

Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disease, characterized by the loss of memory, multiple cognitive impairments and changes in the personality and behavior. Several decades of intense research have revealed that multiple cellular changes are involved in disease process, including synaptic damage, mitochondrial abnormalities and inflammatory responses, in addition to formation and accumulation of amyloid-β (Aβ) and phosphorylated tau. Although tremendous progress has been made in understanding the impact of neurotransmitters in the progression and pathogenesis of AD, we still do not have a drug molecule associated with neurotransmitter(s) that can delay disease process in elderly individuals and/or restore cognitive functions in AD patients. The purpose of our article is to assess the latest developments in neurotransmitters research using cell and mouse models of AD. We also updated the current status of clinical trials using neurotransmitters’ agonists/antagonists in AD.
Keywords: acetylcholine inhibitors, adenosine receptors, Alzheimer’s disease, histaminergic, N-Methyl-D-Aspartate, neurotransmitters
INTRODUCTION
Alzheimer’s disease (AD) is the most common form of dementia, characterized by impairment of cognition, abnormal behavior, and personality changes [1–5]. Histological examination of postmortem brains from AD patients revealed two major pathological hallmarks—intracellular neurofibrillary tangles and extracellular neuritic plaques. AD is also associated with synaptic damage, mitochondrial abnormalities, inflammatory responses, hormonal changes, and cell cycle abnormalities [1, 6–10]. Currently, 5.4 million Americans suffer with AD, including 5.2 million people age 65 and older and 200,000 individuals under the age of 65. One in nine people age 65 and older has AD [7, 11]. Currently, 47 million people live with dementia worldwide, which is more than the population of Spain [12, 13]. This number is projected to increase more than 131 million by 2050. Dementia also has a huge economic impact. According to the World Alzheimer’s Report - 2016, the total estimated worldwide cost of dementia is $818 billion, and it will become a trillion-dollar disease by 2018 [13]. AD is a multifactorial disease (Fig. 1), caused by mutations in amyloid-β protein precursor (AβPP), presenilin 1, and presenilin 2 genes, DNA variants in sortilin related receptor 1, clusterin, complement component receptor 1, CD2AP, CD33, EPHA1, and MS4A4/MS4A6E genes. ApoE 4/4 genotype is a major contributor for late-onset AD, and other contributing factors include aging, lifestyle activities, and traumatic brain injury [1, 7, 14–20]. Researchers have been involved in developing different drug molecules based on cellular changes found in the brains of AD patients and brain tissues from AD mouse models (Figs. 2 and 3) [1, 27, 28–35].
Figure 1.

Different types of pathogenesis mechanisms in Alzheimer’s disease. a) Oxidative stress factors and their role in activating caspase in neuronal death, mitochondrial dysfunction [15–17], DNA damage, and in reducing synaptic plasticity. b) Activation of kinases/proteases by oxidative stress and the formation of Aβ oligomers and the phosphorylation of tau. c) The formation of senile plaques and neurofibrillary tangles, and generation of oxidative stress through various mechanisms [18, 209].
Figure 2.

Cholinergic and glutamatergic neuronal transmission in the healthy and Alzheimer’s disease brains. a) Normal cholinergic and glutamatergic neuronal transmission, b) cholinergic and glutamatergic neuronal transmission in Alzheimer’s disease, c) acetylcholinesterase inhibitor role in cholinergic and glutamatergic neuronal transmission in Alzheimer’s disease.
Figure 3.

Multiple approaches in Alzheimer’s disease therapeutics, such as amyloid-based therapies, tau-based therapies, neurotransmitter-based therapies, signaling-based therapies, inflammation-based therapies, mitochondria-targeted therapeutics, vaccine therapy, and oxidative stress reducers [210, 211].
After several decades of research on AD prevention, the FDA has only approved acetylcholinesterase inhibitors (AChEIs) such as donepezil, galantamine, and rivastigmine, and the N-Methyl-D-Aspartate (NMDA) antagonist, memantine. Tacrine, which is also an AChEI, was discontinued due to the hepatotoxicity in AD patients [21]. The purpose of this article is to highlight the roles of neurotransmitters in AD progression and pathogenesis. We also provide an update on the current status of clinical trials using various types of neurotransmission modulation (Fig. 4).
Figure 4.

Neurotransmitters based therapy in Alzheimer’s disease such as cholinesterase inhibitors, NMDA receptor inhibitors, GABAergic modulators, serotonin receptor modulators, histaminergic modulators, and adenosine receptor modulators [211].
NEUROTRANSMITTERS
Neurotransmitters are endogenous chemical messengers that enable neurotransmission. These neurotransmitters transmit the signals across the synapse (between the neurons) and neuromuscular junctions [22, 23]. Neurotransmitters are usually stored in the synaptic vesicles, beneath the membrane in the axon terminal, and are released into the synapse with the appropriate signal. The released neurotransmitters exhibit their functions to establish connections through the synaptic cleft and bind to their appropriate receptors (Fig. 2). These neurotransmitters are endogenously synthesized from amino acids [22, 23]. Acetylcholine (ACh) is synthesized from serine; dopamine from L-phenyl alanine/L-tyrosine; GABA from glutamate by decarboxylation; serotonin from L-tryptophan; histaminergic from L-histidine; and NMDA from D-aspartic acid and arginine [24, 25]. These are the main neurotransmitters (except dopamine) that play a crucial role in AD pathogenesis. There are other neurotransmitters, such as glutamate, glycine, norepinephrine, epinephrine, melatonin, gastrin, oxytocin, vasopressin, cholecystokinin, neuropeptide Y, and enkephalins, that do not have a significant role in AD pathogenesis and may have a role in causing oxidative stress [25]. These chemical messengers have been reported to be involved in synaptic activities including the required action potential from synaptic vesicles to propagate exocytosis. The released neurotransmitters diffuse on the synaptic cleft and bind to appropriate receptors on the postsynaptic neuronal membrane for potential synaptic transmission [26] (Fig. 2).
Metabotropic and ionotropic are neurotransmitter receptors which are localized to the postsynaptic membrane. Metabotropic receptors are membrane receptors that act through the secondary messenger, whereas ionotropic receptors are ligand-gated channels. In this regard, acetylcholine receptors (AChRs) include a group of metabotropic neurotransmitter receptors (muscarinic AChRs, histamine receptors for histamine, GABAB receptors for GABA, adenosine receptors for adenosine), serotonin receptors (G-protein coupled receptors for serotonin), and a group of ionotropic neurotransmitter receptors (nicotinic AChRs, GABAA& A-ρ receptors also for GABA, and additionally 5-HT) [26–32]. For researchers, these are the major therapeutic targets for AD prevention.
NEUROTRANSMITTER-BASED THERAPEUTICS IN AD
Since 1901, when Dr. Alzheimer observed senile dementia in ‘Auguste Deter’, well over a century ago, there are still no metabolic-based therapies for its prevention, though some may temporarily relieve the symptoms. There are three FDA approved AChEIs, one NMDA antagonist, and one AChEI and NMDA antagonist combination (Table 1 & Fig. 5). There is one drug, tacrine, that has been discontinued due to hepatotoxicity in AD patients [33].
Table 1.
FDA Approved AChEIs and NMDA antagonist in various stages of Alzheimer’s disease
| Drug Name | Brand Name | Applicable Stages of AD | FDA Approved Year |
|---|---|---|---|
| Donepezil (AChE Inhibitor) | Aricept | All | 1996 |
| Galantamine (AChE Inhibitor) | Razadyne | Mild AD to Moderate AD | 2001 |
| Rivastigmine (AChE inhibitor) | Namenda | Moderate AD to Severe AD | 2003 |
| Memantine (NMDA Antagonist) | Exelon | All | 2000 |
| Donepezil + Memantine | Namzaric | Moderate AD to Severe AD | 2014 |
Adapted from Alz.org
Figure 5.
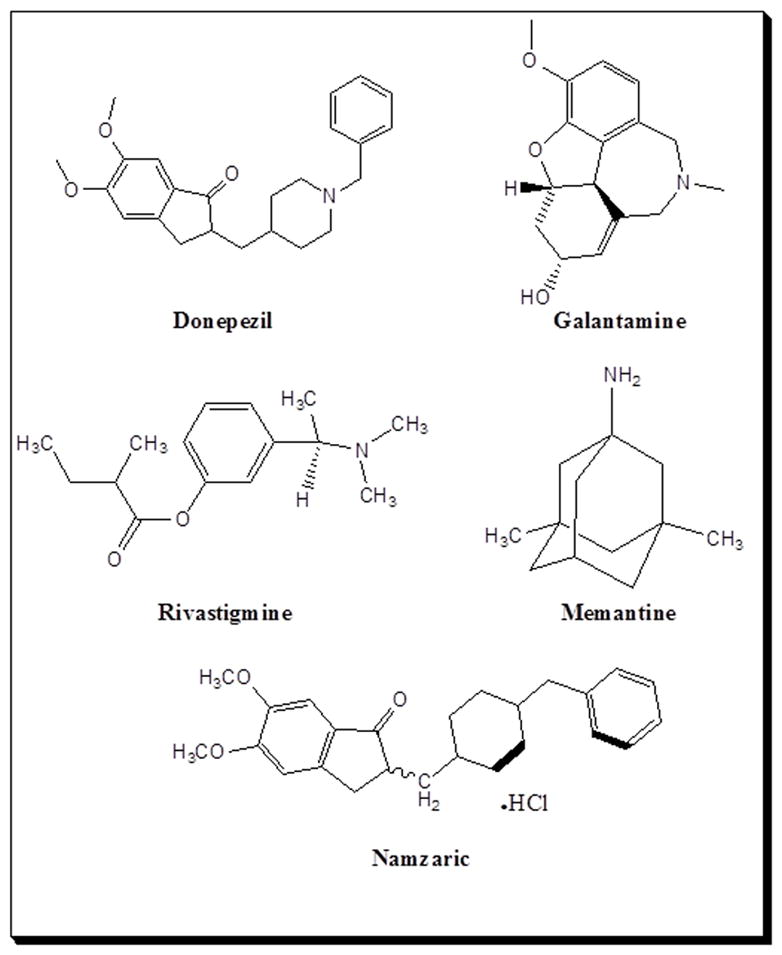
Structures of the FDA-approved acetylcholinesterase inhibitors including donepezil, galantamine, and rivastigmine and the NMDA antagonist, memantine, in Alzheimer’s disease therapy for delaying neuronal death.
Neurotransmission Modulation
Cholinergic neurons such as ACh-producing neurons are mainly involved in the pathogenesis of AD. ACh was the first neurotransmitter discovered in 1920. It is present in the neuromuscular junctions, brain, spinal cord, in the autonomous nervous system, such as ganglia, and postganglionic terminal buttons of the parasympathetic nervous system. Recent literature suggests that ACh is synthesizing from the nucleus basalis of Meynert and the projection from the nucleus basalis provide the primary source of neocortical ACh [34]. On the other hand, cholinergic projections also extend from the medial septum and a diagonal band of Broca to the hippocampus [35, 36]. These cholinergic projections are the primary sources in the production of ACh in the brain. ACh plays a pivotal role in learning and memory as the cortex originates from the basal forebrain, and thus, is involved in memory consolidation in these sites. ACh also regulates cortical structure, cerebral blood dynamics, and the wake-sleep cycle [37, 38]. Although much research has been done on ACh in AD pathogenesis, the precise molecular links are not well understood.
Slotkin et al. found a decrease in choline acetyltransferase activity and, in turn, a decrease in ACh synthesis, and an eventual decrease in ACh uptake by acetylcholine receptors (AChRs) in AD [39, 40]. Loss of memory is one of the major phenotypic changes that occur in AD, and it is due to the loss of AChE from both cholinergic and non-cholinergic neurons of the central nervous system (CNS). However, AChE activity is increased around amyloid plaques. This increase in AChE opens the doors for therapeutic avenues based on AChEIs (Fig. 2C), because recent studies have also shown the role of amyloid-β (Aβ) protein in the expression of AChE [41]. The produced ACh is received by AChRs, which are present on the postsynaptic membrane of target cells (Fig. 2A). These neurotransmitter receptors are nothing but proteins which interact with extracellular signals; ACh thus converts these signals into intracellular effects [42]. ACh neurotransmitter receptors are one of a chief excitatory neurotransmitters [43]. The binding of ACh to AChRs results in a change in membrane potential of the target cell, leading to an action potential in the neuron or muscle cell. The produced ACh is cleared by enzymatic action in the synapse. Normally, when a neurotransmitter binds with the corresponding receptor, the channel opens and allows sodium ions into the cell, which depolarizes the plasma membrane to create the action potential [43, 44]. There are two subtypes of AChRs: excitatory ACh, which is present in the parasympathetic nervous system; and inhibitory ACh, involved in the autonomic ganglionic actions (Fig. 2B).
Along with ACh, glutamate is an ionic form of glutamic acid which is the excitatory neurotransmitter involved in the learning and memory process in the cortex and hippocampus and thus plays a role in long-term potentiation (LTP) and long-term depression (LTD). NMDA is the receptor which is present on the postsynaptic membrane of the neuron. The action of this receptor is modulated by the glutamate neurotransmitter [45–47]. Revett et al. have shown there is an increased presynaptic release of glutamate and there is a failure in re-uptake of released glutamate, which in turn leads to a tonic activation of NMDA receptors, and thus contributes to an excess in AD [48]. Loss of insulin signaling and mitochondrial dysfunction [49] are the contributing factors to glutamate excitotoxicity found in AD neurons [50]. Mitochondrial dysfunction may be due to oxidative stress and/or oxidative insults in AD neurons. Along with ACh and GABA, there are other components such as serotonin and histaminergic neurons also playing a crucial role in the pathogenesis of AD.
Acetylcholinesterase inhibitors (AChEI)
The FDA approved four AChEIs (tacrine, donepezil, rivastigmine, and galantamine) [21] for the treatment of AD. Donepezil (Aricept) was approved in 1996; rivastigmine (Exelon) was approved in 2000; and galantamine (Reminyl) was approved in 2001. Tacrine (Cognex), the first cholinesterase inhibitor, was approved in 1993; however, tacrine is rarely prescribed because of associated side effects, including possible liver damage, and the same fact was mentioned in the World Alzheimer Report 2016 and Alzheimer’s Association fact sheet (Table 1) [13, 51].
AChEIs inhibits the enzyme activity of AChE, which in turn decreases the breakdown of ACh and thus plays the pivotal role in enhancing cholinergic neurotransmission and making them available to the cholinergic AChRs for generating LTP in learning and memory processing. These receptors are expressed both on the principal and inhibitory neurons in most regions of the hippocampus and cortex, and localized to both pre and post-synaptic membranes [52]. Galantamine, an alkaloid, additionally acts as an AChEI that impedes AChE and plays a vital role in the regulation of nicotinic receptor activities. In a randomized, double-blind, placebo-controlled study, galantamine exhibited improved cognitive function in subjects over a 6-month period. Additionally, galantamine had positive effects on subjects’ instrumental and essential activities of daily living, and also delayed the progression of the implicated behavioral symptoms that are associated with the disease process. An open extension of the study occurred for an additional 6 months, with a galantamine dose of 24 mg/day afforded to patients, who completed one of the 6-month placebo-controlled studies. Retention of cognitive function and perseverance of activities essential to the subjects’ daily living were observed at the end of the 12-month study period in those that had been treated with galantamine 24 mg/day at full length of the study. This group significantly retained and exhibited an improvement in cognitive functions at the 12-month mark, in comparison to subjects who had been treated with a placebo for 6 months before being treated with galantamine for the remaining 6 months of the 12-month study. These studies indicate that galantamine postpones the progression of symptoms in AD. Since galantamine shows the greatest benefits when treatment is started early, its long-term benefits may result from an effect on the underlying disease process; such an effect might be mediated by galantamine’s concomitant action on nicotinic receptors. On the other hand, galantamine also possesses agonist activity at the nicotinic α4β2 receptor; however, clinical benefits of galantamine are due to the above mentioned (two) mechanisms [53].
Muscarinic AChRs (mAChRs) have been recently identified by Anisuzzaman et al. to be present in intracellular locations, namely the hippocampal regions of mice, rats, and humans. Transmission of extracellular cholinergic signals into the cytoplasm from the position of these receptors on the cell surface, in addition to contributing uniquely to synaptic plasticity, activates signaling cascades distinct from those of other cell surface receptors. In contrast to the selective incidence of M1-mAChRs on the cell surface of other tissues, intracellular and surface disseminations of M1-mAChRs in the hippocampus and cortex of rats, mice, and humans have been revealed with immunohistochemical observations in radio ligand-binding experiments with cell-permeable and cell-impermeable ligand. Muscarinic binding sites were annulled throughout intracellular locations in M1-mAChR gene knockout mice. Phosphatidylinositol hydrolysis and network associations at theta rhythm were generated by rat hippocampal neurons upon activation of cell surface M1-mAChRs, which enhanced LTP transiently. Moreover, phosphorylation of extracellular regulated kinase 1/2 and a gradual augmentation of LTP resulted from activation of intracellular M1-mAChRs. These results suggest a new mode of cholinergic transmission in the CNS [54] in regulating LTP and synaptic plasticity.
Interestingly, various studies have shown the role of cholinergic neurons in neuronal precursor development and its differentiation. These neurons projecting from the basal forebrain innervate the cerebral cortex during neuronal development [55]. Afferents and their cortical target cells interact and are likely to influence each other during the establishment and refinement of connections. Intracortical cholinergic interneurons similarly have a local effect on cortical circuits. Reduced cholinergic innervation during development therefore leads to reduced cortical thickness and dendritic abnormalities. ACh is also likely to play a critical role in neuronal plasticity. In the hippocampus neurite extension, target selection and synaptogenesis play a role in the maturation of GABAergic synapses during the developmental shift from depolarizing to hyperpolarizing transmission. Thus, the cholinergic transmission also plays an important role in modulating the phosphatidylinositol signals and extracellular regulated kinase pathways in adult neurogenesis [55]. Klugman et al. also suggested that AChEIs reduce oxidative stress and mitochondrial dysfunction in AD [56].
Various meta-analysis studies revealed that AChEIs plays a crucial role in improving cognitive function from mild-to-moderate AD patients and moderate-to-severe AD patients [57, 58]. Three cholinesterase inhibitors such as donepezil, galantamine, and rivastigmine are efficacious for mild to moderate AD. Even though these are the FDA approved, transdermal delivery of these drugs is not approved, as it is attributed to relieving AD symptoms in an efficient manner. The gastrointestinal and other liver-related side effects of these AChEIs are due to an increase in the plasma concentration for a short time interval. A transdermal patch has been developed with rivastigmine. In an open-label, parallel group study in AD patients that were randomized to receive either capsule (1.5–6 mg Q12H, i.e., 3–12 mg/day) or patch (5–20 cm2) in ascending doses through four 14-day periods, Lefevre et al. compared patients’ drug exposure between path and capsule administrations by investigating the pharmacokinetics and pharmacodynamics of rivastigmine and NAP226-90. Plasma butyrylcholinesterase inhibition rose slowly after patch administration; interestingly, two distinct peaks were seen after capsule administration [59]. The subjects also experienced significantly decreased discomfort with patches [60]. In another study, the authors used a novel AChEI molecule, memogain (benzoyl ester of galantamine), an inactive pro-drug of galantamine which has more than 15-fold higher bioavailability in the CNS than the same doses of galantamine that has been approved for the treatment of mild to moderate AD. Compared to presently available drugs, memogain regains its pharmacological activity as a cholinergic enhancer in the brain when it is enzymatically cleaved to galantamine without showing any significant levels of gastrointestinal side effects that are typical for the unmodified drug and other inhibitors of cholinesterase, such as donepezil and rivastigmine. Memogain produced several fold larger cognitive improvement than the same doses of galantamine. These results and other preclinical data revealed that the inactive pro-drug of galantamine, memogain, may prove a prolific and beneficial drug for AD treatment, combining reduced gastrointestinal side effects and considerably higher potency in enhancing cognition, as compared to presently available treatment drugs [61].
Huperzine A is a potent and reversible inhibitor of AChE, and it was initially isolated from a Chinese herb. It has been found to improve cognitive abnormalities in AD animal models and has been used for AD treatment in China. The therapeutic effects of huperzine A include regulation of AβPP metabolism and protecting against Aβ-mediated oxidative stress and apoptosis [62]. ZT-1 (pro-drug of huperzine A) [63] and methanesulfonyl fluoride (SNX-001) [64] has shown admirable therapeutic value in AD in Phase I trial studies.
In AD, postsynaptic M1-AChRs remain unchanged but presynaptic M2-AChRs decrease. There are several M1 partial agonists and allosteric agonists available; these agonists can show their effects on the biological action of AChRs. The M1 partial agonists are mainly AF102B, AF150(S), AF267B, and AF292. There are also some allosteric agonists such as 77-LH-28-1, LY-593093, and Lu AE51090 that have shown significant effects on cholinergic neurotransmission. Recently, ML 169 was reported as an M1 positive allosteric modulator [65]. The M1 agonists have a role in AβPP processing and thus control tau phosphorylation. In 3xTgAD and transgenic mice expressing human Swedish, Dutch, and Iowa triple-mutant AβPP (Tg-SwDI), two widely used animal models, and also with the M(1)R (−/−), it has been revealed by Medeiros et al. that AD-related synaptotoxicity and cognitive impairment through mechanisms that rely on the transcriptional dysregulation of genes required for memory are worsened by M(1)R deletion in the 3xTgAD and Tg-SwDI mice [66]. Depositing of fibrillary Aβ was in observed in cerebrovascular locations in Tg-SwDI mice upon eradication of M1-AChRs alongside an escalation of plaque and tangle levels in brains of 3xTgAD mice [66]. Notably, tau hyperphosphorylation and potentiation of amyloidogenic processing in the mice with AD lacking M1-AChRs were attributed to changes in GSK3β and protein kinase C activities [66]. Finally, deleting the M1-AChRs increased the astrocytic and microglial response associated with Aβ plaques. These results suggest the significant role of M1-AChRs in exacerbating AD-related cognitive decline and provide critical preclinical evidence to justify further development and evaluation of selective M1-AChRs agonists for treating AD [66]. To strengthen these results, AF102B, an M1 partial agonist, significantly lowered cerebrospinal fluid (CSF) Aβ levels in AD patients [67]. In preclinical studies, other partial agonists of M1-AChRs such as AF150(S) and AF267B have yielded productive effects [68–70]. Therapeutics of mixed muscarinic AChRs such as ANAVEX 2–73, which are in phase I/IIa clinical trials, has yielded regression in Aβ-induced toxicity, gradual amelioration of memory deficits [71], and the impediment of GSK3β activation, all together which prevent the phosphorylation of tau [72]. As of now, there are no published research studies that show the pathological role of nicotinic AChRs (nAChRs) in AD [73, 74].
On the other hand, α7 nicotinic agonists have divergent effects, which would be beneficial in some AD patients [69] and require further investigation to understand the role of α7 nicotinic agonists in other dementias. Echeverria and Zeitlin discussed positive modulatory effects of cotinine, a metabolite of nicotine on α7 nAChRs in AD prevention [75]. The neuroprotective action of cotinine and the mechanistic approach to improving memory deficits in AD mice and in AD has been demonstrated. Cotinine improves memory by inhibiting the aggregation of Aβ, the stimulation of Akt (pro-survival factors), and the inhibition of activation GSK3β (pro-apoptotic factor) [75].
Different types of ligands for α7 nAChRs are also currently being studied as potential AD therapeutics [76]. EVP-6124, [(R)-7-chloro-N-quinuclidin-3-yl) benzo[b]thiophene-2-carboxamide], is a novel partial agonist of α7 nAChRs that was evaluated in a cell line and in rats. The results showed improvement in memory performance in rats by potentiating the acetylcholine response of α7 nAChRs and support new therapeutic strategies for the treatment of cognitive impairment [77]. There are other nicotinic agonists, which completed phase II trials, including NCT01073228, NCT01764243, NCT01137526, and NCT01527916; however, MT-4666 was terminated in phase III due to the benefit-risk balance of MT-4666 (data were taken from ClinicalTrials.gov).
Currently, there are several different AChEIs involved in clinical trials: 1) NCT00969696: A phase I AD clinical trial was completed. In this trial, the effects of galantamine on cognitive performance in marijuana users were shown; 2) NCT00880412: This clinical trial determined the clinical safety/tolerability and an exploratory efficacy of EHT 0202 as adjunctive therapy to AChEI in mild to moderate AD. A phase II trial was completed; 3) NCT01908010: This clinical trial describes safety, tolerability, and pharmacokinetics of ABT-354 in up to 20 male and female subjects, between 55 to 90 years of age with mild to moderate AD, given stable doses of AChEIs. A phase II was completed; 4) NCT00987220: This trial hypothesized that the AChEI, donepezil (Aricept), will increase acute CSF ACh levels in healthy volunteers following a 5 mg single dose oral administration. This study has been completed, but there is no further information available; 5) NCT00309725: The objective of this trial was to investigate the effect of galantamine on heart rate and PR interval (the time it takes for the heart’s electrical impulse to get from the atria to the ventricles) during the administration of rapidly increasing doses at the end of a 2-week treatment period with 32 mg per day in patients with AD. It has completed phase III trial; 6) NCT01626391: This trial examines the safety and tolerability of TRx0237 when taken at the same time as AChEIs (i.e., donepezil, galantamine, or rivastigmine) and/or memantine to treat patients with mild to moderate AD and has been terminated for administrative reasons; 7) NCT00348140: This trial is focused on rosiglitazone extended release tablets (RSG-XR) as adjunctive therapy in subjects with mild to moderate AD and has completed phase III trials. This study tests whether RSG XR safely provides clinical benefit to people with mild to moderate AD when combined with one of the currently approved AD medications, Aricept®, Razadyne® or Exelon®. RSG XR is a new approach to AD therapy and this study tests a new way to treat AD by testing whether one’s genetic makeup affects the response to the study drug; 8) NCT00096473: This clinical trial focused on donepezil hydrochloride (Aricept) and was approved to treat symptoms associated with mild to moderate AD. Aricept has been shown to improve cognition and learning abilities in AD patients. The purpose of this study was to further investigate the effectiveness and safety of donepezil in patients with severe AD and it has completed phase III; 9) NCT00369603: The purpose of this study was to determine whether standard medications approved for AD treatment differ in their action on brain function, and while this study was completed, findings are not yet available (data were taken from ClinicalTrials.gov); and 10) NCT00001933: This clinical trial conducted by the National Institute of Neurological Disorders and Stroke (NINDS) aimed to examine the ability of Nefiracetam, a new drug that stimulates acetylcholine, to improve memory, cognition, and routine-living activities of patients with mild to moderate intellectual impairment due to AD, and successfully advanced through phase II clinical trials.
Based on these findings regarding M1 agonists and AChEIs, M1 agonists have more beneficiary relief when compared to AChEIs. M1 agonists could be regarded as disease-modifying agents, as well as provide symptomatic relief in AD. The benefit of AChEIs in the behavioral symptoms of AD and their synergistic role in combination therapy with memantine will be discussed in the following section.
NMDA inhibitors: a glutamatergic neurotransmission system
Glutamatergic neurotransmission is an important mechanism involved in learning and memory; therefore it is involved in establishing LTP, which is affected in AD patients. In AD, Aβ induces oxidative stress, which in turn, deregulates the glutamatergic neurotransmission system, ultimately leading to the failure of its role in cognition, learning, and memory in AD patients [78]. Therefore, therapeutic avenues involving glutamatergic neurotransmission may play a crucial role in AD prevention.
Glutamate is the most abundant neurotransmitter in the CNS and its concentration is regulated by the glutamate cycle between pre- and post-synaptic neurons to establish strong synaptic connections (Fig. 2). Glutamate transporters such as VGLUT1 and VGLUT 2 in presynaptic neurons maintain glutamate levels in the vesicles. During neuronal transmission, the neuronal signal activates synapses and thus favors the release of glutamate into the synaptic cleft. The released glutamate activates different types of glutamate receptors such as NMDA, AMPA, kainite, and metabotropic. NMDA receptors may become active once synchronized with AMPA receptors, which upon activation cause depolarization of the postsynaptic membrane and allow the flow of calcium (Ca2+) ions into the postsynaptic neuron [79]. The activated glutamate receptor activates downstream cellular signaling pathways, while glutamate released in parallel from the synaptic cleft is removed by another set of glutamate transporters, and these are mostly expressed in astrocytes. Once glutamate has completed its function, it is immediately converted to glutamine by glutamine synthetase and returned to the presynaptic neuronal terminals, where, in presynaptic terminals, glutamine converts back to glutamate by glutaminase. During AD progression, this glutamate cycle gets affected and thus causes extracellular accumulation of glutamate and increased NMDA receptor activation, thus leading to excitotoxicity [48]. Excess glutamate causes the activation of NMDA receptors and enhances the production of Aβ and, vice versa, Aβ oligomers binding and activation of NMDA receptors, further substantiating the importance of the glutamatergic system in AD [48, 80].
The main molecule for the NMDA-based therapy in the treatment of AD is memantine, which is the uncompetitive antagonist and used in the treatment of moderate to severe AD in more than 60 countries including the USA and several European countries [81]. Symptomatic effects and tolerability of memantine are due to an affinity with NMDA receptor (IC50 around 1 μM at −70 mV) along with strong voltage dependency. This association resulted from double exponential blocking/unblocking kinetics in AD patients [82–85]. These NMDA beneficial therapeutic and functional properties have been confirmed by various groups in rat hippocampal and cortical neurons by using patch clamp recordings and HEK 293 cells [81, 83, 86–94]. Usually NMDA receptors are blocked by magnesium (Mg+2) ions, but in the presence of a strong synaptic signal, glutamate relieves the Mg+2 from the NMDA receptor channel, thus allowing Ca+2 ions into the post-synaptic neurons. However, in AD, this action is poorly driven due to the accumulation of Aβ peptides. These toxic peptides decrease membrane potential and unblock the Mg+2 ions from NMDA receptors, leading to defective flow of Ca+2 ions into the post-synaptic neurons. Numerous physiological experiments revealed that memantine binds within the ion channel of the NMDA receptor [83, 95] and regulates Ca+2-Mg+2 voltage gradient across the pre- and postsynaptic neurons [83, 84, 96, 97]. However, during normal physiological conditions, the high concentrations of glutamate dissociate memantine from the NMDA receptor channel, leading to normal neurotransmission [84, 96–98]. In the case of AD, memantine may compete with Aβ peptides to bind with the NMDA receptor channel in a normal glutamate cycle. This generates a voltage-dependent gradient that is employed in postsynaptic intracellular signaling events [99]. Furthermore, in vivo and in vitro testing has shown that this effect translates into a reversal of learning impairments induced by overactivation of NMDA receptors [96, 100]. Recent experimental results in animal models revealed that memantine improves spatial learning by protecting neurons from Aβ toxic insults, and thus, synaptic plasticity is preserved by preventing concomitant cognitive deficits [101]. Memantine also has antagonizing effects not only limited to the NMDA receptor channel, but also extended to α7, α4β2 nAChRs, 5-HT3, α3β2, 5-HT2A, dopamine D2 receptors, and histaminergic neurons [21, 102–107]. Evidence of the multiple receptor-binding activities of memantine, the therapeutic activity of this molecule has been noted to be consequent of binding to receptors such as NMDA, AChRs, and AMPA. However, experimental evidence has only reflected the binding of memantine to AChRs and NMDA receptors [108].
Currently, memantine is widely used for treating moderate and severe AD patients with a Mini-Mental State Examination (MMSE) score of less than 15 or less than 20, respectively. But the clinical action of this drug in the early stages of AD is still under investigation [109–113]. Regarding the efficacy of memantine in treating mild AD, a recent meta-analysis study [114] suggests administering memantine alone or in conjunction with cholinesterase inhibitors to assess the potential efficacy of memantine in mild to moderate AD in prospective trials. The possible reason for this is attributed to the early damage of cholinergic neurons when compared with the loss of the glutamatergic system in the progression of AD [115]. Alternatively, the blockage of α7nAchRs by memantine in the early stages of AD would affect neurotransmission [116]. AChEIs and memantine are licensed for the symptomatic treatment of mild-moderate and moderate-severe forms of AD, respectively. High doses of the AChEI, i.e., donepezil, were licensed in the USA for moderate-severe AD, and the association AChEI with memantine was proposed for AD at this stage. Molino et al. [131] reviewed different clinical trials regarding effectiveness of memantine, donepezil, or the two drugs in combination to manage moderate-severe AD. The authors considered double-blind, placebo-controlled, randomized trials, employing memantine, donepezil independently or in conjunction with memantine and compared to placebo treatments in moderately severe AD. The combination of memantine and donepezil treatments yielded no major advantages. Notwithstanding, the conditions of heterogeneity explored by randomized controlled trials in a relatively short observation period (24–52 weeks), and the different cognitive assessment tools used did not permit a proper comparison of different trials [117].
Arguably, this area requires intensive research to figure out when memantine needs to be given to AD patients in the early stage of AD or if it better to avoid memantine treatment in the early stages of AD. Across the world, numerous investigators tried the combination of AChEIs with memantine to treat AD [113, 118–123], and results of these studies suggest that it would be beneficial to treat patients with moderate to severe AD. There is a phase IV clinical trial (NCT00505167) on memantine versus donepezil in the early stages of AD of which glutamatergic hyperstimulation is understood to result in neuronal death in AD, and further reflects memantine as a low-affinity antagonist of NMDA glutamate receptors that confer neuroprotective effects and delay progression of the disease in its early stages. The effects of memantine should be compared to those of donepezil, which is the most prescribed AChE drug. The final results of this study are not yet published on “ClinicalTrials.gov”. There is another clinical trial (NCT00255086) mainly focused on determining if the NMDA receptor antagonist memantine has a neuroprotective effect on magnetic resonance spectroscopic imaging (MRS) measures of brain N-acetyl aspartate (NAA) and magnetic resonance imaging (MRI) volumetric measures of hippocampal volume. In their secondary analyses, they want to determine if measures of clinical stabilization produced by memantine in the treatment of AD parallels stabilization of MRS measures of brain NAA and MRI volumetric measures of hippocampal volume. This clinical trial has finished phase III. SUVN-502 with donepezil and memantine for the treatment of moderate AD, a phase IIa study (NCT02580305), is currently recruiting patients and the study is a proof-of-concept, 26-week, double-blind, multicenter, randomized, parallel group, placebo-controlled study to compare the efficacy and safety of treatment with SUVN-502 to placebo treatment in subjects with moderate AD receiving stable doses of donepezil HCl and memantine HCl. NCT00234637, a completed phase IV trial, examined rivastigmine monotherapy and combination therapy with memantine in patients with moderately severe AD, who failed to benefit from previous AChEI treatment.
Per the Alzheimer’s Association website information, the FDA approved drugs for the treatment of AD (Table 1) include three AChEIs: donepezil (Aricept) is approved to treat all stages of AD; rivastigmine (Exelon) is approved to treat mild to moderate AD; and galantamine (Razadyne) is approved to treat mild to moderate AD (http://www.alz.org/alzheimers_disease_standard_prescriptions.asp).
GABA neurotransmitter modulation
GABA is a neuroinhibitory neurotransmitter in adults. The role of glutamatergic and cholinergic neuronal transmission is widely investigated in AD, but the precise role of GABA neurotransmitters in AD is not well understood. It is demonstrated that the GABA (A) receptor of the GABAergic system is altered in aging and AD. Intriguingly, in AD, there is a compensatory increase in GABA (A) receptor within neurons [124]. Mizukami et al. demonstrated alterations of GABA (A) receptor subunits α1 and β2/3 in AD patients. In another study, reduction in the α1 subunit in the hippocampus is reported [125]. These two studies have shown that the β2/3 subunits are relatively resistant to alteration in AD patients [126]. Ulrich [127] investigated the effect of Aβ peptides on synaptic transmission through the GABA (A) receptor-mediated endocytosis mechanism and found that Aβ may weaken synaptic inhibition through downregulation of GABA (A) receptors and reversed in the presence of GABA (A) receptor agonist (isoguvacine).
In another study, Lagostena et al. found a neuroexcitatory role of GABA in AD11 mice [128]. Genetically engineered transgenic AD11 mice show age-dependent neurodegenerative pathology and failure in cholinergic function, which is similar to the pathology of AD patients. 6-month-old AD11 mice in the presence of the GABA (A) agonist (isoguvacine) have shown significantly increased firing of CA1 principal cells. Thus, these results suggested that chronic nerve growth factor deprivation shifts GABAergic signaling from the hyperpolarizing to the depolarizing direction [128]. Etazolate is a selective GABA (A) modulator that stimulates rat cortical neurons and, in guinea pig brains, produces sAβPPα. Etazolate protected cortical neurons from Aβ toxicity in a dose-dependent manner. When the neuroprotective action of etazolate was blocked with GABA (A) receptor antagonists (causes decreased Aβ toxicity), the neuroprotection was mediated through to GABA (A) receptor signaling. Thus, etazolate shows its neuroprotective effect through sAβPPα induction. Therefore, it indicates GABA (A) receptor signaling is a potential therapeutic approach in the treatment of AD [129]. However, it is not clear what kind of GABA function damage occurs to the neurons/cells. Recently, Jo et al. determined that GABA from reactive astrocytes impairs memory in APP/PS1 mouse models. In this study, the authors found that activated reactive astrocytes produce the inhibitory gliotransmitter GABA with the catalytic action of monoamine oxidase-B (MAOB) and releases through the bestrophin 1 channel [130]. This GABA reduces the spike probability of granule cells by acting on presynaptic GABA receptors in the dentate gyrus. So eventually by controlling GABA production, it can control LTP for learning and memory in the mice. Postmortem AD patients’ brains also have shown significant increases in astrocytic GABA and MAOB expression. These results indicated selective inhibition of astrocytic GABA synthesis or release and may be useful for improving memory in AD [130]. In Parkinson’s disease, Smad3 deficiency as noted in GABA (A) neurotransmission was observed to increase through the inhibition of dentate gyrus LTP [131]. Similarly, alteration of GABA neurotransmission is thought to produce hippocampal cognitive dysfunction in Down’s syndrome patients with AD. Townsend et al. also found indirectly the above fact by α7 nAChR agonist (FRM-17848), which enhanced neural plasticity in the hippocampus via a GABAergic circuit [132].
A comparative analysis of in vivo imaging studies in neuropsychiatric disorders by focusing on GABA (A) receptor function revealed that GABAergic dysfunction may be associated with the disturbances of dopaminergic and serotonergic neurotransmission [133].
A clinical trial (NCT00093951) on SGS742, which is GABA(B) receptor antagonist, in the treatment of patients with mild to moderate AD has completed phase II. In another clinical trial on EHT0202 (etazolate hydrochloride), a GABA (A) receptor modulator was the focus of a 3-month, randomized, placebo-controlled, double-blind study that revealed clinical safety and tolerability of EHT0202 as a primary objective, with secondary endpoints (cognitive function, daily living activities, behavior, caregiver burden, and global functioning). Although this clinical trial has completed phase IIA [134], the long-term benefits of this compound still needs to be investigated on a large scale such as multiple cohort and longitudinal studies. Triazolophthalazine compound selectively reduces the effects of GABA (A) receptors subunit, i.e., Alpha5IA, which plays an important role in hippocampal synaptic plasticity and LTP. GABA (A) receptor alpha5 subtype-selective inverse agonist alpha5IA triazolophthalazine was tested in elderly subjects and alpha5IA agonist was found to not improve performance in a paired-associate learning tasks, but it is able to reverse the ethanol-induced impairment in performance in healthy young normal volunteers. These data demonstrate that in humans, an alpha5-selective inverse agonist may be effective at increasing performance under certain conditions [135].
These incomplete results are not sufficient to recommend the use of GABA modulators and GABA (A) and GABA (B) antagonists in AD therapeutics, and further investigation in large multi-ethnic populations at different stages of AD is required.
Serotonin receptor-mediated AD therapeutics
The serotonergic system is the most abundant monoaminergic system in the CNS when compared with noradrenergic and dopaminergic systems [136, 137]. Serotonin or 5-hydroxytryptamine (5-HT), which is present in CNS, is responsible for pain, feeding, motor functions, sexual functions, emotions, and a sleep-wake cycle [136, 138]. In addition, 5-HT is also involved in cognitive mechanisms such learning and short- and long-term memory, both in the frontal cortex and hippocampus [138]. Reynolds et al. reported the significant loss of 5-HT 4 receptors in hippocampal and cortical neurons of AD patients, which is consistent with cognitive processing [139]. Earlier investigations demonstrated the abundant localization of this receptor in the human caudate nucleus and intermediate density in the hippocampus. A significant decrease in the density of this receptor was seen in the hippocampus of AD patients and suggests an association with this receptor and cholinergic neurons rather than with dopaminergic neurons [139–141]. There are seven types of 5-HT receptors represented by G-protein coupled receptors (5-HTR1, 2, 4–7) and by ligand-gated cation channels (5-HTR3) [136]. In the hippocampus and cortex where learning and memory processes are involved, high levels of 5-HT1A, 5-HT4, 5-HT6, and 5-HT7 receptors were shown [138, 142], but the anatomical distribution of these receptors are different in the cortex and hippocampus on the pre- and postsynaptic membrane [136, 143]. The results from Vilaro et al. [144] suggested localization of 5-HT4 receptors at axons and somato-dendrites. In addition, 5-HT4(b) present in olfactory tubercle, striatum, hippocampus, inferior colliculus, substantia nigra, and parietal cortex with 5-HT4(a) and 5-HT4(e) show a somewhat more restricted distribution. In other regions, such as periaqueductal gray, reticular formation, medial septum, and the diagonal band of Broca had shown 5-HT4(a) + 4(e) and 5-HT4(b) receptors. Stimulation of the 5-HT1A receptor, which is present on the post-synaptic membrane, inhibits cholinergic transmission; interestingly, pre-synaptic 5-HT1A (autoreceptors) exerts a negative feedback on serotonergic transmission [136, 144]. In another study, it was revealed that basal ganglia and hippocampus also have a high density of 5-HT6 receptors [145]. Serotonin receptors co-localize with cholinergic neurons, GABAergic neurons, and glutamatergic neurons, and thus serotonin receptors regulate the above neurotransmitter system [142].
AD has shown a decreased number of serotonergic neurons in the dorsal and the median raphe nuclei [136, 146–150]. These results were further confirmed in postmortem studies on AD-affected brains where there is a reduced density of 5-HT neurons in the raphe nuclei [151]. Abundant Aβ plaques and neurofibrillary tangles in the dorsal and the median raphe nuclei of AD have been associated with a rapid progression of clinical symptoms [148, 152–154]. AD patients with a family history of AD also show raphe pathology for the reduction in the density of 5-HT neurons with the progressive clinical AD pathology [148]. 5-HT neurons are also correlated with age, and there is a greater 5-HT neuronal loss when age advances [149, 155]. It is also true in APPswe/PS1 transgenic mice [156, 157], but not in the triple-transgenic (3xTg-AD) mice.
Based on 5-HT transporters anatomical localization and its function, numerous investigators studied AD therapeutics. 1) Citalopram, selective serotonin reuptake inhibitors (SSRIs) tested on AD patients with behavioral symptoms, showed improved behavioral patterns [158]. 2) In another study where AChEI with monoamine oxidase inhibitor (MAOI) was tested on AD patients with depression, agitation as well as depressive behavior along with episodic memory was improved [159]. 3) Moclobemide, reversible MAOI (400 mg/day), improved depressive behavior in AD patients with depression [160]. 4) AChEI + sertraline, SSRI, (50–200 mg/day), in AD patients with behavioral symptoms showed improved irritability, anxiety, agitation, and affective symptoms, and reduced aggressive behavior [161]. 5) Citalopram, SSRI (5–30 mg/day) tested in AD patients with behavioral symptoms for 36 weeks, showed significant improve in behavioral patterns without sedation [162]. 6) AChEIs + SSRI, (5–50 mg/day) tested on AD patients with depression for 9 months, showed significant preservation of cognitive function along with improved depressive behavior [163].
Currently, various serotonin-mimetic compounds (MAOI+SSRI) are under consideration in AD as monotherapy for treating not only behavioral disturbances and cognitive disturbances but also for reducing pathological changes [136]. The 5-HT1A antagonist lecozotan [164], 5-HT4 agonist, such as PRX-03140, velusetrag, TD-8954, RQ-00000009, SUVN-D1003019, and SUVN-1004028, proved to be safe and also improved cognitive disturbances [165]. In phase II trials, SB-742457 (5-HT6 agonist) with donepezil [166, 167] showed improvement in AD-related behavioral symptoms. In a recent study, a 5-HT6R antagonist, SB271036, rescued memory impairment by attenuating the generation of Aβ through inhibition of γ-secretase activity and the inactivation of astrocytes and microglia in the AD mouse model. In this study, the authors found significant recovery of reduced serotonin levels by SB271036, which was mediated by an indirect regulation of serotonergic neurons via GABA. To support these findings, another serotonin-mimetic compound, fluoxetine, a (SSRI), significantly improved cognitive impairment and behavioral changes [168].
The role of 5-HT7 in AD treatment is still under consideration. However, the clinical use of 5-HT6 ligands is yet to be understood in AD therapeutics.
Histaminergic role in AD therapeutics
Histamine is produced from neurons, mast cells, and circulating basophils by the mediation of Ig-E. Histamine is synthesized from L-histidine by histidine decarboxylase (HDC). The important functions of histamine are mediated through H1, H2, H3, and H4 receptors. All these (G-protein-coupled receptors) GPCRs receptors are mediated while executing their functions. In particular, the H3 receptor plays a pivotal role in the CNS by inhibiting histamine release. The histaminergic system involves cognitive functions along with the sleep-wake cycle, sensory and motor functions, and energy and endocrine homeostasis [169–171]. Histaminergic neurons localize to the hypothalamic tuberomammillary nucleus, and then project to many brain areas, including the basal forebrain, hippocampus, thalamus, cortex, amygdala, basal ganglia, pyramidal cells, raphe nuclei, and substantia nigra. Various studies on the histaminergic system in AD have contradictory results. Few studies reported the elevated histaminergic system in AD [172, 173] with the expected increase in CSF histamine levels in the frontal cortex, basal ganglia, and hippocampus. On the other hand, decreased histamine levels were observed in the hippocampus and both the frontal and the temporal cortex [174, 175] of AD patients.
In a recent study, cognitive deficits were demonstrated along with changes in histaminergic neurotransmission [176, 177]. Higuchi et al. also reported the correlation between H1R binding action and memory loss in AD patients of frontal and temporal regions, where they found decreased expression of H1 receptors [178]. Interestingly, HDC and H1R knockout mice have also shown behavioral and neurochemical alterations [177, 179, 180]. Adult H1R deficient mice also exhibit the severe impairment in learning and memory, thus affecting both spatial and working memory [181, 182], novel objects [183], temporal order memory [184], and the retrieval of formed episodic memories [185]. However, in these H1R deficient mice, there is an increase in ACh concentrations in the frontal cortex and amygdala [183]. Therefore, based on this, researchers investigated H3R antagonists: 1) Haig et al. conducted a randomized study on the efficacy and safety of ABT-288 (H3R antagonist) in mild-to-moderate AD patients and found that ABT-288 had no significant effect on cognitive impairment (assessed using the 13-item Alzheimer’s Disease Assessment Scale-cognitive subscale) when compared to placebo [186]. 2) In another randomized, double-blind, placebo-controlled study on a H3R antagonist (GSK239512), partial beneficial results on cognitive function in mild to moderate AD were found [187]. 3) Previously, another group tested the clinical efficacy of GSK239512 and beneficial effects on measures of attention and memory on a small sample with moderate effect sizes between 0.56 and 1.37 [188]. 4) Some investigators studied H3R modulators for AD behavioral therapy and found an alleviation in disease-related behavioral patterns [189].
H3R antagonists/inverse agonist molecules such as JNJ-10181457, Thioperamide, Clobenpropit, JNJ-5207852, Pitolisant, Spiro fused piperazine amides, ABT-288, GSK189254, SAR110894, and CEP-26401 are able to reverse partial lost cognitive functions in amnesia models of C57BL/6J mice, Wistar rats, Long-Evans rats, CD1/ICR mice, Sprague Dawley rats, CD-1 mice, and lister hooded rats [177]. The first H3R antagonist, i.e., pitolisant, was evaluated in human trials for treating narcolepsy patients [177, 190] and facilitates the consolidation of memory in mice [191, 192]. It is currently in clinical trials and completed phase II trials in schizophrenia patients (NCT00690274). While pitolisant reduces cognitive deficits in schizophrenia, it would be interesting to study these effects in AD patients based on the recent findings of H3R antagonist, i.e., ABT-239, where it reverted the reduced pSer9GSK-3β hippocampal expression in AβPP-overexpressing Tg2576 mice [177, 193, 194]. WakixR is marketed for pitolisant and has been approved by the European Medical Agency in November 2015 for narcolepsy [192].
Recently, Provensi et al. did experiments by combining H3R antagonist (ABT-239) with AChEI (donepezil) in normal and histamine-depleted mice. The effect of the ABT-239 and donepezil was evaluated in the object recognition test and on the levels of GSK-3β phosphorylation. Their results indicate that both donepezil and ABT-239 require the integrity of the brain histaminergic system to exert their procognitive effects [194]. However, all these findings are in the primitive stage, so there is a need to search for H3R antagonists to treat the AD patients in an early stage as the histamine plays a crucial role in oxidative stress and inflammation, and there is also a need to the check the efficacy and tolerability of novel H3R antagonists [195] including pitolisant [191] in AD mouse models and patients.
Adenosine receptor modulation in AD therapeutics
To date, it is known that adenosine is a nucleoside and involved in the production of ATP, but recent research on this nucleoside revealed its role in neurodegeneration through the modulation of inhibitory A1 or facilitatory A2A receptors (A2ARs) for the regulation of synaptic transmission. A1 or A2ARs are located in the synapses of the neocortex and limbic system [196, 197]. A1Rs are involved in the inhibition of glutamate release, NMDA activation, and calcium entry into the neurons, but researchers considered only A2Rs in therapeutics of neurodegenerative diseases because of the rapid downregulation and functional desensitization with neuronal insults of A1Rs [198]. A2ARs can easily show a neuroprotective role against chronic neuronal insults in the adult brain [199, 200], thus increasing the extracellular levels of adenosine [198]. Caffeine is a non-selective A2ARs antagonist and is able to reverse cognitive impairment through a p38 mitogen-activated protein kinase pathway [197] and thus gives neuroprotection in different rodent models [201, 202]. Previously some studies also reported caffeine consumption decreases the incidence of AD [203] and improves memory loss in AD mouse models [204, 205]. In these studies, the protective effects are mediated through A2ARs and prevented the formation of toxic Aβ peptides, thus reducing synaptic toxicity [201].
Recently, Laurent et al. removed the gene encoding for A2AR in THY-Tau22 mice, and then investigated pathological, inflammatory changes, neurotransmitter profile, hippocampal plasticity, and spatial and learning impairments [206]. They found that the deletion of A2ARs protected mice from tau-induced spatial and learning deficits including hippocampal-mediated LTD. The normalization of glutamate/GABA ratio in the hippocampus, reduction in neuroinflammatory markers, and a reduction in tau hyperphosphorylation were also observed. In addition, oral therapy of MSX-3 (A2AR antagonist) significantly improved memory impairments and reduced hyperphosphorylation of tau in THY-Tau22 mice [206]. In another study, Espinosa et al. studied the effect of caffeine on A2ARs in the prevention of memory impairment and neuronal damage in the hippocampus of a rat model of sporadic AD-type dementia. Initially, the authors developed sporadic AD by intracerebroventricular injection of streptozotocin (STZ) [207]. Caffeine consumption, which is an A2ARs antagonist, prevented the STZ-induced memory impairment and neuronal damage [207]. Similarly, Pagnussat et al. studied the effect of caffeine on A2ARs for revoking the cognitive impairments in scopolamine-induced neurodegenerative mice and found the reversal of cognitive deficits with the caffeine [208].
In the clinical trial, NCT01409564, cilostazol augmentation in mild to moderate AD patients with subcortical white matter hyperintensities treated by donepezil was studied. Cilostazol is a cyclic adenosine monophosphate phosphodiesterase 3 inhibitor and was used as an antiplatelet agent in white matter hyperintensities. It also upregulates phosphorylation of cyclic adenosine monophosphate pathway response element binding protein, which plays a crucial role in memory enhancement and synaptic plasticity related to neurodegeneration prevention. This study completed phase IV but an outcome of the study is not available.
All these findings suggested that A2ARs antagonists play a significant role in reversing cognitive impairments in mild AD, but it requires a few meta-analysis and longitudinal studies along with drug trial studies to confirm the reversal of cognitive impairments in sporadic and mild AD.
CONCLUSIONS
All of the above described neurotransmitter(s)-based AD therapeutics were successful in relieving the symptoms of AD. The combination of AChEIs and NMDA-based therapy is successful to some extent, but it is limited to the moderate to severe AD cases. Regarding H3R antagonists, it remains to be determined in clinical trials for histaminergic neurotransmission in AD. Furthermore, refined H3R antagonists are also essentially required to treat AD patients who have other disorders such as epilepsy and schizophrenia. A2ARs antagonists play a significant role in reversing cognitive impairments in mild AD, but additional research is required to prove its efficacy in the early stages of AD.
Acknowledgments
Work presented in this article is supported by the National Institutes of Health grants (AG042178, AG047812) and the Garrison Family Foundation (to P.H.R).
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/16-1118r1).
References
- 1.Kandimalla R, Reddy PH. Multiple faces of dynamin-related protein 1 and its role in Alzheimer’s disease pathogenesis. Biochim Biophys Acta. 2016;1862:814–828. doi: 10.1016/j.bbadis.2015.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kandimalla RJ, Prabhakar S, Binukumar BK, Wani WY, Gupta N, Sharma DR, Sunkaria A, Grover VK, Bhardwaj N, Jain K, Gill KD. Apo-Eε4 allele in conjunction with Aβ42 and tau in CSF: biomarker for Alzheimer’s disease. Curr Alzheimer Res. 2011;8:187–196. doi: 10.2174/156720511795256071. [DOI] [PubMed] [Google Scholar]
- 3.Sharma DR, Wani WY, Sunkaria A, Kandimalla RJ, Verma D, Cameotra SS, Gill KD. Quercetin protects against chronic aluminum-induced oxidative stress and ensuing biochemical, cholinergic, and neurobehavioral impairments in rats. Neurotox Res. 2013;23:336–357. doi: 10.1007/s12640-012-9351-6. [DOI] [PubMed] [Google Scholar]
- 4.Kandimalla RJ, SP, Bk B, Wani WY, Sharma DR, Grover VK, Bhardwaj N, Jain K, Gill KD. Cerebrospinal fluid profile of amyloid β42 (Aβ42), hTau and ubiquitin in North Indian Alzheimer’s disease patients. Neurosci Lett. 2011;487:134–138. doi: 10.1016/j.neulet.2010.06.075. [DOI] [PubMed] [Google Scholar]
- 5.Kandimalla RJ, Wani WY, Binukumar BK, Gill KD. siRNA against presenilin 1 (PS1) down regulates amyloid β42 production in IMR-32 cells. J Biomed Sci. 2012;19:2. doi: 10.1186/1423-0127-19-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lam K, Pan K, Linnekamp JF, Medema JP, Kandimalla R. DNA methylation based biomarkers in colorectal cancer: a systematic review. Biochim Biophys Acta. 2016;1866:106–120. doi: 10.1016/j.bbcan.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 7.Wani WY, Kandimalla RJ, Sharma DR, Kaushal A, Ruban A, Sunkaria A, Vallamkondu J, Chiarugi A, Reddy PH, Gill KD. Cell cycle activation in p21 dependent pathway: An alternative mechanism of organophosphate induced dopaminergic neurodegeneration. Biochim Biophys Acta. 2016 doi: 10.1016/j.bbadis.2016.05.014. [DOI] [PubMed] [Google Scholar]
- 8.Kandimalla R, Vallamkondu J, Corgiat EB, Gill KD. Understanding aspects of aluminum exposure in Alzheimer’s disease development. Brain Pathol. 2016;26:139–154. doi: 10.1111/bpa.12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kandimalla RJ, Anand R, Veeramanikandan R, Wani WY, Prabhakar S, Grover VK, Bharadwaj N, Jain K, Gill KD. CSF ubiquitin as a specific biomarker in Alzheimer’s disease. Curr Alzheimer Res. 2014;11:340–348. doi: 10.2174/1567205011666140331161027. [DOI] [PubMed] [Google Scholar]
- 10.Wang R, Reddy PH. Role of glutamate and NMDA receptors in Alzheimer’s disease. J Alzheimers Dis. 2016 doi: 10.3233/JAD-160763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pugazhenthi S, Qin L, Reddy PH. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim Biophys Acta. 2016 doi: 10.1016/j.bbadis.2016.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alzheimer’s Association. 2016 Alzheimer’s Disease Facts and Figures. 2016 doi: 10.1016/j.jalz.2016.03.001. http://www.alz.org/facts/overview.asp. [DOI] [PubMed]
- 13.Alzheimer’s Disease International. World Alzheimer Report. 2016 https://www.alz.co.uk/research/WorldAlzheimerReport2016.pdf.
- 14.Silva DF, Selfridge JE, Lu J, EL, Roy N, Hutfles L, Burns JM, Michaelis EK, Yan S, Cardoso SM, Swerdlow RH. Bioenergetic flux, mitochondrial mass and mitochondrial morphology dynamics in AD and MCI cybrid cell lines. Hum Mol Genet. 2013;22:3931–3946. doi: 10.1093/hmg/ddt247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu X, Lee HG, Casadesus G, Avila J, Drew K, Perry G, Smith MA. Oxidative imbalance in Alzheimer’s disease. Mol Neurobiol. 2005;31:205–217. doi: 10.1385/MN:31:1-3:205. [DOI] [PubMed] [Google Scholar]
- 16.Moreira PI, Honda K, Liu Q, Santos MS, Oliveira CR, Aliev G, Nunomura A, Zhu X, Smith MA, Perry G. Oxidative stress: the old enemy in Alzheimer’s disease pathophysiology. Curr Alzheimer Res. 2005;2:403–408. doi: 10.2174/156720505774330537. [DOI] [PubMed] [Google Scholar]
- 17.Moreira PI, Siedlak SL, Wang X, Santos MS, Oliveira CR, Tabaton M, Nunomura A, Szweda LI, Aliev G, Smith MA, Zhu X, Perry G. Increased autophagic degradation of mitochondria in Alzheimer disease. Autophagy. 2007;3:614–615. doi: 10.4161/auto.4872. [DOI] [PubMed] [Google Scholar]
- 18.Moreira PI, Santos MS, Oliveira CR, Shenk JC, Nunomura A, Smith MA, Zhu X, Perry G. Alzheimer disease and the role of free radicals in the pathogenesis of the disease. CNS Neurol Disord Drug Targets. 2008;7:3–10. doi: 10.2174/187152708783885156. [DOI] [PubMed] [Google Scholar]
- 19.Moreira PI, Carvalho C, Zhu X, Smith MA, Perry G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim Biophys Acta. 2010;1802:2–10. doi: 10.1016/j.bbadis.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 20.Santos RX, Correia SC, Wang X, Perry G, Smith MA, Moreira PI, Zhu X. Alzheimer’s disease: diverse aspects of mitochondrial malfunctioning. Int J Clin Exp Pathol. 2010;3:570–581. [PMC free article] [PubMed] [Google Scholar]
- 21.Anand R, Gill KD, Mahdi AA. Therapeutics of Alzheimer’s disease: past, present and future. Neuropharmacology. 2014;76(Pt A):27–50. doi: 10.1016/j.neuropharm.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 22.Lodish H, Burke A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J. Molecular Cell Biology. Freeman WH; New York: 2000. [Google Scholar]
- 23.McEnery MW, Siegel RE. Encyclopedia of the Neurological Sciences. 2. Academic Press; Oxford: 2014. Neurotransmitter receptors; pp. 552–564. [Google Scholar]
- 24.Sapolsky R. Biology and Human Behavior: The Neurological Origins of Individuality. McGraw Hill; 2005. [Google Scholar]
- 25.Snyder SH, Innis RB. Peptide neurotransmitters. Annu Rev Biochem. 1979;48:755–782. doi: 10.1146/annurev.bi.48.070179.003543. [DOI] [PubMed] [Google Scholar]
- 26.Whishaw BK, Whishaw IQ. An Introduction to Brain and Behavior. Worth Publishers; New York: 2014. [Google Scholar]
- 27.Tsentsevitsky A, Nurullin L, Nikolsky E, Malomouzh A. Metabotropic and ionotropic glutamate receptors mediate the modulation of acetylcholine release at the frog neuromuscular junction. J Neurosci Res. 2016 doi: 10.1002/jnr.23977. [DOI] [PubMed] [Google Scholar]
- 28.Sheng N, Yang J, Silm K, Edwards RH, Nicoll RA. A slow excitatory postsynaptic current mediated by a novel metabotropic glutamate receptor in CA1 pyramidal neurons. Neuropharmacology. 2016 doi: 10.1016/j.neuropharm.2016.08.028. [DOI] [PubMed] [Google Scholar]
- 29.Corradi J, Bouzat C. Understanding the bases of function and modulation of α7 nicotinic receptors: implications for drug discovery. Mol Pharmacol. 2016;90:288–299. doi: 10.1124/mol.116.104240. [DOI] [PubMed] [Google Scholar]
- 30.Tsamis KI, Mytilinaios DG, Njau SN, Baloyannis SJ. Glutamate receptors in human caudate nucleus in normal aging and Alzheimer’s disease. Curr Alzheimer Res. 2013;10:469–475. doi: 10.2174/1567205011310050002. [DOI] [PubMed] [Google Scholar]
- 31.Birnbaum JH, Bali J, Rajendran L, Nitsch RM, Tackenberg C. Calcium flux-independent NMDA receptor activity is required for Aβ oligomer-induced synaptic loss. Cell Death Dis. 2015;6:e1791. doi: 10.1038/cddis.2015.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee HG, Zhu X, O’Neill MJ, Webber K, Casadesus G, Marlatt M, Raina AK, Perry G, Smith MA. The role of metabotropic glutamate receptors in Alzheimer’s disease. Acta Neurobiol Exp (Wars) 2004;64:89–98. doi: 10.55782/ane-2004-1494. [DOI] [PubMed] [Google Scholar]
- 33.Watkins PB, Zimmerman HJ, Knapp MJ, Gracon SI, Lewis KW. Hepatotoxic effects of tacrine administration in patients with Alzheimer’s disease. JAMA. 1994;271:992–998. [PubMed] [Google Scholar]
- 34.Mesulam MM, Van Hoesen GW. Acetylcholinesterase-rich projections from the basal forebrain of the rhesus monkey to neocortex. Brain Res. 1976;109:152–157. doi: 10.1016/0006-8993(76)90385-1. [DOI] [PubMed] [Google Scholar]
- 35.Squire LR. The organization and neural substrates of human memory. Int J Neurol. 1987;21–22:218–222. [PubMed] [Google Scholar]
- 36.Senut MC, Menetrey D, Lamour Y. Cholinergic and peptidergic projections from the medial septum and the nucleus of the diagonal band of Broca to dorsal hippocampus, cingulate cortex and olfactory bulb: a combined wheatgerm agglutinin-apohorseradish peroxidase-gold immunohistochemical study. Neuroscience. 1989;30:385–403. doi: 10.1016/0306-4522(89)90260-1. [DOI] [PubMed] [Google Scholar]
- 37.Berger-Sweeney J. The cholinergic basal forebrain system during development and its influence on cognitive processes: important questions and potential answers. Neurosci Biobehav Rev. 2003;27:401–411. doi: 10.1016/s0149-7634(03)00070-8. [DOI] [PubMed] [Google Scholar]
- 38.Schliebs R, Arendt T. The significance of the cholinergic system in the brain during aging and in Alzheimer’s disease. J Neural Transm (Vienna) 2006;113:1625–1644. doi: 10.1007/s00702-006-0579-2. [DOI] [PubMed] [Google Scholar]
- 39.Slotkin TA, Seidler FJ, Crain BJ, Bell JM, Bissette G, Nemeroff CB. Regulatory changes in presynaptic cholinergic function assessed in rapid autopsy material from patients with Alzheimer disease: implications for etiology and therapy. Proc Natl Acad Sci U S A. 1990;87:2452–2455. doi: 10.1073/pnas.87.7.2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu Y, Yan J, Zhou P, Li J, Gao H, Xia Y, Wang Q. Neurotransmitter receptors and cognitive dysfunction in Alzheimer’s disease and Parkinson’s disease. Prog Neurobiol. 2012;97:1–13. doi: 10.1016/j.pneurobio.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sberna G, Sáez-Valero J, Beyreuther K, Masters CL, Small DH. The amyloid beta-protein of Alzheimer’s disease increases acetylcholinesterase expression by increasing intracellular calcium in embryonal carcinoma P19 cells. J Neurochem. 1997;69:1177–1184. doi: 10.1046/j.1471-4159.1997.69031177.x. [DOI] [PubMed] [Google Scholar]
- 42.Hucho F, Järv J, Weise C. Substrate-binding sites in acetylcholinesterase. Trends Pharmacol Sci. 1991;12:422–426. doi: 10.1016/0165-6147(91)90621-x. [DOI] [PubMed] [Google Scholar]
- 43.Picciotto MR, Higley MJ, Mineur YS. Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron. 2012;76:116–129. doi: 10.1016/j.neuron.2012.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zolles G, Wagner E, Lampert A, Sutor B. Functional expression of nicotinic acetylcholine receptors in rat neocortical layer 5 pyramidal cells. Cereb Cortex. 2009;19:1079–1091. doi: 10.1093/cercor/bhn158. [DOI] [PubMed] [Google Scholar]
- 45.Köles L, Kató E, Hanuska A, Zádori ZS, Al-Khrasani M, Zelles T, Rubini P, Illes P. Modulation of excitatory neurotransmission by neuronal/glial signalling molecules: interplay between purinergic and glutamatergic systems. Purinergic Signal. 2016;12:1–24. doi: 10.1007/s11302-015-9480-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Do J, Kim JI, Bakes J, Lee K, Kaang BK. Functional roles of neurotransmitters and neuromodulators in the dorsal striatum. Learn Mem. 2012;20:21–28. doi: 10.1101/lm.025015.111. [DOI] [PubMed] [Google Scholar]
- 47.Gal-Ben-Ari S, Rosenblum K. Molecular mechanisms underlying memory consolidation of taste information in the cortex. Front Behav Neurosci. 2011;5:87. doi: 10.3389/fnbeh.2011.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Revett TJ, Baker GB, Jhamandas J, Kar S. Glutamate system, amyloid β peptides and tau protein: functional interrelationships and relevance to Alzheimer disease pathology. J Psychiatry Neurosci. 2013;38:6–23. doi: 10.1503/jpn.110190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beal MF. Mechanisms of excitotoxicity in neurologic diseases. FASEB J. 1992;6:3338–3344. [PubMed] [Google Scholar]
- 50.Novelli A, Reilly JA, Lysko PG, Henneberry RC. Glutamate becomes neurotoxic via the N-methyl-D-aspartate receptor when intracellular energy levels are reduced. Brain Res. 1988;451:205–212. doi: 10.1016/0006-8993(88)90765-2. [DOI] [PubMed] [Google Scholar]
- 51.Alzheimer’s Association. Alzheimer’s disease. 2017 http://www.alz.org/alzheimers_disease_1973.asp.
- 52.Drever BD, Riedel G, Platt B. The cholinergic system and hippocampal plasticity. Behav Brain Res. 2011;221:505–514. doi: 10.1016/j.bbr.2010.11.037. [DOI] [PubMed] [Google Scholar]
- 53.Coyle J, Kershaw P. Galantamine, a cholinesterase inhibitor that allosterically modulates nicotinic receptors: effects on the course of Alzheimer’s disease. Biol Psychiatry. 2001;49:289–299. doi: 10.1016/s0006-3223(00)01101-x. [DOI] [PubMed] [Google Scholar]
- 54.Anisuzzaman AS, Uwada J, Masuoka T, Yoshiki H, Nishio M, Ikegaya Y, Takahashi N, Matsuki N, Fujibayashi Y, Yonekura Y, Momiyama T, Muramatsu I. Novel contribution of cell surface and intracellular M1-muscarinic acetylcholine receptors to synaptic plasticity in hippocampus. J Neurochem. 2013;126:360–371. doi: 10.1111/jnc.12306. [DOI] [PubMed] [Google Scholar]
- 55.Bruel-Jungerman E, Lucassen PJ, Francis F. Cholinergic influences on cortical development and adult neurogenesis. Behav Brain Res. 2011;221:379–388. doi: 10.1016/j.bbr.2011.01.021. [DOI] [PubMed] [Google Scholar]
- 56.Klugman A, Naughton DP, Isaac M, Shah I, Petroczi A, Tabet N. Antioxidant enzymatic activities in Alzheimer’s disease: the relationship to acetylcholinesterase inhibitors. J Alzheimers Dis. 2012;30:467–474. doi: 10.3233/JAD-2012-120124. [DOI] [PubMed] [Google Scholar]
- 57.Li Y, Hai S, Zhou Y, Dong BR. Cholinesterase inhibitors for rarer dementias associated with neurological conditions. Cochrane Database Syst Rev. 2015;3:CD009444. doi: 10.1002/14651858.CD009444.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang J, Yu JT, Wang HF, Meng XF, Wang C, Tan CC, Tan L. Pharmacological treatment of neuropsychiatric symptoms in Alzheimer’s disease: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2015;86:101–109. doi: 10.1136/jnnp-2014-308112. [DOI] [PubMed] [Google Scholar]
- 59.Lefevre G, Sedek G, Jhee SS, Leibowitz MT, Huang HL, Enz A, Maton S, Ereshefsky L, Pommier F, Schmidli H, Appel-Dingemanse S. Pharmacokinetics and pharmacodynamics of the novel daily rivastigmine transdermal patch compared with twice-daily capsules in Alzheimer’s disease patients. Clin Pharmacol Ther. 2008;83:106–114. doi: 10.1038/sj.clpt.6100242. [DOI] [PubMed] [Google Scholar]
- 60.Winblad B, Grossberg G, Frolich L, Farlow M, Zechner S, Nagel J, Lane R. IDEAL: a 6-month, double-blind, placebo-controlled study of the first skin patch for Alzheimer disease. Neurology. 2007;69:S14–22. doi: 10.1212/01.wnl.0000281847.17519.e0. [DOI] [PubMed] [Google Scholar]
- 61.Maelicke A, Hoeffle-Maas A, Ludwig J, Maus A, Samochocki M, Jordis U, Koepke AK. Memogain is a galantamine pro-drug having dramatically reduced adverse effects and enhanced efficacy. J Mol Neurosci. 2010;40:135–137. doi: 10.1007/s12031-009-9269-5. [DOI] [PubMed] [Google Scholar]
- 62.Zhang HY, Yan H, Tang XC. Non-cholinergic effects of huperzine A: beyond inhibition of acetylcholinesterase. Cell Mol Neurobiol. 2008;28:173–183. doi: 10.1007/s10571-007-9163-z. [DOI] [PubMed] [Google Scholar]
- 63.Jia JY, Zhao QH, Liu Y, Gui YZ, Liu GY, Zhu DY, Yu C, Hong Z. Phase I study on the pharmacokinetics and tolerance of ZT-1, a prodrug of huperzine A, for the treatment of Alzheimer’s disease. Acta Pharmacol Sin. 2013;34:976–982. doi: 10.1038/aps.2013.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moss DE, Berlanga P, Hagan MM, Sandoval H, Ishida C. Methanesulfonyl fluoride (MSF): a double-blind, placebo-controlled study of safety and efficacy in the treatment of senile dementia of the Alzheimer type. Alzheimer Dis Assoc Disord. 1999;13:20–25. doi: 10.1097/00002093-199903000-00003. [DOI] [PubMed] [Google Scholar]
- 65.Reid PR, Bridges TM, Sheffler DJ, Cho HP, Lewis LM, Days E, Daniels JS, Jones CK, Niswender CM, Weaver CD, Conn PJ, Lindsley CW, Wood MR. Discovery and optimization of a novel, selective and brain penetrant M1 positive allosteric modulator (PAM): the development of ML169, an MLPCN probe. Bioorg Med Chem Lett. 2011;21:2697–2701. doi: 10.1016/j.bmcl.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Medeiros R, Kitazawa M, Caccamo A, Baglietto-Vargas D, Estrada-Hernandez T, Cribbs DH, Fisher A, LaFerla FM. Loss of muscarinic M1 receptor exacerbates Alzheimer’s disease-like pathology and cognitive decline. Am J Pathol. 2011;179:980–991. doi: 10.1016/j.ajpath.2011.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nitsch RM, Deng M, Tennis M, Schoenfeld D, Growdon JH. The selective muscarinic M1 agonist AF102B decreases levels of total Abeta in cerebrospinal fluid of patients with Alzheimer’s disease. Ann Neurol. 2000;48:913–918. [PubMed] [Google Scholar]
- 68.Fisher A. Cholinergic treatments with emphasis on m1 muscarinic agonists as potential disease-modifying agents for Alzheimer’s disease. Neurotherapeutics. 2008;5:433–442. doi: 10.1016/j.nurt.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fisher A. Cholinergic modulation of amyloid precursor protein processing with emphasis on M1 muscarinic receptor: perspectives and challenges in treatment of Alzheimer’s disease. J Neurochem 120 Suppl. 2012;1:22–33. doi: 10.1111/j.1471-4159.2011.07507.x. [DOI] [PubMed] [Google Scholar]
- 70.Fisher A, Brandeis R, Bar-Ner RH, Kliger-Spatz M, Natan N, Sonego H, Marcovitch I, Pittel Z. AF150(S) and AF267B: M1 muscarinic agonists as innovative therapies for Alzheimer’s disease. J Mol Neurosci. 2002;19:145–153. doi: 10.1007/s12031-002-0025-3. [DOI] [PubMed] [Google Scholar]
- 71.Collina S, Gaggeri R, Marra A, Bassi A, Negrinotti S, Negri F, Rossi D. Sigma receptor modulators: a patent review. Expert Opin Ther Pat. 2013;23:597–613. doi: 10.1517/13543776.2013.769522. [DOI] [PubMed] [Google Scholar]
- 72.Lahmy V, Meunier J, Malmström S, Naert G, Givalois L, Kim SH, Villard V, Vamvakides A, Maurice T. Blockade of Tau hyperphosphorylation and Aβ1–42 generation by the aminotetrahydrofuran derivative ANAVEX2-73, a mixed muscarinic and σ1 receptor agonist, in a nontransgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology. 2013;38:1706–1723. doi: 10.1038/npp.2013.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hernandez CM, Dineley KT. α7 nicotinic acetylcholine receptors in Alzheimer’s disease: neuroprotective, neurotrophic or both? Curr Drug Targets. 2012;13:613–622. doi: 10.2174/138945012800398973. [DOI] [PubMed] [Google Scholar]
- 74.Parri RH, Dineley TK. Nicotinic acetylcholine receptor interaction with beta-amyloid: molecular, cellular, and physiological consequences. Curr Alzheimer Res. 2010;7:27–39. doi: 10.2174/156720510790274464. [DOI] [PubMed] [Google Scholar]
- 75.Echeverria V, Zeitlin R. Cotinine: a potential new therapeutic agent against Alzheimer’s disease. CNS Neurosci Ther. 2012;18:517–523. doi: 10.1111/j.1755-5949.2012.00317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Toyohara J, Hashimoto K. α7 nicotinic receptor agonists: potential therapeutic drugs for treatment of cognitive impairments in schizophrenia and Alzheimer’s disease. Open Med Chem J. 2010;4:37–56. doi: 10.2174/1874104501004010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Prickaerts J, van Goethem NP, Chesworth R, Shapiro G, Boess FG, Methfessel C, Reneerkens OA, Flood DG, Hilt D, Gawryl M, Bertrand S, Bertrand D, König G. EVP-6124, a novel and selective α7 nicotinic acetylcholine receptor partial agonist, improves memory performance by potentiating the acetylcholine response of α7 nicotinic acetylcholine receptors. Neuropharmacology. 2012;62:1099–1110. doi: 10.1016/j.neuropharm.2011.10.024. [DOI] [PubMed] [Google Scholar]
- 78.Butterfield DA, Pocernich CB. The glutamatergic system and Alzheimer’s disease: therapeutic implications. CNS Drugs. 2003;17:641–652. doi: 10.2165/00023210-200317090-00004. [DOI] [PubMed] [Google Scholar]
- 79.Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 80.Dinamarca MC, Ríos JA, Inestrosa NC. Postsynaptic receptors for Amyloid-β oligomers as mediators of neuronal damage in Alzheimer’s disease. Front Physiol. 2012;3:464. doi: 10.3389/fphys.2012.00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gilling KE, Jatzke C, Hechenberger M, Parsons CG. Potency, voltage-dependency, agonist concentration-dependency, blocking kinetics and partial untrapping of the uncompetitive N-methyl-D-aspartate (NMDA) channel blocker memantine at human NMDA (GluN1/GluN2A) receptors. Neuropharmacology. 2009;56:866–875. doi: 10.1016/j.neuropharm.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 82.Rogawski MA. Therapeutic potential of excitatory amino acid antagonists: channel blockers and 2,3-benzodiazepines. Trends Pharmacol Sci. 1993;14:325–331. doi: 10.1016/0165-6147(93)90005-5. [DOI] [PubMed] [Google Scholar]
- 83.Parsons CG, Gruner R, Rozental J, Millar J, Lodge D. Patch clamp studies on the kinetics and selectivity of N-methyl-D-aspartate receptor antagonism by memantine (1-amino-3,5-dimethyladamantan) Neuropharmacology. 1993;32:1337–1350. doi: 10.1016/0028-3908(93)90029-3. [DOI] [PubMed] [Google Scholar]
- 84.Parsons CG, Danysz W, Quack G. Memantine is a clinically well tolerated N-methyl-D-aspartate (NMDA) receptor antagonist--a review of preclinical data. Neuropharmacology. 1999;38:735–767. doi: 10.1016/s0028-3908(99)00019-2. [DOI] [PubMed] [Google Scholar]
- 85.Johnson JW, Kotermanski SE. Mechanism of action of memantine. Curr Opin Pharmacol. 2006;6:61–67. doi: 10.1016/j.coph.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 86.Parsons CG, Quack G, Bresink I, Baran L, Przegalinski E, Kostowski W, Krzascik P, Hartmann S, Danysz W. Comparison of the potency, kinetics and voltage-dependency of a series of uncompetitive NMDA receptor antagonists in vitro with anticonvulsive and motor impairment activity in vivo. Neuropharmacology. 1995;34:1239–1258. doi: 10.1016/0028-3908(95)00092-k. [DOI] [PubMed] [Google Scholar]
- 87.Parsons CG, Panchenko VA, Pinchenko VO, Tsyndrenko AY, Krishtal OA. Comparative patch-clamp studies with freshly dissociated rat hippocampal and striatal neurons on the NMDA receptor antagonistic effects of amantadine and memantine. Eur J Neurosci. 1996;8:446–454. doi: 10.1111/j.1460-9568.1996.tb01228.x. [DOI] [PubMed] [Google Scholar]
- 88.Parsons CG, Hartmann S, Spielmanns P. Budipine is a low affinity, N-methyl-D-aspartate receptor antagonist: patch clamp studies in cultured striatal, hippocampal, cortical and superior colliculus neurones. Neuropharmacology. 1998;37:719–727. doi: 10.1016/s0028-3908(98)00059-8. [DOI] [PubMed] [Google Scholar]
- 89.Bresink I, Benke TA, Collett VJ, Seal AJ, Parsons CG, Henley JM, Collingridge GL. Effects of memantine on recombinant rat NMDA receptors expressed in HEK 293 cells. Br J Pharmacol. 1996;119:195–204. doi: 10.1111/j.1476-5381.1996.tb15971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen HS, Lipton SA. Mechanism of memantine block of NMDA-activated channels in rat retinal ganglion cells: uncompetitive antagonism. J Physiol. 1997;499(Pt 1):27–46. doi: 10.1113/jphysiol.1997.sp021909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Blanpied TA, Boeckman FA, Aizenman E, Johnson JW. Trapping channel block of NMDA-activated responses by amantadine and memantine. J Neurophysiol. 1997;77:309–323. doi: 10.1152/jn.1997.77.1.309. [DOI] [PubMed] [Google Scholar]
- 92.Sobolevsky A, Koshelev S. Two blocking sites of amino-adamantane derivatives in open N-methyl-D-aspartate channels. Biophys J. 1998;74:1305–1319. doi: 10.1016/S0006-3495(98)77844-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sobolevsky AI, Beck C, Wollmuth LP. Molecular rearrangements of the extracellular vestibule in NMDAR channels during gating. Neuron. 2002;33:75–85. doi: 10.1016/s0896-6273(01)00560-8. [DOI] [PubMed] [Google Scholar]
- 94.Losi G, Lanza M, Makovec F, Artusi R, Caselli G, Puia G. Functional in vitro characterization of CR 3394: a novel voltage dependent N-methyl-D-aspartate (NMDA) receptor antagonist. Neuropharmacology. 2006;50:277–285. doi: 10.1016/j.neuropharm.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 95.Chen HS, Pellegrini JW, Aggarwal SK, Lei SZ, Warach S, Jensen FE, Lipton SA. Open-channel block of N-methyl-D-aspartate (NMDA) responses by memantine: therapeutic advantage against NMDA receptor-mediated neurotoxicity. J Neurosci. 1992;12:4427–4436. doi: 10.1523/JNEUROSCI.12-11-04427.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Parsons CG, Danysz W, Dekundy A, Pulte I. Memantine and cholinesterase inhibitors: complementary mechanisms in the treatment of Alzheimer’s disease. Neurotox Res. 2013;24:358–369. doi: 10.1007/s12640-013-9398-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Parsons CG, Stöffler A, Danysz W. Memantine: a NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system--too little activation is bad, too much is even worse. Neuropharmacology. 2007;53:699–723. doi: 10.1016/j.neuropharm.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 98.Frankiewicz T, Parsons CG. Memantine restores long term potentiation impaired by tonic N-methyl-D-aspartate (NMDA) receptor activation following reduction of Mg2+ in hippocampal slices. Neuropharmacology. 1999;38:1253–1259. doi: 10.1016/s0028-3908(99)00060-x. [DOI] [PubMed] [Google Scholar]
- 99.Danysz W, Parsons CG. The NMDA receptor antagonist memantine as a symptomatological and neuroprotective treatment for Alzheimer’s disease: preclinical evidence. Int J Geriatr Psychiatry. 2003;18(Suppl 1):S23–32. doi: 10.1002/gps.938. [DOI] [PubMed] [Google Scholar]
- 100.Zajaczkowski W, Frankiewicz T, Parsons CG, Danysz W. Uncompetitive NMDA receptor antagonists attenuate NMDA-induced impairment of passive avoidance learning and LTP. Neuropharmacology. 1997;36:961–971. doi: 10.1016/s0028-3908(97)00070-1. [DOI] [PubMed] [Google Scholar]
- 101.Miguel-Hidalgo JJ, Alvarez XA, Cacabelos R, Quack G. Neuroprotection by memantine against neurodegeneration induced by beta-amyloid(1–40) Brain Res. 2002;958:210–221. doi: 10.1016/s0006-8993(02)03731-9. [DOI] [PubMed] [Google Scholar]
- 102.Buisson B, Bertrand D. Open-channel blockers at the human alpha4beta2 neuronal nicotinic acetylcholine receptor. Mol Pharmacol. 1998;53:555–563. doi: 10.1124/mol.53.3.555. [DOI] [PubMed] [Google Scholar]
- 103.Maskell PD, Speder P, Newberry NR, Bermudez I. Inhibition of human alpha 7 nicotinic acetylcholine receptors by open channel blockers of N-methyl-D-aspartate receptors. Br J Pharmacol. 2003;140:1313–1319. doi: 10.1038/sj.bjp.0705559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rammes G, Rupprecht R, Ferrari U, Zieglgänsberger W, Parsons CG. The N-methyl-D-aspartate receptor channel blockers memantine, MRZ 2/579 and other amino-alkyl-cyclohexanes antagonise 5-HT(3) receptor currents in cultured HEK-293 and N1E-115 cell systems in a non-competitive manner. Neurosci Lett. 2001;306:81–84. doi: 10.1016/s0304-3940(01)01872-9. [DOI] [PubMed] [Google Scholar]
- 105.Lee RH, Tseng TY, Wu CY, Chen PY, Chen MF, Kuo JS, Lee TJ. Memantine inhibits α3β2-nAChRs-mediated nitrergic neurogenic vasodilation in porcine basilar arteries. PLoS One. 2012;7:e40326. doi: 10.1371/journal.pone.0040326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Motawaj M, Burban A, Davenas E, Arrang JM. Activation of brain histaminergic neurotransmission: a mechanism for cognitive effects of memantine in Alzheimer’s disease. J Pharmacol Exp Ther. 2011;336:479–487. doi: 10.1124/jpet.110.174458. [DOI] [PubMed] [Google Scholar]
- 107.Nakaya K, Nakagawasai O, Arai Y, Onogi H, Sato A, Niijima F, Tan-No K, Tadano T. Pharmacological characterizations of memantine-induced disruption of prepulse inhibition of the acoustic startle response in mice: involvement of dopamine D2 and 5-HT2A receptors. Behav Brain Res. 2011;218:165–173. doi: 10.1016/j.bbr.2010.11.053. [DOI] [PubMed] [Google Scholar]
- 108.Rammes G, Danysz W, Parsons CG. Pharmacodynamics of memantine: an update. Curr Neuropharmacol. 2008;6:55–78. doi: 10.2174/157015908783769671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hellweg R, Wirth Y, Janetzky W, Hartmann S. Efficacy of memantine in delaying clinical worsening in Alzheimer’s disease (AD): responder analyses of nine clinical trials with patients with moderate to severe AD. Int J Geriatr Psychiatry. 2012;27:651–656. doi: 10.1002/gps.2766. [DOI] [PubMed] [Google Scholar]
- 110.McShane R, Areosa Sastre A, Minakaran N. Memantine for dementia. Cochrane Database Syst Rev. 2006:CD003154. doi: 10.1002/14651858.CD003154.pub5. [DOI] [PubMed] [Google Scholar]
- 111.Bakchine S, Loft H. Memantine treatment in patients with mild to moderate Alzheimer’s disease: results of a randomised, double-blind, placebo-controlled 6-month study. J Alzheimers Dis. 2008;13:97–107. doi: 10.3233/jad-2008-13110. [DOI] [PubMed] [Google Scholar]
- 112.Peskind ER, Potkin SG, Pomara N, Ott BR, Graham SM, Olin JT, McDonald S. Memantine treatment in mild to moderate Alzheimer disease: a 24-week randomized, controlled trial. Am J Geriatr Psychiatry. 2006;14:704–715. doi: 10.1097/01.JGP.0000224350.82719.83. [DOI] [PubMed] [Google Scholar]
- 113.Porsteinsson AP, Grossberg GT, Mintzer J, Olin JT Memantine MEM-MD-12 Study Group. Memantine treatment in patients with mild to moderate Alzheimer’s disease already receiving a cholinesterase inhibitor: a randomized, double-blind, placebo-controlled trial. Curr Alzheimer Res. 2008;5:83–89. doi: 10.2174/156720508783884576. [DOI] [PubMed] [Google Scholar]
- 114.Schneider LS, Dagerman KS, Higgins JP, McShane R. Lack of evidence for the efficacy of memantine in mild Alzheimer disease. Arch Neurol. 2011;68:991–998. doi: 10.1001/archneurol.2011.69. [DOI] [PubMed] [Google Scholar]
- 115.Ni R, Marutle A, Nordberg A. Modulation of α7 nicotinic acetylcholine receptor and fibrillar amyloid-β interactions in Alzheimer’s disease brain. J Alzheimers Dis. 2013;33:841–851. doi: 10.3233/JAD-2012-121447. [DOI] [PubMed] [Google Scholar]
- 116.Aracava Y, Pereira EF, Maelicke A, Albuquerque EX. Memantine blocks α7* nicotinic acetylcholine receptors more potently than n-methyl-D-aspartate receptors in rat hippocampal neurons. J Pharmacol Exp Ther. 2005;312:1195–1205. doi: 10.1124/jpet.104.077172. [DOI] [PubMed] [Google Scholar]
- 117.Molino I, Colucci L, Fasanaro AM, Traini E, Amenta F. Efficacy of memantine, donepezil, or their association in moderate-severe Alzheimer’s disease: a review of clinical trials. ScientificWorldJournal. 2013;2013:925702. doi: 10.1155/2013/925702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Atri A, Molinuevo JL, Lemming O, Wirth Y, Pulte I, Wilkinson D. Memantine in patients with Alzheimer’s disease receiving donepezil: new analyses of efficacy and safety for combination therapy. Alzheimers Res Ther. 2013;5:6. doi: 10.1186/alzrt160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dantoine T, Auriacombe S, Sarazin M, Becker H, Pere JJ, Bourdeix I. Rivastigmine monotherapy and combination therapy with memantine in patients with moderately severe Alzheimer’s disease who failed to benefit from previous cholinesterase inhibitor treatment. Int J Clin Pract. 2006;60:110–118. doi: 10.1111/j.1368-5031.2005.00769.x. [DOI] [PubMed] [Google Scholar]
- 120.Howard R, McShane R, Lindesay J, Ritchie C, Baldwin A, Barber R, Burns A, Dening T, Findlay D, Holmes C, Hughes A, Jacoby R, Jones R, Jones R, McKeith I, Macharouthu A, O’Brien J, Passmore P, Sheehan B, Juszczak E, Katona C, Hills R, Knapp M, Ballard C, Brown R, Banerjee S, Onions C, Griffin M, Adams J, Gray R, Johnson T, Bentham P, Phillips P. Donepezil and memantine for moderate-to-severe Alzheimer’s disease. N Engl J Med. 2012;366:893–903. doi: 10.1056/NEJMoa1106668. [DOI] [PubMed] [Google Scholar]
- 121.Lopez OL, Becker JT, Wahed AS, Saxton J, Sweet RA, Wolk DA, Klunk W, Dekosky ST. Long-term effects of the concomitant use of memantine with cholinesterase inhibition in Alzheimer disease. J Neurol Neurosurg Psychiatry. 2009;80:600–607. doi: 10.1136/jnnp.2008.158964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Riepe MW, Adler G, Ibach B, Weinkauf B, Tracik F, Gunay I. Domain-specific improvement of cognition on memantine in patients with Alzheimer’s disease treated with rivastigmine. Dement Geriatr Cogn Disord. 2007;23:301–306. doi: 10.1159/000100875. [DOI] [PubMed] [Google Scholar]
- 123.Tariot PN, Farlow MR, Grossberg GT, Graham SM, McDonald S, Gergel I Memantine Study Group. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291:317–324. doi: 10.1001/jama.291.3.317. [DOI] [PubMed] [Google Scholar]
- 124.Rissman RA, De Blas AL, Armstrong DM. GABA(A) receptors in aging and Alzheimer’s disease. J Neurochem. 2007;103:1285–1292. doi: 10.1111/j.1471-4159.2007.04832.x. [DOI] [PubMed] [Google Scholar]
- 125.Mizukami K, Ikonomovic MD, Grayson DR, Sheffield R, Armstrong DM. Immunohistochemical study of GABAA receptor alpha1 subunit in the hippocampal formation of aged brains with Alzheimer-related neuropathologic changes. Brain Res. 1998;799:148–155. doi: 10.1016/s0006-8993(98)00437-5. [DOI] [PubMed] [Google Scholar]
- 126.Mizukami K, Ikonomovic MD, Grayson DR, Rubin RT, Warde D, Sheffield R, Hamilton RL, Davies P, Armstrong DM. Immunohistochemical study of GABA(A) receptor beta2/3 subunits in the hippocampal formation of aged brains with Alzheimer-related neuropathologic changes. Exp Neurol. 1997;147:333–345. doi: 10.1006/exnr.1997.6591. [DOI] [PubMed] [Google Scholar]
- 127.Ulrich D. Amyloid-β impairs synaptic inhibition via GABA(A) receptor endocytosis. J Neurosci. 2015;35:9205–9210. doi: 10.1523/JNEUROSCI.0950-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lagostena L, Rosato-Siri M, D’Onofrio M, Brandi R, Arisi I, Capsoni S, Franzot J, Cattaneo A, Cherubini E. In the adult hippocampus, chronic nerve growth factor deprivation shifts GABAergic signaling from the hyperpolarizing to the depolarizing direction. J Neurosci. 2010;30:885–893. doi: 10.1523/JNEUROSCI.3326-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Marcade M, Bourdin J, Loiseau N, Peillon H, Rayer A, Drouin D, Schweighoffer F, Désiré L. Etazolate, a neuroprotective drug linking GABA(A) receptor pharmacology to amyloid precursor protein processing. J Neurochem. 2008;106:392–404. doi: 10.1111/j.1471-4159.2008.05396.x. [DOI] [PubMed] [Google Scholar]
- 130.Jo S, Yarishkin O, Hwang YJ, Chun YE, Park M, Woo DH, Bae JY, Kim T, Lee J, Chun H, Park HJ, Lee DY, Hong J, Kim HY, Oh SJ, Park SJ, Lee H, Yoon BE, Kim Y, Jeong Y, Shim I, Bae YC, Cho J, Kowall NW, Ryu H, Hwang E, Kim D, Lee CJ. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat Med. 2014;20:886–896. doi: 10.1038/nm.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Muñoz MD, Antolin-Vallespin M, Tapia-González S, Sánchez-Capelo A. Smad3 deficiency inhibits dentate gyrus LTP by enhancing GABAA neurotransmission. J Neurochem. 2016;137:190–199. doi: 10.1111/jnc.13558. [DOI] [PubMed] [Google Scholar]
- 132.Townsend M, Whyment A, Walczak JS, Jeggo R, van den Top M, Flood DG, Leventhal L, Patzke H, Koenig G. α7 nAChR agonist enhances neural plasticity in the hippocampus via a GABAergic circuit. J Neurophysiol. 2016;116:2663–2675. doi: 10.1152/jn.00243.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nikolaus S, Hautzel H, Müller HW. Focus on GABA(A) receptor function. A comparative analysis of in vivo imaging studies in neuropsychiatric disorders. Nuklearmedizin. 2014;53:227–237. doi: 10.3413/Nukmed-0647-14-03. [DOI] [PubMed] [Google Scholar]
- 134.Vellas B, Sol O, Snyder PJ, Ousset PJ, Haddad R, Maurin M, Lemarié JC, Désiré L, Pando MP EHT0202/002 study group. EHT0202 in Alzheimer’s disease: a 3-month, randomized, placebo-controlled, double-blind study. Curr Alzheimer Res. 2011;8:203–212. doi: 10.2174/156720511795256053. [DOI] [PubMed] [Google Scholar]
- 135.Atack JR. Preclinical and clinical pharmacology of the GABAA receptor alpha5 subtype-selective inverse agonist alpha5IA. Pharmacol Ther. 2010;125:11–26. doi: 10.1016/j.pharmthera.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 136.Rodriguez JJ, Noristani HN, Verkhratsky A. The serotonergic system in ageing and Alzheimer’s disease. Prog Neurobiol. 2012;99:15–41. doi: 10.1016/j.pneurobio.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 137.Sirviö J, Riekkinen P, Jr, Jäkälä P, Riekkinen PJ. Experimental studies on the role of serotonin in cognition. Prog Neurobiol. 1994;43:363–379. doi: 10.1016/0301-0082(94)90060-4. [DOI] [PubMed] [Google Scholar]
- 138.Cifariello A, Pompili A, Gasbarri A. 5-HT(7) receptors in the modulation of cognitive processes. Behav Brain Res. 2008;195:171–179. doi: 10.1016/j.bbr.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 139.Reynolds GP, Mason SL, Meldrum A, De Keczer S, Parnes H, Eglen RM, Wong EH. 5-Hydroxytryptamine (5-HT)4 receptors in post mortem human brain tissue: distribution, pharmacology and effects of neurodegenerative diseases. Br J Pharmacol. 1995;114:993–998. doi: 10.1111/j.1476-5381.1995.tb13303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Waeber C, Sebben M, Grossman C, Javoy-Agid F, Bockaert J, Dumuis A. [3H]-GR113808 labels 5-HT4 receptors in the human and guinea-pig brain. Neuroreport. 1993;4:1239–1242. doi: 10.1097/00001756-199309000-00007. [DOI] [PubMed] [Google Scholar]
- 141.Doménech T, Beleta J, Fernández AG, Gristwood RW, Cruz Sánchez F, Tolosa E, Palacios JM. Identification and characterization of serotonin 5-HT4 receptor binding sites in human brain: comparison with other mammalian species. Brain Res Mol Brain Res. 1994;21:176–180. doi: 10.1016/0169-328x(94)90392-1. [DOI] [PubMed] [Google Scholar]
- 142.King MV, Marsden CA, Fone KC. A role for the 5-HT(1A), 5-HT4 and 5-HT6 receptors in learning and memory. Trends Pharmacol Sci. 2008;29:482–492. doi: 10.1016/j.tips.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 143.Chalmers DT, Watson SJ. Comparative anatomical distribution of 5-HT1A receptor mRNA and 5-HT1A binding in rat brain--a combined in situ hybridisation/in vitro receptor autoradiographic study. Brain Res. 1991;561:51–60. doi: 10.1016/0006-8993(91)90748-k. [DOI] [PubMed] [Google Scholar]
- 144.Vilaró MT, Cortés R, Mengod G. Serotonin 5-HT4 receptors and their mRNAs in rat and guinea pig brain: distribution and effects of neurotoxic lesions. J Comp Neurol. 2005;484:418–439. doi: 10.1002/cne.20447. [DOI] [PubMed] [Google Scholar]
- 145.Marazziti D, Baroni S, Pirone A, Giannaccini G, Betti L, Schmid L, Vatteroni E, Palego L, Borsini F, Bordi F, Piano I, Gargini C, Castagna M, Catena-Dell’osso M, Lucacchini A. Distribution of serotonin receptor of type 6 (5-HT6) in human brain post-mortem. A pharmacology, autoradiography and immunohistochemistry study. Neurochem Res. 2012;37:920–927. doi: 10.1007/s11064-011-0684-y. [DOI] [PubMed] [Google Scholar]
- 146.Aletrino MA, Vogels OJ, Van Domburg PH, Ten Donkelaar HJ. Cell loss in the nucleus raphes dorsalis in Alzheimer’s disease. Neurobiol Aging. 1992;13:461–468. doi: 10.1016/0197-4580(92)90073-7. [DOI] [PubMed] [Google Scholar]
- 147.Chen CP, Eastwood SL, Hope T, McDonald B, Francis PT, Esiri MM. Immunocytochemical study of the dorsal and median raphe nuclei in patients with Alzheimer’s disease prospectively assessed for behavioural changes. Neuropathol Appl Neurobiol. 2000;26:347–355. doi: 10.1046/j.1365-2990.2000.00254.x. [DOI] [PubMed] [Google Scholar]
- 148.Halliday GM, McCann HL, Pamphlett R, Brooks WS, Creasey H, McCusker E, Cotton RG, Broe GA, Harper CG. Brain stem serotonin-synthesizing neurons in Alzheimer’s disease: a clinicopathological correlation. Acta Neuropathol. 1992;84:638–650. doi: 10.1007/BF00227741. [DOI] [PubMed] [Google Scholar]
- 149.Kovacs GG, Klöppel S, Fischer I, Dorner S, Lindeck-Pozza E, Birner P, Bötefür IC, Pilz P, Volk B, Budka H. Nucleus-specific alteration of raphe neurons in human neurodegenerative disorders. Neuroreport. 2003;14:73–76. doi: 10.1097/00001756-200301200-00014. [DOI] [PubMed] [Google Scholar]
- 150.Yamamoto T, Hirano A. Nucleus raphe dorsalis in Alzheimer’s disease: neurofibrillary tangles and loss of large neurons. Ann Neurol. 1985;17:573–577. doi: 10.1002/ana.410170608. [DOI] [PubMed] [Google Scholar]
- 151.Lyness SA, Zarow C, Chui HC. Neuron loss in key cholinergic and aminergic nuclei in Alzheimer disease: a meta-analysis. Neurobiol Aging. 2003;24:1–23. doi: 10.1016/s0197-4580(02)00057-x. [DOI] [PubMed] [Google Scholar]
- 152.Curcio CA, Kemper T. Nucleus raphe dorsalis in dementia of the Alzheimer type: neurofibrillary changes and neuronal packing density. J Neuropathol Exp Neurol. 1984;43:359–368. doi: 10.1097/00005072-198407000-00001. [DOI] [PubMed] [Google Scholar]
- 153.Ebinger G, Bruyland M, Martin JJ, Herregodts P, Cras P, Michotte Y, Gommé L. Distribution of biogenic amines and their catabolites in brains from patients with Alzheimer’s disease. J Neurol Sci. 1987;77:267–283. doi: 10.1016/0022-510x(87)90128-6. [DOI] [PubMed] [Google Scholar]
- 154.German DC, White CL, 3rd, Sparkman DR. Alzheimer’s disease: neurofibrillary tangles in nuclei that project to the cerebral cortex. Neuroscience. 1987;21:305–312. doi: 10.1016/0306-4522(87)90123-0. [DOI] [PubMed] [Google Scholar]
- 155.Hendricksen M, Thomas AJ, Ferrier IN, Ince P, O’Brien JT. Neuropathological study of the dorsal raphe nuclei in late-life depression and Alzheimer’s disease with and without depression. Am J Psychiatry. 2004;161:1096–1102. doi: 10.1176/appi.ajp.161.6.1096. [DOI] [PubMed] [Google Scholar]
- 156.Bernedo V, Insua D, Suárez ML, Santamarina G, Sarasa M, Pesini P. Beta-amyloid cortical deposits are accompanied by the loss of serotonergic neurons in the dog. J Comp Neurol. 2009;513:417–429. doi: 10.1002/cne.21985. [DOI] [PubMed] [Google Scholar]
- 157.Liu Y, Yoo MJ, Savonenko A, Stirling W, Price DL, Borchelt DR, Mamounas L, Lyons WE, Blue ME, Lee MK. Amyloid pathology is associated with progressive monoaminergic neurodegeneration in a transgenic mouse model of Alzheimer’s disease. J Neurosci. 2008;28:13805–13814. doi: 10.1523/JNEUROSCI.4218-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Nyth AL, Gottfries CG. The clinical efficacy of citalopram in treatment of emotional disturbances in dementia disorders. A Nordic multicentre study. Br J Psychiatry. 1990;157:894–901. doi: 10.1192/bjp.157.6.894. [DOI] [PubMed] [Google Scholar]
- 159.Schechter LE, Smith DL, Rosenzweig-Lipson S, Sukoff SJ, Dawson LA, Marquis K, Jones D, Piesla M, Andree T, Nawoschik S, Harder JA, Womack MD, Buccafusco J, Terry AV, Hoebel B, Rada P, Kelly M, Abou-Gharbia M, Barrett JE, Childers W. Lecozotan (SRA-333): a selective serotonin 1A receptor antagonist that enhances the stimulated release of glutamate and acetylcholine in the hippocampus and possesses cognitive-enhancing properties. J Pharmacol Exp Ther. 2005;314:1274–1289. doi: 10.1124/jpet.105.086363. [DOI] [PubMed] [Google Scholar]
- 160.Roth M, Mountjoy CQ, Amrein R. Moclobemide in elderly patients with cognitive decline and depression: an international double-blind, placebo-controlled trial. Br J Psychiatry. 1996;168:149–157. doi: 10.1192/bjp.168.2.149. [DOI] [PubMed] [Google Scholar]
- 161.Finkel SI, Mintzer JE, Dysken M, Krishnan KR, Burt T, McRae T. A randomized, placebo-controlled study of the efficacy and safety of sertraline in the treatment of the behavioral manifestations of Alzheimer’s disease in outpatients treated with donepezil. Int J Geriatr Psychiatry. 2004;19:9–18. doi: 10.1002/gps.998. [DOI] [PubMed] [Google Scholar]
- 162.Siddique H, Hynan LS, Weiner MF. Effect of a serotonin reuptake inhibitor on irritability, apathy, and psychotic symptoms in patients with Alzheimer’s disease. J Clin Psychiatry. 2009;70:915–918. doi: 10.4088/JCP.08m04828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Rozzini L, Chilovi BV, Conti M, Bertoletti E, Zanetti M, Trabucchi M, Padovani A. Efficacy of SSRIs on cognition of Alzheimer’s disease patients treated with cholinesterase inhibitors. Int Psychogeriatr. 2010;22:114–119. doi: 10.1017/S1041610209990184. [DOI] [PubMed] [Google Scholar]
- 164.Patat A, Parks V, Raje S, Plotka A, Chassard D, Le Coz F. Safety, tolerability, pharmacokinetics and pharmacodynamics of ascending single and multiple doses of lecozotan in healthy young and elderly subjects. Br J Clin Pharmacol. 2009;67:299–308. doi: 10.1111/j.1365-2125.2008.03348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Shen F, Smith JA, Chang R, Bourdet DL, Tsuruda PR, Obedencio GP, Beattie DT. 5-HT(4) receptor agonist mediated enhancement of cognitive function in vivo and amyloid precursor protein processing in vitro: A pharmacodynamic and pharmacokinetic assessment. Neuropharmacology. 2011;61:69–79. doi: 10.1016/j.neuropharm.2011.02.026. [DOI] [PubMed] [Google Scholar]
- 166.Maher-Edwards G, Zvartau-Hind M, Hunter AJ, Gold M, Hopton G, Jacobs G, Davy M, Williams P. Double-blind, controlled phase II study of a 5-HT6 receptor antagonist, SB-742457, in Alzheimer’s disease. Curr Alzheimer Res. 2010;7:374–385. doi: 10.2174/156720510791383831. [DOI] [PubMed] [Google Scholar]
- 167.Maher-Edwards G, Dixon R, Hunter J, Gold M, Hopton G, Jacobs G, Hunter J, Williams P. SB-742457 and donepezil in Alzheimer disease: a randomized, placebo-controlled study. Int J Geriatr Psychiatry. 2011;26:536–544. doi: 10.1002/gps.2562. [DOI] [PubMed] [Google Scholar]
- 168.Yun HM, Park KR, Kim EC, Kim S, Hong JT. Serotonin 6 receptor controls Alzheimer’s disease and depression. Oncotarget. 2015;6:26716–26728. doi: 10.18632/oncotarget.5777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Naddafi F, Mirshafiey A. The neglected role of histamine in Alzheimer’s disease. Am J Alzheimers Dis Other Demen. 2013;28:327–336. doi: 10.1177/1533317513488925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Motawaj M, Burban A, Davenas E, Gbahou F, Faucard R, Morisset S, Arrang JM. The histaminergic system: a target for innovative treatments of cognitive deficits. Therapie. 2010;65:415–422. doi: 10.2515/therapie/2010058. [DOI] [PubMed] [Google Scholar]
- 171.Shan L, Bao AM, Swaab DF. The human histaminergic system in neuropsychiatric disorders. Trends Neurosci. 2015;38:167–177. doi: 10.1016/j.tins.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 172.Cacabelos R, Yamatodani A, Niigawa H, Hariguchi S, Tada K, Nishimura T, Wada H, Brandeis L, Pearson J. Brain histamine in Alzheimer’s disease. Methods Find Exp Clin Pharmacol. 1989;11:353–360. [PubMed] [Google Scholar]
- 173.Fernández-Novoa L, Cacabelos R. Histamine function in brain disorders. Behav Brain Res. 2001;124:213–233. doi: 10.1016/s0166-4328(01)00215-7. [DOI] [PubMed] [Google Scholar]
- 174.Mazurkiewicz-Kwilecki IM, Nsonwah S. Changes in the regional brain histamine and histidine levels in postmortem brains of Alzheimer patients. Can J Physiol Pharmacol. 1989;67:75–78. doi: 10.1139/y89-013. [DOI] [PubMed] [Google Scholar]
- 175.Panula P, Rinne J, Kuokkanen K, Eriksson KS, Sallmen T, Kalimo H, Relja M. Neuronal histamine deficit in Alzheimer’s disease. Neuroscience. 1998;82:993–997. doi: 10.1016/s0306-4522(97)00353-9. [DOI] [PubMed] [Google Scholar]
- 176.Shan L, Bossers K, Unmehopa U, Bao AM, Swaab DF. Alterations in the histaminergic system in Alzheimer’s disease: a postmortem study. Neurobiol Aging. 2012;33:2585–2598. doi: 10.1016/j.neurobiolaging.2011.12.026. [DOI] [PubMed] [Google Scholar]
- 177.Zlomuzica A, Dere D, Binder S, De Souza Silva MA, Huston JP, Dere E. Neuronal histamine and cognitive symptoms in Alzheimer’s disease. Neuropharmacology. 2016;106:135–145. doi: 10.1016/j.neuropharm.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 178.Higuchi M, Yanai K, Okamura N, Meguro K, Arai H, Itoh M, Iwata R, Ido T, Watanabe T, Sasaki H. Histamine H(1) receptors in patients with Alzheimer’s disease assessed by positron emission tomography. Neuroscience. 2000;99:721–729. doi: 10.1016/s0306-4522(00)00230-x. [DOI] [PubMed] [Google Scholar]
- 179.Schneider EH, Neumann D, Seifert R. Modulation of behavior by the histaminergic system: lessons from H(1)R-and H(2)R-deficient mice. Neurosci Biobehav Rev. 2014;42:252–266. doi: 10.1016/j.neubiorev.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 180.Schneider EH, Neumann D, Seifert R. Modulation of behavior by the histaminergic system: lessons from HDC-, H3R- and H4R-deficient mice. Neurosci Biobehav Rev. 2014;47:101–121. doi: 10.1016/j.neubiorev.2014.07.020. [DOI] [PubMed] [Google Scholar]
- 181.Ambrée O, Buschert J, Zhang W, Arolt V, Dere E, Zlomuzica A. Impaired spatial learning and reduced adult hippocampal neurogenesis in histamine H1-receptor knockout mice. Eur Neuropsychopharmacol. 2014;24:1394–1404. doi: 10.1016/j.euroneuro.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 182.Zlomuzica A, Ruocco LA, Sadile AG, Huston JP, Dere E. Histamine H1 receptor knockout mice exhibit impaired spatial memory in the eight-arm radial maze. Br J Pharmacol. 2009;157:86–91. doi: 10.1111/j.1476-5381.2009.00225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zlomuzica A, Viggiano D, De Souza Silva MA, Ishizuka T, Gironi Carnevale UA, Ruocco LA, Watanabe T, Sadile AG, Huston JP, Dere E. The histamine H1-receptor mediates the motivational effects of novelty. Eur J Neurosci. 2008;27:1461–1474. doi: 10.1111/j.1460-9568.2008.06115.x. [DOI] [PubMed] [Google Scholar]
- 184.Zlomuzica A, Dere D, Dere E. The histamine H1 receptor and recollection-based discrimination in a temporal order memory task in the mouse. Pharmacol Biochem Behav. 2013;111:58–63. doi: 10.1016/j.pbb.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 185.Dere E, Zlomuzica A, Viggiano D, Ruocco LA, Watanabe T, Sadile AG, Huston JP, De Souza-Silva MA. Episodic-like and procedural memory impairments in histamine H1 Receptor knockout mice coincide with changes in acetylcholine esterase activity in the hippocampus and dopamine turnover in the cerebellum. Neuroscience. 2008;157:532–541. doi: 10.1016/j.neuroscience.2008.09.025. [DOI] [PubMed] [Google Scholar]
- 186.Haig GM, Pritchett Y, Meier A, Othman AA, Hall C, Gault LM, Lenz RA. A randomized study of H3 antagonist ABT-288 in mild-to-moderate Alzheimer’s dementia. J Alzheimers Dis. 2014;42:959–971. doi: 10.3233/JAD-140291. [DOI] [PubMed] [Google Scholar]
- 187.Grove RA, Harrington CM, Mahler A, Beresford I, Maruff P, Lowy MT, Nicholls AP, Boardley RL, Berges AC, Nathan PJ, Horrigan JP. A randomized, double-blind, placebo-controlled, 16-week study of the H3 receptor antagonist, GSK239512 as a monotherapy in subjects with mild-to-moderate Alzheimer’s disease. Curr Alzheimer Res. 2014;11:47–58. doi: 10.2174/1567205010666131212110148. [DOI] [PubMed] [Google Scholar]
- 188.Nathan PJ, Boardley R, Scott N, Berges A, Maruff P, Sivananthan T, Upton N, Lowy MT, Nestor PJ, Lai R. The safety, tolerability, pharmacokinetics and cognitive effects of GSK239512, a selective histamine H3 receptor antagonist in patients with mild to moderate Alzheimer’s disease: a preliminary investigation. Curr Alzheimer Res. 2013;10:240–251. doi: 10.2174/1567205011310030003. [DOI] [PubMed] [Google Scholar]
- 189.Medhurst AD, Roberts JC, Lee J, Chen CP, Brown SH, Roman S, Lai MK. Characterization of histamine H3 receptors in Alzheimer’s Disease brain and amyloid over-expressing TASTPM mice. Br J Pharmacol. 2009;157:130–138. doi: 10.1111/j.1476-5381.2008.00075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Schwartz JC. The histamine H3 receptor: from discovery to clinical trials with pitolisant. Br J Pharmacol. 2011;163:713–721. doi: 10.1111/j.1476-5381.2011.01286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Brabant C, Charlier Y, Tirelli E. The histamine H3-receptor inverse agonist pitolisant improves fear memory in mice. Behav Brain Res. 2013;243:199–204. doi: 10.1016/j.bbr.2012.12.063. [DOI] [PubMed] [Google Scholar]
- 192.Sadek B, Saad A, Sadeq A, Jalal F, Stark H. Histamine H3 receptor as a potential target for cognitive symptoms in neuropsychiatric diseases. Behav Brain Res. 2016;312:415–430. doi: 10.1016/j.bbr.2016.06.051. [DOI] [PubMed] [Google Scholar]
- 193.Bitner RS, Markosyan S, Nikkel AL, Brioni JD. In-vivo histamine H3 receptor antagonism activates cellular signaling suggestive of symptomatic and disease modifying efficacy in Alzheimer’s disease. Neuropharmacology. 2011;60:460–466. doi: 10.1016/j.neuropharm.2010.10.026. [DOI] [PubMed] [Google Scholar]
- 194.Provensi G, Costa A, Passani MB, Blandina P. Donepezil, an acetylcholine esterase inhibitor, and ABT-239, a histamine H3 receptor antagonist/inverse agonist, require the integrity of brain histamine system to exert biochemical and procognitive effects in the mouse. Neuropharmacology. 2016;109:139–147. doi: 10.1016/j.neuropharm.2016.06.010. [DOI] [PubMed] [Google Scholar]
- 195.Chen PY, Tsai CT, Ou CY, Hsu WT, Jhuo MD, Wu CH, Shih TC, Cheng TH, Chung JG. Computational analysis of novel drugs designed for use as acetylcholinesterase inhibitors and histamine H3 receptor antagonists for Alzheimer’s disease by docking, scoring and de novo evolution. Mol Med Rep. 2012;5:1043–1048. doi: 10.3892/mmr.2012.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM. Adenosine and brain function. Int Rev Neurobiol. 2005;63:191–270. doi: 10.1016/S0074-7742(05)63007-3. [DOI] [PubMed] [Google Scholar]
- 197.Canas PM, Porciúncula LO, Cunha GM, Silva CG, Machado NJ, Oliveira JM, Oliveira CR, Cunha RA. Adenosine A2A receptor blockade prevents synaptotoxicity and memory dysfunction caused by beta-amyloid peptides via p38 mitogen-activated protein kinase pathway. J Neurosci. 2009;29:14741–14751. doi: 10.1523/JNEUROSCI.3728-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.de Mendonça A, Sebastião AM, Ribeiro JA. Adenosine: does it have a neuroprotective role after all? Brain Res Brain Res Rev. 2000;33:258–274. doi: 10.1016/s0165-0173(00)00033-3. [DOI] [PubMed] [Google Scholar]
- 199.Cunha RA. Neuroprotection by adenosine in the brain: From A(1) receptor activation to A (2A) receptor blockade. Purinergic Signal. 2005;1:111–134. doi: 10.1007/s11302-005-0649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Chen JF, Sonsalla PK, Pedata F, Melani A, Domenici MR, Popoli P, Geiger J, Lopes LV, de Mendonça A. Adenosine A2A receptors and brain injury: broad spectrum of neuroprotection, multifaceted actions and “fine tuning” modulation. Prog Neurobiol. 2007;83:310–331. doi: 10.1016/j.pneurobio.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 201.Cunha RA. Caffeine, adenosine receptors, memory and Alzheimer disease. Med Clin (Barc) 2008;131:790–795. doi: 10.1016/s0025-7753(08)75506-4. [DOI] [PubMed] [Google Scholar]
- 202.Takahashi RN, Pamplona FA, Prediger RD. Adenosine receptor antagonists for cognitive dysfunction: a review of animal studies. Front Biosci. 2008;13:2614–2632. doi: 10.2741/2870. [DOI] [PubMed] [Google Scholar]
- 203.Maia L, de Mendonça A. Does caffeine intake protect from Alzheimer’s disease? Eur J Neurol. 2002;9:377–382. doi: 10.1046/j.1468-1331.2002.00421.x. [DOI] [PubMed] [Google Scholar]
- 204.Arendash GW, Schleif W, Rezai-Zadeh K, Jackson EK, Zacharia LC, Cracchiolo JR, Shippy D, Tan J. Caffeine protects Alzheimer’s mice against cognitive impairment and reduces brain beta-amyloid production. Neuroscience. 2006;142:941–952. doi: 10.1016/j.neuroscience.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 205.Dall’Igna OP, Fett P, Gomes MW, Souza DO, Cunha RA, Lara DR. Caffeine and adenosine A(2a) receptor antagonists prevent beta-amyloid (25–35)-induced cognitive deficits in mice. Exp Neurol. 2007;203:241–245. doi: 10.1016/j.expneurol.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 206.Laurent C, Burnouf S, Ferry B, Batalha VL, Coelho JE, Baqi Y, Malik E, Mariciniak E, Parrot S, Van der Jeugd A, Faivre E, Flaten V, Ledent C, D’Hooge R, Sergeant N, Hamdane M, Humez S, Müller CE, Lopes LV, Buée L, Blum D. A2A adenosine receptor deletion is protective in a mouse model of Tauopathy. Mol Psychiatry. 2016;21:149. doi: 10.1038/mp.2014.151. [DOI] [PubMed] [Google Scholar]
- 207.Espinosa J, Rocha A, Nunes F, Costa MS, Schein V, Kazlauckas V, Kalinine E, Souza DO, Cunha RA, Porciúncula LO. Caffeine consumption prevents memory impairment, neuronal damage, and adenosine A2A receptors upregulation in the hippocampus of a rat model of sporadic dementia. J Alzheimers Dis. 2013;34:509–518. doi: 10.3233/JAD-111982. [DOI] [PubMed] [Google Scholar]
- 208.Pagnussat N, Almeida AS, Marques DM, Nunes F, Chenet GC, Botton PH, Mioranzza S, Loss CM, Cunha RA, Porciúncula LO. Adenosine A(2A) receptors are necessary and sufficient to trigger memory impairment in adult mice. Br J Pharmacol. 2015;172:3831–3845. doi: 10.1111/bph.13180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Bonda DJ, Wang X, Lee HG, Smith MA, Perry G, Zhu X. Neuronal failure in Alzheimer’s disease: a view through the oxidative stress looking-glass. Neurosci Bull. 2014;30:243–252. doi: 10.1007/s12264-013-1424-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Bonda DJ, Wang X, Perry G, Nunomura A, Tabaton M, Zhu X, Smith MA. Oxidative stress in Alzheimer disease: a possibility for prevention. Neuropharmacology. 2010;59:290–294. doi: 10.1016/j.neuropharm.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 211.Moreira PI, Zhu X, Nunomura A, Smith MA, Perry G. Therapeutic options in Alzheimer’s disease. Expert Rev Neurother. 2006;6:897–910. doi: 10.1586/14737175.6.6.897. [DOI] [PubMed] [Google Scholar]