

Abstract
Background
Methylated gene markers have shown promise in predicting breast cancer outcomes and treatment response. We evaluated whether baseline and change in tissue and serum methylation would predict pathological complete response (pCR) in patients with HER2-negative early breast cancer undergoing preoperative chemotherapy.
Methods
The TBCRC008 trial investigated pCR following 12 weeks of preoperative carboplatin and albumin-bound paclitaxel + vorinostat/placebo (n=62). We measured methylation of a 10 gene panel by quantitative multiplex methylation-specific polymerase chain reaction (QM-MSP) and expressed results as cumulative methylation index (CMI). We evaluated association between CMI level (baseline, day 15 [D15], and change) and pCR using univariate and multivariable logistic regression models controlling for treatment and hormone receptor (HR) status, and performed exploratory subgroup analyses.
Results
In univariate analysis, one log unit increase in tissue CMI levels at D15 was associated with 40% lower chance of obtaining pCR (odds ratio, OR=0.60, 95% CI 0.37-0.97; p=0.037). Subgroup analyses suggested a significant association between tissue D15 CMI levels and pCR in vorinostat-treated (OR 0.44 [0.20, 0.93], p=0.03), but not placebo-treated patients.
Conclusion
In this first study investigating the predictive role of tissue and serum CMI in patients with early breast cancer, we demonstrate that high D15 tissue CMI levels may predict poor response. Larger studies and improved analytical procedures to detect methylated gene markers in early stage breast cancer are needed.
Keywords: Preoperative chemotherapy, breast cancer, methylation, cMethDNA, biomarkers
Introduction
Long term outcomes for the majority of patients with breast cancer are excellent, due to early detection and the widespread availability of local and systemic therapies [1]. Nonetheless, some patients will experience recurrence of their breast cancer despite standard treatment approaches. Others will have an excellent prognosis, and may be over treated [2]. The identification of biomarkers that are prognostic and predictive of response to therapy is therefore of the utmost importance in the management of this disease [3-5]. Potential biomarkers may be identified through collection of baseline and serial blood and tumor samples incorporated as part of prospectively designed studies.
Aberrant methylation of DNA is characteristic of breast cancer, where hypermethylation of hundreds of cancer specific genes provides a rich source of biomarkers of detection, prognosis and prediction of response to therapy [6-9]. Hypermethylation often results in silencing of important genes such as tumor suppressor genes and those involved with growth regulation that are active in non-cancerous breast tissue. Various quantitative techniques [10-12], including quantitative multiplex methylation-specific PCR (QM-MSP) [13-16], have the ability to robustly detect gene methylation in tumor samples. Tumor tissue methylation has shown promise as a biomarker to predict response to therapy and to prognosticate outcome of disease. In patients with glioma treated with preoperative temozolomide, tissue MGMT methylation predicted favorable progression-free survival (PFS, p < 0.0001) [17]. Fiegl et al suggested that NEUROD1 methylation is a chemosensitivity marker in estrogen receptor-negative breast cancer [18]. Other studies in breast cancer have been performed in locally advanced disease to investigate pharmaco-epigenetic effects following preoperative chemotherapy [19, 20].
Our group and others have recently identified methylated cell-free DNA in serum, shed presumably by circulating or dying tumor cells, as a promising prognostic biomarker, and a predictive biomarker of response to treatment in breast cancer (cMethDNA) [21, 22] and other methylation assays [23-27]. Unlike technologies used by others, cMethDNA is a quantitative multiplex methylation-specific PCR assay consisting of a panel of up to 10 novel known breast cancer hypermethylated markers. It uses spiked standards for each gene, provides absolute quantitation, and consistently detects cell-free tumor-specific DNA in peripheral blood of patients with metastatic breast cancer with high sensitivity and specificity [21]. In a small validation clinical study of patients with metastatic breast cancer initiating a new course of treatment (n=140), the assay predicted patient response to chemotherapy early in the course of treatment, and prognosticated outcome [22]. Although established in patients with metastatic breast cancer, cMethDNA has not been previously evaluated in the early breast cancer setting.
We hypothesized that both tissue and serum methylation would be valuable predictive biomarkers in earlier stages of the disease. Specifically, we hypothesized that baseline and change in tumor tissue (QM-MSP) and serum methylation (cMethDNA) would predict pathologic complete response (pCR) in women receiving preoperative chemotherapy. To test this hypothesis, we performed an exploratory planned biomarker study in women with HER2-negative breast cancer who received preoperative carboplatin and albumin-bound paclitaxel with or without the histone deacetylase (HDAC) inhibitor, vorinostat, through the prospective TBCRC008 clinical trial [28].
Materials and Methods
Clinical Trial Design
TBCRC008 was a multicenter placebo-controlled trial that compared pCR following 12 weeks of preoperative chemotherapy with or without vorinostat in 62 patients with HER2-negative breast cancer (Figure 1) [28]. Participants received 12 weeks of preoperative carboplatin (AUC 2 weekly) and nab-paclitaxel (100 mg/m2 weekly) with vorinostat (400 mg oral daily, days 1-3 of every 7-day period) or a matched placebo. The primary end point was pCR, defined as no viable invasive cancer in the breast and axilla.
Figure 1. TBCRC008 Study Schema.
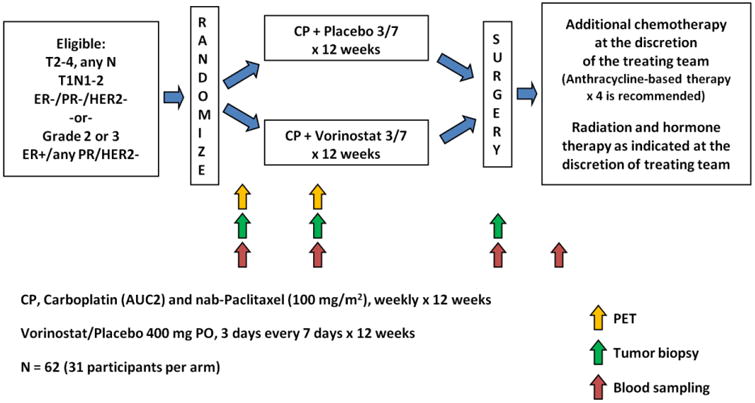
Eligible participants for TBCRC008 were women 18 years of age or older, with histologically-proven infiltrating carcinoma of the breast who presented with operable, clinical stage T1c, N1-3 or T2-4 lesions, any N; and M0. Tumors must have been HER2-negative and grade 2 or 3, with any estrogen (ER) or progesterone receptor (PR)-status.
After a brief phase I run-in portion, participants in the phase II portion of the study were randomly assigned 1:1 to receive carboplatin and nab-paclitaxel with vorinostat or placebo using permuted block randomization, stratified by hormone receptor status (ER and PR < 1% vs. ER or PR ≥1%) [28]. Additional non-study chemotherapy (doxorubicin and cyclophosphamide, AC) was allowed per treating physician discretion prior to definitive surgery, for patients with incomplete response or disease progression on study treatment. Women signed an informed consent approved by the Institutional Review Boards of participating institutions.
Sample Collection and Methylation Analysis
A study-specific core biopsy and blood sampling (serum) were obtained at baseline and 15 days after the first treatment (D15), preferably about 4 hours after taking vorinostat/placebo and prior to receiving carboplatin and nab-paclitaxel. Core biopsy specimens were suspended in 10% buffered formalin, and subsequently embedded in paraffin. Serum samples were processed and stored at -80° C or below prior to analysis. Tissue and serum DNA was extracted and then treated with sodium bisulfite as previously described [13, 21]. Methylation was measured using CMI in quantitative multiplex methylation-specific polymerase chain reaction assays, cMethDNA assay for serum [21] and QM-MSP for tissue [13]. The cumulative methylation index (CMI) was calculated as a sum of all gene-specific methylation indexes within a panel of 10 genes which included: HIST1H3C, AKR1B1, GPX7, HOXB4, TMEFF2, RASGRF2, COL6A2, ARHGEF7, TM6SF1, and RASSF1A. These 10 genes were selected in a previous study following genome wide methylome analysis (Illumina Infinium HumanMethylation27 BeadChip) in tissue and serum, as they were frequently highly methylated in breast tumors of all stages and in serum from patients with metastatic breast cancer. These genes were unmethylated or methylated at low levels in normal breast tissue [29] and in cell-free circulating serum DNA of normal individuals [21].
Statistical Analysis
The objectives of this pre-planned exploratory analysis were to evaluate: 1) the association of baseline methylation (tissue and serum) with pCR; 2) the association of change (baseline versus D15) in methylation (tissue and serum) with pCR; 3) the effect of vorinostat and other clinical variables on the association between methylation status and pCR [30].
The analysis population included all patients for whom complete CMI data were available (Table 1). Only samples where methylation results were available for all genes were evaluated. CMI at baseline and D15 were natural log-transformed as log (Baseline CMI+1) and log (D15 CMI+1), respectively. Change in CMI was defined as log (D15 CMI+1/Baseline CMI+1).
Table 1. Univariate analysis of association of CMI levels with pCR.
| CMI* | Tissue | Serum | ||||
|---|---|---|---|---|---|---|
| n | OR (95% CI) | P-value | n | OR (95% CI) | P-value | |
| Baseline | 58 | 0.73 (0.49, 1.09) | 0.129 | 59 | 1.17 (0.82, 1.67) | 0.400 |
| D15 | 50 | 0.60 (0.37, 0.97) | 0.037 | 61 | 0.9 (0.57, 1.42) | 0.652 |
| Change from baseline | 48 | 0.52 (0.26, 1.05) | 0.069 | 58 | 0.83 (0.57, 1.21) | 0.324 |
Natural log transformed
Change from baseline, change of CMI defined as log (D15 CMI+1/Baseline CMI+1); n, number of available samples; CI, confidence interval; CMI, cumulative methylation index; D15, 15 days after the first treatment; OR, odds ratio
Distributions of CMI levels at baseline and D15 were evaluated with descriptive statistics, and compared using Wilcoxon matched-pairs signed rank test. The difference in tissue CMI levels at D15 by treatment arm and by pCR status was compared using nonparametric Mann-Whitney U test. Furthermore, associations between CMI level (baseline, D15, change) and pCR were evaluated using univariate as well as multivariable logistic regression models controlling for treatment arm and hormone receptor status. Additional subgroup analyses were performed to assess the association of CMI level with pCR using logistic regression models, stratified by ER status and treatment arm, respectively.
All tests were considered statistically significant at p<0.05. No multiplicity adjustment was made. Analyses were performed using SAS 9.4 (SAS Institute, Cary, NC) and GraphPad Prism (version 5.00 for Windows, GraphPad Software, San Diego, CA, www.graphpad.com).
Results
Patient Characteristics
Demographic and clinical variables from TBCRC008 have been previously published and are summarized in Supplementary Table 1 [28]. Approximately 60% of participants had ER/PR-positive tumors and 60% had positive axillary lymph nodes at study entry. pCR data were available for 60 of 62 patients for this analysis. Two patients with unknown response were considered non-responders following the intent-to-treat principle. Overall pCR was 27% and was similar in both arms (29% placebo vs. 26% vorinostat). AC chemotherapy was administered prior to definitive surgery in 18 patients deemed to have incomplete response or disease progression on study treatment.
Patient Tissue and Serum Methylation
Available tissue methylation data were evaluated in 58 (baseline), 50 (D15) and 48 (both) patients, and serum methylation data were available in 59 (baseline), 61 (D15), 58 (both) patients (Figure 2). Median tissue CMI levels were 50 (range 0, 417) at baseline and 25 (range 0, 163) at D15, and median CMI change within individuals between baseline and D15 was 14 (range -310, 69). Median serum CMI levels were 3 (range 0, 124) at baseline and 4 (range 0, 90) at D15, and median CMI change was 0 (range -120, 29) (Figure 2). Within individuals, we observed significant changes in tumor content (% tumor in biopsy section, p<0.0001; paired T-test) and tumor methylation (CMI, p<0.0001; Wilcoxon matched pairs test) from baseline to D15 (Figure 3).
Figure 2. Tissue and Serum DNA Methylation.
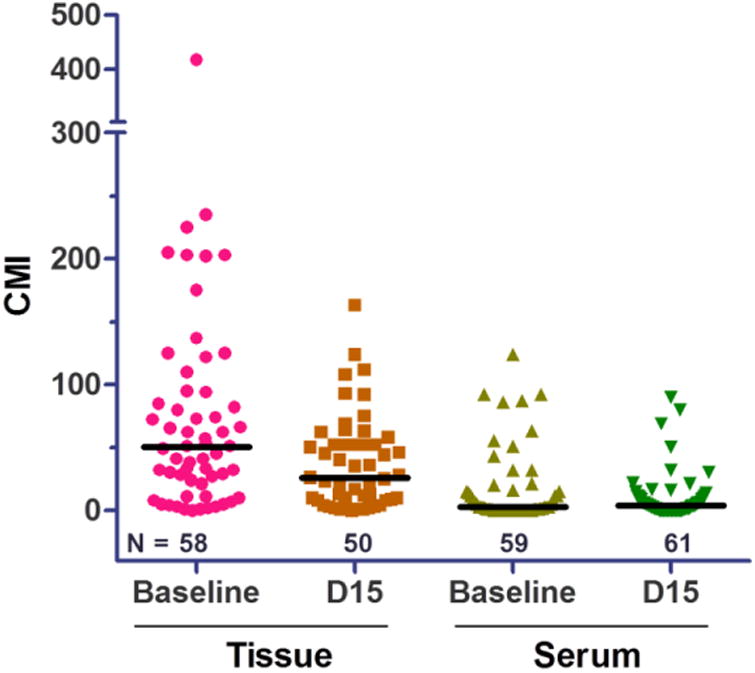
QM-MSP and cMethDNA analyses were performed on tissue (n=58 baseline and 50 D15) and serum (n=59 baseline and 61 D15) samples, respectively. Scatter plots show CMI methylation levels as the sum of methylation for each marker in 10-gene panel. Median methylation is indicated (bar) and only samples with methylation values for all 10 genes were evaluated.
Figure 3. Tumor Content and DNA Methylation of Tumors in Vorinostat vs Placebo Arms.
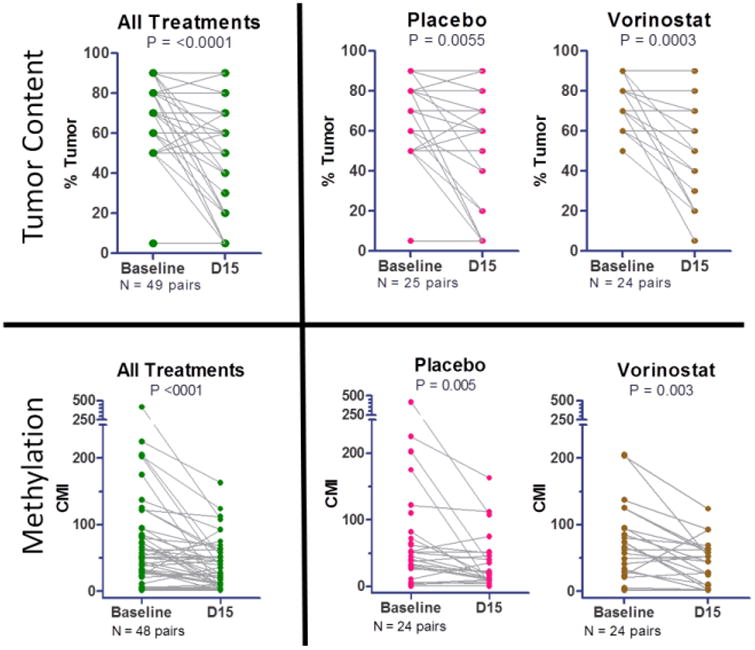
Tumor content in paired samples of the same individual. Change between baseline and D15 tumor content (% tumor in biopsy section) and methylation was quantified using Wilcoxon matched pairs test. Significant reductions in both tumor content and methylation were observed at D15 within the study population (“All patients”), as well as vorinostat and placebo groups. Only samples having methylation values for all 10 genes were evaluated.
Associations Between CMI Level and Response to Therapy
Baseline methylation was not associated with pCR for either tissue or serum. Tissues from patients who achieved pCR had significantly lower methylation at D15 compared to those with no pCR (p=0.0143) (Figure 4A). Supported by the univariate analysis, a one log unit increase in tissue CMI levels at D15 was associated with a 40% lower chance of obtaining a pCR (odds ratio, OR=0.60, 95% CI 0.37-0.97; p=0.037) (Table 1). A similar association was not observed for serum methylation (Table 1).
Figure 4. Tissue CMI at D15 by pCR Status and Treatment Arm.
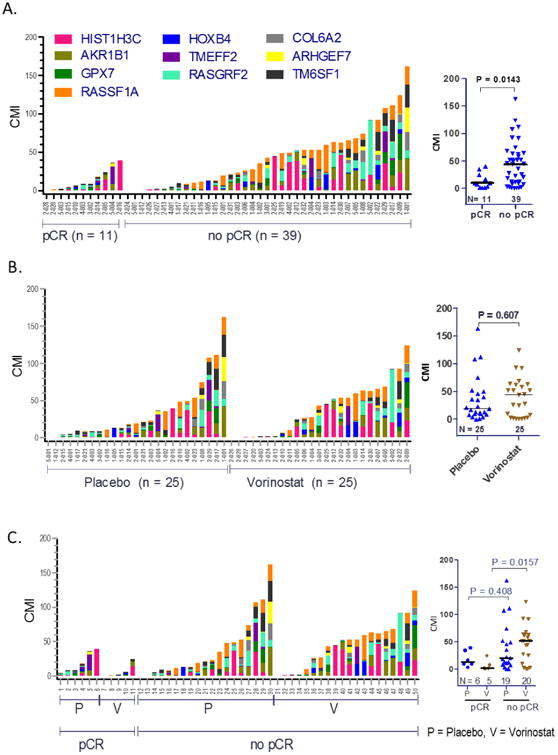
CMI of the 10-gene panel is indicated by scatter plots for each treatment group (median shown as bar). For each individual sample (x-axis) the bar graphs indicate the level of CMI (height of the bar, y-axis) and the relative methylation of each gene (shown by colored segment). A. D15 methylation levels of tissue samples in pCR versus no pCR groups. B. D15 methylation levels of tissue and serum samples in vorinostat (V) versus placebo (P) groups. C. D15 methylation levels of tissue samples grouped pCR versus no pCR, ± vorinostat. Only samples having methylation values for all 10 genes were evaluated (nonparametric Mann-Whitney U test).
The distribution of tissue CMI at D15 was further evaluated in vorinostat-treated patients compared to patients receiving placebo. Within individuals, we observed significant decrease in tumor content (p<0.0001; paired T-test) and in tumor methylation (LogCMI, p<0.0001; paired T-test) from baseline to D15 irrespective of treatment assignment (Figure 3). No significant difference was observed in D15 tissue CMI between treatment arms (tissue: p=0.607; Figure 4B, Figure S1). Tumor methylation levels of individual genes did not significantly differ between vorinostat and placebo groups, either at baseline or on D15 (Figure S1).
Interestingly, D15 tumors from individuals who achieved pCR after receiving vorinostat had significantly lower CMI than those that failed to achieve pCR (p=0.0157). This trend appeared to be less profound in tumors from patients in the placebo arm that did or did not achieve pCR (p=0.408) (Figure 4C, Supplementary Table 2), suggesting a potential interaction between the treatment effect and D15 methylation in tumors.
Multivariable analysis adjusting for treatment arm and hormone receptor status did not show a statistically significant association of tissue or serum CMI with pCR, although a similar trend in magnitude towards association with pCR was observed for D15 and change in tumor tissue CMI (OR 0.67 and 0.57, p=0.129 and 0.147 respectively) (Table 2a). Additional multivariable analyses also failed to demonstrate a statistically significant association of tissue or serum CMI with pCR (Table 2b and 2c).
Table 2. Multivariable analysis of association of CMI levels with pCR.
| a: Multivariable analysis adjusting for treatment arm and hormone receptor status (HR), by time point and by tissue and serum | ||||
|---|---|---|---|---|
| Variable | Tissue | Serum | ||
| OR (95% CI) | P-value | OR (95% CI) | P-value | |
| Baseline CMI | 0.83 (0.53, 1.30) | 0.418 | 1.1 (0.75, 1.63) | 0.615 |
| Vorinostat vs. placebo | 0.48 (0.12, 1.86) | 0.287 | 0.8 (0.23, 2.77) | 0.721 |
| HR+ vs. TNBC | 0.13 (0.03, 0.53) | 0.004 | 0.17 (0.05, 0.6) | 0.006 |
| Variable | Tissue | Serum | ||
| OR (95% CI) | P-value | OR (95% CI) | P-value | |
| D15 CMI | 0.67 (0.39, 1.13) | 0.129 | 0.85 (0.51, 1.42) | 0.542 |
| Vorinostat vs. placebo | 0.75 (0.17, 3.29) | 0.703 | 0.68 (0.19, 2.43) | 0.555 |
| HR+ vs. TNBC | 0.28 (0.06, 1.25) | 0.096 | 0.16 (0.05, 0.57) | 0.005 |
| Variable | Tissue | Serum | ||
| OR (95% CI) | P-value | OR (95% CI) | P-value | |
| CMI change from baseline | 0.57 (0.27, 1.22) | 0.147 | 0.85 (0.57, 1.27) | 0.431 |
| Vorinostat vs. placebo | 0.60 (0.13, 2.81) | 0.519 | 0.69 (0.2, 2.41) | 0.559 |
| H+ vs. TNBC | 0.26 (0.06, 1.2) | 0.084 | 0.19 (0.05, 0.66) | 0.009 |
| b: Multivariable analysis adjusting for nodal status and hormone receptor status, by time point and by tissue and serum | ||||
| Variable | Tissue | Serum | ||
| OR (95% CI) | P-value | OR (95% CI) | P-value | |
| Baseline CMI | 1.07 (0.62, 1.86) | 0.802 | 1.29 (0.83, 2) | 0.26 |
| Node + vs. - | 0.08 (0.02, 0.42) | 0.003 | 0.16 (0.04, 0.65) | 0.011 |
| HR+ vs. TNBC | 0.14 (0.03, 0.69) | 0.016 | 0.2 (0.05, 0.77) | 0.019 |
| Variable | Tissue | Serum | ||
| OR (95% CI) | P-value | OR (95% CI) | P-value | |
| D15 CMI | 0.74 (0.43, 1.27) | 0.27 | 0.87 (0.52, 1.43) | 0.575 |
| Node + vs. - | 0.15 (0.03, 0.77) | 0.023 | 0.2 (0.05, 0.74) | 0.016 |
| HR+ vs. TNBC | 0.26 (0.05, 1.31) | 0.104 | 0.2 (0.05, 0.74) | 0.016 |
| Variable | Tissue | Serum | ||
| OR (95% CI) | P-value | OR (95% CI) | P-value | |
| CMI change from baseline | 0.48 (0.22, 1.06) | 0.069 | 0.68 (0.42, 1.1) | 0.119 |
| Node + vs. - | 0.05 (0.01, 0.43) | 0.006 | 0.14 (0.03, 0.64) | 0.011 |
| HR+ vs. TNBC | 0.21 (0.03, 1.27) | 0.088 | 0.21 (0.05, 0.8) | 0.023 |
| c: Multivariable analysis adjusting for tumor stage and hormone receptor status, by time point and by tissue and serum | ||||
| Variable | Tissue | Serum | ||
| OR (95% CI) | P-value | OR (95% CI) | P-value | |
| Baseline CMI | 0.87 (0.55, 1.36) | 0.537 | 1.18 (0.8, 1.76) | 0.408 |
| Stage III vs. II | 0.52 (0.13, 2.15) | 0.368 | 0.41 (0.1, 1.72) | 0.225 |
| HR+ vs. TNBC | 0.15 (0.04, 0.59) | 0.007 | 0.18 (0.05, 0.64) | 0.008 |
| Variable | Tissue | Serum | ||
| OR (95% CI) | P-value | OR (95% CI) | P-value | |
| D15 CMI | 0.69 (0.41, 1.15) | 0.155 | 0.94 (0.57, 1.53) | 0.796 |
| Stage III vs. II | 0.75 (0.15, 3.71) | 0.725 | 0.37 (0.08, 1.61) | 0.183 |
| HR+ vs. TNBC | 0.28 (0.06, 1.28) | 0.1 | 0.18 (0.05, 0.66) | 0.009 |
| Variable | Tissue | Serum | ||
| OR (95% CI) | P-value | OR (95% CI) | P-value | |
| CMI change from baseline | 0.58 (0.28, 1.2) | 0.141 | 0.84 (0.56, 1.25) | 0.39 |
| Stage III vs. II | 0.62 (0.12, 3.16) | 0.564 | 0.36 (0.08, 1.57) | 0.173 |
| HR+ vs. TNBC | 0.25 (0.05, 1.17) | 0.079 | 0.2 (0.06, 0.73) | 0.015 |
CMI levels were natural log transformed.
Change from baseline, change of CMI defined as log (D15 CMI+1/Baseline CMI+1); CMI, cumulative methylation index; D15, 15 days after the first treatment; HR, hormone receptor PR; TNBC, triple-negative breast cancer; OR, odds ratio; CI, confidence interval;
Subgroup analyses (univariate) suggested a significant association between tissue CMI levels at D15 and pCR in the group treated with vorinostat [tissue OR: 0.44 (0.20, 0.93), p=0.03; serum OR 0.37 (0.13, 1.07), p=0.07] but not in the placebo group (Supplementary Table 3c, 3d). There was no significant association between CMI levels and pCR in patients with hormone receptor-positive breast cancer versus those with triple-negative disease (Supplementary Table 3a, 3b).
Discussion
In a planned exploratory biomarker analysis from the prospectively designed preoperative TBCRC008 trial, we have evaluated for the first time the predictive role of tissue and serum CMI in patients with early stage HER2-negative breast cancer treated with preoperative chemotherapy with or without an HDAC inhibitor. We have demonstrated in univariate analysis that an increase in tissue CMI level at D15 was associated with poor response to this therapy and may potentially have a predictive role in the preoperative setting. Our data suggest that lack of an early and robust tumor response to preoperative therapy will result in a greater burden of DNA methylation at this D15 timepoint, i.e., an association of DNA methylation as early as D15 with pCR.
Epigenetic alterations, including abnormal methylation of DNA in the promoter region of important genes, are prevalent in breast cancers and often result in silencing of gene expression [31]. This knowledge has prompted investigation into their clinical significance and whether they can be manipulated to improve patient outcomes [32]. While a few studies have reported the utility of single methylated markers in tissue or serum in predicting response to chemotherapy in both the preoperative and adjuvant setting, there have been no follow up validation studies to further support their findings.
DNA methylation in breast cancer gene promoters has been evaluated by QM-MSP in archival tumor and blood samples [22, 23, 33-37]. Genome-wide methylation array analysis has also been performed on 103 primary invasive breast cancers and on 21 normal breast samples, using the Illumina Infinium HumanMethylation27 array [29]. A higher frequency of methylation was noted in hormone receptor-positive tumors when compared to HR-negative tumors. The hypermethylated loci in ER-negative tumors, however, were closer to the transcriptional start sites, perhaps indicating a tighter control of transcriptional repression. These studies indicate that methylation of promoter regions of breast cancer-related genes may have utility as potential prognostic biomarkers in breast cancer.
Patients, advocates and physicians are increasingly aware of the importance of obtaining tumor biopsies, including serial samples, for correlative analyses with the hope of individualizing therapy for future patients. Blood-based biomarkers, however, may offer a non-invasive and more convenient way to assess prognosis or monitor therapy, which may be more acceptable to patients. cMethDNA underwent preliminary validation in a multisite prospective study TBCRC005 using serum samples from 141 women at baseline, at week 4, and at first restaging. A CMI was generated on the basis of 6 of the 10 genes tested. In multivariable analyses, an increase in the CMI from baseline to week 4 was associated with worse PFS (p < 0.001) and progressive disease at first restaging (p< 0.001). CMI at week 4 was a strong predictor of PFS, even in the presence of circulating tumor cells (p=0.004). Thus, serum cMethDNA assay can detect advanced breast cancer, may serve as a prognostic biomarker, and has the potential to monitor tumor burden and treatment response in patients with advanced breast cancer [21]. Based on these promising results, we were keen to investigate this blood-based assay in our early breast cancer dataset as described above. Alternative approaches which continue to be studied in the early breast cancer setting include the use of plasma tumor DNA (ptDNA) to predict prognosis and response to therapy [38, 39].
Strengths of our study include the prospective design of the TBCRC008 preoperative clinical trial, with a preplanned exploratory analysis to evaluate the predictive value of serum and tumor methylation. All patients in the study (n=62) underwent baseline image-guided tumor biopsy, with the majority (59/62) also undergoing a short-interval follow up biopsy at D15 [28]. Alongside successful serial collection of blood samples, this study highlights the feasibility of such sample collection for correlative analyses in the preoperative setting in patients with early breast cancer. Limitations of this analysis include the small sample size which may limit the interpretation of our positive findings regarding the association of tissue CMI levels at D15 and response to this therapy, as well as the extent to which interaction between tissue CMI levels at D15 and treatment response to preoperative therapy may be assessed.
Additional larger prospective studies will be needed to further define the role of gene methylation signatures in predicting clinical outcome and response to systemic therapy in patients with breast cancer, and indeed other tumor types. Much of the focus to date has been on the advanced breast cancer setting, where indeed ongoing analyses using prospectively collected serum will attempt to answer these important questions in regards to the cMethDNA assay. Based on our observation that there was a significant association between tissue CMI levels at D15 and pCR in those treated with vorinostat but not placebo, consideration should be given for investigating more potent demethylating agents in this setting. Investigations should also consider additional clinical scenarios such as the early detection of recurrent breast cancer after primary surgery. Finally, the development of more sensitive assays which can detect lower levels of DNA methylation in the early breast cancer setting are clearly needed. Close collaboration between basic scientists, clinical investigators and experts in biomarker development is essential moving forward in order to further personalize treatment approaches for patients with breast cancer.
Supplementary Material
Supplementary Figure 1: Quantitative Gene Methylation by Treatment Arm. Scatter plots indicate methylation levels in tissues from individuals treated either with vorinostat (V) or placebo (P) in addition to standard therapy. Cumulative methylation index (CMI) was calculated for tissue samples in which PCR-amplification was successful for all 10 genes in the panel. CMI = the sum of individual gene methylation within the 10 gene-panel (possible 1000 CMI units, 100% × 10 genes). Gene methylation is defined as % methylation = (# copies methylated DNA/# total copies unmethylated DNA + methylated DNA) (100). The median is shown by bar and significance was calculated using the Mann-Whitney U test.
Supplementary Table 1: Patient characteristics
Supplementary Table 2: Tissue CMI at D15 by pCR status and treatment arm
Supplementary Table 3 (a-d): Subgroup Analyses
Supplementary Table 3a: Univariate analysis of association of CMI levels with pCR in HR+ patients (n=38)
Supplementary Table 3b: Univariate analysis of association of CMI levels with pCR in TNBC patients (n=24)
Supplementary Table 3c: Univariate analysis of association of CMI levels with pCR in the vorinostat arm (n=31)
Supplementary Table 3d: Univariate analysis of association of CMI levels with pCR in the placebo arm (n=31)
Acknowledgments
We thank the patients who participated in this study. SPORE in breast cancer (P50 CA88843), TBCRC and its foundation partners (The AVON Foundation, The Breast Cancer Research Foundation and Susan G. Komen for the Cure), Abraxis Bioscience, Merck Oncology, and the Cindy Rosencrans Fund for Triple Negative Breast Cancer Research for generous funding, TBCRC participating site investigators, research nurses, and study coordinators. The staff of the Breast and Ovarian Cancer Program and Avon Breast Center at Johns Hopkins.
Footnotes
Prior Presentation: Connolly RM, Fackler MJ, Zhang Z, Xian Z, Goetz MP, Boughey JC, Walsh B, Carpenter J, Storniolo AM, Watkins S, Gabrielson E, Sukumar S, Stearns V. Tumor and serum DNA methylation in women receiving preoperative chemotherapy (PST) with or without vorinostat in primary operable HER2-negative breast cancer in TBCRC008. SABCS 2016 (P1-09-15)
Compliance with ethical standards: TBCRC008 is registered on clinical trials.gov (NCT00616967). Women enrolling in the study signed an informed consent approved by the Institutional Review Boards of participating institutions.
Conflicts of interest: VS has received research grants from Merck, Celgene Corporation, Abbvie, Pfizer, Novartis, Medimmune, and Puma Biotechnology. RC has received research grants from Novartis, Puma Biotechnology, Genentech, Merrimack, Clovis, Merck. AMS has received consulting fees from Eli Lilly and Co., and Pfizer. SS has received research grants from AVON Foundation, grants, licensing/royalty fees and consulting fees from Cepheid for QM-MSP and cMethDNA assays. MJF has received licensing/royalty fees and consulting fees from Cepheid. No other authors have disclosed any conflicts of interest.
References
- 1.Stearns V, Zhou Q, Davidson NE. Epigenetic regulation as a new target for breast cancer therapy. Cancer Invest. 2007;25(8):659–665. doi: 10.1080/07357900701719234. [DOI] [PubMed] [Google Scholar]
- 2.Mukhtar RA, Wong JM, Esserman LJ. Preventing Overdiagnosis and Overtreatment: Just the Next Step in the Evolution of Breast Cancer Care. J Natl Compr Canc Netw. 2015;13(6):737–743. doi: 10.6004/jnccn.2015.0088. [DOI] [PubMed] [Google Scholar]
- 3.Abramovitz M, Krie A, Dey N, De P, Williams C, Leyland-Jones B. Identifying biomarkers to select patients with early breast cancer suitable for extended adjuvant endocrine therapy. Curr Opin Oncol. 2016;28(6):461–468. doi: 10.1097/CCO.0000000000000324. [DOI] [PubMed] [Google Scholar]
- 4.Lee JS, Magbanua MJ, Park JW. Circulating tumor cells in breast cancer: applications in personalized medicine. Breast Cancer Res Treat. 2016;160(3):411–424. doi: 10.1007/s10549-016-4014-6. [DOI] [PubMed] [Google Scholar]
- 5.Selli C, Dixon JM, Sims AH. Accurate prediction of response to endocrine therapy in breast cancer patients: current and future biomarkers. Breast Cancer Res. 2016;18(1):118. doi: 10.1186/s13058-016-0779-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Day TK, Bianco-Miotto T. Common gene pathways and families altered by DNA methylation in breast and prostate cancers. Endocr Relat Cancer. 2013;20(5):R215–232. doi: 10.1530/ERC-13-0204. [DOI] [PubMed] [Google Scholar]
- 7.Gyorffy B, Bottai G, Fleischer T, Munkacsy G, Budczies J, Paladini L, Borresen-Dale AL, Kristensen VN, Santarpia L. Aberrant DNA methylation impacts gene expression and prognosis in breast cancer subtypes. Int J Cancer. 2016;138(1):87–97. doi: 10.1002/ijc.29684. [DOI] [PubMed] [Google Scholar]
- 8.Van De Voorde L, Speeckaert R, Van Gestel D, Bracke M, De Neve W, Delanghe J, Speeckaert M. DNA methylation-based biomarkers in serum of patients with breast cancer. Mutat Res. 2012;751(2):304–325. doi: 10.1016/j.mrrev.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 9.Wittenberger T, Sleigh S, Reisel D, Zikan M, Wahl B, Alunni-Fabbroni M, Jones A, Evans I, Koch J, Paprotka T, et al. DNA methylation markers for early detection of women's cancer: promise and challenges. Epigenomics. 2014;6(3):311–327. doi: 10.2217/epi.14.20. [DOI] [PubMed] [Google Scholar]
- 10.Shim J, Humphreys GI, Venkatesan BM, Munz JM, Zou X, Sathe C, Schulten K, Kosari F, Nardulli AM, Vasmatzis G, et al. Detection and quantification of methylation in DNA using solid-state nanopores. Sci Rep. 2013;3:1389. doi: 10.1038/srep01389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yong WS, Hsu FM, Chen PY. Profiling genome-wide DNA methylation. Epigenetics Chromatin. 2016;9:26. doi: 10.1186/s13072-016-0075-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lizardi PM, Yan Q, Wajapeyee N. DNA Bisulfite Sequencing for Single-Nucleotide-Resolution DNA Methylation Detection. Cold Spring Harb Protoc. 2016 doi: 10.1101/pdb.prot094839. [DOI] [PubMed] [Google Scholar]
- 13.Fackler MJ, McVeigh M, Mehrotra J, Blum MA, Lange J, Lapides A, Garrett E, Argani P, Sukumar S. Quantitative multiplex methylation-specific PCR assay for the detection of promoter hypermethylation in multiple genes in breast cancer. Cancer Res. 2004;64(13):4442–4452. doi: 10.1158/0008-5472.CAN-03-3341. [DOI] [PubMed] [Google Scholar]
- 14.Stearns V, Fackler MJ, Hafeez S, Bujanda ZL, Chatterton RT, Jacobs LK, Khouri NF, Ivancic D, Kenney K, Shehata C, et al. Gene Methylation and Cytological Atypia in Random Fine-Needle Aspirates for Assessment of Breast Cancer Risk. Cancer Prev Res (Phila) 2016;9(8):673–682. doi: 10.1158/1940-6207.CAPR-15-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fackler MJ, Rivers A, Teo WW, Mangat A, Taylor E, Zhang Z, Goodman S, Argani P, Nayar R, Susnik B, et al. Hypermethylated genes as biomarkers of cancer in women with pathologic nipple discharge. Clin Cancer Res. 2009;15(11):3802–3811. doi: 10.1158/1078-0432.CCR-08-1981. [DOI] [PubMed] [Google Scholar]
- 16.Fackler MJ, Malone K, Zhang Z, Schilling E, Garrett-Mayer E, Swift-Scanlan T, Lange J, Nayar R, Davidson NE, Khan SA, et al. Quantitative multiplex methylation-specific PCR analysis doubles detection of tumor cells in breast ductal fluid. Clin Cancer Res. 2006;12(11 Pt 1):3306–3310. doi: 10.1158/1078-0432.CCR-05-2733. [DOI] [PubMed] [Google Scholar]
- 17.Everhard S, Kaloshi G, Criniere E, Benouaich-Amiel A, Lejeune J, Marie Y, Sanson M, Kujas M, Mokhtari K, Hoang-Xuan K, et al. MGMT methylation: a marker of response to temozolomide in low-grade gliomas. Ann Neurol. 2006;60(6):740–743. doi: 10.1002/ana.21044. [DOI] [PubMed] [Google Scholar]
- 18.Fiegl H, Jones A, Hauser-Kronberger C, Hutarew G, Reitsamer R, Jones RL, Dowsett M, Mueller-Holzner E, Windbichler G, Daxenbichler G, et al. Methylated NEUROD1 promoter is a marker for chemosensitivity in breast cancer. Clin Cancer Res. 2008;14(11):3494–3502. doi: 10.1158/1078-0432.CCR-07-4557. [DOI] [PubMed] [Google Scholar]
- 19.Dejeux E, Ronneberg JA, Solvang H, Bukholm I, Geisler S, Aas T, Gut IG, Borresen-Dale AL, Lonning PE, Kristensen VN, et al. DNA methylation profiling in doxorubicin treated primary locally advanced breast tumours identifies novel genes associated with survival and treatment response. Mol Cancer. 2010;9:68. doi: 10.1186/1476-4598-9-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klajic J, Busato F, Edvardsen H, Touleimat N, Fleischer T, Bukholm I, Borresen-Dale AL, Lonning PE, Tost J, Kristensen VN. DNA methylation status of key cell-cycle regulators such as CDKNA2/p16 and CCNA1 correlates with treatment response to doxorubicin and 5-fluorouracil in locally advanced breast tumors. Clin Cancer Res. 2014;20(24):6357–6366. doi: 10.1158/1078-0432.CCR-14-0297. [DOI] [PubMed] [Google Scholar]
- 21.Fackler MJ, Lopez Bujanda Z, Umbricht C, Teo WW, Cho S, Zhang Z, Visvanathan K, Jeter S, Argani P, Wang C, et al. Novel methylated biomarkers and a robust assay to detect circulating tumor DNA in metastatic breast cancer. Cancer Res. 2014;74(8):2160–2170. doi: 10.1158/0008-5472.CAN-13-3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Visvanathan K, Fackler MS, Zhang Z, Lopez-Bujanda ZA, Jeter SC, Sokoll LJ, Garrett-Mayer E, Cope LM, Umbricht CB, Euhus DM, et al. Monitoring of Serum DNA Methylation as an Early Independent Marker of Response and Survival in Metastatic Breast Cancer: TBCRC 005 Prospective Biomarker Study. J Clin Oncol. 2017;35(7):751–758. doi: 10.1200/JCO.2015.66.2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Avraham A, Uhlmann R, Shperber A, Birnbaum M, Sandbank J, Sella A, Sukumar S, Evron E. Serum DNA methylation for monitoring response to neoadjuvant chemotherapy in breast cancer patients. Int J Cancer. 2012;131(7):E1166–1172. doi: 10.1002/ijc.27526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kristiansen S, Jorgensen LM, Hansen MH, Nielsen D, Soletormos G. Concordance of Hypermethylated DNA and the Tumor Markers CA 15-3, CEA, and TPA in Serum during Monitoring of Patients with Advanced Breast Cancer. Biomed Res Int. 2015;2015:986024. doi: 10.1155/2015/986024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kristiansen S, Nielsen D, Soletormos G. Detection and monitoring of hypermethylated RASSF1A in serum from patients with metastatic breast cancer. Clin Epigenetics. 2016;8:35. doi: 10.1186/s13148-016-0199-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu L, Sun L, Li C, Li X, Zhang Y, Yu Y, Xia W. Quantitative detection of methylation of FHIT and BRCA1 promoters in the serum of ductal breast cancer patients. Biomed Mater Eng. 2015;26(Suppl 1):S2217–2222. doi: 10.3233/BME-151527. [DOI] [PubMed] [Google Scholar]
- 27.Xia B, Shan M, Wang J, Zhong Z, Geng J, He X, Vu T, Zhang D, Pang D. Homeobox A11 hypermethylation indicates unfavorable prognosis in breast cancer. Oncotarget. 2017;8(6):9794–9805. doi: 10.18632/oncotarget.14216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Connolly RM, Leal JP, Goetz MP, Zhang Z, Zhou XC, Jacobs LK, Mhlanga J, O JH, Carpenter J, Storniolo AM, et al. TBCRC 008: early change in 18F-FDG uptake on PET predicts response to preoperative systemic therapy in human epidermal growth factor receptor 2-negative primary operable breast cancer. J Nucl Med. 2015;56(1):31–37. doi: 10.2967/jnumed.114.144741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fackler MJ, Umbricht CB, Williams D, Argani P, Cruz LA, Merino VF, Teo WW, Zhang Z, Huang P, Visvananthan K, et al. Genome-wide methylation analysis identifies genes specific to breast cancer hormone receptor status and risk of recurrence. Cancer Res. 2011;71(19):6195–6207. doi: 10.1158/0008-5472.CAN-11-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McShane LM, Hayes DF. Publication of tumor marker research results: the necessity for complete and transparent reporting. J Clin Oncol. 2012;30(34):4223–4232. doi: 10.1200/JCO.2012.42.6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Widschwendter M, Jones PA. DNA methylation and breast carcinogenesis. Oncogene. 2002;21(35):5462–5482. doi: 10.1038/sj.onc.1205606. [DOI] [PubMed] [Google Scholar]
- 32.Connolly R, Stearns V. Epigenetics as a therapeutic target in breast cancer. J Mammary Gland Biol Neoplasia. 2012;17(3-4):191–204. doi: 10.1007/s10911-012-9263-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Groot JS, Moelans CB, Elias SG, Jo Fackler M, van Domselaar R, Suijkerbuijk KP, Witkamp AJ, Sukumar S, van Diest PJ, van der Wall E. DNA promoter hypermethylation in nipple fluid: a potential tool for early breast cancer detection. Oncotarget. 2016;7(17):24778–24791. doi: 10.18632/oncotarget.8352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kajabova V, Smolkova B, Zmetakova I, Sebova K, Krivulcik T, Bella V, Kajo K, Machalekova K, Fridrichova I. RASSF1A Promoter Methylation Levels PositivelyCorrelate with Estrogen Receptor Expression in Breast Cancer Patients. TranslOncol. 2013;6(3):297–304. doi: 10.1593/tlo.13244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sebova K, Zmetakova I, Bella V, Kajo K, Stankovicova I, Kajabova V, Krivulcik T, Lasabova Z, Tomka M, Galbavy S, et al. RASSF1A and CDH1 hypermethylation as potential epimarkers in breast cancer. Cancer Biomark. 2011;10(1):13–26. doi: 10.3233/CBM-2012-0230. [DOI] [PubMed] [Google Scholar]
- 36.Suijkerbuijk KP, Pan X, van der Wall E, van Diest PJ, Vooijs M. Comparison of different promoter methylation assays in breast cancer. Anal Cell Pathol (Amst) 2010;33(3):133–141. doi: 10.3233/ACP-CLO-2010-0542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Swift-Scanlan T, Vang R, Blackford A, Fackler MJ, Sukumar S. Methylated genes in breast cancer: associations with clinical and histopathological features in a familial breast cancer cohort. Cancer Biol Ther. 2011;11(10):853–865. doi: 10.4161/cbt.11.10.15177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beaver JA, Jelovac D, Balukrishna S, Cochran RL, Croessmann S, Zabransky DJ, Wong HY, Valda Toro P, Cidado J, Blair BG, et al. Detection of cancer DNA in plasma of patients with early-stage breast cancer. Clin Cancer Res. 2014;20(10):2643–2650. doi: 10.1158/1078-0432.CCR-13-2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garcia-Murillas I, Schiavon G, Weigelt B, Ng C, Hrebien S, Cutts RJ, Cheang M, Osin P, Nerurkar A, Kozarewa I, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med. 2015;7(302):302ra133. doi: 10.1126/scitranslmed.aab0021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Quantitative Gene Methylation by Treatment Arm. Scatter plots indicate methylation levels in tissues from individuals treated either with vorinostat (V) or placebo (P) in addition to standard therapy. Cumulative methylation index (CMI) was calculated for tissue samples in which PCR-amplification was successful for all 10 genes in the panel. CMI = the sum of individual gene methylation within the 10 gene-panel (possible 1000 CMI units, 100% × 10 genes). Gene methylation is defined as % methylation = (# copies methylated DNA/# total copies unmethylated DNA + methylated DNA) (100). The median is shown by bar and significance was calculated using the Mann-Whitney U test.
Supplementary Table 1: Patient characteristics
Supplementary Table 2: Tissue CMI at D15 by pCR status and treatment arm
Supplementary Table 3 (a-d): Subgroup Analyses
Supplementary Table 3a: Univariate analysis of association of CMI levels with pCR in HR+ patients (n=38)
Supplementary Table 3b: Univariate analysis of association of CMI levels with pCR in TNBC patients (n=24)
Supplementary Table 3c: Univariate analysis of association of CMI levels with pCR in the vorinostat arm (n=31)
Supplementary Table 3d: Univariate analysis of association of CMI levels with pCR in the placebo arm (n=31)