

Abstract
Background
Ascochyta blight, caused by the fungus Ascochyta lentis, is one of the most destructive lentil diseases worldwide, resulting in over $16 million AUD annual loss in Australia alone. The use of resistant cultivars is currently considered the most effective and environmentally sustainable strategy to control this disease. However, little is known about the genes and molecular mechanisms underlying lentil resistance against A. lentis.
Results
To uncover the genetic basis of lentil resistance to A. lentis, differentially expressed genes were profiled in lentil plants during the early stages of A. lentis infection. The resistant ‘ILL7537’ and susceptible ‘ILL6002’ lentil genotypes were examined at 2, 6, and 24 h post inoculation utilising high throughput RNA-Sequencing. Genotype and time-dependent differential expression analysis identified genes which play key roles in several functions of the defence response: fungal elicitors recognition and early signalling; structural response; biochemical response; transcription regulators; hypersensitive reaction and cell death; and systemic acquired resistance. Overall, the resistant genotype displayed an earlier and faster detection and signalling response to the A. lentis infection and demonstrated higher expression levels of structural defence-related genes.
Conclusions
This study presents a first-time defence-related transcriptome of lentil to A. lentis, including a comprehensive characterisation of the molecular mechanism through which defence against A. lentis is induced in the resistant lentil genotype.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4488-1) contains supplementary material, which is available to authorized users.
Keywords: Lentil, Lens culinaris, Ascochyta lentis, RNA sequencing and transcriptome analysis, De novo assembly, Fabaceae, Defence response
Background
Lentil (Lens culinaris ssp. culinaris) is a rich source of protein, minerals and vitamins; and thus plays a staple food role in the diets of vegetarian, vegan and low meat consuming communities. Due to exponential population growth in regions where lentil is a main staple, annual global production has drastically risen, from 0.85 to 5.03 Mt during the last five decades [1]. However, global production and quality is substantially impacted by Ascochyta blight, caused by the necrotrophic fungus Ascochyta lentis [2]. Together with the cost of management through fungicides, this pathogen is responsible for an annual estimated loss of $16.2 million AUD in Australia alone [3, 4].
Much research has been conducted to understand A. lentis epidemiology, diagnostics, lifecycle, survival and chemical susceptibility [2, 5–8]. This information, together with the adoption of high yielding resistant cultivars, provides the most environmentally friendly and economic strategy for disease management [3]. Relatively few genotypes, containing simply inherited ‘resistance’ to A. lentis, have been employed widely in resistance breeding programs on a global scale [8–10]. The lentil industry in Australia is reliant on A. lentis resistance from three main sources; two Canadian cultivars cv. Northfield and cv. Indianhead and the landrace ILL7537, all underpinned by one or two major resistance genes. Recently, the widely adopted resistance derived from cv. Northfield (ILL5588), under the control of one [10] or two dominant genes [11], seems to have been eroded through increased pathogen aggressiveness [12]. It is likely that other major resistances may also be under threat through selective adaptation of the pathogen population, and therefore there is an urgent need to understand the key functional genes employed by resistant genotypes to strategically improve the longevity of the defence mechanisms available. The initial step towards this is to identify and characterise the genes involved, however, there is currently limited information on the lentil genome or its interaction with A. lentis.
Some initial efforts have been made to explore the genomic and molecular aspects involved in defence to A. lentis. Comparative gene expression analysis with a boutique microarray, comprising a limited number of defence-related cDNA probes sourced from other leguminosae species, revealed several genes important in the early resistance reaction of the resistant lentil accession ILL7537 to A. lentis at 2, 6, and 24 h post inoculation (hpi) [13]. The suit of differentially expressed (DE) genes uncovered confirmed the biological significance of the early stages in the A. lentis–lentil interaction; representative of pathogen recognition (2 hpi), induced defence responses (6 hpi), and necrotic structural defence reactions (24 hpi) [7, 8]. In particular, serine/threonine protein kinases were reported to be a key component of the signalling mechanism required to activate downstream A. lentis defence responses, which included an hypersensitive reaction [13]. Histopathology research of A. lentis infection and disease progress observed major changes in the lentil physiology at 2, 6, and 24 hpi and depicted those as important checkpoints in the defence response of lentil to A. lentis [8]. Lentil plants detect A. lentis attack as soon as they come in contact at the host surface or in minutes of invasion. This mainly occurs between 2-6 hpi (first/early phase of oxidative burst) as previously reported [8]. These rapid events are transcription-independent, cause morphological and physiological changes in the infected cells and their surroundings and further transcriptional and post-translational activation of transcription factors takes place. Secondly, a sustained oxidative burst phase that occurs hours after pathogen attack usually associated with the establishment of the defences and the hypersensitive response is carried out. In lentil plants this occurred between 20 and 24 hpi, which may act as a signal for gene activation resulting in secretion of fungal penetration-inhibitory substances into the surrounding plant cell wall to arrest further penetration and spread [8].
Although a good foundation, these results were greatly limited by their dependence on homologous sequences previously discovered as important in other species and pathosystems. Therefore, there remains a large knowledge gap regarding which genes/functional alleles are involved in the early defence pathways of recognition, and biochemical and physiological defence responses in the lentil–A. lentis interaction. To bridge this gap, an in-depth molecular study of the interaction is required.
Next generation sequencing and more specifically RNA-Sequencing (RNA-Seq) has become a popular and comprehensively informative approach to monitor wide transcriptional changes during host-pathogen interactions [14–17]. Recently, an RNA-Seq approach was employed to characterise the functional defence response genes of faba bean to A. fabae and these included phytoalexins (Dihydrofla-vonol-4-reductase), a chitin elicitor-binding protein (CEBiP), jasmonate O-methyltransferase and an F-box/leucine-rich repeat (LRR) protein, as well as several pathogenesis-related (PR) proteins [17]. Likewise, RNA-Seq revealed that protein kinases such as receptor-like kinases, PR protein classes (2-9, except PR7), diterpene phytoalexin biosynthesis genes, and WRKY transcription factors were involved in the defence of rice to Ustilaginoidea virens [15].
A few RNA-seq studies were conducted recently to assemble the expressed transcriptomes of lentil, which provided a good reference for the genes expected to be expressed throughout various tissues and genotypes. However, these focused on different developmental stages and toxic tolerance [18] and marker development [19, 20] and none of them covered the transcriptional changes that occur during a pathogen attack and its counteract defence response. A targeted RNA-Seq approach during lentil–A. lentis interactions would be beneficial in better understanding the molecular defence responses of lentil to Ascochyta blight.
Thus, the aims of this study are to use RNA-Seq to identify the genes and gene functions, and predict the molecular pathways employed by a resistant lentil accession in the early recognition and defence to an aggressive isolate of A. lentis.
Methods
Bioassay and RNA extraction
Seedlings of lentil genotypes ILL7537 (resistant to A. lentis [8]) and ILL6002 (susceptible to A. lentis [8]) were grown in a controlled-environment growth room at 20 °C ± 2 °C with 12/12 h dark/light lengths. Three replicates (pots) of five seedlings per 7 cm diameter pot were grown in a light commercial pine bark soil for each of the time points assessed. At 14 d after sowing, seedlings were sprayed until run-off with a 1×105 suspension of A. lentis ALP2 isolate condiospores [12] or water as a negative control, according to the method described by Davidson et al. [12]. All seedlings were then placed in the dark for 48 h within a plastic box and adequate humidity was maintained to encourage fungal growth and germination. During this period, the seedlings were harvested and pooled from each pot (replicate) at 2, 6, and 24 hpi. This provided triplicate biological representative reactions for each genotype, at each time point and from both fungal and water inoculated treatments (Fig. 1). Another pot of five seedlings of each genotype that had been inoculated with the isolate was left unharvested and allowed to grow in the growth room at 20 °C ± 2 °C with 12/12 h dark/light lengths to confirm visible disease symptoms after 7−10 d.
Fig. 1.
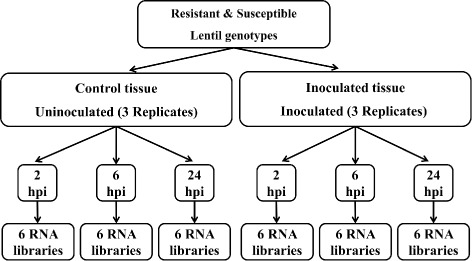
Experimental design of the Ascochyta lentis resistant (ILL7537) and susceptible (ILL6002) lentil genotypes used for RNA extraction
At each time of collection, the five seedlings of each replicate were combined and instantly frozen in liquid N2. Total RNA was extracted from the whole seedling bulks using the RNeasy plant mini kit along with DNase treatment, according to the manufacturer’s instruction (QIAGEN, Germany). RNA quality and quantity were determined with an Experion RNA analysis kit (Bio-Rad, USA) (Table 1). Subsequently, 6 μg total RNA of each sample were diluted in 50 μL RNAse-free H2O and used for cDNA library preparation and transcriptome profiling.
Table 1.
RNA sample details including quality and concentration measurements
| Sample ID | HPIa | Cultivar | Treatmentb | Replicate | RQIc | RNA conc. (ng/ μL) |
|---|---|---|---|---|---|---|
| 1C | 2 | ILL6002 | Mock | 1 | 9.5 | 504.93 |
| 2C | 2 | ILL6002 | Mock | 2 | 9.5 | 477.45 |
| 3C | 2 | ILL6002 | Mock | 3 | 9.4 | 359.48 |
| 4C | 2 | ILL6002 | Treated | 1 | 9.4 | 390.33 |
| 5C | 2 | ILL6002 | Treated | 2 | 9.4 | 461.47 |
| 6C | 2 | ILL6002 | Treated | 3 | 9.1 | 214.97 |
| 7C | 2 | ILL7537 | Mock | 1 | 9.5 | 316.84 |
| 8C | 2 | ILL7537 | Mock | 2 | 9.4 | 348.37 |
| 9C | 2 | ILL7537 | Mock | 3 | 9.6 | 281.67 |
| 10C | 2 | ILL7537 | Treated | 1 | 9.9 | 325.65 |
| 11C | 2 | ILL7537 | Treated | 2 | 9.4 | 317.97 |
| 12C | 2 | ILL7537 | Treated | 3 | 9.6 | 211.23 |
| 13C | 6 | ILL6002 | Mock | 1 | 10.0 | 263.26 |
| 14C | 6 | ILL6002 | Mock | 2 | 10.0 | 359.14 |
| 15C | 6 | ILL6002 | Mock | 3 | 10.0 | 157.02 |
| 16C | 6 | ILL6002 | Treated | 1 | 10.0 | 165.98 |
| 17C | 6 | ILL6002 | Treated | 2 | 10.0 | 236.68 |
| 18C | 6 | ILL6002 | Treated | 3 | 10.0 | 260.98 |
| 19C | 6 | ILL7537 | Mock | 1 | 10.0 | 210.47 |
| 20C | 6 | ILL7537 | Mock | 2 | 10.0 | 222.57 |
| 21C | 6 | ILL7537 | Mock | 3 | 10.0 | 296.11 |
| 22C | 6 | ILL7537 | Treated | 1 | 10.0 | 275.24 |
| 23C | 6 | ILL7537 | Treated | 2 | 10.0 | 221.47 |
| 24C | 6 | ILL7537 | Treated | 3 | 10.0 | 275.41 |
| 25C | 24 | ILL6002 | Mock | 1 | 9.7 | 511.10 |
| 26C | 24 | ILL6002 | Mock | 2 | 9.7 | 303.83 |
| 27C | 24 | ILL6002 | Mock | 3 | 9.6 | 467.35 |
| 28C | 24 | ILL6002 | Treated | 1 | 9.6 | 407.76 |
| 29C | 24 | ILL6002 | Treated | 2 | 9.5 | 336.80 |
| 30C | 24 | ILL6002 | Treated | 3 | 9.7 | 263.37 |
| 31C | 24 | ILL7537 | Mock | 1 | 9.8 | 338.72 |
| 32C | 24 | ILL7537 | Mock | 2 | 9.7 | 340.26 |
| 33C | 24 | ILL7537 | Mock | 3 | 9.7 | 550.03 |
| 34C | 24 | ILL7537 | Treated | 1 | 9.8 | 317.72 |
| 35C | 24 | ILL7537 | Treated | 2 | 9.8 | 414.79 |
| 36C | 24 | ILL7537 | Treated | 3 | 9.8 | 481.03 |
aHours post inoculation
bSample treatment: Treated – inoculation with highly pathogenic isolate of A. lentis ALP2; Mock – water spray as a negative control
cRNA quality indicator (RQI), as determined by an Experion RNA Analysis System (Bio-Rad)
Transcriptome profiling
Library preparation and RNA-Sequencing
Library preparation and sequencing were performed at the Pangenomics Laboratory, RMIT University, Bundoora, following the methods described in the Ion Proton user’s guide (Thermofisher Scientific, USA). Briefly, mRNA was isolated from the total RNA using Dynabeads mRNA Purification Kit (Thermofisher Scientific, USA). This was followed by enzymatic fragmentation of mRNA to create short reads, <300 base pairs (bp), suitable for the Ion Proton sequencer. The cDNA was synthesised using reverse transcription and a unique barcode was attached to the fragments of each library. The RNA-Seq libraries were prepared using Ion Total RNA-Seq Kit v2 (Thermofisher Scientific, USA) according to the manufacturer’s instructions. Finally, four RNA-Seq libraries were multiplexed and loaded on an Ion ProtonTM chip for sequencing. The resulting raw RNA-Seq reads were deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA study accession number: SRP075524).
Assembly
Short read sequences from the RNA-Seq were downloaded and processed on the Griffith University ‘Gowonda’ High Performance Computing Cluster using Linux command-line operations. Preliminary quality check of the reads was performed using FastQC (v0.11.2), followed by 3’ and 5’ ends quality trimming and adaptor removal by Trimmomatic (v0.32 [21]), with stringency parameters of SLIDINGWINDOW:4:10 MINLEN:36. The clean and trimmed reads were then de novo assembled using Trinity (r20140717 [22]), to establish the full lentil defence-response transcriptome. Assessment of the assembled transcripts was performed by calculating and plotting an ExN50 value against a fraction of the most highly expressed transcripts (Ex). This plot enabled identification of the assembly saturation point, at which the maximum length of N50 was obtained, after removal of the transcripts with minor contribution to the total expression, which are often associated with assembly errors [22, 23].
Gene and protein annotations
Open reading frames (ORFs) were predicted from the assembled transcripts using TransDecoder (r20140704); an ORF was considered as complete by the presence of a starting methionine amino acid and an ending stop codon. Transcripts and predicted peptides were annotated by sequence alignment similarity search (BLAST 2.3.0+ [24]) to protein databases (NCBI nr, UniProt, Swiss-Prot and KOBAS, e-value <1e-5), and by hidden Markov models protein domain identification (HMMER3.1b [25]) against the HMMER/Pfam protein database (v28.0 [26]). Annotated ORFs were classified into taxonomy groups by extracting the species from their top scoring Blast result. Higher taxonomy levels (family, phyla) were inferred from the NCBI Taxonomy database using the taxize R package [27, 28]. Based on these annotations, Gene Ontology (GO [29]) and Kyoto Encyclopedia of Genes and Genomes (KEGG [30, 31]) terms were assigned to each putative protein. Since L. culinaris is not included in the KEGG database of species-specific terms, KEGG orthology terms were used to match a function to each ORF. Furthermore, predictions of putative secretory signal peptides (SignalP v4.1 [32]) and trans-membrane topology (TMHMM v2.0 [33]) were performed. The resulting annotation output files were further processed and cleaned to remove duplicates, select best matching annotation and identify errors or missing values.
To determine how inclusive the assembly was of full length genes, coverage levels of the reconstructed protein-coding ORFs were calculated from the UniRef90 Blast results, as described in the Trinity documentation (https://github.com/trinityrnaseq/trinityrnaseq/wiki/Counting-Full-Length-Trinity-Transcripts). A similar assessment of the representation of single-copy conserved plant orthologues was performed using BUSCO [34].
Differential gene expression
The number of reads that mapped to each ORF was estimated by the super-efficient and alignment-free software, kallisto [35]. Preliminary exploratory data analysis of the estimated counts of each sample was performed by variance-stabilising transformation of the raw counts, followed by computing and plotting a between-sample distance matrix and principle coordinate analysis to identify sample-related biases.
The estimated counts were normalised using the Trimmed Mean of M-values (TMM), a normalisation method implemented in the edgeR Bioconductor package (http://bioconductor.org/packages/release/bioc/html/edgeR.html), to account for differences in library size between samples [36, 37]. To represent count data variability, standard error values were calculated per gene, based on triplicate TMM counts (n =3) at each genotype/time point, with the exception of the reference genes, where SE were calculated based on all samples and across all experimental groups (excluding the outlier sample 4C, n =35). TMM counts of selected defence-related transcript are provided in Additional file 1.
The estimated counts were further analysed by edgeR to identify statistically significant DE ORFs between the experimental groups [38, 39]. Raw p-values were adjusted for multiple comparisons by the Benjamini-Hochberg procedure, which controls the false discovery rate [40]. Transcripts were considered to be DE with a |log2FC|>1.5 (positive or negative for either over- or under-expression respectively), and an adjusted p-value <0.05.
Gene set enrichment analysis of the DE genes was then performed for the GO and KEGG annotations to determine over-represented functional pathways (with a p-value <0.01 and p-value <0.001, respectively) at each comparison level (for specific genotype, sampling time and treatment combination). Each pathway was further categorised into one of GO’s functional groups (biological processes, cell cycle or molecular function) or KEGG’s functional groups (metabolism, environmental information processing, organismal systems, cellular processes or genetic information processing). The analysis was performed using a custom-written R script (https://github.com/IdoBar/Trinotate_GSEA_plotteR), utilising the goseq R package [41].
Quantitative reverse transcription PCR
Real time quantitative reverse transcription PCR (RT-qPCR) was used to validate the expression patterns of selected genes. Three biological replicates from inoculated resistant and susceptible plants at 2, 6, and 24 hpi were collected, instantly frozen in liquid N2 and stored at −80 °C until RNA extraction. Samples were pulverised while frozen in liquid N2 using a mortar and pestle and total RNA was extracted using NucleoSpin Ⓡ RNA Plant kit along with DNase treatment, according to the manufacturer’s instruction (Macherey Nagel, Germany). Quality and quantity of the total RNA for each sample were determined using gel electrophoresis, NanoDropTM (Thermo Fisher Scientific, USA) and Qubit (Invitrogen, USA). 0.7 μg of total RNA from each sample was used to synthesise cDNA using PrimeScriptTM RT reagent Kit (TaKaRa Bio, Japan), incorporating an additional gDNA removal step. The RT-qPCR was performed in the CFX96 TouchTM Real-Time PCR Detection System (Bio-Rad, USA).
Five antifungal compounds and transcriptional regulators involved in defence were chosen as target genes for the assay: RBP, PR2, PR10, PGIP, ARP and DELLA; and PP2A as a reference gene. Primers were designed from the transcriptome sequences for each gene and were tested to ensure acceptable amplification efficiency, specificity, consistency and detection range, based on serial dilution standard curves and melt curves. Amplification efficiencies for each gene were calculated from the coefficients of linear regression equations fitted to the Window-of-Linearity phase of each reaction of the main RT-qPCR assay. The calculated efficiencies were then averaged across all reactions of each gene, as implemented in LinRegPCR (v2017.1) [42, 43]. Details of the primers used for the RT-qPCR assay are listed in Table 2. Each RT-qPCR reaction contained 2 μl of cDNA template (diluted 1:25 from the synthesis reaction), 10 μl SYBR Ⓡ Premix Ex TaqTM (TaKaRa Bio, Japan) and a final primer concentration of 1.6 μM in a final volume of 20 μl. The reactions were performed using the following cycle conditions: an initial 95 °C for 2 min, followed by 38 cycles of 95 °C for 10 s, 50–57 °C for 30 s (depending on the empirically determined optimal melting temperature for each primer pair), 72 °C for 15 s, and a final 5 min extension at 72 °C. All reactions were performed in three technical replicates for each biological sample (n =3) at each time point. Inter-run calibrators reactions were included in each plate using a pooled cDNA as template for each of the three reference genes.
Table 2.
Genes and primers used in qRT-PCR assay
| Target gene | Gene function | Primer sequences (5’-3’) | Amplification efficiencya | Tm (°C) |
|---|---|---|---|---|
| DELLA | SAR signalling | F: GTCTTCTAATTCAAACCA | 1.926 ± 0.027 | 53 |
| R: ATATCTGTTTACCCAAGTAA | ||||
| RBP-hnRNP | Transcriptional factor | F: GAGAAAGATATTTGTTGGAG | 1.947 ± 0.024 | 51 |
| R: TGATCGTACATTACTACAACA | ||||
| PGIP | Anti fungal compound | F: TGAAGGTGATGCTTCTATGCT | 2.013 ± 0.026 | 53 |
| R: GACTCACATCCAACGTTGCT | ||||
| PR2 | Anti fungal compound | F: GGCATGCTGGGAAACAATCT | 2.018 ± 0.023 | 52 |
| R: TGGCACACCTAACATGAGCT | ||||
| PR10 | Anti fungal compound | F: TGGCACTTCTGCTGTTAGATGGAC | 2.017 ± 0.083 | 50 |
| R: GGTAATCCATCCAGCCATTTGGAG | ||||
| PP2A | Reference gene | F: GCCTCATTTGCAGCTGGTTT | 2.002 ± 0.025 | 53 |
| R: TACTCTCGTTCTAGGGTCCT |
aMean and standard deviation of the amplification efficiency for each gene were calculated from the coefficients of linear regression models fitted to each reaction (value of 2 equivalents to 100% efficiency, see [43])
Amplification data (Cq values) were normalised between plates using Factor-qPCR (v2016.0) [44] which estimates between-plates correction factors based on the inter-run calibrators. DE ratio of each gene between the resistant and susceptible genotypes at each time point was then calculated with the Relative Expression Software Tool (RESTⒸ 2009 [45]), considering the amplification efficiency of each gene [46, 47]. Statistical significance of the DE genes was tested by a randomisation test incorporated in RESTⒸ 2009 and were considered as significant with a p-value <0.05.
Data analysis
The annotation and expression data files were then combined and loaded onto a lightweight, standalone relational SQLite database (http://www.sqlite.org/), using the scripts provided in the Trinotate pipeline (v3.0.1; https://trinotate.github.io/). This allowed for a fast and easy retrieval of sequences, annotation and expression data using any combination of conditional filtering and ordering. A complete bioinformatics data processing and analysis workflow is presented in Fig. 2.
Fig. 2.
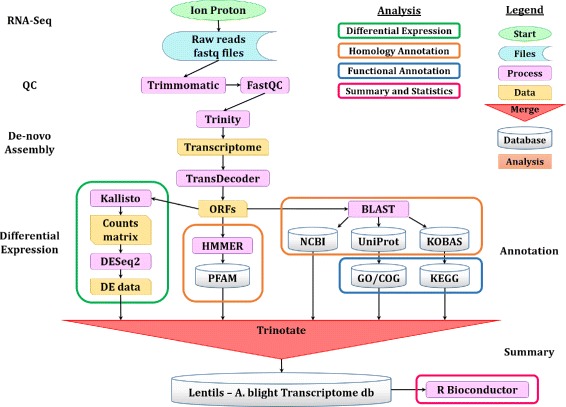
Bioinformatics flowchart of tools and methods used to process and analyse the RNA-Sequencing data and produce the transcriptome
Statistical analysis and additional data summarising were performed using the R statistical programming language (v3.2.5 [48]). Specifically, relevant data was retrieved from the SQLite database and pre-processed using the dplyr package, and then analysed using various tools from the Bioconductor [49] and the Comprehensive R Archive Network (CRAN, https://cran.r-project.org/) R repositories.
Results
RNA-Sequencing and assembly
All RNA extracts were of high quality, with an average RNA quality indicator of 9.72, as determined on the Experion (Table 1). A total number of 7.25×108 reads with an average length of 90 bp were produced by the Ion Proton RNA sequencing platform. Of the total number of bases sequenced across all reads (7.90×1010 bp), 13% ± 3% in average were trimmed to remove adapters and low quality base calls in each read file. In addition, reads which were too short after adapter trimming and/or had an average low quality bases were dropped, resulting in a total of 6.47×108 clean reads, comprised of 6.86×1010 bp high quality bases. Detailed trimming statistics of each read file are provided in Additional file 2. The defence-related transcriptome of lentil was then de novo assembled to 317,412 transcripts (total length of 1.45×108 bp), which were grouped into 256,326 trinity ‘genes’, with an N50 = 497 bp. Plotting the ExN50 value against varying levels of cumulative transcript expression (Ex) identified a saturation point of the assembly at 96% of the total expression, giving an improved E96N50 of 827 bp and reducing the effective contig count to 44,007 (Fig. 3). Detailed statistics of the transcriptome assembly are provided in Table 3.
Fig. 3.
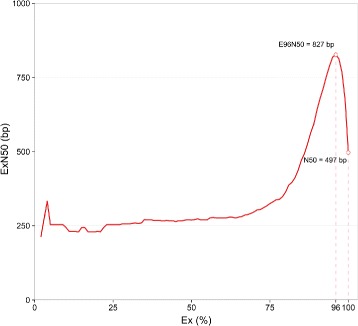
Expression-dependant N50 (ExN50), as calculated against a fraction of the total expressed data (Ex). ExN50 at the point of assembly saturation (96%) and traditional N50 are highlighted
Table 3.
Lentil–A. lentis transcriptome assembly details
| Feature | Value |
|---|---|
| Assembled contigs (n) | 317,412 |
| Longest contig (bp) | 11,358 |
| Mean contig (bp) | 457 |
| Contig N50 (bp) | 497 |
| Contig N90 (bp) | 236 |
| Contig E96N50 (bp) | 827 |
| E96 contigs (n) | 44,007 |
| Total contig length (bp) | 1.45×108 |
| Assembly GC content (%) | 41.74 |
| Total reads (n) | 7.247×108 |
| Avg. sequence retention ratea (%) | 89.63 |
aRaw sequenced reads which passed quality control measures as determined by FastQC
Annotation
Multiple ORFs were predicted from each transcript, to a total number of 106,754. Close to 30% of the predicted ORFs presented a minimum of 100 amino acids, a starting codon for Methionine and an ending stop codon and were therefore predicted as ‘complete’. Another 31% of the ORFs were predicted to contain a partial 5’, more than double of those with partial 3’, most likely due to the poly(A) enrichment stage in the library preparation process, which is reported to introduce a bias towards the 3’ end of the transcripts [50].
Both the assembled transcripts and predicted ORFs were annotated to known genes, using NCBI’s comprehensive nucleotide (nt) and protein (nr) databases respectively, resulting in significant matches for 70% of the transcripts and over 62% of the ORFs. Taxonomic analysis of the Blast matches revealed that 91.1% of the ORFs matched previously annotated plant genes, 97.6% of those from the legume family (Fabaceae); another 6.5% matched fungi proteins (Ascomycota, mainly from the Didymellaceae family); and another small fraction (1.8%) matched bacterial genes (Fig. 4).
Fig. 4.
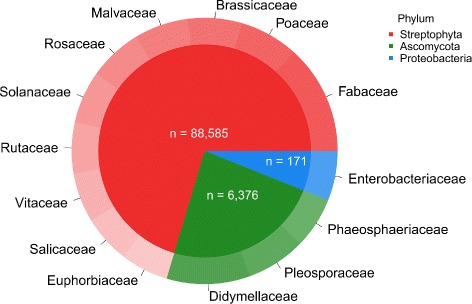
Taxonomy distribution of significant* Blast matches. Annotation was considered as significant with a BitScore >100, pie slices are calculated in logarithmic scale to assist in visualisation
Close to 25% of the Blast annotated ORFs were predicted to cover more than 80% of their target gene’s full length sequence, thus providing strong evidence that the transcriptome assembly contained a reasonable coverage of the genes in the tissues. A similar conclusion arose from the BUSCO analysis, identifying 379 complete single-copy, 339 duplicated and 176 fragmented conserved plant orthologues in the assembly, out of 956 orthologue groups in total (overall coverage of 93.5%).
Additional annotation of protein domains was performed against the Pfam database, assigning significant domains to 56% of the ORFs. Functional annotation of the putative ORFs using GO and KEGG terms, found a significant match to 37 and 33% of the ORFs, respectively, thus adding another layer of annotation at the molecular pathway level. Transmembrane structure and signal peptide predictions were performed as well and added to the complete ORFs annotation database (full details in Table 4).
Table 4.
Transcript and open reading frame annotation
| Annotation | Occurrences (#) | Rate (%) |
|---|---|---|
| Transcripts:a | 317,412 | 100 |
| NCBI nta | 223,246 | 70.3 |
| UniRef90a | 138,131 | 43.5 |
| ORFs:b | 106,754 | 100 |
| NCBI nra | 93,077 | 62.4 |
| SwissProta | 52,452 | 49.1 |
| Pfamc | 60,404 | 56.6 |
| KEGGa | 39,919 | 37.4 |
| GOa | 34,937 | 32.7 |
aBlast based annotation was considered as significant with a BitScore >100
bORFs predicted by TransDecoder
cHMM-predicted Pfam protein domains were considered as significant with FullSeqScore and FullDomainScore >20
Differential gene expression
Exploratory data analysis
An exploratory data analysis of the estimated counts of reads that mapped to each ORF clearly showed that the main difference between the samples were derived from the genotype variable, which explained 40% of the total detected variation. Sampling time (2, 6, and 24 hpi) contributed another 17% of the variability (Fig. 5).
Fig. 5.
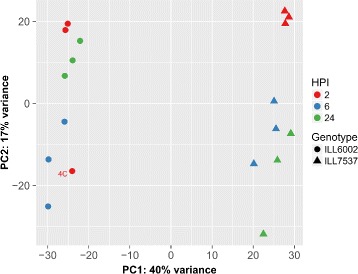
Principal component analysis of the variance-stabilized estimated raw counts. Samples are categorized by Genotype (as marker shapes) and Hours post inoculation (HPI, marker colour)
The three replicates of each sampling group clustered well together, with the exception of sample 4C of the susceptible genotype (ILL6002, round markers in Fig. 5), which clustered with the 6 hpi samples of the same genotype. This sample was identified as an outlier and was excluded from downstream DE analysis.
Gene set enrichment analysis
The number of putative genes (based on ORF prediction) that were DE, with |log2FC|>1.5 and FDR <0.05, was determined within each comparison, demonstrating once more that the most noticeable DE was found between the resistant genotype and the susceptible one, in particular at 24 hpi, with 2617 DE genes (Fig. 6). A substantial number of genes (507) were commonly DE between the two genotypes, regardless of sampling time. The DE genes were then grouped into functional pathways by their assigned GO and KEGG terms. Comparison of GO enrichment analysis of DE genes in the resistant genotype between 2, 6, and 24 hpi identified high representation of over-expressed genes involved in pathogen recognition, signalling at 2 hpi compared with 6 hpi. In contrast, high proportions of genes associated with anti-fungal compounds, plant cell wall organisation and construction, as well as transcriptional regulators, were observed at 6 hpi (Fig. 7a). At 24 hpi, the majority of DE gene were associated with regulatory functions in plant stress tolerance, antimicrobial compounds and photosynthesis pathways (Fig. 7b).
Fig. 6.
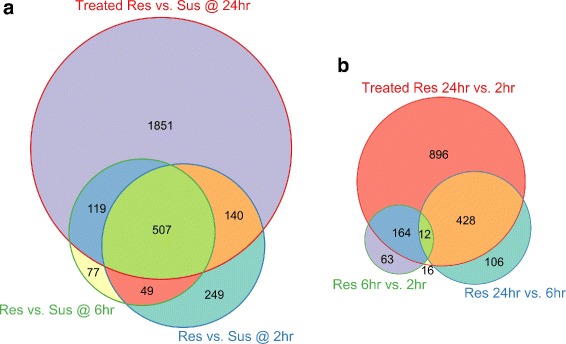
The number of unique and common differentially expressed genes between the subgroups of inoculated samples (time post inoculation and genotype). Comparison of inoculated resistant vs. susceptible genotypes at each time point (2 hpi, 6 hpi and 24 hpi, a); and within the inoculated resistant (ILL7537) genotype samples between the different time points (b). Circle area is plotted to scale (Euler diagram) when geometrically possible
Fig. 7.
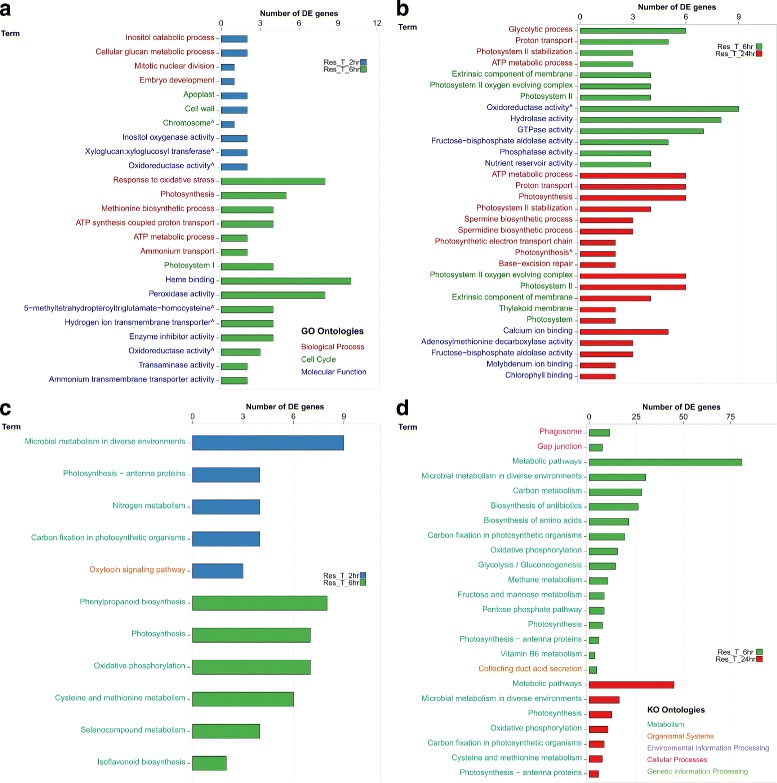
GO and KEGG pathway enrichment analysis, based on over-expressed DE genes at each time point (2, 6, and 24 hpi) in the resistant lentil genotype ILL7537. GO pathway enrichment at 2 and 6 hpi (a) and 6 and 24 hpi (b); KEGG pathway enrichment at 2 and 6 hpi (c) and 6 and 24 hpi (d)
KO pathway enrichment analysis of the same time points provided a similar picture to the GO enrichment analysis (Fig. 7c-d). High representation of genes involved in microbial, carbon and nitrogen metabolism and signalling pathways were identified at 2 hpi; a substantial number of genes involved in metabolic pathways, photosynthesis, and to a lesser extent, defence response genes at 6 hpi and an increased enrichment of photosynthesis related genes at 24 hpi.
Primary defence response (2 hpi)
Analysis of DE transcripts among the resistant (ILL7537) and susceptible (ILL6002) genotypes at 2 hpi, compared to those at 6 and 24 hpi identified transcripts with matching annotation to several gene families. Specifically, genes from the protein kinase-like family, known to be involved in pathogen recognition and early stage of signalling [51], were moderately over-expressed at 2 hpi (Fig. 8a). A member of this family is the LRR receptor-like kinase (LRR-RK), which demonstrated its highest expression levels in the resistant genotype at 2 hpi, with TMM = 40 and a gradual decrease down to TMM = 7.3 at 24 hpi (log2FC=2.45). The expression of LRR-RK at 2 hpi in ILL6002 was lower than in ILL7537 (TMM = 20.9), however, it then increased dramatically at 6 hpi (TMM = 73), before decaying back to base levels at 24 hpi, as in ILL7537 (Fig. 8a).
Fig. 8.
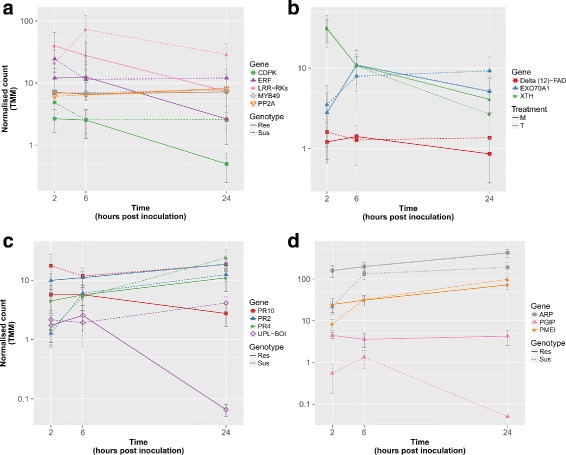
Expression levels of selected genes with exceptional DE trends in the earlier stages of the defence response to A. lentis in ILL7537 and ILL6002 over 2, 6, and 24 hpi. Expression levels of the following genes are presented: CDPK, ERF and LRR-RK, with PP2A and MYB49 as examples of stable reference genes (a); Delta (12)-FAD, EXO70A1 and XTH (b); PR genes and UPL-BOI (c); ARP, PGIP and PMEI (d). A full line represents the expression level in the resistant genotype ILL7537 and the dashed line represents the expression level of the gene in the susceptible genotype ILL6002. Y-axis is in logarithmic scale, error bars represent standard error values between replicates
Calmodulin domain protein kinase-like (CDPK) was also moderately expressed at 2 hpi and 6 hpi, with a TMM of 2.6, dropping at 24 hpi to just under 0.5 at a log2FC of 2.5 in the resistant genotype (Fig. 8a). Slightly higher expression levels were noticed in the susceptible genotype at 2 hpi (TMM = 4.8), dropping to a stable level of 2.5 at 6 and 24 hpi. Interestingly, a very similar expression pattern, with approximately 5x times higher TMM values compared with CDPK, was found for Ethylene-responsive transcription factor (ERF) across all time points and genotypes (Fig. 8a).
Xyloglucan endotransglucosylase/hydrolase (XTH), encoding an enzyme involved in cell wall elongation and restructuring [52], was most highly expressed at 2 hpi in the susceptible genotype, with TMM = 61 (Fig. 8). XTH expression levels then rapidly decreased at 6 hpi to 15.3 and then more gradually to 5.4 at 24 hpi (log2FC of 2 and 3.5, respectively). A similar expression pattern was observed in the resistant genotype, at approximately half the levels shown in the susceptible genotype at each time point (Fig. 8b).
A unique expression pattern was identified for Exocyst subunit 70A1 (EXO70A1), a structural gene involved in papilla formation [53]. This gene was expressed at moderate levels (TMM = 3.47) at 2 hpi in the resistant genotype and then increased at 6 hpi and slightly more at 24 hpi. In contrast, in the susceptible genotype, EXO70A1 was expressed at low levels at 2 hpi (TM <1), with a steep incline at 6 hpi to TMM = 12.8, superseding the expression levels at the resistant genotype, before declining again at 24 hpi to TMM of 2.5, with a log2FC of 4.44 (Fig. 8b). Delta (12)-FAD demonstrated modest expression levels of just over 1 TMM in the resistant genotype, but was still up-regulated comparing its expression in the susceptible lentil genotype, which was negligible (Fig. 8b).
A noticeable expression pattern was observed for pathogenesis related proteins PR-2–O-glycosyl hydrolase family 17 (PR2) and PR-4–Thaumatin-like (PR4) proteins. PR2 was over-expressed in ILL7537 at all time points, from which the most significant differential over-expression between the genotypes occurred at 2 hpi with a log2FC of 3. PR4 demonstrated similar expression to PR2 at 2 hpi with a log2FC of 1.65 between the resistant and susceptible genotypes, however, its expression in the susceptible genotype then increased to match the same expression level as the resistant genotype at 6 hpi and even further at 24 hpi (Fig. 8c).
Plant invertase pectin methylesterase inhibitor (PMEI) was slightly over-expressed in comparison to the susceptible genotype at 2 hpi, with a log2FC of 1.6, followed by a gradual increase in expression at 6 hpi and 24 hpi in both genotypes (Fig. 8d). Polygalacturonase inhibitor (PGIP), which encodes a plant extracellular leucine-rich repeat protein [54], was over-expressed in the resistant genotype at 2 hpi with a TMM of 4.3 and log2FC of 3.14 in comparison with the susceptible genotype. Another fungal inhibitor with high expression across all experimental groups is the Auxin-repressed protein (ARP). ARP was found to be DE between the genotypes at 2 hpi, with a normalised count of 158 in the resistant genotype compared with 21.6 in the susceptible, resulting in a log2FC of 2.9 (Fig. 8d).
Secondary defence responses (6 hpi)
Differential gene expression of resistant genotype at 6 hpi compared to 2 and 24 hpi and to susceptible genotype at the same time-point revealed additional functional genes. A significant increase in the expression from 2 to 6 hpi was noticed in the susceptible genotype for the anti-fungal genes mentioned at the primary defence stage (PMEI, PGIP, ARP; Fig. 8d). An expression pattern similar to that of the anti-fungal genes mentioned above was observed for Laccase diphenol oxidase (PPOl), showing a slight up-regulation at 6 hpi in the susceptible genotype, with a normalised count of 3 and log2FC of 3.2 compared to 2 hpi and overall up-regulation in the resistant genotype, mainly at 2 hpi (Fig. 9a).
Fig. 9.
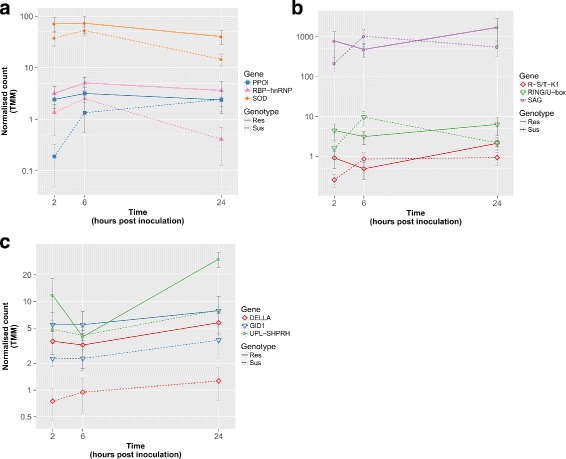
Expression levels of selected genes with exceptional DE trends in the earlier stages of the defence response to A. lentis in ILL7537 and ILL6002 over 2, 6, and 24 hpi. Expression levels of the following genes are presented: R-S/T-K1, RING/U-box and SAG (a); R-S/T-K1, RING/U-box and SAG (b); DELLA, GID1, NB-ARC and UPL-SHPRH (c). A full line represents the expression level in the resistant genotype ILL7537 and the dashed line represents the expression level of the gene in the susceptible genotype ILL6002. Y-axis is in logarithmic scale, error bars represent standard error values between replicates
Bet v 1 domain PR-10 (PR10) protein demonstrated quite an opposite expression pattern in the resistant and the susceptible genotypes. In the susceptible genotype, PR10 was fairly highly expressed at 2 and 24 hpi with TMM of 17.3 and 18.3, dropping slightly to 11.6 at 6 hpi (Fig. 8c). In contrast, the expression of PR10 in the resistant genotype was highest at 2 hpi (TMM = 5.6), before dropping to half of that at 24 hpi, with overall lower expression levels than in the susceptible genotype at all time points, with log2FC of 2.7, 2.1 and 2.8 at 2, 6, and 24 hpi, respectively. Quite similar expression pattern was observed for Botrytis susceptible interactor E3 ubiquitin protein ligase (UPL-BOI), with a peak in its expression at 6 hpi in the resistant genotype with TMM of 2.5, dropping to almost non-existent levels at 24 hpi. In contrast to the resistant genotype, UPL-BOI was up-regulated at 24 hpi with a TMM = 4 (Fig. 8c).
Superoxide dismutase (SOD) showed high expression levels at 2 and 6 hpi in the resistant genotype with TMM >70, dropping to TMM = 40 at 24 hpi, with matching trend at the susceptible genotype, though with lower expression levels (log2FC of about 1.5 at 24 hpi) (Fig. 9a). An RNA binding motif of the heterogeneous nuclear ribonucleoproteins class (RBP-hnRNP) also showed its highest expression at 6 hpi in ILL7537, with a TMM of 5. RBP-hnRNP showed significant over-expression in the resistant genotype at all time points, but chiefly at 24 hpi with a log2FC of 3.3 (Fig. 9a).
Tertiary defence responses (24 hpi)
The expression levels of senescence-associated gene (SAG) were very high, peaking at 24 hpi in the resistant genotype with a normalized count of 1714 and log2FC of 3 compared with the susceptible one at the same time point (Fig. 9b). Interestingly, the expression of SAG in the susceptible genotype was a mirroring image of the resistant one, with completely opposite pattern, showing lowest expression levels at 2 hpi (TMM = 213), increasing to a maximum at 6 hpi (TMM >1000) and dropping again at 24 hpi to a TMM of approximately 550. Two other genes, the E3 Ubiquitin ligase RING/U-box and Receptor-like Serine/Threonine kinase 1 (R-S/T-K1), expressed with exactly the same pattern of the resistant vs. susceptible genotypes as SAG, however, with much lower expression levels (Fig. 9b).
Gibberellin signalling DELLA protein and Gibberellin receptor GID1 exhibited very similar expression trends: a gradual increase in expression with time and over-expression in the resistant genotype, with an average log2FC across all time points of 2.13 and 1.3, respectively (Fig. 9c). GID1 expression was approximately 2.5x higher than that of DELLA at all time points. The expression of NB-ARC domain disease resistance protein demonstrated similar trend, gradually increasing throughout the experiment and up-regulated in the resistant genotype across all time points, mainly at 24 hpi with a TMM of 5 compared to 2 in the susceptible genotype and a log2FC of 1.85 (Fig. 9b). The E3 Ubiquitin ligase SHPRH also demonstrated similar gradual increase in expression in the susceptible genotype, with moderate expression levels at 24 hpi with a TMM of 8, compared to its much elevated expression in the resistant genotype at the same time point, with a TMM of 30, giving a log2FC of 1.9.
Reference genes
A number of genes showed stable expression patterns at moderate levels across all time points and experimental groups assessed. A Myb-related transcription factor-like protein (MYB49) showed the most stable expression, with an average TMM normalised count of 7.00 ± 0.38, followed by a protein phosphatase 2A (PP2A) and a P72 DEAD box, displaying average TMM of 6.90 ± (44) and 8.3 ± (5), respectively (Fig. 8a). These would potentially be useful for future reference-based comparisons of DE of specific gene target studies, such as RT-qPCR.
RT-qPCR validation
The DE patterns of five genes involved in the lentil defence to A. lentis were validated using RT-qPCR and compared to those obtained from the RNA-Seq transcriptome analysis. PP2A demonstrated a constant expression across all time points and both genotypes, with a standard deviation of just ±0.62 in its Cq values, similar to its RNA-Seq derived expression, described in the previous section and therefore it was used as a reference gene in the RT-qPCR analysis.
The target genes, encoding antifungal compounds and transcriptional regulators, consisted of PR2, PR10, RBP, DELLA and PGIP and the DE of each gene between the resistant (ILL7537) and susceptible (ILL6002) genotypes was measured at each time point. The DE results revealed similar expression patterns to those measured by RNA-Seq across the three time points, with significant over-expression at the ILL7537 genotype, with the exception of PR10 (under-expressed in ILL7537 according to the RNA-Seq analysis), whose expression was slightly over-expressed at 6 hpi and slightly under-expressed (although not significantly) at 2 and 24 hpi (Fig. 10).
Fig. 10.
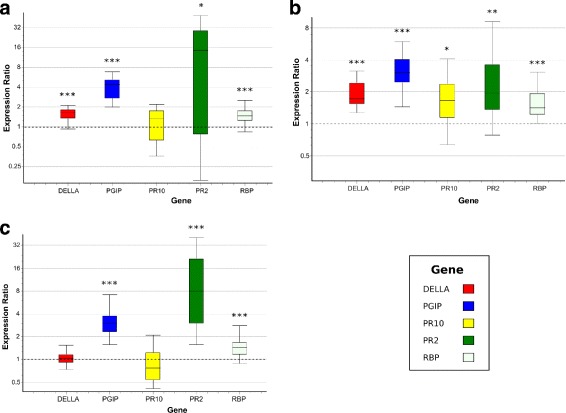
Expression ratios between resistant (ILL7537) and susceptible (ILL6002) lentil genotypes, measured by RT-qPCR. Expression ratios of selected defence-related genes at 2, 6, and 24 hpi (a, b, c, respectively). Asterisks denote statistic significance (DE ≠ 1), with the following p-values: * <0.05, ** <0.01, *** <0.005
Discussion
Exploratory data analysis
The clear separation of the resistant genotype samples from the susceptible genotype samples supports the reported genetic distance between the genotypes [55]. Other than the outlier sample (4C), all samples clustered along with their respective replicates, validating the reproducibility of the assay and the different expression patterns of each experimental group.
Gene set enrichment analysis
Significant enrichment of metabolic regulation pathways was observed and these included changes to photosynthesis genes. This enrichment is likely to be directly related to the bioassay method, as both treated (inoculated with fungus) and control (sprayed with H2O only) plants were placed in darkness after fungal inoculation for 24 h in order to enhance pathogen germination on the plant surface [56, 57]. In addition to the darkness treatment, A. lentis causes severe leaf and stem lesions and eventually wilting, which adversely affects the photosynthesis capabilities of the plant and therefore would have contributed to changes in photosynthesis related transcripts [58, 59].
Gene expression
The DE defence-related genes characterised in this study could be divided into six groups, based on their function and timing of expression relevant to the defence against the A. lentis infection.
Recognition and early signalling
In the primary resistance response, two protein kinases were identified to play key roles in pathogen recognition and early signalling: a leucine-rich repeat receptor kinase (LRR-RK) and a calmodulin domain protein kinase (CDPK). The leucine-rich repeat motif in LRR-RK and serine/threonine kinase-like domain in CDPK are known to be involved in pathogen invasion recognition and signalling, respectively, to trigger the defence response during host-pathogen interaction [15, 51, 60, 61].
In a related defence-response pathway, activation of ethylene response factor (ERF) positively regulates the expression of Ca2+/Calmodulin-dependant protein kinase (SlCCaMK), recently described as a key signalling gene in resistance of tomato to Sclerotinia sclerotiorum [62, 63]. Considering that CDPK demonstrated an expression pattern highly matching that of ERF, along with its sequence and structural similarity to CCaMK [64, 65], this suggests the CDPK-like transcript detected in the present study as a key early signalling molecule in lentil, following the recognition of A. lentis invasion. The LRR-RK, which showed similar expression pattern to ERF and CDPK in the resistant genotype, is likely to trigger the lentil CDPK-like gene for downstream signalling by activating ERF [51, 61, 66]. The reduced levels of LRR-RK that were observed in the susceptible genotype (ILL6002) at this early stage, followed by an increase in its expression at later stages, in correlation with elevated levels of ERF and CDPK, suggests a late and over-stressed defence response in this genotype.
Structural defence response
Subsequent to early signalling, structural and biochemical responses to the invading A. lentis hyphae were detected. As a physical barrier, lentils often accumulate structural compounds at the point of penetration, also known as papilla formation [7, 8] (Fig. 11). Accumulation of xyloglucan endotransglucosylase/hydrolase (XTH) and its function in elongation and restructure of cell walls as part of a physical barrier, was reported in the response of tomato to Cuscuta reflexa [67]. Detection of elevated transcript levels of XTH at 2 hpi in both genotypes in the current study, suggests early response to the inoculation and preparation for papilla formation. One more structural gene, encoding laccase diphenol oxidase (PPOl), was over-expressed in the resistant genotype at 2 hpi and to a lesser extent at 6 hpi, the timing of actual papilla formation [7]. PPOl prevents in vivo pathogen spread [68] by cross-linking cell wall polymers and triggering a hypersensitive response through production of free radicals [69]. Another gene associated with papilla formation in response to spore germination is the gene encoding for Exocyst subunit 70A1 family protein (Exo70A1), through its cellular polarity regulating function [53, 70, 71]. The under-expression of Exo70A1 in the susceptible genotype at 2 hpi was compensated by a steep incline in expression at 6 hpi. The expression levels of both Exo70A1 and PPOl were low at 2 hpi in the ILL6002 susceptible genotype and then increased to “catch up” with the resistant genotype only at 6 hpi, suggesting a delayed recognition and response to A. lentis compared to ILL7537.
Fig. 11.
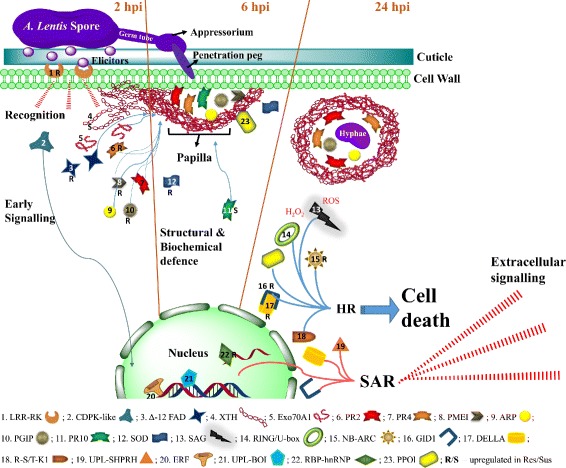
Defence-related molecules involved in response of lentil to A. lentis during the first 24 h
Another gene which was over-expressed in the resistant genotype and practically missing from the susceptible one, thus seemingly playing a part in the structural defence response to A. lentis, is Delta (12)-FAD. FAD proteins are essential for maintaining cellular function and influence a variety of processes such as the regulation of membrane fatty acid profiles in different tissues, different developmental stages, and in response to abiotic and biotic stresses [72]. Accumulation of Delta (12)-FAD mRNA was previously demonstrated in parsley cells following treatment by a fungal elicitor, Pep25, and was reported to be involved in the complex defence response by reinforcing existing cell walls [73].
Biochemical defence response
During a biochemical defence response to A. lentis, lentils use anti-fungal compounds including pathogenesis-related (PR) proteins and reactive oxygen species (ROS), as was previously described [8, 13]. In the present study, members of three families of PR proteins were identified to be significantly DE in ILL7537 in response to A. lentis when compared to ILL6002. The three PR protein families include: PR2, 4 and 10 (Fig. 11 and Table 5). PR2 proteins catalyse the hydrolytic cleavage of 1,3- β-D-glucosidic linkages in β-1,3-glucans present in the fungal cell walls, and cause cell lysis and death in fungi [74]; and were shown to play a vital role in defence against pathogenic fungi such as Fusarium oxysporum in chickpea [75]. PR2 protein is involved not only in hydrolysis of fungal-cell components, but it also releases elicitors from the walls of fungi, which in turn may be recognised by plant receptor molecules and stimulate various downstream signalling and defence responses [76]. PR4 protein disrupts fungal cell polarity and inhibits its growth, reacting with nascent chitin at the hyphal tip [13] and its involvement in lentil defence against A. lentis was previously characterised in some depth [77].
Table 5.
Key genes in lentil defence-response to A. lentis
| Functional groups | Defence gene name | Gene abbreviation | Dominant genotypea |
|---|---|---|---|
| Recognition: Receiving & signalling molecules | Calmodulin domain protein kinase-like | CDPK-like | |
| LRR receptor-like kinase | LRR-RK | ILL6002 | |
| Receptor-like serine/ threonine protein kinase 1 | R-S/T-K1 | ||
| NB-ARC domain disease resistance protein | NB-ARC | ILL7537 | |
| Botrytis susceptible interactor E3 ubiquitin protein ligase | UPL-BOI | ||
| Early structural response: Papilla formation | Exocyst subunit 70A1 protein | EXO70A1 | |
| Laccase diphenol oxidase | PPOl | ||
| Delta (12)-fatty-acid desaturase | Delta (12)-FAD | ILL7537 | |
| Xyloglucan endotransglucosylase/ hydrolase | XTH | ILL6002 | |
| Early Biochemical response: Pathogenesis-related & anti-fungal compounds | PR protein 2 – O-glycosyl hydrolase | PR2 | ILL7537 |
| PR protein 4 – Thaumatin family | PR4 | ||
| PR protein 10 – Bet V I type | PR10 | ILL6002 | |
| Plant invertase pectin methylesterase inhibitor | PMEI | ||
| Auxin-repressed protein | ARP | ILL7537 | |
| Polygalacturonase inhibitor | PGIP | ILL7537 | |
| Superoxide dismutase | SOD | ILL7537 | |
| Regulation of defence-related transcripts: DNA & RNA binding proteins | Ethylene response factor | ERF | |
| Heterogeneous nuclear ribonucleoprotein RNA binding motif | RBP-hnRNP | ILL7537 | |
| Senescence associated gene | SAG | ||
| Hypersensitive response: Cell death & SAR | RING/U-box E3 ubiquitin protein ligase | RING/ U-box | |
| Gibberellin signalling DELLA protein | DELLA | ILL7537 | |
| Gibberellin receptor | GID1 | ILL7537 | |
| E3 ubiquitin-protein ligase SHPRH | UPL-SHPRH |
aDominant genotype was specified for genes whose expression was generally up-regulated at all time points in one of the genotypes: ILL7537 – Resistant, ILL6002 – Susceptible
In contrast, PR10, which was over-expressed in the susceptible genotype at all time points, is known to exhibit RNase activity on invading intracellular fungal hyphae [78] and was shown to accumulate and correlate with increasing fungal biomass [79]. Therefore, the expression of PR10 indicates that the susceptible genotype was challenged by higher fungal load throughout the experiment. The Botrytis susceptible1 interactor (UPL-BOI) gene showed a similar expression trend to that of PR10. UPL-BOI is an E3 ubiquitin protein ligase, which regulates pathogen resistance responses in Arabidopsis to Botrytis cinerea [80]. Therefore, UPL-BOI may be involved in expression regulation of PR10 in lentil in response to A. lentis.
Other DE genes with known antifungal activity were plant invertase pectin methylesterase inhibitor (PMEI), polygalacturonase inhibitor (PGIP), and auxin-repressed protein (ARP), all significantly up-regulated at 2 hpi in ILL7537 following A. lentis inoculation. PMEI reduces the susceptibility of the plant wall to fungal endopolygalacturonases and was previously reported to aid in Arabidopsis defence to B. cinerea and Pectobacterium carotovorum [81, 82], in wheat defence to Bipolaris sorokiniana and F. graminearum [83] and in pepper defence to Xanthomonascampestris pv. vesicatoria [84]. Similarly, PGIP limits the destructive potential of fungal polygalacturonases through specific binding and inhibition of them [85]. PGIP also increases the production of oligogalacturonides, leading to the accumulation of phytoalexin, an antibiotic, in plant tissue [86], as reported in tomato defence to B. cinerea [87] and Lathyrus sativus defence to Aspergillus niger and Rhizopus spp. [88]. Meanwhile, ARP inhibits pathogens by either producing auxin or manipulating host auxin [89] and was shown to be involved in rice defence to Magnaporthe grisea and Striga hermonthica [90]. Together, PMEI, PGIP and ARP, thus appear to operate in the lentil ILL7537 resistant genotype to control the spread and growth of A. lentis in the early stages following invasion.
Hypersensitive reaction and cell death
A hypersensitive reaction in the infected plant is triggered by an oxidative burst and is characterised by an increase in free radicals that leads to localised cell death. As previously determined through histopathological and molecular studies, this defence response is likely to be important in resistant lentil genotypes [7, 8, 13]. This was further demonstrated in the current study through DE of the senescence associated gene (SAG), which highly over-expressed at 24 hpi in ILL7537. SAGs are induced by free radicals, such as ROS and H2O2, leading to a programmed cell death [91, 92] and have been implicated in the defence of Arabidopsis to several biotrophic pathogens [93]. This is the first report of SAG involvement in defence to a necrotrophic pathogen. A similar expression trend was observed for the gene encoding NB-ARC domain disease resistance protein (NB-ARC), a domain of NB-LRRs that regulates signal transduction leading from recognition to hypersensitive response-signalling and cell death [94, 95]. The gene encoding RING/U-box protein (RING/U-box), which is another E3 ubiquitin ligase, was also significantly over-expressed at the same time points as SAG and NB-ARC. RING/U-box proteins are involved in the hypersensitive defence response of tomato to Phytophthora infestans [96] and in pathogen-instigated programmed cell death of Tobacco [97]. Therefore, it can be assumed that SAG, NB-ARC and RING/U-box are key control genes for the hypersensitive response of lentil to A. lentis, triggered in an effort to contain the invading pathogen.
Following the hypersensitive response, the plant activates mechanisms to protect its healthy cells from further damage, as demonstrated by the over-expression of superoxide dismutase (SOD) in ILL7537 genotype and its elevated expression levels at 6 hpi. This serves to regulate the redox status of the plant cells and protect from the oxidative burst and generated ROS [98].
Systemic acquired resistance (SAR)
Systemic acquired resistance (SAR) is the long distance signalling of pathogen recognition and defence induced by signal molecules and plant hormones. This process may confer long-lasting protection against an invading pathogen and together with the hypersensitive reaction, signal the last stage of early defence responses [99]. In the defence transcriptome of lentil to A. lentis, three putative SAR-associated genes, Gibberellin signalling DELLA protein (DELLA), Gibberellin receptor (GID1) and an E3 ubiquitin ligase (UPL-SHPRH), had similar DE patterns with highest transcription levels observed at 24 hpi and general over-expression in ILL7537. DELLA proteins promote defence to necrotrophic fungal pathogens by activating jasmonic acid/ethylene-dependent defence responses [100] and by regulating ROS levels [100, 101]. GID1 binds to DELLA, which then leads to ubiquitination and degradation of DELLA during SAR signalling [102, 103]. Meanwhile, E3 UPLs are involved in recognition, signalling, hypersensitive reaction and cell death mechanisms in Arabidopsis [104, 105] and rice [106–109]. Therefore, considering their expression trends in previous and current studies, we suggest that DELLA, GID1 and UPL-SHPRH proteins are involved in SAR signalling in the early lentil defence mechanisms to A. lentis.
Transcription regulators
Since the differences observed between the resistant and the susceptible genotypes in the current study were transcriptomic changes, it is also useful to observe changes in the expression of transcription regulators. Therefore, a DE DNA binding transcription factor was identified, encoding Ethylene response factor (ERF). ERF stimulates the expression of PR proteins gene promoters and as discussed earlier, it increases the expression level of early signalling molecules [62, 64, 66]. The decline in ERF’s expression in the resistant genotype at 24 hpi, may indicate the success of the early defence responses mentioned previously for ILL7537, whereas the late response of ILL6002 required continuous elevated expression of the defence-related proteins.
Moreover, an RNA binding protein of the heterogeneous nuclear ribonucleoproteins (RBP-hnRNPs) class, up-regulated in ILL7537, is a member of an RNA binding transcriptional factors family that regulate post-transcriptional gene expression [110]. These RNA binding transcriptional factors play key role in regulating transcription of genes in response to biotic and a-biotic stress in plants [111, 112]. The hnRNP-like protein AtGRP7 have been shown to play a regulatory role on SOD and ARP in Arabidopsis, by affecting the processing of their regulatory microRNAs [113]. AtGRP7 was further suggested to be involved in defence against fungal and bacterial infections in Arabidopsis by interacting with specific LRR-RKs [114]. The hnRNP that was identified in this study was up-regulated in ILL7537, in a pattern matching that of SOD and ARP (Figs. 8 and 9), suggesting it performs a similar function to AtGRP7.
Reference genes and RT-qPCR validation
Protein phosphatase 2A (PP2A) and P72 DEAD box RNA helicase have previously been used as reference genes for relative DE analysis in plant-pathogen interaction studies [115, 116]. Their stable expression across all the treatments and samples in the current study, along with the RT-qPCR DE results of the selected defence genes, which overall conform to those found in the RNA-Seq analysis, validate the reliability and reproducibility of the RNA-Seq quantification and downstream DE analysis.
MYB49, whose exact function is yet to be discovered, is a member of a diverse family of DNA-binding transcription factors [117, 118]. Considering its most stable expression in our data, which supersedes PP2A and P72 DEAD box, it should also be considered as a reference gene in similar plant-pathogen DE studies.
Conclusions
The results of the current study are highly concordant with the physiology of the interaction between lentil and A. lentis and similar pathosystems [7]. Overall representation of the molecules involved in the defence response of lentil to A. lentis during the first 24 h is summarised in Table 5 and Fig. 11.
The majority of time-dependant DE defence-related genes between the ILL7537 resistant genotype and the ILL6002 susceptible genotype were found at 2 hpi, suggesting that the resistant genotype demonstrated an earlier and faster detection and signalling response to the A. lentis infection, thus being better prepared molecularly to deploy critical defence response proteins. In addition, overall higher expression levels of structural defence response genes were found in the resistant genotype regardless of the time post inoculation, indicating an innate ability to form stronger structural barriers against the fungus compared with the susceptible genotype.
The information provided by this study further extends the available knowledge of lentil resistance to A. lentis infection and may assist in future efforts to identify and develop additional resistant cultivars and management strategies, thereby reducing the losses caused by the pathogen.
Additional files
Trimmed Mean of M-values (TMM) of the estimated expression of selected defence-related genes in lentil–A. lentis transcriptome. Treatment – Mock (M) for H2O control, Treatment (T) for ALP2 A. lentis inoculation. HPI – Hours post inoculation. Norm_TMM_count – Trimmed Mean of M-values of the raw estimated counts, as calculated by edgeR. Count_SE – Standard error values per gene at each genotype/time point, calculated from TMM counts of subgroup replicates (n =3). (XLSX 28 kb)
Detailed information and statistics of read and base trimming per sample read file. (XLSX 17 kb)
Acknowledgements
We would like to acknowledge the Griffith University ‘Gowonda’ High Performance Computing Cluster for kindly providing the infrastructure and computing resources for the NGS data analysis.
Funding
This research was funded by the following Grains Research Development Corporation (GRDC) projects: #CUR00014, “New technologies and biological concepts for pre-breeding resistance to the Ascochyta blight disease of pea, chickpea, lentil and faba bean”; and #CUR00023, “Ascochyta blight of pulses - integrating development of novel selection methods, mining germplasm for resistance and pathogen surveillance”. The work was also supported in part by the Australian National Health and Medical Research Council grants (#1059775 and #1083450) to YZ and Griffith University’s International Postgraduate Scholarship (GUIPRS) to MK. The funding bodies did not play active part in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript, but rather provided financial support to cover costs of salaries (IB), stipends (MK), bioassays and sequencing.
Availability of data and materials
The data supporting the conclusions of this article (raw RNA-Seq reads) are available in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA): SRP075524, https://trace.ncbi.nlm.nih.gov/Traces/sra/?study=SRP075524.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
- bp
Base pairs (measuring unit)
- DE
Differential expression/differentially expressed
- FC
Fold change
- FDR
False discovery rate
- GO
Gene Ontology
- hpi
Hours post inoculation (measuring unit)
- KEGG
Kyoto encyclopedia of genes and genomes
- NCBI
National Center for Biotechnology Information
- ORF
Open reading frame
- PCR
Polymerase chain reaction
- PR
Pathogenesis-related
- QC
Quality check
- RNA-Seq
RNA-sequencing
- ROS
Reactive oxygen species
- RQI
RNA quality indicator
- RT-qPCR
Quantitative reverse transcription PCR
- SRA
Sequence read archive
- TMM
Trimmed mean of m-values
Ethics approval and consent to participate
Plants used in this study were grown from seeds sourced from the University of Melbourne collection. Sampling of plant material was performed in compliance with institutional guidelines. No further approvals, licences or permissions were required since no sampling was conducted from wild and/or native flora.
Consent for publication
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4488-1) contains supplementary material, which is available to authorized users.
Authors’ contributions
MK conducted sample collection and preparation for RNA-Seq, wrote the manuscript and analysed the data. IB and PJW processed and analysed the sequencing data and assisted in drafting the manuscript. GS conducted the bioassays and prepared the RNA libraries. VB and NM prepared the cDNA libraries and performed the RNA-Sequencing. YY and YZ contributed to the RNA-Seq data analysis. SHB provided useful insights while writing the manuscript. RF conceived of the study, participated in its design, assisted in data analysis and helped to draft the manuscript. All authors have read and approved the final manuscript.
Contributor Information
Mahsa Khorramdelazad, Email: mahsa.khorramdelazad@griffithuni.edu.au.
Ido Bar, Email: i.bar@griffith.edu.au.
Paul Whatmore, Email: Paul.Whatmore@nifes.no.
Gabrielle Smetham, Email: gsmetham@yahoo.com.au.
Vijay Bhaaskaria, Email: s3410221@student.rmit.edu.au.
Yuedong Yang, Email: yuedong.yang@griffith.edu.au.
Shahla Hosseini Bai, Email: s.hosseini-bai@griffith.edu.au.
Nitin Mantri, Email: nitin.mantri@rmit.edu.au.
Yaoqi Zhou, Email: yaoqi.zhou@gmail.com.
Rebecca Ford, Email: Rebecca.ford@griffith.edu.au.
References
- 1.FAOSTAT . Food and Agriculture Organization of the United Nations (FAO) statistical yearbook 2014. Statistical yearbook. Rome: Fisheries and Aquaculture Department, Food and Agriculture Organization (FAO) of the United Nations; 2014. [Google Scholar]
- 2.Nene YL, Hanounik SB, Qureshi SH, Sen B. Fungal and bacterial foliar diseases of pea, lentil, faba bean and chickpea. In: Summerfield RJE, editor. World Crops: Cool Season Food Legumes: A Global Perspective of the Problems and Prospects for Crop Improvement in Pea, Lentil, Faba Bean and Chickpea. Dordrecht: Springer Netherlands; 1988. [Google Scholar]
- 3.Taylor PWJ, Ford R. Diagnostics, genetic diversity and pathogenic variation of ascochyta blight of cool season food and feed legumes. Eur J Plant Pathol. 2007;119(1):127–33. doi: 10.1007/s10658-007-9177-x. [DOI] [Google Scholar]
- 4.Murray GM, Brennan JP. The Current and Potential Costs from Diseases of Pulse Crops in Australia. Canberra: Grains Research and Development Corporation; 2012. [Google Scholar]
- 5.Galloway J, MacLeod WJ, Lindbeck KD. Formation of Didymella lentis, the teleomorph of Ascochyta lentis, on lentil stubble in the field in Victoria and Western Australia. Australas Plant Pathol. 2004;33(3):449–50. doi: 10.1071/AP04033. [DOI] [Google Scholar]
- 6.Nasir M, Bretag TW. Pathogenic variability in australian isolates of Ascochyta lentis. Australas Plant Pathol. 1997;26(4):217–20. doi: 10.1071/AP97036. [DOI] [Google Scholar]
- 7.Roundhill SJ, Fineran BA, Cole ALJ, Ingerfeld M. Structural aspects of Ascochyta Blight of lentil. Can J Bot. 1995;73(3):485–97. doi: 10.1139/b95-049. [DOI] [Google Scholar]
- 8.Sambasivam P, Taylor PWJ, Ford R. Pathogenic variation and virulence related responses of Ascochytalentis on lentil. Eur J Plant Pathol. 2016:1–13. 10.1007/s10658-016-0999-2.
- 9.Ahmad M, Russell AC, McNeil DL. Identification and genetic characterization of different resistance sources to Ascochyta blight within the genus lens. Euphytica. 1997;97(3):311–5. doi: 10.1023/A:1003095423132. [DOI] [Google Scholar]
- 10.Ford R, Pang EC, Taylor PWJ. Genetics of resistance to ascochyta blight (Ascochyta lentis) of lentil and the identification of closely linked rapd markers. Theor Appl Genet. 1999;98:93–8. doi: 10.1007/s001220051044. [DOI] [Google Scholar]
- 11.Ye G, McNeil DL, Hill GD. Breeding for resistance to lentil Ascochyta blight. Plant Breed. 2002;121:185–91. doi: 10.1046/j.1439-0523.2002.00705.x. [DOI] [Google Scholar]
- 12.Davidson J, Smetham G, Russ MH, McMurray L, Rodda M, Krysinska-Kaczmarek M, Ford R. Changes in Aggressiveness of the Ascochyta lentis Population in Southern Australia. Front Plant Sci. 2016; 7. 10.3389/fpls.2016.00393. [DOI] [PMC free article] [PubMed]
- 13.Mustafa BM, Coram TE, Pang ECK, Taylor PWJ, Ford R. A cDNA microarray approach to decipher lentil (Lens culinaris) responses to Ascochyta lentis. Australas Plant Pathol. 2009;38(6):617–31. doi: 10.1071/AP09048. [DOI] [Google Scholar]
- 14.Gusberti M, Gessler C, Broggini GA. RNA-seq analysis reveals candidate genes for ontogenic resistance in malus-venturia pathosystem. PLoS ONE. 2013;8(11):78457. doi: 10.1371/journal.pone.0078457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han Y, Zhang K, Yang J, Zhang N, Fang A, Zhang Y, Liu Y, Chen Z, Hsiang T, Sun W. Differential expression profiling of the early response to Ustilaginoidea virens between false smut resistant and susceptible rice varieties. BMC Genomics. 2015;16:955. doi: 10.1186/s12864-015-2193-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nanoth Vellichirammal N, Wang H, Eyun SI, Moriyama EN, Coates BS, Miller NJ, Siegfried BD. Transcriptional analysis of susceptible and resistant european corn borer strains and their response to Cry1F protoxin. BMC Genomics. 2015;16:558. doi: 10.1186/s12864-015-1751-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ocaña S, Seoane P, Bautista R, Palomino C, Claros GM, Torres AM, Madrid E. Large-scale transcriptome analysis in faba bean (Vicia faba L,) under Ascochyta fabae infection. PLOS ONE. 2015;10(8):0135143. doi: 10.1371/journal.pone.0135143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sudheesh S, Verma P, Forster JW, Cogan NOI, Kaur S. Generation and Characterisation of a Reference Transcriptome for Lentil (Lens culinaris Medik.)Int J Mol Sci. 2016; 17(11). 10.3390/ijms17111887. [DOI] [PMC free article] [PubMed]
- 19.Sharpe AG, Ramsay L, Sanderson LA, Fedoruk MJ, Clarke WE, Li R, Kagale S, Vijayan P, Vandenberg A, Bett KE. Ancient orphan crop joins modern era: Gene-based SNP discovery and mapping in lentil. BMC Genomics. 2013;14:192. doi: 10.1186/1471-2164-14-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaur S, Cogan NO, Pembleton LW, Shinozuka M, Savin KW, Materne M, Forster JW. Transcriptome sequencing of lentil based on second-generation technology permits large-scale unigene assembly and SSR marker discovery. BMC Genomics. 2011;12:265. doi: 10.1186/1471-2164-12-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, MacManes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, LeDuc RD, Friedman N, Regev A. De novo transcript sequence reconstruction from RNA-seq: reference generation and analysis with Trinity. Nat Protoc. 2013;8(8):1494–512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henschel R, Lieber M, Wu LS, Nista PM, Haas BJ, LeDuc RD. Proceedings of the 1st Conference of the Extreme Science and Engineering Discovery Environment: Bridging from the eXtreme to the Campus and Beyond. XSEDE ’12. New York: ACM; 2012. Trinity RNA-seq assembler performance optimization. [Google Scholar]
- 24.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7(10):1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer ELL, Tate J, Punta M. Pfam: the protein families database. Nucleic Acids Res. 2014;42(D1):222–30. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chamberlain SA, Szöcs E. Taxize: Taxonomic search and retrieval in R. F1000 Research. 2013; 2. 10.12688/f1000research.2-191.v2. [DOI] [PMC free article] [PubMed]
- 28.Chamberlain S, Szöcs E. taxize: taxonomic search and retrieval in R [version 2; referees: 3 approved]. F1000Research [Internet]. 2013; 2. Available from: http://openr.es/24v. [DOI] [PMC free article] [PubMed]
- 29.GO Consortium Gene ontology annotations and resources. Nucleic Acids Res. 2013;41(D1):530–5. doi: 10.1093/nar/gks1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2011;40(D1):109–14. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, Kong L, Gao G, Li CY, Wei L. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;39(suppl 2):316–22. doi: 10.1093/nar/gkr483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8(10):785–6. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 33.Krogh A, Larsson B, von Heijne G. Sonnhammer ELL. Predicting transmembrane protein topology with a hidden markov model: application to complete genomes. J Mol Biol. 2001;305(3):567–80. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 34.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–2. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 35.Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34(5):525–7. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 36.Dillies MA, Rau A, Aubert J, Hennequet-Antier C, Jeanmougin M, Servant N, Keime C, Marot G, Castel D, Estelle J, Guernec G, Jagla B, Jouneau L, Laloë D, Gall CL, Schaëffer B, Crom SL, Guedj M, Jaffrézic F. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief Bioinform. 2013;14(6):671–83. doi: 10.1093/bib/bbs046. [DOI] [PubMed] [Google Scholar]
- 37.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Y, Lun ATL, Smyth GK. Differential Expression Analysis of Complex RNA-seq Experiments Using edgeR. In: Datta S, Nettleton D, editors. Statistical Analysis of Next Generation Sequencing Data. Frontiers in Probability and the Statistical Sciences. Switzerland: Springer International Publishing; 2014. [Google Scholar]
- 39.McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40(10):4288–97. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J R Stat Soc Ser B Methodol. 1995;57(1):289–300. [Google Scholar]
- 41.Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11(2):14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruijter JM, Pfaffl MW, Zhao S, Spiess AN, Boggy G, Blom J, et al. Evaluation of qPCR curve analysis methods for reliable biomarker discovery: Bias, resolution, precision, and implications. Methods. 2013;59:32–46. doi: 10.1016/j.ymeth.2012.08.011. [DOI] [PubMed] [Google Scholar]
- 43.Ruijter JM, Ramakers C, Hoogaars WMH, Karlen Y, Bakker O, van den Hoff MJB, et al. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009;37:e45. doi: 10.1093/nar/gkp045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruijter JM, Ruiz Villalba A, Hellemans J, Untergasser A, van den Hoff MJB. Removal of between-run variation in a multi-plate qPCR experiment. Biomolecular Detection and Quantification. 2015;5:10–4. doi: 10.1016/j.bdq.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (RESTⒸ) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:research0034.1–11. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ihaka R, Gentleman R. R: A Language for Data Analysis and Graphics. J Comput Graph Stat. 1996;5:299–314. [Google Scholar]
- 49.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Z, Gerstein M, Snyder M. RNA-Seq: A revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morris ER, Walker JC. Receptor-like protein kinases: the keys to response. Curr Opin Plant Biol. 2003;6(4):339–42. doi: 10.1016/S1369-5266(03)00055-4. [DOI] [PubMed] [Google Scholar]
- 52.Cosgrove DJ. Growth of the plant cell wall. Nat Rev Mol Cell Biol. 2005;6(11):850–61. doi: 10.1038/nrm1746. [DOI] [PubMed] [Google Scholar]
- 53.žárský V, Kulich I, Fendrych M, Pecenková T. Exocyst complexes multiple functions in plant cells secretory pathways. Curr Opin Plant Biol. 2013;16(6):726–33. doi: 10.1016/j.pbi.2013.10.013. [DOI] [PubMed] [Google Scholar]
- 54.Di Matteo A, Bonivento D, Tsernoglou D, Federici L, Cervone F. Polygalacturonase-inhibiting protein (PGIP) in plant defence: a structural view. Phytochemistry. 2006;67(6):528–33. doi: 10.1016/j.phytochem.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 55.Lombardi M, Materne M, Cogan NOI, Rodda M, Daetwyler HD, Slater AT, Forster JW, Kaur S. Assessment of genetic variation within a global collection of lentil (Lens culinaris Medik,) cultivars and landraces using SNP markers. BMC Genetics. 2014;15:150. doi: 10.1186/s12863-014-0150-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Darko E, Heydarizadeh P, Schoefs B, Sabzalian MR. Photosynthesis under artificial light: The shift in primary and secondary metabolism. Phil Trans R Soc B: Biol Sci. 2014;369:1640. doi: 10.1098/rstb.2013.0243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Florian A, Nikoloski Z, Sulpice R, Timm S, Araújo WL, Tohge T, Bauwe H, Fernie AR. Analysis of Short-Term Metabolic Alterations in Arabidopsis Following Changes in the Prevailing Environmental Conditions. Mol Plant. 2014;7(5):893–911. doi: 10.1093/mp/ssu008. [DOI] [PubMed] [Google Scholar]
- 58.Garry G, Jeuffroy MH, Ney B, Tivoli B. Effects of Ascochyta blight (Mycosphaerella pinodes) on the photosynthesizing leaf area and the photosynthetic efficiency of the green leaf area of dried-pea (Pisum sativum) Plant Pathol. 1998;47(4):473–9. doi: 10.1046/j.1365-3059.1998.00259.x. [DOI] [Google Scholar]
- 59.Goodwin PH. Effect of common bacterial blight on leaf photosynthesis of bean. Can J Plant Pathol. 1992;14(3):203–6. doi: 10.1080/07060669209500875. [DOI] [Google Scholar]
- 60.Song WY, Wang GL, Chen LL, Kim HS, Pi LY, Holsten T, Gardner J, Wang B, Zhai WX, Zhu LH. A receptor kinase-like protein encoded by the rice disease resistance gene, Xa21. Science. 1995;270(5243):1804–6. doi: 10.1126/science.270.5243.1804. [DOI] [PubMed] [Google Scholar]
- 61.Afzal AJ, Wood AJ, Lightfoot DA. Plant receptor-like serine threonine kinases: Roles in signaling and plant defense. Mol Plant-Microbe Interact. 2008;21(5):507–17. doi: 10.1094/MPMI-21-5-0507. [DOI] [PubMed] [Google Scholar]
- 62.Romeis T, Ludwig AA, Martin R, Jones JDG. Calcium-dependent protein kinases play an essential role in a plant defence response. EMBO J. 2001;20(20):5556–567. doi: 10.1093/emboj/20.20.5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Romeis T, Herde M. From local to global: CDPKs in systemic defense signaling upon microbial and herbivore attack. Curr Opin Plant Biol. 2014;20:1–10. doi: 10.1016/j.pbi.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 64.Wang JP, Munyampundu JP, Xu YP, Cai XZ. Phylogeny of plant calcium and calmodulin-dependent protein kinases (CCaMKs) and functional analyses of tomato CCaMK in disease resistance. Front Plant Sci. 2015; 6. 10.3389/fpls.2015.01075. [DOI] [PMC free article] [PubMed]
- 65.Wang JP, Xu YP, Munyampundu JP, Liu TY, Cai XZ. Calcium-dependent protein kinase (CDPK) and CDPK-related kinase (CRK) gene families in tomato: Genome-wide identification and functional analyses in disease resistance. Mol Gen Genomics. 2016;291(2):661–76. doi: 10.1007/s00438-015-1137-0. [DOI] [PubMed] [Google Scholar]
- 66.Ludwig AA, Saitoh H, Felix G, Freymark G, Miersch O, Wasternack C, Boller T, Jones JDG, Romeis T. Ethylene-mediated cross-talk between calcium-dependent protein kinase and MAPK signaling controls stress responses in plants. Proc Natl Acad Sci USA. 2005;102(30):10736–41. doi: 10.1073/pnas.0502954102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Albert M, Werner M, Proksch P, Fry SC, Kaldenhoff R. The cell wall-modifying xyloglucan endotransglycosylase/hydrolase LeXTH1 is expressed during the defence reaction of tomato against the plant parasite Cuscuta reflexa. Plant Biol (Stuttgart, Germany) 2004;6(4):402–7. doi: 10.1055/s-2004-817959. [DOI] [PubMed] [Google Scholar]
- 68.Walker JRL, Ferrar PH. Diphenol oxidases, enzyme-catalysed browning and plant disease resistance. Biotechnol Genet Eng Rev. 1998;15(1):457–98. doi: 10.1080/02648725.1998.10647966. [DOI] [PubMed] [Google Scholar]
- 69.Sklodowska M, Gajewska E, Kuzniak E, Wielanek M, Mikicinski A, Sobiczewski P. Antioxidant profile and polyphenol oxidase activities in apple leaves after Erwinia amylovora infection and pretreatment with a benzothiadiazole-type resistance inducer (BTH) J Phytopathol. 2011;159(7-8):495–504. doi: 10.1111/j.1439-0434.2011.01793.x. [DOI] [Google Scholar]
- 70.Kulich I, Cole R, Drdová E, Cvrcková F, Soukup A, Fowler J, žárský V. Arabidopsis exocyst subunits SEC8 and EXO70A1 and exocyst interactor ROH1 are involved in the localized deposition of seed coat pectin. New Phytol. 2010;188(2):615–25. doi: 10.1111/j.1469-8137.2010.03372.x. [DOI] [PubMed] [Google Scholar]
- 71.Pecenková T, Hála M, Kulich I, Kocourková D, Drdová E, Fendrych M, Toupalová H, Zársky V. The role for the exocyst complex subunits Exo70B2 and Exo70H1 in the plant-pathogen interaction. J Exp Bot. 2011;62(6):2107–16. doi: 10.1093/jxb/erq402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang D, Pirtle IL, Park SJ, Nampaisansuk M, Neogi P, Wanjie SW, Pirtle RM, Chapman KD. Identification and expression of a new delta-12 fatty acid desaturase (FAD2-4) gene in upland cotton and its functional expression in yeast and Arabidopsis thaliana plants. Plant Physiol Biochem. 2009;47(6):462–71. doi: 10.1016/j.plaphy.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 73.Kirsch C, Hahlbrock K, Somssich IE. Rapid and transient induction of a parsley microsomal [delta]12 fatty acid desaturase mRNA by fungal elicitor. Plant Physiol. 1997;115(1):283–9. doi: 10.1104/pp.115.1.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Datta SK, Muthukrishnan S. Pathogenesis-Related Proteins in Plants. Florida: CRC Press; 1999. [Google Scholar]
- 75.Saikia R, Singh BP, Kumar R, Arora DK. Detection of pathogenesis-related proteins-chitinase and β-1,3-glucanase in induced chickpea. Current Science (India) 2005;89:659–63. [Google Scholar]
- 76.Ren YY, West CA. Elicitation of diterpene biosynthesis in rice (Oryza sativa L.) by chitin. Plant Physiol. 1992;99(3):1169–78. doi: 10.1104/pp.99.3.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vaghefi N, Mustafa B, Dulal N, Selby-Pham J, Taylor P, Ford R. A novel pathogenesis-related protein (LcPR4a) from lentil, and its involvement in defence against Ascochyta lentis. Phytopathol Mediterr. 2013;52(1):192–201. [Google Scholar]
- 78.Liu JJ, Ekramoddoullah AKM. The family 10 of plant pathogenesis-related proteins: Their structure, regulation, and function in response to biotic and abiotic stresses. Physiol Mol Plant Pathol. 2006;68(1-3):3–13. doi: 10.1016/j.pmpp.2006.06.004. [DOI] [Google Scholar]
- 79.Castro A, Vidal S, Ponce de León I. Moss Pathogenesis-Related-10 Protein Enhances Resistance to Pythium irregulare in Physcomitrella patens and Arabidopsis thaliana. Front Plant Sci. 2016; 7. 10.3389/fpls.2016.00580. [DOI] [PMC free article] [PubMed]
- 80.Luo H, Laluk K, Lai Z, Veronese P, Song F, Mengiste T. The arabidopsis Botrytis Susceptible 1 interactor defines a subclass of RING E3 ligases that regulate pathogen and stress responses. Plant Physiol. 2010;154(4):1766–82. doi: 10.1104/pp.110.163915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Raiola A, Lionetti V, Elmaghraby I, Immerzeel P, Mellerowicz EJ, Salvi G, Cervone F, Bellincampi D. Pectin methylesterase is induced in Arabidopsis upon infection and is necessary for a successful colonization by necrotrophic pathogens. Mol Plant-Microbe Interact. 2010;24(4):432–40. doi: 10.1094/MPMI-07-10-0157. [DOI] [PubMed] [Google Scholar]
- 82.Lionetti V, Raiola A, Camardella L, Giovane A, Obel N, Pauly M, Favaron F, Cervone F, Bellincampi D. Overexpression of pectin methylesterase inhibitors in arabidopsis restricts fungal infection by Botrytis cinerea. Plant Physiol. 2007;143(4):1871–80. doi: 10.1104/pp.106.090803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Volpi C, Janni M, Lionetti V, Bellincampi D, Favaron F, D’Ovidio R. The ectopic expression of a pectin methyl esterase inhibitor increases pectin methyl esterification and limits fungal diseases in wheat. Mol Plant-Microbe Interact. 2011;24(9):1012–19. doi: 10.1094/MPMI-01-11-0021. [DOI] [PubMed] [Google Scholar]
- 84.An SH, Sohn KH, Choi HW, Hwang IS, Lee SC, Hwang BK. Pepper pectin methylesterase inhibitor protein CaPMEI1 is required for antifungal activity, basal disease resistance and abiotic stress tolerance. Planta. 2008;228(1):61–78. doi: 10.1007/s00425-008-0719-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lorenzo GD, D’Ovidio R, Cervone F. The role of polygalacturonase-inhibiting proteins (pgips) in defense against pathogenic fungi. Annu Rev Phytopathol. 2001;39(1):313–35. doi: 10.1146/annurev.phyto.39.1.313. [DOI] [PubMed] [Google Scholar]
- 86.Cervone F, Hahn MG, De Lorenzo G, Darvill A, Albersheim P. Host-Pathogen Interactions. Plant Physiol. 1989;90(2):542–8. doi: 10.1104/pp.90.2.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Powell ALT, van Kan J, ten Have A, Visser J, Greve LC, Bennett AB, Labavitch JM. Transgenic expression of pear PGIP in tomato limits fungal colonization. Mol Plant-Microbe Interact. 2000;13(9):942–50. doi: 10.1094/MPMI.2000.13.9.942. [DOI] [PubMed] [Google Scholar]
- 88.Tamburino R, Chambery A, Parente A, Di Maro A. A novel polygalacturonase-inhibiting protein (PGIP) from Lathyrus sativus L, seeds. Protein Peptide Lett. 2012;19(8):820–5. doi: 10.2174/092986612801619561. [DOI] [PubMed] [Google Scholar]
- 89.Wang D, Pajerowska-Mukhtar K, Culler AH, Dong X. Salicylic acid inhibits pathogen growth in plants through repression of the auxin signaling pathway. Curr Biol. 2007;17(20):1784–90. doi: 10.1016/j.cub.2007.09.025. [DOI] [PubMed] [Google Scholar]
- 90.Ghanashyam C, Jain M. Role of auxin-responsive genes in biotic stress responses. Plant Signal Behav. 2009;4(9):846–8. doi: 10.4161/psb.4.9.9376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lim PO, Kim HJ, Nam HG. Leaf senescence. Annu Rev Plant Biol. 2007;58(1):115–36. doi: 10.1146/annurev.arplant.57.032905.105316. [DOI] [PubMed] [Google Scholar]
- 92.Zimmermann P, Zentgraf U. The correlation between oxidative stress and leaf senescence during plant development. Cell Mol Biol Lett. 2005;10(3):515–34. [PubMed] [Google Scholar]
- 93.Feys BJ, Wiermer M, Bhat RA, Moisan LJ, Medina-Escobar N, Neu C, Cabral A, Parker JE. Arabidopsis SENESCENCE-ASSOCIATED GENE101 stabilizes and signals within an ENHANCED DISEASE SUSCEPTIBILITY1 complex in plant innate immunity. Plant Cell. 2005;17(9):2601–13. doi: 10.1105/tpc.105.033910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ooijen GV, Burg HAVD, Cornelissen BJC, Takken FLW. Structure and function of resistance proteins in Solanaceous plants. Annu Rev Phytopathol. 2007;45(1):43–72. doi: 10.1146/annurev.phyto.45.062806.094430. [DOI] [PubMed] [Google Scholar]
- 95.Takken FL, Albrecht M, Tameling WI. Resistance proteins: molecular switches of plant defence. Curr Opin Plant Biol. 2006;9(4):383–90. doi: 10.1016/j.pbi.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 96.Ni X, Tian Z, Liu J, Song B, Xie C. Cloning and molecular characterization of the potato RING finger protein gene StRFP1 and its function in potato broad-spectrum resistance against Phytophthora infestans. J Plant Physiol. 2010;167(6):488–96. doi: 10.1016/j.jplph.2009.10.019. [DOI] [PubMed] [Google Scholar]
- 97.Yang CW, González-Lamothe R, Ewan RA, Rowland O, Yoshioka H, Shenton M, Ye H, O’Donnell E, Jones JDG, Sadanandom A. The E3 ubiquitin ligase activity of arabidopsis PLANT U-BOX17 and its functional tobacco homolog ACRE276 are required for cell death and defense. The Plant Cell. 2006;18(4):1084–98. doi: 10.1105/tpc.105.039198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lightfoot DJ, Mcgrann GRD, Able AJ. The role of a cytosolic superoxide dismutase in barley-pathogen interactions. Mol Plant Pathol. 2016. 10.1111/mpp.12399. [DOI] [PMC free article] [PubMed]
- 99.Fu ZQ, Dong X. Systemic acquired resistance: turning local infection into global defense. Annu Rev Phytopathol. 2013;64:839–63. doi: 10.1146/annurev-arplant-042811-105606. [DOI] [PubMed] [Google Scholar]
- 100.Bari R, Jones JDG. Role of plant hormones in plant defence responses. Plant Mol Biol. 2008;69(4):473–88. doi: 10.1007/s11103-008-9435-0. [DOI] [PubMed] [Google Scholar]
- 101.Achard P, Renou JP, Berthomé R, Harberd NP, Genschik P. Plant DELLAs restrain growth and promote survival of adversity by reducing the levels of reactive oxygen species. Curr Biol. 2008;18(9):656–60. doi: 10.1016/j.cub.2008.04.034. [DOI] [PubMed] [Google Scholar]
- 102.Griffiths J, Murase K, Rieu I, Zentella R, Zhang ZL, Powers SJ, Gong F, Phillips AL, Hedden P, Sun T. -p, Thomas SG. Genetic characterization and functional analysis of the GID1 gibberellin receptors in Arabidopsis. The Plant Cell. 2006;18(12):3399–414. doi: 10.1105/tpc.106.047415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tanaka N, Matsuoka M, Kitano H, Asano T, Kaku H, Komatsu S. gid1, a gibberellin-insensitive dwarf mutant, shows altered regulation of probenazole-inducible protein (PBZ1) in response to cold stress and pathogen attack. Plant Cell Environ. 2006;29(4):619–31. doi: 10.1111/j.1365-3040.2005.01441.x. [DOI] [PubMed] [Google Scholar]
- 104.Bao Z, Yang H, Hua J. Perturbation of cell cycle regulation triggers plant immune response via activation of disease resistance genes. Proc Natl Acad Sci. 2013;110(6):2407–12. doi: 10.1073/pnas.1217024110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhou J, Lu D, Xu G, Finlayson SA, He P, Shan L. The dominant negative arm domain uncovers multiple functions of PUB13 in Arabidopsis immunity, flowering, and senescence. J Exp Bot. 2015;66(11):3353–66. doi: 10.1093/jxb/erv148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu J, Li W, Ning Y, Shirsekar G, Cai Y, Wang X, Dai L, Wang Z, Liu W, Wang GL. The U-Box E3 Ligase SPL11/PUB13 is a convergence point of defense and flowering signaling in plants. Plant Physiol. 2012;160(1):28–37. doi: 10.1104/pp.112.199430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ning Y, Shi X, Wang R, Fan J, Park CH, Zhang C, Zhang T, Ouyang X, Li S, Wang GL. OsELF3-2, an ortholog of Arabidopsis ELF3, interacts with the E3 Ligase APIP6 and negatively regulates immunity against Magnaporthe oryzae in rice. Mol Plant. 2015;8(11):1679–82. doi: 10.1016/j.molp.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 108.Park CH, Chen S, Shirsekar G, Zhou B, Khang CH, Songkumarn P, Afzal AJ, Ning Y, Wang R, Bellizzi M, Valent B, Wang GL. The Magnaporthe oryzae effector AvrPiz-t targets the RING E3 Ubiquitin Ligase APIP6 to suppress pathogen-associated molecular pattern-triggered immunity in rice. The Plant Cell. 2012;24(11):4748–62. doi: 10.1105/tpc.112.105429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang J, Qu B, Dou S, Li L, Yin D, Pang Z, Zhou Z, Tian M, Liu G, Xie Q. The E3 ligase OsPUB15 interacts with the receptor-like kinase PID2 and regulates plant cell death and innate immunity. BMC Plant Biol. 2015;15:49. doi: 10.1186/s12870-015-0442-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Maris C, Dominguez C, Allain FH-T. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 2005;272(9):2118–31. doi: 10.1111/j.1742-4658.2005.04653.x. [DOI] [PubMed] [Google Scholar]
- 111.Lorković ZJ. Role of plant RNA-binding proteins in development, stress response and genome organization. Trends Plant Sci. 2009;14(4):229–36. doi: 10.1016/j.tplants.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 112.Mazzucotelli E, Mastrangelo AM, Crosatti C, Guerra D, Stanca AM, Cattivelli L. Abiotic stress response in plants: When post-transcriptional and post-translational regulations control transcription. Plant Sci. 2008;174(4):420–31. doi: 10.1016/j.plantsci.2008.02.005. [DOI] [Google Scholar]
- 113.Köster T, Meyer K, Weinholdt C, Smith LM, Lummer M, Speth C, Grosse I, Weigel D, Staiger D. Regulation of pri-miRNA processing by the hnRNP-like protein AtGRP7 in Arabidopsis. Nucleic Acids Res. 2014; 716. 10.1093/nar/gku716. [DOI] [PMC free article] [PubMed]
- 114.Nicaise V, Joe A, Jeong B. -r, Korneli C, Boutrot F, Westedt I, Staiger D, Alfano JR, Zipfel C. Pseudomonas HopU1 modulates plant immune receptor levels by blocking the interaction of their mRNAs with GRP7. EMBO J. 2013;32(5):701–12. doi: 10.1038/emboj.2013.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Petriccione M, Mastrobuoni F, Zampella L, Scortichini M. Reference gene selection for normalization of RT-qPCR gene expression data from Actinidia deliciosa leaves infected with Pseudomonas syringae pv. actinidiae. Sci Rep. 2015;5:16961. doi: 10.1038/srep16961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Štajner N, Cregeen S, Javornik B. Evaluation of Reference Genes for RT-qPCR Expression Studies in Hop (Humulus lupulus L,) during Infection with Vascular Pathogen Verticillium albo-atrum. PLoS ONE. 2013;8(7):68228. doi: 10.1371/journal.pone.0068228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ambawat S, Sharma P, Yadav NR, Yadav RC. MYB transcription factor genes as regulators for plant responses: An overview. Physiol Mol Biol Plants. 2013;19(3):307–21. doi: 10.1007/s12298-013-0179-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Martin C, Paz-Ares J. MYB transcription factors in plants. Trends Genet TIG. 1997;13(2):67–73. doi: 10.1016/S0168-9525(96)10049-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Trimmed Mean of M-values (TMM) of the estimated expression of selected defence-related genes in lentil–A. lentis transcriptome. Treatment – Mock (M) for H2O control, Treatment (T) for ALP2 A. lentis inoculation. HPI – Hours post inoculation. Norm_TMM_count – Trimmed Mean of M-values of the raw estimated counts, as calculated by edgeR. Count_SE – Standard error values per gene at each genotype/time point, calculated from TMM counts of subgroup replicates (n =3). (XLSX 28 kb)
Detailed information and statistics of read and base trimming per sample read file. (XLSX 17 kb)
Data Availability Statement
The data supporting the conclusions of this article (raw RNA-Seq reads) are available in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA): SRP075524, https://trace.ncbi.nlm.nih.gov/Traces/sra/?study=SRP075524.