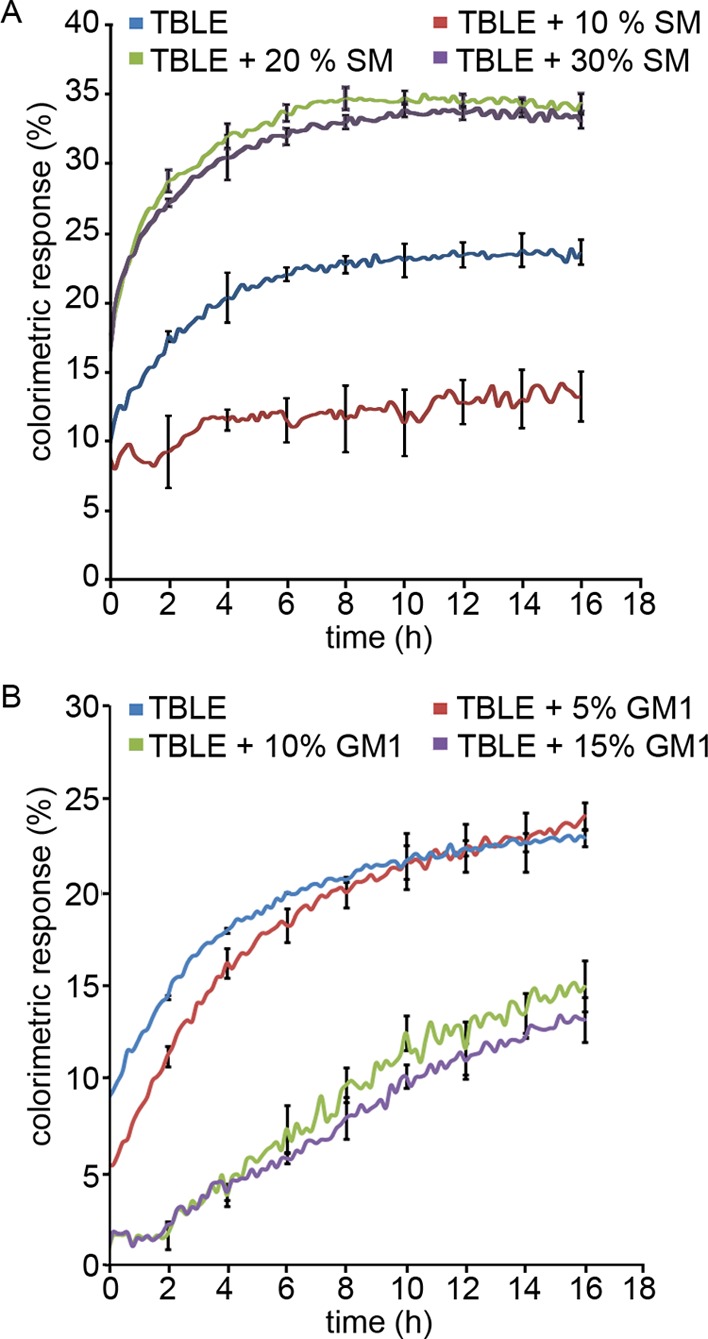
Percent colorimetric response (% CR) of TBLE/ polydiacetylene (PDA) vesicles containing various amounts of exogenous (A) SM or (B) GM1 upon exposure to htt-exon1(51Q) plotted as a function of time. The error bars indicate 1 standard deviation (n = 3).
SM and GM1 Modulate the Morphological Changes in Lipid Bilayers Associated with Exposure to htt
We investigated the influence of SM or GM1 on the aggregation of htt on lipid membranes using in-solution AFM. Supported lipid bilayers on mica were systematically produced with varying lipid contents. These lipid bilayers included pure TBLE, TBLE + 10 wt % SM, TBLE + 20 wt % SM, TBLE + 5 wt % GM1, and TBLE + 10 wt % GM1. Such supported lipid bilayers are known to retain several important properties associated with lipid membranes, such as lateral fluidity,76 and similar systems have been used as model surfaces to study protein aggregation with lipid membranes.33,41−43,45,77−82 Pure TBLE bilayers and those with exogenously added SM or GM1 were all smooth in appearance [root mean square (rms) roughness less than 0.2 nm], with no indication of distinct micron-scale lipid domains or phase separation prior to exposure to htt-exon1(51Q) (Figure 5A), and were stable with no induced morphological changes for at least 8 h under continuous AFM imaging. Having established that supported bilayers appropriate for this study could be made from the different lipid mixtures, these model bilayers were systematically exposed to freshly prepared aliquots of htt-exon1(51Q) at a final protein concentration of 20 μM in the AFM fluid cell and continuously imaged using AFM in-solution, allowing for the direct tracking of protein aggregation on the bilayer. Only continuous TBLE bilayers (40 × 40 μm in size), as assessed by AFM imaging, were exposed to htt-exon1(51Q), and observations were limited to these verified regions of the continuous bilayer.
Figure 5.

(A) AFM height images taken in-solution of continuous, supported lipid bilayers of TBLE, TBLE + 10% SM, TBLE + 20% SM, TBLE + 5% GM1, and TBLE + 10% GM1 prior to exposure to any htt-exon1(51Q). (B) Sequential AFM height images taken in-solution of supported TBLE bilayers exposed to htt-exon1(51Q). Blue rectangles identify the same region of the surface in the sequential images. (C) Zoomed-in AFM images demonstrating the rough, grainy morphological changes induced in a pure TBLE bilayer by htt-exon1(51Q). Blue arrows indicate oligomeric aggregates. (D) Height and volume histograms of oligomeric aggregates observed on TBLE bilayers presented as a function of time. To ease visualization, the histograms were normalized for each time point by setting the value of the most common height or volume to 1.
Consistent with our previous studies,45 pure TBLE bilayers exposed to 20 μM solutions of htt-exon1(51Q) quickly (within 1 h) develop regions characterized by extensive roughness (rms roughness greater than 1.0 nm) and appearance of discrete, oligomeric htt aggregates (Figure 5B). These regions of increased roughness expand in area and become increasingly rougher with time (observed for up to 5 h). Previous reports have correlated these morphological changes with altered mechanical properties of the lipid bilayer and membrane leakage.42,45,46 Although the number of oligomers observed on the surface increased with time, these oligomers appeared to be stable (Figure 5C,D). That is, specific individual oligomers could be identified in subsequent images without large changes in their size. As a population, the average size of oligomers did not change with time (a mode of 4–5 nm in height and ∼22 000 nm3 in volume that was uncorrected for the size of the AFM probe) (Figure 5D). Collectively, these observations suggest that the TBLE bilayer promotes and stabilizes these specific oligomeric species. htt fibrils were not observed on lipid bilayers during the time course of the experiment. The same htt-exon1(51Q) construct has been shown to readily form fibrils over the same time course under the same preparation conditions and concentration in the absence of lipids.83 The lack of fibril formation on the pure TBLE bilayer suggests that the lipid system either promotes the formation of aggregates off the pathway to fibrils or that aggregate intermediate oligomers are stabilized by interaction with the membrane.
When TBLE bilayers were enriched with 10 or 20% SM, the morphological changes associated with exposure to htt-exon1(51Q) were altered (Figure 6A,B). When TBLE was enriched with 10% SM, the regions of the bilayer altered were smaller in area and had a smoother, plateau-like appearance. These regions increased in height with time. These regions were also not associated with the abundant formation of oligomeric aggregates. With the addition of 20% SM to TBLE bilayers, regions of increased roughness that were associated with the appearance of small oligomers initially developed; however, the morphology of these regions evolved with time to eventually form plateau-like domains that extended 10–16 nm above the surface, corresponding to the height of several lipid bilayers. These plateau-like domains are clearly distinguishable in phase-contrast AFM imaging, suggesting that they are mechanically different than the unaffected regions of the bilayer (Figure 6C). Fewer htt oligomers were observed on bilayers that contained 20% exogenous SM compared with pure TBLE. Oligomers observed on bilayers with the 20% extra SM were initially slightly smaller (a mode of 2–3 nm in height and ∼20 000 nm3 in uncorrected volume) than the stable oligomers associated with aggregation of htt on pure TBLE bilayers (Figure 6D). After 2 h, the oligomers observed on lipid bilayers with 20% SM increased in size, but were still slightly smaller than those observed on pure TBLE.
Figure 6.

(A) Sequential AFM height images taken in-solution of supported TBLE bilayers enriched with either 10 or 20% SM exposed to htt-exon1(51Q). Blue rectangles identify the same region of the surface in the sequential images. (B) Zoomed-in AFM images demonstrating the morphological changes induced in TBLE bilayers enriched in SM. Blue arrows indicate oligomeric aggregates. Green lines correspond to the height profiles presented directly below the images. The lower right corners of the images for the 1 h time point are presented at the same height scale as the 3 h images for easier comparison. (C) AFM image split into height and phase data of a region of a TBLE + 20% SM bilayer exposed to htt-exon1(51Q). (D) Height and volume histograms of oligomeric aggregates observed on TBLE bilayers enriched with 20% SM, presented as a function of time. To ease visualization, the histograms were normalized for each time point by setting the value of the most common height or volume to 1.
When TBLE bilayers were enriched with 5 or 10% GM1, the resulting morphological change associated with exposure to htt-exon1(51Q) was similar to that observed on pure TBLE bilayers; however, the area of the bilayer altered decreased with increasing GM1 content (Figure 7A,B) parallel to the lipid monolayer insertion and TBLE/PDA binding assays, which indicated decreased htt insertion/association at a higher GM1 concentration. Patches of increased bilayer roughness that were highly associated with the appearance of htt oligomers developed, but these areas developed more slowly with increasing GM1 content. The number of oligomers increased with time on bilayers with both 5 or 10% enrichment with GM1, and these oligomers were morphologically indistinguishable from those observed in pure TBLE bilayers (a mode of 4–5 nm in height and ∼22 000 nm3 in uncorrected volume; Figure 7B,C). The oligomers were also stable as individual oligomers could be observed in subsequent images for several hours, and their dimensions did not appreciably change. After 5 h of exposure to htt-exon1(51Q), some fibrils could be observed in one trial with TBLE + 5% GM1 (Figure 7A).
Figure 7.

(A) Sequential AFM height images taken in-solution of supported TBLE bilayers enriched with either 5 or 10% GM1 exposed to htt-exon1(51Q). Blue rectangles identify the same region of the surface in the sequential images. Black arrows indicate fibrillar aggregates. (B) Zoomed-in AFM images demonstrating the rough, grainy morphological changes induced in TBLE bilayers enriched in GM1. Blue arrows indicate oligomeric aggregates. (C) Height and volume histograms of oligomeric aggregates observed on TBLE bilayers enriched with GM1 presented as a function of time. To ease visualization, the histograms were normalized for each time point by setting the value of the most common height or volume to 1.
To quantify the interaction of htt-exon1(51Q) with the supported bilayers containing various amounts of SM or GM1 as observed by AFM, the percentage of the surface that displayed any altered morphology and the rms roughness of those regions were determined as a function of time via image analysis (Figure 8). Despite the differences in the developed morphology associated with exposure to htt-exon1(51Q), with the addition of 10% SM, the percent area of the total bilayer altered was smaller compared with that observed with pure TBLE bilayers; but the addition of 20% SM resulted in an increased area of the bilayer with an altered morphology. This relative area of altered morphology observed by AFM was consistent with our PDA assay, which measures interfacial disruption induced by htt-exon1(51Q). With the addition of GM1, there was no significant change in the area of the bilayer surface displaying altered morphology until enrichment with 10% GM1, when the modified surface area was lessened, and there was no significant change in the rms roughness of these regions compared with the pure TBLE controls.
Figure 8.

Percent area of disrupted bilayer morphology and increased rms roughness associated with exposure of lipid bilayers containing various amounts of (A,B) SM or (C,D) GM1 to htt-exon1(51Q), presented as a function of time. The error bars indicate 1 standard deviation over three separate experiments.
Role of htt/Lipid Interactions in HD Pathogenesis
The interaction of htt with membranous surfaces may mediate pathogenesis in several ways. These include the direct promotion or stabilization of specific aggregate species,42 compromised mechanical integrity of cellular and subcelluar membranes,36,41,44,46,47 and altered trafficking of htt within the cell.21 Several lines of evidence point to a prominent toxic role for oligomeric species,84−86 and lipids may promote the formation and stability of specific oligomeric species. Such a scenario is consistent with our direct observation of stable oligomers that formed on the various bilayers studied here and previous reports indicating that synthetic polyQ peptides aggregate differently on bilayer surfaces compared with mica substrates.42 If specific htt conformations or aggregates play a pivotal role in HD, understanding how specific lipid components may shift the relative abundance of disease relevant conformations may be critical. Although enriching TBLE bilayers with GM1 reduced the total insertion of the peptide into lipid monolayers and the accumulation of htt on lipid bilayers, it did not result in a significant reduction of membrane permeabilization in our study, suggesting that reduced susceptibility of membranes to htt-induced disruption may not contribute to the protective effect associated with GM1 replacement in HD models. As GM1 modulates the action of numerous cellular receptors, GM1 deficits associated with HD interrupt the processes associated with these receptors that can be rescued by restoring GM1 levels. In addition, GM1 treatments were found to enhance htt phosphorylation in mice, which is known to reduce htt-related toxicity.56,61
Previously, we reported that enriching TBLE bilayers with cholesterol resulted in large plateau-like domains upon exposure to htt-exon1(51Q) of the bilayer, similar to those seen in SM-doped membranes; however, these cholesterol-enriched membranes were resistant to htt-induced permeabilization in contrast to SM-enriched ones.45 One key difference between these two systems is that the plateau-like regions that developed in the bilayers enriched with SM grew to be thicker and mechanically different (based on phase imaging) compared to the unaffected regions of the bilayer. In the case of membranes enriched with cholesterol, oligomeric aggregates of htt were not observed to form on the membrane surface at any time to an appreciable extent.45 With this in mind, it is interesting to note that enrichment with SM (of at least 20%) and GM1 here resulted in htt-induced membrane permeabilization (Figure 3) to a similar or greater extent compared with pure TBLE bilayers and that oligomers (although sometimes transient) were observed under all of these conditions (Figures 5–7). Collectively, these observations suggest that the formation of oligomers may play a key role in the ability of htt to induce membrane permeabilization. These findings also justify the need for the analysis of how the altered membrane composition affects the htt peptide structure upon binding.
Conclusions
Lipid interactions are associated with a number of wild-type functions attributed to htt. Membrane damage and altered lipid homeostasis are the biochemical features of HD, and this htt-induced damage to membranes represents a potential toxic mechanism in HD, as a similar membrane disruption has been associated with a number of other amyloid-forming proteins. As a result, determining the factors that regulate the affinity of htt for membranes is critical not only for understanding the normal functions of htt but also for identifying ways to modify htt–lipid interactions for potential therapeutic strategies. In this study, we investigated the role of sphingolipids, SM and GM1, in modulating the interaction with and subsequent aggregation of model htt peptides (both synthetic and recombinant) on TBLE lipid membranes and the associated consequences. The exogenous addition of SM or GM1 to TBLE lipid monolayers reduced the insertion of htt, likely due to the increased extent of the sphingosine backbone hydrogen-bonding network within the membrane. The extent of htt-induced vesicle permeabilization was not affected by the addition of GM1, whereas increasing the SM content enhanced the susceptibility of vesicles to htt-induced permeability; though less htt was inserted into the membranes containing a higher concentration of sphingolipid, the disruption induced by htt caused more content leakage. In agreement with the lipid monolayer insertion experiments, the total interaction of htt with bilayers enriched with GM1 was reduced, as measured by both a colorimetric assay and AFM. Although the extent of bilayer alteration caused by exposure to htt was reduced with increasing GM1 content, the actual morphological features associated with the affected regions of the bilayer, including oligomer formation, were consistent with those observed on pure TBLE. The extent of htt/lipid interaction varied greatly with the SM content, and the induced morphological changes of the bilayer were distinct in comparison with those observed on pure TBLE bilayers. These observations suggest that sphingolipid composition strongly influences the material properties of the lipid membrane, which affects htt binding and aggregation, and that subsequent oligomer formation on the interface may correlate with membrane permeabilization.
Methods
Peptide Preparation
A synthetic, model htt peptide consisting of the Nt17 domain, 35 glutamines (sufficient for aggregation),87 a polyproline (polyP) domain 10 residues in length, and 2 C-terminal lysines, referred to as Nt17-Q35-P10-KK, was obtained via custom synthesis (Keck Biotechnology Resource Laboratory, New Haven, Connecticut). Lysines were added to enhance solubility. Established protocols for disaggregation and solubilization of the peptide were used.88 In short, the peptide was dissolved overnight in a 1:1 mixture of TFA (Acros Organics) and hexafluoroisopropanol (Acros Organics) at 0.5 mg/mL. A stream of N2 was used to evaporate the solvent, and the residual solvent was removed using a Vacufuge concentrator for 3 h (Eppendorf, Hauppauge, NY). The resulting peptide films, stored at −20 °C, were then dissolved in ultrapure water adjusted to pH 3 with TFA at a concentration of 200 μM. This solution was subsequently diluted in PBS (10 mM phosphate, 140 mM NaCl, 2.7 mM KCl) (Life Technologies) to a final concentration of 20 μM and a final pH of 7.3.
Purification of Glutathione S-transferase (GST)–htt-Exon1 Fusion Proteins
GST–htt-exon1 fusion proteins containing 51 repeat glutamines were purified, as previously described.89 Briefly, the GST–htt fusion proteins were expressed by induction in Escherichia coli with isopropyl β-d-thiogalactoside at 30 °C for 4 h, and the cells were lysed with lysozyme (0.5 mg/mL). Fusion proteins were purified from the lysate with a GST affinity column via liquid chromatography (LPLC, BioRad). Relevant fractions were identified by ultraviolet absorption and confirmed by gel electrophoresis. Before use in any assay, the solutions of fusion proteins were centrifuged at 20 000g for 30 min at 4 °C to remove preexisting aggregates. GST was cleaved by Factor Xa (Promega, Madison, WI), and this cleavage initiates aggregation. Experiments were carried out in buffer A (50 mM Tris-HCl, pH 7.0, 150 mM NaCl). To ensure efficient GST cleavage, the GST–htt-exon1 fusion protein and the respective cleavage agents were incubated for 1 h on ice prior to their addition to any assay.
Langmuir Trough Configuration
All lipid monolayer surface pressure versus molecular area isotherms and peptide insertion experiments were performed in a Teflon Langmuir trough with symmetric barriers (large inverted microscopy model, NIMA Technologies, Coventry, England). A stationary Wilmhelmy balance (NIMA Technologies, Coventry, England) was used to measure the surface pressure at the air/buffer interface. All measurements were collected at 30 ± 0.5 °C, and the trough temperature was maintained with a circulating heated water bath (Isotemp 3016D water circulator, Thermo Fisher Scientific).
Lateral Compression Experiments
Lipid monolayer isotherms were conducted to characterize monolayer phase behavior as a function of lipid-packing density and membrane composition. Stock TBLE (Avanti Polar Lipids) (extract lipid headgroup composition: 9.6 wt % phosphatidylcholine, 16.7 wt % phosphatidylethanolamine, 1.6 wt % phosphatidylinositol, 10.6 wt % phosphatidylserine, 2.8 wt % phosphatidic acid, and 58.7 wt % unknown) was obtained in chloroform and used without further purification. Because of the uncharacterized composition of TBLE, an average TBLE molecular weight of 850 g/mol was assumed. SM (brain, porcine; Avanti Polar Lipids) was received in powder form and was dissolved in chloroform (Sigma-Aldrich). In the SM brain extract, the amide-linked fatty acid bound to the sphingosine backbone varies with the following composition: C16:0 2%, C18:0 50%, C20:0 5%, C22:0 7%, C24:0 5%, C24:1 21%, and others 10%. GM1 ganglioside (brain, ovine, sodium salt; Avanti Polar Lipids) was received in powdered form and dissolved in 8:2 chloroform/methanol. TBLE was mixed in different ratios with SM or GM1 to a total spreading concentration of 0.4 mg/mL and stored in glass vials at −20 °C. The lipid monolayer was formed by dropwise addition of the spreading solution on the PBS (pH = 7.3) surface; the solvent was allowed to evaporate for 15 min. Barriers compressed the monolayer at 6 cm2/min, and isotherm measurements in the form of surface pressure (mN/m) versus mean area per lipid molecule (Å2/molecule) were taken at 1 s intervals until monolayer collapse.
Measuring Peptide Insertion into a Lipid Monolayer
Insertion experiments were performed to quantify the Nt17-Q35-P10-KK peptide insertion into pure TBLE and mixed TBLE/SM and TBLE/GM1 monolayers. The lipid monolayer was compressed to a surface pressure of 30 mN/m to match the lateral density of a cellular bilayer,63,64 and then, the barriers were switched to feedback mode to maintain a constant surface pressure by adjusting the monolayer surface area. Lyophilized peptide was rehydrated in TFA/PBS, as described above, and immediately injected beneath the compressed monolayer to obtain a 900 nM final peptide concentration in the subphase. After 10 min, if no increase in the average area per molecule to signify peptide insertion was observed, the maintained surface pressure was reduced 4 mN/m and the process was repeated down to a minimum of 14 mN/m. Percent insertion of the peptide at each surface pressure was determined as a % increase in the area [(Afinal – Ainit)/Ainit × 100].
Vesicle Preparation for Leakage Assay
LUVs of TBLE, TBLE/SM, and TBLE/GM1 were prepared to determine internal content leakage caused by htt peptide insertion. Lipid films, evaporated from stock solution with a stream of N2, were hydrated in 70 mM calcein (Sigma) in 10 mM Tris, pH = 7.3 via 5 min of vortexing and 5 min of sonication. The vesicles were treated with 7 freeze–thaw cycles in ethanol/dry ice and warm water baths and then extruded 11 times through a 100 nm-pore polycarbonate filter (Avanti). The excess dye was removed with size exclusion chromatography using Sephadex G-50 beads (Sigma-Aldrich). Vesicle diameter size uniformity (120–160 nm depending on vesicle composition) was confirmed with dynamic light scattering [Zetasizer ZS90 (Malvern Worcestershire, UK)]. Lipid concentration was determined by a phosphorus assay adapted from Anderson and Rouser.90,91 Briefly, a lipid fraction (500 μL) and 8.9 N H2SO4 (225 μL) were vortexed and heated at 210 °C for 20 min. After cooling, 6% H2O2 (75 μL) was added and heated for an additional 25 min at 210 °C until the solution was colorless. After cooling, 1.95 mL of ultrapure water, 5% molybdic acid (225 μL), and 10% ascorbic acid (225 μL) were added, and the solution was placed in a boiling water bath for 10 min. Absorbance at 820 nm was measured using a Jasco V-550 UV/visible spectrophotometer (Jasco, Easton, MD) and compared to a KH2PO4 reference plot to calculate the final lipid concentration.
Vesicle Leakage Assay
TBLE LUVs with exogenous SM (0–30 wt %) or GM1 (0–15 wt %) were exposed to the Nt17-Q35-P10-KK peptide in a concentration range, resulting in peptide/lipid molar ratios ranging from 1:67 to 1:1.7 at 25 °C. Lyophilized peptides were hydrated as outlined above and used promptly to minimize the presence of nonmonomeric species. Fluorescence at 515 nm (495 nm excitation) from calcein was measured prior to peptide exposure and 3 min after peptide administration using a RF-1501 spectrofluorometer (Shimadzu, Columbia, MD). Vesicles were then completely lysed using 1% Triton X-100 (Sigma) to measure the maximum calcein fluorescence. Vesicle leakage caused by the peptide was calculated by
| 1 |
where If is the fluorescent intensity after peptide addition, II is the initial fluorescence of the LUVs, and IT is the total fluorescence intensity after complete lysis.
TBLE/PDA Lipid-Binding Assay
TBLE/PDA assays were performed using previously reported protocols.75,92 In summary, diacetylene monomers 10,12-tricosadiynoic acid (GFS Chemicals, Columbus, OH), TBLE, and an appropriate amount of either SM or GM1 were dissolved in a solution of 1:1 chloroform/ethanol. Total lipid and PDA was always mixed in a 2:3 molar ratio. The solution was evaporated off in a rotary evaporator. The resulting films were resuspended in Tris-buffered saline, heated to 70 °C, sonicated for 5 min at 100 W using a sonic dismembrator (FisherSci), and stored at 4 °C overnight to ensure self-assembly of the vesicles. Polymerization of the diacetylene monomers was accomplished by irradiation at 254 nm with 7 lumens for 10 min (room temperature with stirring), which results in a blue color. Polymerized TBLE/PDA vesicles were exposed to htt-exon1(51Q) at a final concentration of 20 μM. TBLE/PDA vesicle samples were also exposed to 1× buffer A (negative control) or NaOH (pH 12) (positive control). All experiments were performed in triplicate in a 96-well format, and the colorimetric response at 500 and 640 nm was recorded for 16 h with an Infinite M1000 Pro plate reader (TECAN, Switzerland) at 25 °C. A quantitative value representative of the blue-to-red color transition was obtained by determining the % CR, defined as75,92
| 2 |
where PB is the red/blue ratio of absorbance (A) defined as Ablue/(Ablue + Ared). Ablue is the absorbance at ∼640 nm (absorbance at this wavelength range results in a blue color), Ared is the absorbance at ∼500 nm, PB0 is the red/blue ratio of the control sample (before induction of color change), and PBI is the value obtained for the vesicle solution after addition of peptides.
AFM Imaging Conditions
In-solution AFM experiments were performed with a Nanoscope V MultiMode scanning probe microscope (Bruker, Santa Barbara, CA) equipped with a closed-loop “vertical engage” J-scanner and a sealed tapping fluid cell. Images were acquired with silicon cantilevers (VISTAprobes) with spring constants of ∼0.1 N/m, scan rates of ∼2 Hz, and drive frequencies ranging from ∼8 to 10 kHz. AFM images were processed and analyzed using MATLAB and its image processing toolbox (Mathworks, Natick, MA), as previously described.83
Preparation of Supported Bilayers for AFM Experiments
TBLE, SM, and GM1 (Avanti Polar Lipids) were dissolved in chloroform (Fisher Scientific) and mixed at appropriate ratios to have samples of pure TBLE and TBLE spiked with specific weight percentages of exogenously added SM or GM1. The chloroform was evaporated off using a Vacufuge concentrator (Eppendorf, Hauppauge, NY). The resulting lipid films were resuspended in buffer A (pH 7.3) at 0.5 mg/mL. To form vesicles, these lipid suspensions underwent five sequential freeze–thaw cycles, followed by 15 min of sonication in a bath sonicator. The resulting vesicle solutions (35 μL) were injected into the AFM fluid cell. The formation of a continuous bilayer on mica via vesicle fusion was observed by continual AFM imaging, and all AFM experiments were performed between 23 and 25 °C. The fluid cell was flushed to remove vesicles remaining in solution. htt-exon1(51Q) was then injected into the cell to a final concentration of 20 μM, and the protein was present for the entirety of the experiment.
Acknowledgments
This work was supported by the National Institutes of Health grant R15NS090380. This work was supported, in part, by a grant to the Gettysburg College from the Howard Hughes Medical Institute through the Precollege and Undergraduate Science Education Program. S.L.F. thanks the Gettysburg College for the financial support.
Glossary
Abbreviations
- AFM
atomic force microscopy
- chol
choleseterol
- HD
Huntington disease
- HFIP
hexafluoroisopropanol
- htt
huntingtin
- GM1
ganglioside GM1
- GST
glutathione S-transferase
- LPLC
low pressure liquid chromatography
- LUVs
Large unilamellar vesicles
- % CR
% colorimetric response
- PBS
phosphate-buffered saline
- PDA
polydiacetylene
- polyQ
polyglutamine
- polyP
polyproline
- Nt17
the first 17 amino acids of htt
- SM
sphingomyelin
- TBLE
total brain lipid extract
- TFA
trifluoroacetic acid
Author Contributions
J.L. and S.L.F. conceived and coordinated the study and wrote the paper. M.C. and X.G. performed all in-solution AFM experiments on htt-exon1(51Q) and lipid bilayers and purified GST–htt-exon1(51Q). S.L.F. and W.A.C. performed Langmuir trough experiments and vesicle leakage assays with Nt17-Q35-P10-KK. M.C. and P.J. performed the TBLE/PDA lipid-binding assays and assisted in purifying GST–htt-exon1(51Q). All authors analyzed the results and approved the final version of the manuscript.
The authors declare no competing financial interest.
References
- The Huntington’s Disease Collaborative Research Group A Novel Gene Containing a Trinucleotide Repeat That Is Expanded and Unstable on Huntington’s Disease Chromosomes. Cell 1993, 72, 971–983. 10.1016/0092-8674(93)90585-E. [DOI] [PubMed] [Google Scholar]
- DiFiglia M.; Sapp E.; Chase K. O.; Davies S. W.; Bates G. P.; Vonsattel J. P.; Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Legleiter J.; Mitchell E.; Lotz G. P.; Sapp E.; Ng C.; DiFiglia M.; Thompson L. M.; Muchowski P. J. Mutant Huntingtin Fragments Form Oligomers in a Polyglutamine Length-dependent Manner in Vitro and in Vivo. J. Biol. Chem. 2010, 285, 14777–14790. 10.1074/jbc.m109.093708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X.; Greiner E. R.; Mishra R.; Kodali R.; Osmand A.; Finkbeiner S.; Steffan J. S.; Thompson L. M.; Wetzel R.; Yang X. W. Serines 13 and 16 Are Critical Determinants of Full-Length Human Mutant Huntingtin Induced Disease Pathogenesis in HD Mice. Neuron 2009, 64, 828–840. 10.1016/j.neuron.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel R. Physical Chemistry of Polyglutamine: Intriguing Tales of a Monotonous Sequence. J. Mol. Biol. 2012, 421, 466–490. 10.1016/j.jmb.2012.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker J. L.; Zareie M. H.; Fong H.; Sarikaya M.; Muchowski P. J. Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nat. Struct. Mol. Biol. 2004, 11, 1215–1222. 10.1038/nsmb860. [DOI] [PubMed] [Google Scholar]
- Hoop C. L.; Lin H.-K.; Kar K.; Magyarfalvi G.; Lamley J. M.; Boatz J. C.; Mandal A.; Lewandowski J. R.; Wetzel R.; van der Wel P. C. A. Huntingtin exon 1 fibrils feature an interdigitated β-hairpin–based polyglutamine core. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 1546–1551. 10.1073/pnas.1521933113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penney J. B.; Vonsattel J.-P.; MacDonald M. E.; Gusella J. F.; Myers R. H. CAG repeat number governs the development rate of pathology in Huntington’s disease. Ann. Neurol. 1997, 41, 689–692. 10.1002/ana.410410521. [DOI] [PubMed] [Google Scholar]
- Ravina B.; Romer M.; Constantinescu R.; Biglan K.; Brocht A.; Kieburtz K.; Shoulson I.; McDermott M. P. The relationship between CAG repeat length and clinical progression in Huntington’s disease. Mov. Disord. 2008, 23, 1223–1227. 10.1002/mds.21988. [DOI] [PubMed] [Google Scholar]
- Snell R. G.; Macmillan J. C.; Cheadle J. P.; Fenton I.; Lazarou L. P.; Davies P.; Macdonald M. E.; Gusella J. F.; Harper P. S.; Shaw D. J. Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington’s disease. Nat. Genet. 1993, 4, 393–397. 10.1038/ng0893-393. [DOI] [PubMed] [Google Scholar]
- Kim Y. J.; Yi Y.; Sapp E.; Wang Y.; Cuiffo B.; Kegel K. B.; Qin Z.-H.; Aronin N.; DiFiglia M. Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington’s disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 12784–12789. 10.1073/pnas.221451398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratovitski T.; Gucek M.; Jiang H.; Chighladze E.; Waldron E.; D’Ambola J.; Hou Z.; Liang Y.; Poirier M. A.; Hirschhorn R. R.; Graham R.; Hayden M. R.; Cole R. N.; Ross C. A. Mutant huntingtin N-terminal fragments of specific size mediate aggregation and toxicity in neuronal cells. J. Biol. Chem. 2009, 284, 10855–10867. 10.1074/jbc.m804813200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies S. W.; Turmaine M.; Cozens B. A.; DiFiglia M.; Sharp A. H.; Ross C. A.; Scherzinger E.; Wanker E. E.; Mangiarini L.; Bates G. P. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997, 90, 537–548. 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- Landles C.; Sathasivam K.; Weiss A.; Woodman B.; Moffitt H.; Finkbeiner S.; Sun B.; Gafni J.; Ellerby L. M.; Trottier Y.; Richards W. G.; Osmand A.; Paganetti P.; Bates G. P. Proteolysis of mutant huntingtin produces an exon 1 fragment that accumulates as an aggregated protein in neuronal nuclei in Huntington disease. J. Biol. Chem. 2010, 285, 8808–8823. 10.1074/jbc.m109.075028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudou F.; Humbert S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. 10.1016/j.neuron.2016.02.003. [DOI] [PubMed] [Google Scholar]
- Atwal R. S.; Truant R. A stress sensitive ER membrane-association domain in Huntingtin protein defines a potential role for Huntingtin in the regulation of autophagy. Autophagy 2008, 4, 91–93. 10.4161/auto.5201. [DOI] [PubMed] [Google Scholar]
- Gauthier L. R.; Charrin B. C.; Borrell-Pagès M.; Dompierre J. P.; Rangone H.; Cordelières F. P.; De Mey J.; MacDonald M. E.; Leßmann V.; Humbert S.; Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004, 118, 127–138. 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Gunawardena S.; Her L.-S.; Brusch R. G.; Laymon R. A.; Niesman I. R.; Gordesky-Gold B.; Sintasath L.; Bonini N. M.; Goldstein L. S. B. Disruption of axonal transport by loss of huntingtin or expression of pathogenic PolyQ proteins in Drosophila. Neuron 2003, 40, 25–40. 10.1016/s0896-6273(03)00594-4. [DOI] [PubMed] [Google Scholar]
- Lee W.-C. M.; Yoshihara M.; Littleton J. T. Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington’s disease. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 3224–3229. 10.1073/pnas.0400243101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal A.; Severin F.; Lommer B.; Shevchenko A.; Zerial M. Huntingtin–HAP40 complex is a novel Rab5 effector that regulates early endosome motility and is up-regulated in Huntington’s disease. J. Cell Biol. 2006, 172, 605–618. 10.1083/jcb.200509091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwal R. S.; Xia J.; Pinchev D.; Taylor J.; Epand R. M.; Truant R. Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum. Mol. Genet. 2007, 16, 2600–2615. 10.1093/hmg/ddm217. [DOI] [PubMed] [Google Scholar]
- Hilditch-Maguire P.; Trettel F.; Passani L. A.; Auerbach A.; Persichetti F.; MacDonald M. E. Huntingtin: an iron-regulated protein essential for normal nuclear and perinuclear organelles. Hum. Mol. Genet. 2000, 9, 2789–2797. 10.1093/hmg/9.19.2789. [DOI] [PubMed] [Google Scholar]
- Vidal R.; Caballero B.; Couve A.; Hetz C. Converging Pathways in the Occurrence of Endoplasmic Reticulum (ER) Stress in Huntingtons Disease. Curr. Mol. Med. 2011, 11, 1–12. 10.2174/156652411794474419. [DOI] [PubMed] [Google Scholar]
- Suopanki J.; Gotz C.; Lutsch G.; Schiller J.; Harjes P.; Herrmann A.; Wanker E. E. Interaction of huntingtin fragments with brain membranes—clues to early dysfunction in Huntington’s disease. J. Neurochem. 2006, 96, 870–884. 10.1111/j.1471-4159.2005.03620.x. [DOI] [PubMed] [Google Scholar]
- Kegel K. B.; Kim M.; Sapp E.; McIntyre C.; Castaño J. G.; Aronin N.; DiFiglia M. Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J. Neurosci. 2000, 20, 7268–7278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kegel K. B.; Sapp E.; Alexander J.; Valencia A.; Reeves P.; Li X.; Masso N.; Sobin L.; Aronin N.; DiFiglia M. Polyglutamine expansion in huntingtin alters its interaction with phospholipids. J. Neurochem. 2009, 110, 1585–1597. 10.1111/j.1471-4159.2009.06255.x. [DOI] [PubMed] [Google Scholar]
- Kegel K. B.; Sapp E.; Yoder J.; Cuiffo B.; Sobin L.; Kim Y. J.; Qin Z.-H.; Hayden M. R.; Aronin N.; Scott D. L.; Isenberg G.; Goldmann W. H.; DiFiglia M. Huntingtin associates with acidic phospholipids at the plasma membrane. J. Biol. Chem. 2005, 280, 36464–36473. 10.1074/jbc.m503672200. [DOI] [PubMed] [Google Scholar]
- Kegel K. B.; Schewkunow V.; Sapp E.; Masso N.; Wanker E. E.; DiFiglia M.; Goldmann W. H. Polyglutamine expansion in huntingtin increases its insertion into lipid bilayers. Biochem. Biophys. Res. Commun. 2009, 387, 472–475. 10.1016/j.bbrc.2009.07.039. [DOI] [PubMed] [Google Scholar]
- Kegel-Gleason K. B. Huntingtin Interactions with Membrane Phospholipids: Strategic Targets for Therapeutic Intervention?. J. Huntington’s Dis. 2013, 2, 239–250. 10.3233/JHD-130068. [DOI] [PubMed] [Google Scholar]
- Michalek M.; Salnikov E. S.; Bechinger B. Structure and Topology of the Huntingtin 1–17 Membrane Anchor by a Combined Solution and Solid-State NMR Approach. Biophys. J. 2013, 105, 699–710. 10.1016/j.bpj.2013.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalek M.; Salnikov E. S.; Werten S.; Bechinger B. Membrane Interactions of the Amphipathic Amino Terminus of Huntingtin. Biochemistry 2013, 52, 847–858. 10.1021/bi301325q. [DOI] [PubMed] [Google Scholar]
- Arndt J. R.; Chaibva M.; Legleiter J. The emerging role of the first 17 amino acids of huntingtin in Huntington’s disease. Biomol. Concepts 2015, 6, 33–46. 10.1515/bmc-2015-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaibva M.; Jawahery S.; Pilkington A. W.; Arndt J. R.; Sarver O.; Valentine S.; Matysiak S.; Legleiter J. Acetylation within the First 17 Residues of Huntingtin Exon 1 Alters Aggregation and Lipid Binding. Biophys. J. 2016, 111, 349–362. 10.1016/j.bpj.2016.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Toro D.; Xifró X.; Pol A.; Humbert S.; Saudou F.; Canals J. M.; Alberch J. Altered cholesterol homeostasis contributes to enhanced excitotoxicity in Huntington’s disease. J. Neurochem. 2010, 115, 153–167. 10.1111/j.1471-4159.2010.06912.x. [DOI] [PubMed] [Google Scholar]
- Karasinska J. M.; Hayden M. R. Cholesterol metabolism in Huntington disease. Nat. Rev. Neurol. 2011, 7, 561–572. 10.1038/nrneurol.2011.132. [DOI] [PubMed] [Google Scholar]
- Liu K.-Y.; Shyu Y.-C.; Barbaro B. A.; Lin Y.-T.; Chern Y.; Thompson L. M.; Shen C.-K. J.; Marsh J. L. Disruption of the nuclear membrane by perinuclear inclusions of mutant huntingtin causes cell-cycle re-entry and striatal cell death in mouse and cell models of Huntington’s disease. Hum. Mol. Genet. 2015, 24, 1602–1616. 10.1093/hmg/ddu574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Difiglia M.; Sapp E.; Chase K.; Schwarz C.; Meloni A.; Young C.; Martin E.; Vonsattel J.-P.; Carraway R.; Reeves S. A.; Boyce F. M.; Aronin N. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron 1995, 14, 1075–1081. 10.1016/0896-6273(95)90346-1. [DOI] [PubMed] [Google Scholar]
- Gutekunst C. A.; Levey A. I.; Heilman C. J.; Whaley W. L.; Yi H.; Nash N. R.; Rees H. D.; Madden J. J.; Hersch S. M. Identification and localization of huntingtin in brain and human lymphoblastoid cell lines with anti-fusion protein antibodies. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 8710–8714. 10.1073/pnas.92.19.8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutekunst C. A.; Li S. H.; Yi H.; Mulroy J. S.; Kuemmerle S.; Jones R.; Rye D.; Ferrante R. J.; Hersch S. M.; Li X. J. Nuclear and neuropil aggregates in Huntington’s disease: Relationship to neuropathology. J. Neurosci. 1999, 19, 2522–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia A.; Reeves P. B.; Sapp E.; Li X.; Alexander J.; Kegel K. B.; Chase K.; Aronin N.; DiFiglia M. Mutant huntingtin and glycogen synthase kinase 3-β accumulate in neuronal lipid rafts of a presymptomatic knock-in mouse model of Huntington’s disease. J. Neurosci. Res. 2010, 88, 179–190. 10.1002/jnr.22184. [DOI] [PubMed] [Google Scholar]
- Burke K. A.; Hensal K. M.; Umbaugh C. S.; Chaibva M.; Legleiter J. Huntingtin disrupts lipid bilayers in a polyQ-length dependent manner. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 1953–1961. 10.1016/j.bbamem.2013.04.025. [DOI] [PubMed] [Google Scholar]
- Burke K. A.; Kauffman K. J.; Umbaugh C. S.; Frey S. L.; Legleiter J. The Interaction of Polyglutamine Peptides with Lipid Membranes Is Regulated by Flanking Sequences Associated with Huntingtin. J. Biol. Chem. 2013, 288, 14993–15005. 10.1074/jbc.m112.446237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke K. A.; Yates E. A.; Legleiter J. Biophysical insights into how surfaces, including lipid membranes, modulate protein aggregation related to neurodegeneration. Front. Neurol. 2013, 4, 17. 10.3389/fneur.2013.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho C. S.; Khadka N. K.; She F.; Cai J.; Pan J. Polyglutamine aggregates impair lipid membrane integrity and enhance lipid membrane rigidity. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 661–670. 10.1016/j.bbamem.2016.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X.; Campbell W. A.; Chaibva M.; Jain P.; Leslie A. E.; Frey S. L.; Legleiter J. Cholesterol Modifies Huntingtin Binding to, Disruption of, and Aggregation on Lipid Membranes. Biochemistry 2016, 55, 92–102. 10.1021/acs.biochem.5b00900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke K. A.; Yates E. A.; Legleiter J. Amyloid-Forming Proteins Alter the Local Mechanical Properties of Lipid Membranes. Biochemistry 2013, 52, 808–817. 10.1021/bi301070v. [DOI] [PubMed] [Google Scholar]
- Ueda M.; Li S.; Itoh M.; Wang M.-x.; Hayakawa M.; Islam S.; Tana; Nakagawa K.; Chen H.; Nakagawa T. Expanded polyglutamine embedded in the endoplasmic reticulum causes membrane distortion and coincides with Bax insertion. Biochem. Biophys. Res. Commun. 2016, 474, 259–263. 10.1016/j.bbrc.2016.04.034. [DOI] [PubMed] [Google Scholar]
- Heipertz R.; Pilz H.; Scholz W. The fatty acid composition of sphingomyelin from adult human cerebral white matter and changes in childhood, senium and unspecific brain damage. J. Neurol. 1977, 216, 57–65. 10.1007/bf00312817. [DOI] [PubMed] [Google Scholar]
- Goebel H. H.; Heipertz R.; Scholz W.; Iqbal K.; Tellez-Nagel I. Juvenile Huntington chorea: clinical, ultrastructural, and biochemical studies. Neurology 1978, 28, 23. 10.1212/wnl.28.1.23. [DOI] [PubMed] [Google Scholar]
- Skene D. J.; Middleton B.; Fraser C. K.; Pennings J. L. A.; Kuchel T. R.; Rudiger S. R.; Bawden C. S.; Morton A. J. Metabolic profiling of presymptomatic Huntington’s disease sheep reveals novel biomarkers. Sci. Rep. 2017, 7, 43030. 10.1038/srep43030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirhaji L.; Milani P.; Leidl M.; Curran T.; Avila-Pacheco J.; Clish C. B.; White F. M.; Saghatelian A.; Fraenkel E. Revealing disease-associated pathways by network integration of untargeted metabolomics. Nat. Methods 2016, 13, 770–776. 10.1038/nmeth.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherrett J. R.; Brown B. L. Erythrocyte glycolipids in Huntington’s chorea. Neurology 1969, 19, 489. 10.1212/wnl.19.5.489. [DOI] [PubMed] [Google Scholar]
- Desplats P. A.; Denny C. A.; Kass K. E.; Gilmartin T.; Head S. R.; Sutcliffe J. G.; Seyfried T. N.; Thomas E. A. Glycolipid and ganglioside metabolism imbalances in Huntington’s disease. Neurobiol. Dis. 2007, 27, 265–277. 10.1016/j.nbd.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D. H.; Wang L.; Pittock P.; Lajoie G.; Whitehead S. N. Increased Expression of GM1 Detected by Electrospray Mass Spectrometry in Rat Primary Embryonic Cortical Neurons Exposed to Glutamate Toxicity. Anal. Chem. 2016, 88, 7844–7852. 10.1021/acs.analchem.6b01940. [DOI] [PubMed] [Google Scholar]
- Schneider J. S.; Seyfried T. N.; Choi H.-S.; Kidd S. K. Intraventricular Sialidase Administration Enhances GM1 Ganglioside Expression and Is Partially Neuroprotective in a Mouse Model of Parkinson’s Disease. PLoS One 2015, 10, e0143351 10.1371/journal.pone.0143351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maglione V.; Marchi P.; Di Pardo A.; Lingrell S.; Horkey M.; Tidmarsh E.; Sipione S. Impaired Ganglioside Metabolism in Huntington’s Disease and Neuroprotective Role of GM1. J. Neurosci. 2010, 30, 4072–4080. 10.1523/jneurosci.6348-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Pardo A.; Amico E.; Maglione V. Impaired Levels of Gangliosides in the Corpus Callosum of Huntington Disease Animal Models. Front. Neurosci. 2016, 10, 457. 10.3389/fnins.2016.00457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzler M.; Gan L.; Mazarei G.; Graham R. K.; Liu L.; Bissada N.; Lu G.; Leavitt B. R.; Hayden M. R. Phosphorylation of huntingtin at Ser421 in YAC128 neurons is associated with protection of YAC128 neurons from NMDA-mediated excitotoxicity and is modulated by PP1 and PP2A. J. Neurosci. 2010, 30, 14318–14329. 10.1523/jneurosci.1589-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson L. M.; Aiken C. T.; Kaltenbach L. S.; Agrawal N.; Illes K.; Khoshnan A.; Martinez-Vincente M.; Arrasate M.; O’Rourke J. G.; Khashwji H.; Lukacsovich T.; Zhu Y.-Z.; Lau A. L.; Massey A.; Hayden M. R.; Zeitlin S. O.; Finkbeiner S.; Green K. N.; LaFerla F. M.; Bates G.; Huang L.; Patterson P. H.; Lo D. C.; Cuervo A. M.; Marsh J. L.; Steffan J. S. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J. Cell Biol. 2009, 187, 1083–1099. 10.1083/jcb.200909067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warby S. C.; Doty C. N.; Graham R. K.; Shively J.; Singaraja R. R.; Hayden M. R. Phosphorylation of huntingtin reduces the accumulation of its nuclear fragments. Mol. Cell. Neurosci. 2009, 40, 121–127. 10.1016/j.mcn.2008.09.007. [DOI] [PubMed] [Google Scholar]
- Di Pardo A.; Maglione V.; Alpaugh M.; Horkey M.; Atwal R. S.; Sassone J.; Ciammola A.; Steffan J. S.; Fouad K.; Truant R.; Sipione S. Ganglioside GM1 induces phosphorylation of mutant huntingtin and restores normal motor behavior in Huntington disease mice. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 3528–3533. 10.1073/pnas.1114502109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrotra A.; Sood A.; Sandhir R. Mitochondrial modulators improve lipid composition and attenuate memory deficits in experimental model of Huntington’s disease. Mol. Cell. Biochem. 2015, 410, 281–292. 10.1007/s11010-015-2561-5. [DOI] [PubMed] [Google Scholar]
- Seelig A. Local anesthetics and pressure: a comparison of dibucaine binding to lipid monolayers and bilayers. Biochim. Biophys. Acta, Biomembr. 1987, 899, 196–204. 10.1016/0005-2736(87)90400-7. [DOI] [PubMed] [Google Scholar]
- Blume A. A comparative study of the phase transitions of phospholipid bilayers and monolayers. Biochim. Biophys. Acta, Biomembr. 1979, 557, 32–44. 10.1016/0005-2736(79)90087-7. [DOI] [PubMed] [Google Scholar]
- Simons K.; Ikonen E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Vaknin D.; Kelley M. S.; Ocko B. M. Sphingomyelin at the air–water interface. J. Chem. Phys. 2001, 115, 7697–7704. 10.1063/1.1406501. [DOI] [Google Scholar]
- Frey S. L.; Chi E. Y.; Arratia C.; Majewski J.; Kjaer K.; Lee K. Y. C. Condensing and fluidizing effects of ganglioside GM1 on phospholipid films. Biophys. J. 2008, 94, 3047–3064. 10.1529/biophysj.107.119990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cametti C.; De Luca F.; Macrì M. A.; Maraviglia B.; Misasi R.; Sorice M.; Pavan A.; Garofalo T.; Pontieri G. M.; Bordi F.; Zimatore G. Influence of different glycosphingolipids on the conductometric properties of a model phospholipid membrane system. Colloids Surf., B 1996, 7, 39–46. 10.1016/0927-7765(96)01280-5. [DOI] [Google Scholar]
- Frey S. L.; Lee K. Y. C. Number of sialic acid residues in ganglioside headgroup affects interactions with neighboring lipids. Biophys. J. 2013, 105, 1421–1431. 10.1016/j.bpj.2013.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins E. B.; Miller C. E.; Liao W.-P.; Kuhl T. L. Equilibrium or Quenched: Fundamental Differences between Lipid Monolayers, Supported Bilayers, and Membranes. ACS Nano 2014, 8, 3181–3191. 10.1021/nn4052953. [DOI] [PubMed] [Google Scholar]
- Mino N.; Tamura H.; Ogawa K. Analysis of color transitions and changes on Langmuir-Blodgett films of a polydiacetylene derivative. Langmuir 1991, 7, 2336–2341. 10.1021/la00058a060. [DOI] [Google Scholar]
- Okada S.; Peng S.; Spevak W.; Charych D. Color and Chromism of Polydiacetylene Vesicles. Acc. Chem. Res. 1998, 31, 229–239. 10.1021/ar970063v. [DOI] [Google Scholar]
- Porat Y.; Kolusheva S.; Jelinek R.; Gazit E. The human islet amyloid polypeptide forms transient membrane-active prefibrillar assemblies. Biochemistry 2003, 42, 10971–10977. 10.1021/bi034889i. [DOI] [PubMed] [Google Scholar]
- Kolusheva S.; Shahal T.; Jelinek R. Cation-Selective Color Sensors Composed of Ionophore–Phospholipid–Polydiacetylene Mixed Vesicles. J. Am. Chem. Soc. 2000, 122, 776–780. 10.1021/ja9924288. [DOI] [Google Scholar]
- Sokolovski M.; Sheynis T.; Kolusheva S.; Jelinek R. Membrane interactions and lipid binding of casein oligomers and early aggregates. Biochim. Biophys. Acta, Biomembr. 2008, 1778, 2341–2349. 10.1016/j.bbamem.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Groves J. T.; Ulman N.; Boxer S. G. Micropatterning fluid lipid bilayers on solid supports. Science 1997, 275, 651–653. 10.1126/science.275.5300.651. [DOI] [PubMed] [Google Scholar]
- Yip C. M.; Darabie A. A.; McLaurin J. Aβ42-Peptide Assembly on Lipid Bilayers. J. Mol. Biol. 2002, 318, 97–107. 10.1016/s0022-2836(02)00028-1. [DOI] [PubMed] [Google Scholar]
- Yip C. M.; Elton E. A.; Darabie A. A.; Morrison M. R.; McLaurin J. Cholesterol, a modulator of membrane-associated A β-fibrillogenesis and neurotoxicity. J. Mol. Biol. 2001, 311, 723–734. 10.1006/jmbi.2001.4881. [DOI] [PubMed] [Google Scholar]
- Yip C. M.; McLaurin J. Amyloid-β peptide assembly: A critical step in fibrillogenesis and membrane disruption. Biophys. J. 2001, 80, 1359–1371. 10.1016/s0006-3495(01)76109-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaibva M.; Burke K. A.; Legleiter J. Curvature Enhances Binding and Aggregation of Huntingtin at Lipid Membranes. Biochemistry 2014, 53, 2355–2365. 10.1021/bi401619q. [DOI] [PubMed] [Google Scholar]
- Legleiter J.; Fryer J. D.; Holtzman D. M.; Kowalewski T. The Modulating Effect of Mechanical Changes in Lipid Bilayers Caused by ApoE-Containing Lipoproteins on A β Induced Membrane Disruption. ACS Chem. Neurosci. 2011, 2, 588–599. 10.1021/cn2000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates E. A.; Owens S. L.; Lynch M. F.; Cucco E. M.; Umbaugh C. S.; Legleiter J. Specific Domains of A β Facilitate Aggregation on and Association with Lipid Bilayers. J. Mol. Biol. 2013, 425, 1915–1933. 10.1016/j.jmb.2013.03.022. [DOI] [PubMed] [Google Scholar]
- Burke K. A.; Godbey J.; Legleiter J. Assessing mutant huntingtin fragment and polyglutamine aggregation by atomic force microscopy. Methods 2011, 53, 275–284. 10.1016/j.ymeth.2010.12.028. [DOI] [PubMed] [Google Scholar]
- Lotz G. P.; Legleiter J. The role of amyloidogenic protein oligomerization in neurodegenerative disease. J. Mol. Med. 2013, 91, 653–664. 10.1007/s00109-013-1025-1. [DOI] [PubMed] [Google Scholar]
- Nucifora L. G.; Burke K. A.; Feng X.; Arbez N.; Zhu S.; Miller J.; Yang G.; Ratovitski T.; Delannoy M.; Muchowski P. J.; Finkbeiner S.; Legleiter J.; Ross C. A.; Poirier M. A. Identification of Novel Potentially Toxic Oligomers Formed in Vitro from Mammalian-derived Expanded huntingtin Exon-1 Protein. J. Biol. Chem. 2012, 287, 16017–16028. 10.1074/jbc.m111.252577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisbani G.; Cicchetti F. An in vitro perspective on the molecular mechanisms underlying mutant huntingtin protein toxicity. Cell Death Dis. 2012, 3, e382 10.1038/cddis.2012.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur A. K.; Jayaraman M.; Mishra R.; Thakur M.; Chellgren V. M.; Byeon I.-J. L.; Anjum D. H.; Kodali R.; Creamer T. P.; Conway J. F.; Gronenborn A. M.; Wetzel R. Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat. Struct. Mol. Biol. 2009, 16, 380–389. 10.1038/nsmb.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Wetzel R. Solubilization and disaggregation of polyglutamine peptides. Protein Sci. 2001, 10, 887–891. 10.1110/ps.42301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchowski P. J.; Schaffar G.; Sittler A.; Wanker E. E.; Hayer-Hartl M. K.; Hartl F. U. Hsp70 and Hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 7841–7846. 10.1073/pnas.140202897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson R. L.; Davis S. An organic phosphorus assay which avoids the use of hazardous perchloric acid. Clin. Chim. Acta 1982, 121, 111–116. 10.1016/0009-8981(82)90216-9. [DOI] [PubMed] [Google Scholar]
- Rouser G.; Fleischer S.; Yamamoto A. Two dimensional thin layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids 1970, 5, 494–496. 10.1007/bf02531316. [DOI] [PubMed] [Google Scholar]
- Zheng F.; Wu Z.; Chen Y. A quantitative method for the measurement of membrane affinity by polydiacetylene-based colorimetric assay. Anal. Biochem. 2012, 420, 171–176. 10.1016/j.ab.2011.09.026. [DOI] [PubMed] [Google Scholar]