


Abstract
Profiling of proteome dynamics is crucial for understanding cellular behavior in response to intrinsic and extrinsic stimuli and maintenance of homeostasis. Over the last 20 years, mass spectrometry (MS) has emerged as the most powerful tool for large-scale identification and characterization of proteins. Bottom-up proteomics, the most common MS-based proteomics approach, has always been challenging in terms of data management, processing, analysis and visualization, with modern instruments capable of producing several gigabytes of data out of a single experiment. Here, we present ProteoSign, a freely available web application, dedicated in allowing users to perform proteomics differential expression/abundance analysis in a user-friendly and self-explanatory way. Although several non-commercial standalone tools have been developed for post-quantification statistical analysis of proteomics data, most of them are not end-user appealing as they often require very stringent installation of programming environments, third-party software packages and sometimes further scripting or computer programming. To avoid this bottleneck, we have developed a user-friendly software platform accessible via a web interface in order to enable proteomics laboratories and core facilities to statistically analyse quantitative proteomics data sets in a resource-efficient manner. ProteoSign is available at http://bioinformatics.med.uoc.gr/ProteoSign and the source code at https://github.com/yorgodillo/ProteoSign.
INTRODUCTION
Bottom-up MS-based proteomics has been established during the past decade as the definitive technique for profiling the protein content of complex biological matrices at a global scale (1). In spite of continuous software development in supporting technology, end-user solutions for proteomics data processing, analysis and visualization have lagged behind. In differential proteomics, the acquisition speed of current state-of-the-art instrumentation enables extensive experiment replication, thus allowing for the utilisation of statistical tools for rigorous assessment of data quality and biological variability. The statistical analysis is conducted at the end of the proteomics data processing workflow (2) and until recently, the supporting software development focus has mostly been given to upstream, more fundamental stages of the workflow, such as raw data processing, protein identification (database search) and quantification.
To the authors’ knowledge, not a single non-commercial software solution currently exists for running the entire data processing workflow, the most comprehensive being those that reach the stage of protein quantification. The statistical analysis of protein abundance measurements is usually performed using specialized software and to our view, none provides a high level of automation for wide range of experimental data sets, usually requiring either a lot of user input, or manual preparation and manipulation of the input data, or the input of a statistician or even programming skills.
Hereby we present ProteoSign, an open source web-based platform for protein differential expression/abundance analysis. ProteoSign is specifically designed to serve the needs of any end-user in a friendly and appealing way. Further to our previously developed in-house software (3), ProteoSign utilizes the well-established Linear Models For Microarray Data (LIMMA) (4) methodology in order to statistically assess the difference in abundance of proteins between two or more proteome states. Similar applications analyzing microarray data are also available (e.g. CARMAweb (5)).
The software accepts as input proteomics quantification data mainly produced by either MaxQuant (6) or Proteome Discoverer (Thermo Scientific), both serving as the most popular proteomics data processing desktop applications. The quantification data can originate from label-free or labeled experiments, currently supporting SILAC (7), pulsed SILAC (8), iTRAQ (9), TMTs (10) and dimethyl labeling (11). Label-swap replication is also supported.
Through a simple, four-step wizard, the user begins the analysis and within several minutes (see Table 1), ProteoSign presents a set of publication-quality key data plots in an automated way.
Table 1. Experimental information and running time evaluation regarding the demo data sets available on ProteoSign. MQ: MaxQuant, PD: Proteome Discoverer.
| Data set name and PRIDE ID | Data set size (MB) | Biological conditions | Biological replicates | Technical replicates | Fractionation | Total number of samples | Running time (min) | Publication reference |
|---|---|---|---|---|---|---|---|---|
| SILAC 2-plex (MQ) PXD001909 | 122 | 2 | 3 | 2 | Yes | 72 | <1 | (18) |
| SILAC 2-plex (MQ) large PXD000778 | 787 | 2 | 4 | 6 | Yes | 240 | 6 | (19) |
| SILAC 2-plex (PD) large PXD000778 | 1100 | 2 | 4 | 6 | Yes | 40 | 4 | (19) |
| Label-Free (MQ) large PXD004124 | 1070 | 2 | 2 | 3 | Yes | 108 | 7 | (20) |
| TMT (MQ) PXD002622 | 62 | 2 | 5 | 0 | Yes | 50 | 2 | (21) |
| TMT (PD) PXD002622 | 109 | 2 | 5 | 0 | Yes | 50 | 2 | (21) |
| iTRAQ (PD) PXD004869 | 684 | 4 | 2 | 0 | Yes | 42 | 12 | - |
| pSILAC 3-plex (MQ) PXD001976 | 336 | 2 | 6 | 0 | Yes | 120 | 3 | (22) |
| pSILAC 3-plex (PD) PXD001976 | 831 | 2 | 6 | 0 | Yes | 120 | 7 | (22) |
| Dimethyl 2-plex (PD) large PXD002073 | 1505 | 2 | 3 | 0 | Yes | 36 | 9 | (23) |
SOFTWARE DESCRIPTION AND METHODS
General design and implementation
ProteoSign comes with a frontend web interface, whereas all calculations are computed in a remote server (see below for server details). The frontend is written in HTML and JavaScript and consists of a welcome page, a help page and a further five pages designed to guide the user through the process of data uploading and analysis in an intuitive and interactive way. ProteoSign's backend is written in PHP and R and manages the data uploading and analysis processes, as well as the results visualization and downloading processes. The frontend has been tested in all major modern internet browsers such as Mozilla Firefox, Google Chrome, Apple Safari, Opera and Microsoft Edge. Similarly, the backend has been tested with over 40 different proteomics data sets. For demonstration purposes, 10 representative, publicly available (through PRIDE) (12) proteomics data sets can be found on the web site. Table 1 provides information on the demo data sets available in ProteoSign's web page. A high-level schematic view of the platform is depicted in Figure 1. In sections to follow, details on individual parts of the figure are provided when relevant.
Figure 1.

Software architecture and information flow of the ProteoSign web server. Users can upload MaxQuant (MQ) or Proteome Discoverer (PD) output files on the server via a simple web interface. A set of PHP scripts manage the uploading of the user's input data files, the definition of various experimental parameters, communication with the data analysis R module (blue box) and downloading of the results.
Format of input data
ProteoSign currently accepts quantified differential proteomics data, produced with either Proteome Discoverer (PD) 1.3+ or MaxQuant (MQ) 1.3.0.5+. Data can originate from labeled or label-free bottom-up experiments. The experiments can comprise any number of biological conditions and replicates, as well as chromatographic/electrophoretic fractions.
ProteoSign requires that the data produced by all LC–MS/MS runs during the experiment (fractions and replicates) must be processed simultaneously with PD or MQ, producing this way a single output file in the case of PD, referred to as a ‘consensus report’, and two output files in the case of MQ. In the case of PD, the ProteoSign input data file is produced by exporting the peptide-spectrum matches (PSMs) information from a consensus report via the PD desktop application. Detailed instructions on how to produce the PSMs file can be found in the website's help page. In the case of MQ-processed data, ProteoSign requires two output files that are automatically generated during processing. These files are located in the MQ output directory and are always named ‘evidence.txt’ and ‘proteinGroups.txt’.
In summary, ProteoSign prompts the user to either upload a single PSMs file for the case of PD or ‘evidence.txt’ and ‘proteinGroups.txt’ for the case of MQ.
Experimental parameters
After all input data files have been uploaded, the user, through a two-step procedure, is requested to provide information regarding the proteomics experiment, namely the replication and biological conditions details. In the first step, the user is presented with a list of all LC-MS/MS runs performed during the experiment and is requested to assign to each one of them an experimental structure coordinate. This is a pair of numbers specifying the biological and technical replicate respectively. In the case of label-free data, the experimental condition is assigned here also. In the second step, the user (i) chooses between biological conditions to cross compare and (ii) enters descriptive information regarding the experiment.
In addition, the user can optionally instruct a special kind of filtering on the data set, before submitting it for statistical analysis. The term ‘quantitation filtering’ is specifically designed for application to pulsed SILAC data but can also be applied to the data produced by any of the supported precursor ion level labeling techniques, such as SILAC and dimethyl. Its purpose is to improve the quality of the results in pulsed SILAC data by possibly excluding contaminating proteins from the statistical analysis. Such proteins for example are the ones that were not pulse-labeled. This way, removing relatively highly abundant background species is crucial, as this can have a negative effect on the statistical analysis. The filtering is mainly achieved in two ways: (i) by optionally removing from the data set peptides with non-pulsed, ‘background’ label, also known as ‘singlets’, or (ii) by removing proteins that have been detected solely with background peptides.
Moreover, users are able to modify default data analysis and algorithmic parameters if desired. These include (a) the adjusted P-value used to differentiate between regulated and non-regulated proteins and (b) the pair of parameters used to disqualify proteins that were not reproducibly quantified from the statistical analysis. These are the number of unique peptides a protein was identified from and the number of biological replicates in which these unique peptides were found.
Finally, in order to save time, users can download their experimental parameters of preference in a text file and reuse them in a different session.
Data analysis procedure and results
After experimental parameters have been defined, the user submits the data for analysis to the core backend R module (see Figure 1) and waits for the results. The core module performs the following operations:
Calculate proteins’ intensities through summation of peptide intensities (13): For each protein i, in (biological or technical) replicate r and condition (or label) j calculate its intensity
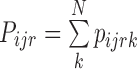 , where p is the peptide intensity given by MQ or PD and N is the total number of unique peptides quantified for protein i. Notably, ProteoSign relies completely on the peptide false discovery rate (FDR) calculated by MQ or PD, thus users do not have control over the number of protein identifications.
, where p is the peptide intensity given by MQ or PD and N is the total number of unique peptides quantified for protein i. Notably, ProteoSign relies completely on the peptide false discovery rate (FDR) calculated by MQ or PD, thus users do not have control over the number of protein identifications.Remove ‘noisy’ proteins: Disqualify proteins that were not quantified with at least m different peptides in at least n biological replicates. For example, with m = n = 2 (default value), given the sets
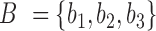 and T
and T 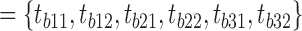 of biological and technical replicates respectively for an experiment where each of the 3 biological replicates was measured twice, a protein quantified with 2 unique peptides in tb11 and tb12 but with one peptide in the rest would be disqualified. Likewise, if there were not any technical replication (
of biological and technical replicates respectively for an experiment where each of the 3 biological replicates was measured twice, a protein quantified with 2 unique peptides in tb11 and tb12 but with one peptide in the rest would be disqualified. Likewise, if there were not any technical replication (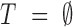 ), a protein quantified with 1 unique peptide in each biological replicate would be disqualified.
), a protein quantified with 1 unique peptide in each biological replicate would be disqualified.Log2-transform the protein intensities and perform quantile normalization across replicates and conditions via the normalizeBetweenArrays function, which is available from the LIMMA R library as part of Bioconductor (14).
Fit a linear model for each protein via the lmFit function (LIMMA) taking into account the estimated correlation (calculated via the duplicateCorrelation function (LIMMA)) between technical replicates, if any.
Compute the contrasts from the model fit and summary statistics via the contrasts.fit and eBayes functions (LIMMA).
- For each combination of conditions/labels, generate the following protein intensities data plots:
- A volcano plot: average log2(intensity ratio) against –log10(P-value).
- A value-ordered ratio plot: protein ID against log2(intensity ratio).
- A matrix of scatterplots and linear regression lines for each combination of replicate log2(intensity ratio).
- An MA plot: average log2(intensity ratio) against average log2(intensity).
- In addition, generate the following plots:
- A histogram of average log2(intensity ratio).
- Boxplots of log2(intensity) for each technical replicate before and after normalization.
- Create a ZIP archive comprising the following files and make it available to the user for downloading:
- A PDF file for each of the plots mentioned in 6.
- A single PDF file comprising the plots mentioned in 7.
- A file comprising all the information generated by the analysis in a tabular format.
- A trimmed version of the aforementioned file comprising just the statistically significant results.
- A file listing the proteins identified in each biological replicate.
- A file listing the proteins that satisfied the reproducibility criterion (see 2.) for each biological replicate.
- Seven intermediate files generated during different stages of the analysis and kept for diagnostic purposes: a design matrix file, a contrasts file, a blocking variable file, two protein intensities files (early- and late-stage versions), an early version of the results file and a log file comprising messages generated by the R module.
- A generated R source script together with a binary data file which can be used to regenerate customized versions of the data plots.
- A text file containing the user parameters of the session for future use.
- A ‘README’ text file describing the content of all files within the ZIP archive.
Figure 2 shows the four data plots previously described (operation 6), generated by ProteoSign for one of the available demo data sets. Differentially expressed proteins (adjusted P-value < 0.05) are colour-coded.
Figure 2.

Representative plots generated by ProteoSign from the 4-replicate SILAC data set of Tian et al. (19). Average log2(protein intensity ratio) against –log10(P-value), also known as volcano plot (A), protein ID versus log2(protein intensity ratio), also known as value-ordered ratio plot (B), scatter plots between replicate log2(protein intensity ratio) along with fitted linear regression lines and corresponding (with respect to the matrix diagonal) Pearson correlation coefficients (C) and average log2(protein intensity ratio) against average log2(protein intensity), also known as MA plot (D).
COMPARISON WITH OTHER TOOLS
In order to highlight ProteoSign's advantages and its contribution in the field, in Table 2 we provide an overview of the strengths and the weaknesses of other available proteomic differential expression analysis tools. We emphasize on the availability of distinguishing key features and user requirements and we show how ProteoSign can complement and outperform existing state-of-the-art applications.
Table 2. Key features and requirements of current dedicated proteomics software offering differential protein expression analysis. DIA: data-independent acquisition; MS: mass spectrometry.
| Features | Requirements | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Software name | Source code open | Web server | GUI | Free | Statistical skills | Programming skills | Manual input data preparation | Experimental data restriction(s) | Publication reference |
| DAPAR/ProStaR | Yes | Yes | Yes | Yes | Yes | No | Yes | Label-free only | (17) |
| Msstats | Yes | No | No | Yes | Yes | Yes | Yes | — | (15) |
| Msstats/Skyline | Yes | No | Yes | Yes | No | No | No | Targeted and DIA MS only | (24) |
| InfernoRDN | Yes | No | Yes | Yes | Yes | No | Yes | — | (25) |
| Scaffold Q+S | No | No | Yes | No | No | No | No | SILAC only | |
| Perseus | No | No | Yes | Yes | Yes | No | No | — | (26) |
| HiQuant | No | No | Yes | Yes | Yes | No | Yes | — | (27) |
| Rover | Yes | No | Yes | Yes | No | No | No | — | (28) |
| PIQMIe | Yes | Yes | Yes | Yes | No | No | No | Precursor ion only | (16) |
| MSqRob | Yes | No | Yes | Yes | Yes | No | No | Label-free only | (29) |
For example, many tools are not open-source, a great limiting factor for their evolution and improvement via contributions and customizations by the community, while others are commercial. In addition, for many other tools, the input of a statistician is mandatory to understand its functionalities, whereas in more extreme cases programming skills are required, thus making familiarity with such tools discouraging (e.g. Msstats (15)). Even in cases where any of the aforementioned expertise is necessary, reformatting and preparation of the input data is most of the times unavoidable. Such preparation can vary from completely manual editing with the use of in-house scripts or spreadsheets to semi-automated preparation using data conversion tools. Another important element is the support of a wide range of experimental setups. Often, we find tools, which lack important functionality and come with many limitations like for example supporting only the SILAC technique and ignoring others. For example, while ProteoSign accepts both precursor ion, isobaric labeling and label-free data, PIQMIe (16) accepts solely precursor ion data that were processed exclusively by MQ whereas DAPAR/ProStaR (17) supports label-free experimental data only and requires advanced statistical skills to operate. In order to cater for a greater pool of users, ProteoSign accepts both MQ and PD data.
Finally, in our opinion, moving away from standalone applications and offering solutions as web services is a very important step forward for the broader community. This way, we eliminate uncertainty regarding supporting packages, stringent installations and input data reformatting, thus making such analyses more appealing to non-experts. To do that, we utilize a client-server architecture to cope with performance and calculations and we aim in increasing the usability by simultaneously reducing the costs and the resources required for purchasing and maintaining high-end computer infrastructure. To our knowledge, none of the currently available tools provides the level of automation and usability offered by ProteoSign. This makes it unique in its field and an ideal resource for post-quantification statistical analysis of protein abundance.
PERFORMANCE
We have empirically tested and evaluated the performance of ProteoSign. We report all our benchmarks in Table 1.
FUTURE DIRECTIONS
ProteoSign was designed to reach the broader community varying from individual end-users to bigger proteomics core facilities by requiring minimal training and user intervention. There are, however, many features that could be added to extend its functionality. Enrichment with other statistical methods for determining differential expression, more advanced visualizations, support of open Proteomics Standards Initiative (PSI) standard file format for quantification data, analysis at the peptide-level for post-translational modification (PTM)-centred experiments (e.g. phosphoproteomics), analysis of time-series data, pathway analysis and functional annotations are key points to be implemented in the next versions. At the moment, ProteoSign is offered as a web application, but we are planning to expose a REST API to make its routines programmatically accessible.
CONCLUSION
Due to the rapid advances of MS instrumentation, an exponential growth of large-scale quantitative proteomics studies has been observed during the past decade. The demands for high-performance data processing and proteomics analysis software are today higher than ever, as an increasing number of research groups have the capacity and capability to generate large data sets every day. To this end, many tools try to address this problem, but familiarity with them is often a bottleneck because of their steep learning curve. Due to its automation and simplicity, ProteoSign is trying to fulfil this demand in the proteomics field and smoothly bridge the gap between analysis, statistics and visualization. It offers a user-friendly, web based interface, dedicated in protein differential expression analysis and it is our hope that ProteoSign will be a protagonist in the ongoing and future research of detecting key proteins in health and disease.
SERVER INFORMATION
The web server is a Dell PowerEdge R720xd machine running Ubuntu Linux (kernel 3.2) with 128 GB RAM and two Intel Xeon E5-2650 processors clocked at 2GHz.
ACKNOWLEDGEMENTS
We would like to thank Charles P. for his valuable comments and suggestions in developing the ProteoSign web interface.
FUNDING
Wellcome Trust [WT094296MA and EU-FP7 ‘Sybilla’ number 201106 to O.A.]; EU-FP7 project InnovCrete. Funding for open access charge: Institute of Molecular Biology and Biotechnology, Foundation of Research and Technology - Hellas (IMBB-FORTH).
Conflict of interest statement. None declared.
REFERENCES
- 1. Nilsson T., Mann M., Aebersold R., Yates J.R. 3rd, Bairoch A., Bergeron J.J.. Mass spectrometry in high-throughput proteomics: ready for the big time. Nat. Methods. 2010; 7:681–685. [DOI] [PubMed] [Google Scholar]
- 2. Cappadona S., Baker P.R., Cutillas P.R., Heck A.J., van Breukelen B.. Current challenges in software solutions for mass spectrometry-based quantitative proteomics. Amino Acids. 2012; 43:1087–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Howden A.J., Geoghegan V., Katsch K., Efstathiou G., Bhushan B., Boutureira O., Thomas B., Trudgian D.C., Kessler B.M., Dieterich D.C. et al. . QuaNCAT: quantitating proteome dynamics in primary cells. Nat. Methods. 2013; 10:343–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Smyth G.K. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Applic. Genet. Mol. Biol. 2004; 3, doi:10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 5. Rainer J., Sanchez-Cabo F., Stocker G., Sturn A., Trajanoski Z.. CARMAweb: comprehensive R- and bioconductor-based web service for microarray data analysis. Nucleic Acids Res. 2006; 34:W498–W503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cox J., Mann M.. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008; 26:1367–1372. [DOI] [PubMed] [Google Scholar]
- 7. Ong S.E., Blagoev B., Kratchmarova I., Kristensen D.B., Steen H., Pandey A., Mann M.. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics: MCP. 2002; 1:376–386. [DOI] [PubMed] [Google Scholar]
- 8. Schwanhausser B., Gossen M., Dittmar G., Selbach M.. Global analysis of cellular protein translation by pulsed SILAC. Proteomics. 2009; 9:205–209. [DOI] [PubMed] [Google Scholar]
- 9. Ross P.L., Huang Y.N., Marchese J.N., Williamson B., Parker K., Hattan S., Khainovski N., Pillai S., Dey S., Daniels S. et al. . Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics: MCP. 2004; 3:1154–1169. [DOI] [PubMed] [Google Scholar]
- 10. Thompson A., Schafer J., Kuhn K., Kienle S., Schwarz J., Schmidt G., Neumann T., Johnstone R., Mohammed A.K., Hamon C.. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003; 75:1895–1904. [DOI] [PubMed] [Google Scholar]
- 11. Hsu J.L., Huang S.Y., Chow N.H., Chen S.H.. Stable-isotope dimethyl labeling for quantitative proteomics. Anal. Chem. 2003; 75:6843–6852. [DOI] [PubMed] [Google Scholar]
- 12. Vizcaino J.A., Cote R.G., Csordas A., Dianes J.A., Fabregat A., Foster J.M., Griss J., Alpi E., Birim M., Contell J. et al. . The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013; 41:D1063–D1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carrillo B., Yanofsky C., Laboissiere S., Nadon R., Kearney R.E.. Methods for combining peptide intensities to estimate relative protein abundance. Bioinformatics (Oxford, England). 2010; 26:98–103. [DOI] [PubMed] [Google Scholar]
- 14. Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J. et al. . Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004; 5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Choi M., Chang C.Y., Clough T., Broudy D., Killeen T., MacLean B., Vitek O.. MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics (Oxford, England). 2014; 30:2524–2526. [DOI] [PubMed] [Google Scholar]
- 16. Kuzniar A., Kanaar R.. PIQMIe: a web server for semi-quantitative proteomics data management and analysis. Nucleic Acids Res. 2014; 42:W100–W106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wieczorek S., Combes F., Lazar C., Giai Gianetto Q., Gatto L., Dorffer A., Hesse A.M., Coute Y., Ferro M., Bruley C. et al. . DAPAR & ProStaR: software to perform statistical analyses in quantitative discovery proteomics. Bioinformatics (Oxford, England). 2017; 33:135–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carter D.M., Westdorp K., Noon K.R., Terhune S.S.. Proteomic identification of nuclear processes manipulated by cytomegalovirus early during infection. Proteomics. 2015; 15:1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tian R., Alvarez-Saavedra M., Cheng H.Y., Figeys D.. Uncovering the proteome response of the master circadian clock to light using an AutoProteome system. Mol. Cell. Proteomics: MCP. 2011; 10, doi:10.1074/mcp.M110.007252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Suarez-Cortes P., Sharma V., Bertuccini L., Costa G., Bannerman N.L., Sannella A.R., Williamson K., Klemba M., Levashina E.A., Lasonder E. et al. . Comparative proteomics and functional analysis reveal a role of Plasmodium falciparum osmiophilic bodies in malaria parasite transmission. Mol. Cell. Proteomics: MCP. 2016; 15:3243–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stewart P.A., Parapatics K., Welsh E.A., Muller A.C., Cao H., Fang B., Koomen J.M., Eschrich S.A., Bennett K.L., Haura E.B.. A pilot proteogenomic study with data integration identifies MCT1 and GLUT1 as prognostic markers in lung adenocarcinoma. PLoS One. 2015; 10:e0142162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hunten S., Kaller M., Drepper F., Oeljeklaus S., Bonfert T., Erhard F., Dueck A., Eichner N., Friedel C.C., Meister G. et al. . p53-regulated networks of protein, mRNA, miRNA, and lncRNA expression revealed by integrated pulsed stable isotope labeling with amino acids in cell culture (pSILAC) and next generation sequencing (NGS) analyses. Mol. Cell. Proteomics: MCP. 2015; 14:2609–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Elkon R., Loayza-Puch F., Korkmaz G., Lopes R., van Breugel P.C., Bleijerveld O.B., Altelaar A.F., Wolf E., Lorenzin F., Eilers M. et al. . Myc coordinates transcription and translation to enhance transformation and suppress invasiveness. EMBO Rep. 2015; 16:1723–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Broudy D., Killeen T., Choi M., Shulman N., Mani D.R., Abbatiello S.E., Mani D., Ahmad R., Sahu A.K., Schilling B. et al. . A framework for installable external tools in Skyline. Bioinformatics (Oxford, England). 2014; 30:2521–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Polpitiya A.D., Qian W.J., Jaitly N., Petyuk V.A., Adkins J.N., Camp D.G. 2nd, Anderson G.A., Smith R.D.. DAnTE: a statistical tool for quantitative analysis of -omics data. Bioinformatics (Oxford, England). 2008; 24:1556–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J.. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016; 13:731–740. [DOI] [PubMed] [Google Scholar]
- 27. Bryan K., Jarboui M.A., Raso C., Bernal-Llinares M., McCann B., Rauch J., Boldt K., Lynn D.J.. HiQuant: rapid postquantification analysis of large-scale MS-generated proteomics data. J. Proteome Res. 2016; 15:2072–2079. [DOI] [PubMed] [Google Scholar]
- 28. Colaert N., Helsens K., Impens F., Vandekerckhove J., Gevaert K.. Rover: a tool to visualize and validate quantitative proteomics data from different sources. Proteomics. 2010; 10:1226–1229. [DOI] [PubMed] [Google Scholar]
- 29. Goeminne L.J., Gevaert K., Clement L.. Peptide-level robust ridge regression improves estimation, sensitivity, and specificity in data-dependent quantitative label-free shotgun proteomics. Mol. Cell. Proteomics: MCP. 2016; 15:657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]