


Abstract
Much remains unknown about the progression and heterogeneity of mutational processes in different cancers and their diagnostic and clinical potential. A growing body of evidence supports mutation rate dependence on the local DNA sequence context for various types of mutations. We propose several tools for the analysis of cancer context-dependent mutations, which are implemented in an online computational framework MutaGene. The framework explores DNA context-dependent mutational patterns and underlying somatic cancer mutagenesis, analyzes mutational profiles of cancer samples, identifies the combinations of underlying mutagenic processes including those related to infidelity of DNA replication and repair machinery, and various other endogenous and exogenous mutagenic factors. As a result, the combination of mutagenic processes can be identified in any query sample with subsequent comparison to mutational profiles derived from malignant and benign samples. In addition, mutagen or cancer-specific mutational background models are applied to calculate expected DNA and protein site mutability to decouple relative contributions of mutagenesis and selection in carcinogenesis, thus elucidating the site-specific driving events in cancer. MutaGene is freely available at https://www.ncbi.nlm.nih.gov/projects/mutagene/.
INTRODUCTION
Cancer genomics studies have revealed high intra- and inter-tumor phenotypic and genetic heterogeneity (1–3). This may be the consequence of various forms of infidelity of DNA replication and repair machinery, differences in tumor micro-environments and various other endogenous and exogenous mutagenic factors. A growing body of evidence supports mutation rate dependence on the local DNA sequence context for various types of mutations (4) and sequence motifs (5). Several DNA context-dependent mutational patterns have been reported that are characteristic for a particular cancer type, tissue (6–11) or mutagen (12–16): UV light, chemical agents, aberrant activity of APOBEC/Activation Induced Deaminase (AID)-family cytidine deaminases and defective mismatch repair or other factors. In addition, sequence dependent structural and thermodynamic properties of the DNA molecule may also affect the DNA repair and replication, and therefore mutagenesis (17,18). Relative contributions of extrinsic and intrinsic factors to DNA mutagenesis have long been debated and the consensus view suggests that many different mutagenic processes have cumulatively shape the observed somatic mutational profiles in cancer.
DNA context-dependent mutational patterns have been analyzed previously (10,17,19–23). Some of these studies examined local DNA sequence contexts around mutated sites for several thousands of cancer genomes and exomes and reported 5 to 30 pervasive mutational signatures (21). It was suggested that these signatures could correspond to the underlying processes of mutagenesis and the etiology of some of them was characterized. Moreover, several computational tools were created to derive signatures from the cancer genomics data (24–28). Nonetheless, there is still a limited understanding of extracted signatures and their clinical potential.
Identification of cancer driver genes and mutations is one of the central problems in cancer research. This calls for further advances in computational techniques to more precisely predict the effects of cancer mutations on protein stability, binding and function (29,30). Statistical models accounting for differential transition and transversion mutation frequencies, kataegis and naïve estimates of the background somatic mutation rates have been used in several driver prediction methods (31). In order to leverage an exponential growth of cancer sequencing data and existing evidence of dependence of mutation rate on cancer type and local DNA sequence contexts (32), it is necessary to explicitly integrate the context-dependent mutations into the cancer specific mutational models to reduce false positive rates in driver gene and driver mutation predictions (33,34).
Here we propose several methods for the analysis of context-dependent mutations, which are implemented in an online computational framework MutaGene (https://www.ncbi.nlm.nih.gov/projects/mutagene/). MutaGene constructs DNA context-dependent mutational profiles and derives signatures from major cancer whole exome and genome sequencing studies available through the International Cancer Genome Consortium (ICGC) and the Cancer Genome Atlas (TCGA) repositories. Mutational profiles are categorized based on cancer type and primary tumor sites and are normalized by removing the bias from mutational hotspots with recurring mutations. Mutational profiles from the human germline SNPs and benign tissue samples from cancer patients are examined as well. Individual cancer samples are further analyzed in terms of their underlying mutagenesis to explore within and between cancer heterogeneity.
The proposed methods can analyze any given set of mutations, determine the contributions of predefined annotated mutational signatures and identify the cancer type, primary tumor site and cohorts of patients with similar mutagenic processes. This can be interpreted in terms of malfunctions in DNA damage repair mechanisms and exposure to mutagens, for further analyzes with regard to survival, treatment prognosis and drug response. Finally, for any gene or genomic region, MutaGene can apply context-dependent mutational profiles and individual signatures to calculate the background DNA mutability and amino acid substitution rates expected from a given underlying mutagenesis process and not affected by selection. The background mutability can be compared to the observed frequencies of mutations in cancer patients that allows to link cancer genotype with the phenotype to decipher relative roles of mutagenesis and selection in carcinogenesis.
MATERIALS AND METHODS
Data sources for extracting cancer mutations
In order to avoid a bias toward more frequently sequenced genes or mutations identified by genotyping we only considered single base substitutions in protein-coding genomic loci from the whole genome and whole exome-sequenced samples originating from ICGC (35) projects, TCGA (36) and the Pediatric Cancer Genome Project (Supplementary Figure S1). We relied upon annotations from The Catalogue of Somatic Mutations in Cancer (COSMIC) release v75 (37) that curates mutations from these sources and verifies whether mutations are somatic, discarding all mutations with an ‘unknown somatic status’. Currently, MutaGene includes 9450 cancer samples from 37 projects with 1,139,534 non-recurring mutations (Figures 1B and S2). Additionally, we included 1,953 somatic mutations identified in 70 benign TCGA samples (38) and common germline variants with no clinical association from the dbSNP database (39).
Figure 1.

Exploring and comparing context-dependent mutational profiles in various cancer types. (A) Mutational profiles of pan-cancer somatic mutations, germline mutations (single nucleotide polymorphisms) and somatic mutations found in benign tissues in cancer patients. (B) The list of fingerprints of mutational profiles of 37 cancer types defined based on large scale whole-genome and whole-exome projects. (C) Similarity matrix calculated by different distance measures between cancer-specific mutational profiles and clustering of cancer types according to similarities of their profiles using cosine metric. (D–F) 2D profiles of colon and gastric cancers illustrate within-cancer heterogeneity, where each line corresponds to the tumor sample and each column to the type of context-dependent mutation. (E) Comparison of relative frequencies of mutation types between colon and gastric cancers on log scale.
Construction of context-dependent mutational profiles
We identified the DNA sequences of transcripts affected by mutations using GRCh38 reference human genome assembly. The nucleotide context of a mutation was defined as the neighboring nucleotides in 5΄ and 3΄ directions from the mutated nucleotide according to the transcript sequence. Altogether, six substitution types (C→A, C→T, C→G, T→A, T→C, T→G) in 16 possible 5΄3΄ contexts result in 96 context-dependent mutation types: 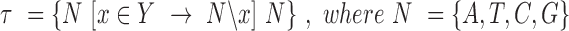 . Given the number of observed mutations
. Given the number of observed mutations 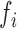 for each type
for each type 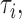 a mutational profile can be represented as a probability mass function of a multinomial distribution for all possible context-dependent mutation types
a mutational profile can be represented as a probability mass function of a multinomial distribution for all possible context-dependent mutation types 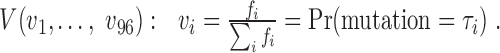 Recurring mutations observed at the same genomic location in multiple patients (Figures S2 and S3) were counted only once since these mutations and sites might be under selection (hotspots).
Recurring mutations observed at the same genomic location in multiple patients (Figures S2 and S3) were counted only once since these mutations and sites might be under selection (hotspots).
Derivation of mutational signatures
Cancer specific context-dependent mutational profile is a manifestation of different mutational processes. These processes may have distinct etiology and may result in distinct sets of mutations in characteristic DNA sequence contexts, so called context-dependent mutational signatures (21). Previously, the numeric deconvolution of matrices of mutational profiles of cancer samples (Figure 2A) was obtained using the non-negative matrix factorization (NMF) method (28,40–42). The major goal of this procedure was to obtain the underlying mutational signatures (Figure 2B) and represent cancer samples in terms of exposure to these signatures (Figure 2C). Mutational signatures should ideally represent distinct uncorrelated context-dependent mutational processes that can be further annotated in terms of their etiology. Sparseness of mutational signatures becomes an important aspect when it comes to annotation of signatures. Assuming that a limited number of mutational processes affects each cancer sample, the exposure matrix also has to be sparse. Therefore we applied non-smooth (ns)NMF method (43) with sparse random initialization allowing to obtain sparse solutions for both signature and exposure matrices (Supplementary Figures S7C and S7D) while avoiding high correlation between the signatures (Supplementary Figure S7B).
Figure 2.

Decomposition of mutational profiles of pan-cancer samples into mutational signatures. (A) The matrix of mutational profiles of pan-cancer samples 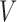 ; (B) Ten mutational signatures MutaGene10 in matrix
; (B) Ten mutational signatures MutaGene10 in matrix 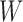 annotated by the corresponding mutagenic processes and (C) exposure matrix
annotated by the corresponding mutagenic processes and (C) exposure matrix 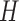 representing relative contributions of mutational processes (represented by ten signatures) in each tumor sample. The matrices are transposed for visualization purpose. Matrices
representing relative contributions of mutational processes (represented by ten signatures) in each tumor sample. The matrices are transposed for visualization purpose. Matrices 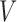 and
and 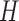 are heat maps where pixel intensity indicates the frequency, whereas in matrix
are heat maps where pixel intensity indicates the frequency, whereas in matrix 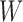 the values of row vectors as shown as bar plots.
the values of row vectors as shown as bar plots.
Given n cancer samples, we represented context dependent mutations (m = 96) in these samples as a non-negative matrix 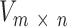 . NMF finds two non-negative matrices
. NMF finds two non-negative matrices 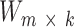 and
and 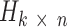 for a given number of components k, so that
for a given number of components k, so that 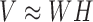 . Matrix
. Matrix 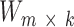 contains k mutational signatures and matrix
contains k mutational signatures and matrix 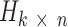 describes exposures of n samples to k mutational processes defined by signatures in a matrix
describes exposures of n samples to k mutational processes defined by signatures in a matrix 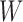 . In case of non-smooth NMF decomposition method (nsNMF) (43), the problem is formulated as
. In case of non-smooth NMF decomposition method (nsNMF) (43), the problem is formulated as 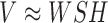 , where a square non-negative smoothing matrix
, where a square non-negative smoothing matrix 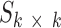 allows reconstructing into globally sparse solutions. Figure 2 and Supplementary Figures S5 and S6 illustrate the deconvolution for NMF and nsNMF methods.
allows reconstructing into globally sparse solutions. Figure 2 and Supplementary Figures S5 and S6 illustrate the deconvolution for NMF and nsNMF methods.
We analyzed the reproducibility of deconvolution results by calculating a cophenetic correlation coefficient of consensus matrix 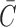 (see Supplementary Data). Previously it was used to determine an optimal number of components in NMF deconvolution as a point where this coefficient begins to decrease (42). A consensus matrix
(see Supplementary Data). Previously it was used to determine an optimal number of components in NMF deconvolution as a point where this coefficient begins to decrease (42). A consensus matrix 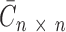 is an average of connectivity matrices produced as a result of multiple runs of NMF or nsNMF algorithms. A connectivity matrix
is an average of connectivity matrices produced as a result of multiple runs of NMF or nsNMF algorithms. A connectivity matrix 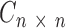 is calculated based on the exposure matrix
is calculated based on the exposure matrix 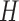 and shows if samples are exposed to the same dominating mutagenic process. Supplementary Figure S7A shows that cophenetic correlation coefficient decreases after five and ten components when applying NMF and nsNMF respectively. Moreover, NMF deconvolution with more than ten signatures/components resulted in highly correlated signatures with correlation coefficients larger than 0.6 (Supplementary Figures S7B and S13). Therefore, we used nsNMF decompositions with five and ten components and obtained two MutaGene5 and MutaGene10 signature sets listed and annotated on the MutaGene website.
and shows if samples are exposed to the same dominating mutagenic process. Supplementary Figure S7A shows that cophenetic correlation coefficient decreases after five and ten components when applying NMF and nsNMF respectively. Moreover, NMF deconvolution with more than ten signatures/components resulted in highly correlated signatures with correlation coefficients larger than 0.6 (Supplementary Figures S7B and S13). Therefore, we used nsNMF decompositions with five and ten components and obtained two MutaGene5 and MutaGene10 signature sets listed and annotated on the MutaGene website.
Identification of mutational profiles, signatures and mutagenic processes for a query set of mutations
MutaGene tools determine the mutational DNA context according to the reference human genome assembly and construct a query mutational profile for any given set of mutations. The query profile can be compared to the collection of profiles in the MutaGene database using the ‘Identify’ tool, which ranks the query mutational profile by its distance to the MutaGene profiles using the k-nearest neighbors classifier and distance measures listed in Supplementary Data. MutaGene also provides Naïve Bayes, random forest and linear support vector machines (SVM) classifiers pre-trained with 4-fold cross-validation, treating different cancer types and primary tumor sites as classification labels. Performance evaluation results are shown in Supplementary Figures S8–11. In addition, contributions (exposures)  of pre-annotated signature sets
of pre-annotated signature sets 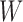 (MutaGene5, MutaGene10 and COSMIC30) for a sample query profile
(MutaGene5, MutaGene10 and COSMIC30) for a sample query profile 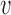 are calculated by solving Wh = v with a non-negative constrained least squares method.
are calculated by solving Wh = v with a non-negative constrained least squares method.
Analysis of mutability
A mutational background model represented by the mutational profile or mutational signature can be applied to any protein-coding gene sequence or any other genomic region in order to calculate the DNA mutability expected for each particular site. DNA mutability of a base 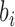 in a trinucleotide context
in a trinucleotide context 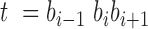 is calculated using the total number of mutations
is calculated using the total number of mutations 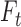 for a given trinucleotide t according to the context-dependent mutational profile
for a given trinucleotide t according to the context-dependent mutational profile 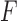 . Given the number of samples included in the mutational profile
. Given the number of samples included in the mutational profile 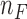 and the number of trinucleotides t in a diploid human exome
and the number of trinucleotides t in a diploid human exome 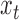 according to the reference genome assembly GRCh38, one can define mutability as
according to the reference genome assembly GRCh38, one can define mutability as 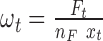 . The number of trinucleotides in the human exome is calculated for each nucleotide base considering its trinucleotide context. In case when mutability is calculated using signatures, mutation rate
. The number of trinucleotides in the human exome is calculated for each nucleotide base considering its trinucleotide context. In case when mutability is calculated using signatures, mutation rate 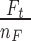 is set to 120, which corresponds to the pan-cancer average of the number of mutations per exome (Supplementary Figure S4). MutaGene also allows to specify an arbitrary mutation rate for mutability calculations. Mutability is estimated per Megabase per sample (Figure 3). Mutability of a codon
is set to 120, which corresponds to the pan-cancer average of the number of mutations per exome (Supplementary Figure S4). MutaGene also allows to specify an arbitrary mutation rate for mutability calculations. Mutability is estimated per Megabase per sample (Figure 3). Mutability of a codon 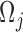 is calculated as the sum of mutabilities of the three nucleotides comprising the codon
is calculated as the sum of mutabilities of the three nucleotides comprising the codon 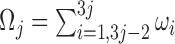 . Amino acid substitutions corresponding to each mutation are calculated by translating the mutated codon using a standard codon table. The relative propensities of all types of mutations in a codon
. Amino acid substitutions corresponding to each mutation are calculated by translating the mutated codon using a standard codon table. The relative propensities of all types of mutations in a codon 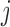 including missense, silent and nonsense mutations sum up to
including missense, silent and nonsense mutations sum up to 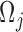 .
.
Figure 3.

Analysis of mutability in DNA and protein sequences. Two examples showing the results of mutability calculations using the pan-cancer mutational profile: (A) a sequence fragment of TP53 gene and protein with Arg273 interacting with the DNA (red arrow) and Arg283 not in direct contact with the DNA (blue arrow) and (B) mutability of site Leu858 (red arrow) in epidermal growth factor receptor (EGFR) gene and protein using a pan-cancer mutational profile. Expected DNA mutability depending on the local sequence context, depicted as a green line, is scaled per Megabase DNA per cancer sample. The heatmap shows expected mutabilities for each DNA position, where the color encodes silent, missense and nonsense nucleotide substitutions and color intensity represents the scale of mutability values. Expected mutability is translated onto the protein level (shown in orange line for each codon) and the heatmap below shows the mutability values for each amino acid substitution. Yellow circles indicate mutations observed in cancer patients, with height of the pins showing relative numbers of observed mutations in log scale. Note that in sites pointed by arrows expected mutability and observed mutation frequencies show the opposite trends. Such maps are generated by MutaGene ‘Analyze mutability’ tool and are designed for interactive use, where clicking on a circle shows a distribution of observed mutations over cancer types.
RESULTS
Exploring the diversity of mutational profiles in human cancer
A context-dependent mutational profile is a results of contributions of different DNA context-dependent processes characteristic for a given cancer sample. Importantly, mutational profile is calculated solely based on the types of nucleotide substitutions and their context, regardless of the chromosome location and gene type. Mutational profiles are represented as probability mass functions in order to emphasize the relative mutational preferences, because an absolute value of mutation rate (Supplementary Figure S2) is not necessarily directly associated with the underlying mutagenic processes and may be determined by the number of replications, cell divisions and other factors (44). Moreover, some sites (hotspots) may harbor several hundreds of mutations from different samples (45) and mainly represent mutations offering selective advantage to the clone. In fact, we found that about eight percent of all cancer mutations were recurring. Proportions of recurring mutations varied depending on cancer type reaching up to 30% of recurring mutations in some cases (Figures S3). To avoid biases caused by selection acting upon particular genomic sites, mutational profiles have been derived by counting mutations only once and excluding recurring mutations (Figure S14).
A collection of mutational profiles in MutaGene allows to explore the diversity of mutagenic processes in different cancers and tissues (Figure 1B). In Figure 1A, a pan-cancer somatic mutational profile is shown along with the profiles of germline mutations obtained from human SNPs and somatic mutations found in benign human tissue samples. Pan-cancer and most cancer specific mutational profiles (Figure 1B) contain a dominating C→T mutations in the NpCpG context, which is also characteristic for germline mutational profile, pointing to striking similarities between the accumulation of mutations in tumors and in germline cells. The mutation rate at nucleotide C in the CpG dinucleotide context associated with methylation was previously found to be much larger than that of other sites (46). The mutational profile of benign pan-tissue somatic mutations allows distinguishing cancer-specific somatic patterns and highlights the differences between benign and malignant tissues, particularly in T→G and C→A transversions.
Figure 1C shows a heat map representing the pairwise comparisons of all cancer-specific mutational profiles. This comparison reveals inter-cancer similarities in terms of their mutational profiles. Namely, the similarities have been detected between lung and oral carcinomas; breast, bladder and cervical carcinomas; liver and kidney carcinomas; and a large group of blood, brain, gastric and colorectal cancers. However, while mutational profiles of different cancer types can be very similar, as in the case of colon and gastric adenocarcinomas (cosine similarity of 0.98) (Figure 1E), mutational profiles of individual samples within the same cancer type may reveal large heterogeneity. In Figure 1D and 1F individual cancer samples are ordered based on the distances between their mutational profiles, shown as bands on the heat maps, indicating that within cancer types these differences may be more pronounced than between cancer types. Understanding of cancer genetic heterogeneity is deeply rooted in our understanding of the underlying mutagenic processes and will be explored in the following sections.
Analysis of query mutational profiles
MutaGene can analyze any set of mutations from one or several cancer samples to identify cancers of unknown primary tumor site, to detect the most likely mutagenic process and to distinguish tumorigenic from benign mutation sets (Supplementary Figure S12). First, a mutational profile is calculated for a query sample of interest. Next, MutaGene compares the query profile to the collection of profiles and signatures in the MutaGene database and calculates the contributions of different annotated mutagenic processes to the mutational profile of interest (exposures). We thoroughly assessed the performance of cancer type and primary tumor site identification with a cross-validation benchmarks, in particular the dependence of its accuracy on the number of mutations in the query sample. We found that the average accuracy of primary site identification ranges from 38 to 85% (Supplementary Figure S9D). According to our benchmarks, random forest classification method outperformed multinomial Naïve Bayes and SVM classifier with a linear kernel (Supplementary Figures S9, S10), therefore random forest is used in MutaGene by default. For cancer types that have similar mutational profiles (Figure 1C), it could be sufficient to identify a correct cancer or tissue type within the top two or three matches. Using this more relaxed criterion, the average accuracy of cancer type prediction using random forest classifier increases from 66 to 90%, if we consider top three matches (Supplementary Figure S10B). For primary site prediction, the same approach would show a boost in accuracy from 72 to 92% (Supplementary Figure S10E).
Per-class performance analysis shows (Supplementary Figure S11A and Table ST1) that some cancer types, such as pancreatic cancer, breast cancer and renal adenocarcinoma are not predicted correctly as a top-matching hit. Due to within-cancer heterogeneity, some samples belonging to lung squamous cell carcinoma were attributed to lung adenocarcinoma (LUAD), however almost all LUAD samples were correctly identified. Regarding the primary tumor sites, esophagus and pancreas are the most problematic sites, where the classifier incorrectly attributes most of the samples to colon and stomach because mutational profiles of stomach and colorectal samples are practically indistinguishable. However, despite high heterogeneity, MutaGene correctly identifies the primary site for almost all liver, lung and colorectal samples (Supplementary Figure S11B and Table ST2). Therefore, we show that it is possible to identify cancer types and primary sites for a given cancer sample with sufficient accuracy using only the information about its mutational profile. This analysis uncovers and illustrates diagnostic potential of context-dependent mutational profiles, however in practical diagnostic applications it may be necessary to combine mutational profile with other types of data such as presence/absence of mutations in certain genes, copy number variations, gene expression and DNA methylation.
Estimating the background DNA and protein site mutability
MutaGene provides background mutational models in the form of cancer-specific mutational profiles or mutagen-specific signatures that can be used to calculate the number of mutations expected as a result of underlying mutagenesis, not affected by selection pressure in somatic cells. The site mutability (see ‘Materials and Methods’ for definition) can be estimated for each genomic site thus allowing to compare relative mutabilities of different sites between each other and simultaneously relate them to the frequencies of mutations observed in certain sites in cancer patients. For protein-coding sequences MutaGene calculates the rates of expected amino acid substitutions for each codon thus taking into account the local DNA context, the nucleotides surrounding the codons of each amino acid. Figure 3A shows the DNA and protein mutability for a fragment of gene TP53 calculated using pan-cancer mutational profile. This figure shows two sites: R273 that is involved in DNA binding (red arrow) and R283 site (blue arrow) that is not directly involved in binding of DNA. There is experimental evidence (47) that any missense mutations in codons of DNA-binding arginine result in a loss of function, whereas many amino acid substitutions of another arginine can be tolerated (48). These two arginines have different mutability values since mutability depends on both codons and nucleotides surrounding these codons. Particularly interesting is that the key position involved in interactions with DNA (R273) has the highest numbers of observed mutations in cancer patients, however its mutability is much lower than that of other arginine (R283) that is not involved in DNA interactions. Consistent with this, Figure 3B shows a very low expected mutability of an oncogene epidermal growth factor receptor (EGFR) L858 site (red arrow) although it is frequently mutated in cancer. Respectively, expected mutabilities of adjacent codons, that are supposedly not under selection in cancer, are high. In general, by comparing observed frequencies to expected mutabilities one can potentially get important clues about the potential cancer driving events.
DISCUSSION
Cancers are notorious for their intra- and inter-tumor functional and genetic heterogeneity, which imposes difficulties in terms of cancer type classification and targeted drug therapy. Exploring the heterogeneity of cancer in terms of mutagenic processes is not trivial. First, mutational profiles of cancer samples with only a few mutations could be too sparse and not well defined. Second, mutational profiles of samples represent a combination of different mutational signatures and processes, many of which remain uncharacterized (21,22,49). Third, mutational processes may act independently, but their signatures may be overlapping, for instance the signature of somatic hypermutation enzyme, AID, as we identified recently, overlaps with the CpG methylation site (50). Finally, mutational profiles and signatures are intended to represent the context-dependent propensities determined by the underlying background mutagenic processes rather than selection and a signal coming from selection processes is hard to eliminate. The evolution of cancer is largely driven by somatic mutations and clonal selection of these mutations (51); it is therefore important to decouple mutagenesis from selection in order to characterize driving events in tumor evolution. Mutagenesis can be affected by the local DNA sequence context around the mutated site and therefore sequence context should be accounted for in estimating the mutational probability and mutation rate at any given site. Context-dependent mutational models allow MutaGene to calculate the expected background mutability of nucleotide and protein sites, thereby linking processes operating at the DNA level to the protein phenotype. The choice of mutational model is crucial and the expected mutability may largely depend on the background model. Additionally, considering mutational hotspots and excluding recurring mutations that may be subject to selection is important for calculating the accurate background mutational model.
In addition to histological characterization of a cancer sample, methods of molecular diagnostics are aimed toward correct and timely diagnosis and the optimal choice of personalized treatment for a cancer patient. Currently these methods are mostly relying on biomarkers related to differential gene expression, methylation, copy number variation and by the presence or absence of mutations in certain genes. However, much remains unknown about the mutational processes operating at the level of DNA in any given cancer patient or sample. Identification of the underlying mutational processes can improve molecular subtype classification, particularly in cancers with high heterogeneity. Identification of cancer type and primary site is also important for free-floating DNA blood samples and metastatic cancer samples, where the original tumor site may be unknown. Additionally, it may help to identify the actual source of tumor in case of a metastatic sample. Mutational studies in cell culture, viral and animal models may also require a comparison to the reference human datasets using MutaGene. Coupled to the analysis of clinical features, such as drug response, resistance and survival for different cohorts of patients with similar mutational profiles mutational analysis with MutaGene server provides an additional factor to consider in explaining cancer heterogeneity.
AVAILABILITY
MutaGene is freely available at https://www.ncbi.nlm.nih.gov/projects/mutagene/.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Michael Lynch, Lyudmil Alexandrov, Nir Ben-Tal, Gerhard Manning and Marie Evangelista for helpful discussions and Janet Coleman for proofreading.
Author Contributions: A.G. and A.R.P. designed the analysis and wrote the paper. A.G. developed the framework. S.L.R, M.L., Q.X.S. and I.B.R. applied the framework to the analysis of cancer genomes. All authors approved the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural Research Programs of the National Library of Medicine; National Institutes of Health. Funding for open access charge: National Library of Medicine.
Conflict of interest statement. None declared.
REFERENCES
- 1. Lawrence M.S., Stojanov P., Polak P., Kryukov G.V., Cibulskis K., Sivachenko A., Carter S.L., Stewart C., Mermel C.H., Roberts S.A. et al. . Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013; 499:214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res. 2012; 72:4875–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Andor N., Graham T.A., Jansen M., Xia L.C., Aktipis C.A., Petritsch C., Ji H.P., Maley C.C.. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat. Med. 2016; 22:105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Francioli L.C., Polak P.P., Koren A., Menelaou A., Chun S., Renkens I. Genome of the Netherlands, C. . Genome of the Netherlands, C. van Duijn C.M., Swertz M., Wijmenga C., van Ommen G. et al. . Genome-wide patterns and properties of de novo mutations in humans. Nat. Genet. 2015; 47:822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sharp N.P., Agrawal A.F.. Low genetic quality alters key dimensions of the mutational spectrum. PLoS Biol. 2016; 14:e1002419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pfeifer G.P., Hainaut P.. On the origin of G→T transversions in lung cancer. Mutat. Res. 2003; 526:39–43. [DOI] [PubMed] [Google Scholar]
- 7. Boutros P.C., Fraser M., Harding N.J., de Borja R., Trudel D., Lalonde E., Meng A., Hennings-Yeomans P.H., McPherson A., Sabelnykova V.Y. et al. . Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat. Genet. 2015; 47:736–745. [DOI] [PubMed] [Google Scholar]
- 8. Alexandrov L.B., Nik-Zainal S., Siu H.C., Leung S.Y., Stratton M.R.. A mutational signature in gastric cancer suggests therapeutic strategies. Nat. Commun. 2015; 6:8683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kandoth C., McLellan M.D., Vandin F., Ye K., Niu B., Lu C., Xie M., Zhang Q., McMichael J.F., Wyczalkowski M.A. et al. . Mutational landscape and significance across 12 major cancer types. Nature. 2013; 502:333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nik-Zainal S., Alexandrov L.B., Wedge D.C., Van Loo P., Greenman C.D., Raine K., Jones D., Hinton J., Marshall J., Stebbings L.A. et al. . Mutational processes molding the genomes of 21 breast cancers. Cell. 2012; 149:979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schulze K., Imbeaud S., Letouze E., Alexandrov L.B., Calderaro J., Rebouissou S., Couchy G., Meiller C., Shinde J., Soysouvanh F. et al. . Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015; 47:505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brash D.E. UV signature mutations. Photochem. Photobiol. 2015; 91:15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Poon S.L., Huang M.N., Choo Y., McPherson J.R., Yu W., Heng H.L., Gan A., Myint S.S., Siew E.Y., Ler L.D. et al. . Mutation signatures implicate aristolochic acid in bladder cancer development. Genome Med. 2015; 7:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Langie S.A., Koppen G., Desaulniers D., Al-Mulla F., Al-Temaimi R., Amedei A., Azqueta A., Bisson W.H., Brown D.G., Brunborg G. et al. . Causes of genome instability: the effect of low dose chemical exposures in modern society. Carcinogenesis. 2015; 36:S61–S88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roberts S.A., Lawrence M.S., Klimczak L.J., Grimm S.A., Fargo D., Stojanov P., Kiezun A., Kryukov G.V., Carter S.L., Saksena G. et al. . An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013; 45:970–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sammalkorpi H., Alhopuro P., Lehtonen R., Tuimala J., Mecklin J.P., Jarvinen H.J., Jiricny J., Karhu A., Aaltonen L.A.. Background mutation frequency in microsatellite-unstable colorectal cancer. Cancer Res. 2007; 67:5691–5698. [DOI] [PubMed] [Google Scholar]
- 17. Sung W., Ackerman M.S., Gout J.F., Miller S.F., Williams E., Foster P.L., Lynch M.. Asymmetric context-dependent mutation patterns revealed through mutation-accumulation experiments. Mol. Biol. Evol. 2015; 32:1672–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bauer N.C., Corbett A.H., Doetsch P.W.. The current state of eukaryotic DNA base damage and repair. Nucleic Acids Res. 2015; 43:10083–10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Siepel A., Haussler D.. Phylogenetic estimation of context-dependent substitution rates by maximum likelihood. Mol. Biol. Evol. 2004; 21:468–488. [DOI] [PubMed] [Google Scholar]
- 20. Pfeifer G.P., Besaratinia A.. Mutational spectra of human cancer. Hum. Genet. 2009; 125:493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Borresen-Dale A.L. et al. . Signatures of mutational processes in human cancer. Nature. 2013; 500:415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Helleday T., Eshtad S., Nik-Zainal S.. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014; 15:585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rogozin I.B., Babenko V.N., Milanesi L., Pavlov Y.I.. Computational analysis of mutation spectra. Brief Bioinform. 2003; 4:210–227. [DOI] [PubMed] [Google Scholar]
- 24. Petljak M., Alexandrov L.B.. Understanding mutagenesis through delineation of mutational signatures in human cancer. Carcinogenesis. 2016; 37:531–540. [DOI] [PubMed] [Google Scholar]
- 25. Hollstein M., Alexandrov L.B., Wild C.P., Ardin M., Zavadil J.. Base changes in tumour DNA have the power to reveal the causes and evolution of cancer. Oncogene. 2016; 36:158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liberzon A., Birger C., Thorvaldsdottir H., Ghandi M., Mesirov J.P., Tamayo P.. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015; 1:417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ardin M., Cahais V., Castells X., Bouaoun L., Byrnes G., Herceg Z., Zavadil J., Olivier M.. MutSpec: a Galaxy toolbox for streamlined analyses of somatic mutation spectra in human and mouse cancer genomes. BMC Bioinformatics. 2016; 17:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gehring J.S., Fischer B., Lawrence M., Huber W.. SomaticSignatures: inferring mutational signatures from single-nucleotide variants. Bioinformatics. 2015; 31:3673–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li M., Kales S.C., Ma K., Shoemaker B.A., Crespo-Barreto J., Cangelosi A.L., Lipkowitz S., Panchenko A.R.. Balancing protein stability and activity in cancer: a new approach for identifying driver mutations affecting CBL ubiquitin ligase activation. Cancer Res. 2016; 76:561–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li M., Goncearenco A., Panchenko A.R.. Annotating mutational effects on proteins and protein interactions: designing novel and revisiting existing protocols. Methods Mol. Biol. 2017; 1550:235–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McFarland C.D., Korolev K.S., Kryukov G.V., Sunyaev S.R., Mirny L.A.. Impact of deleterious passenger mutations on cancer progression. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:2910–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chang M.T., Asthana S., Gao S.P., Lee B.H., Chapman J.S., Kandoth C., Gao J., Socci N.D., Solit D.B., Olshen A.B. et al. . Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 2016; 34:155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Carter H., Chen S., Isik L., Tyekucheva S., Velculescu V.E., Kinzler K.W., Vogelstein B., Karchin R.. Cancer-specific high-throughput annotation of somatic mutations: computational prediction of driver missense mutations. Cancer Res. 2009; 69:6660–6667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gonzalez-Perez A., Mustonen V., Reva B., Ritchie G.R., Creixell P., Karchin R., Vazquez M., Fink J.L., Kassahn K.S. International Cancer Genome Consortium Mutation, P. and Consequences Subgroup of the Bioinformatics Analyses Working, G. et al. . Computational approaches to identify functional genetic variants in cancer genomes. Nat. Methods. 2013; 10:723–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cancer Genome Atlas Research, N. Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M.. The Cancer Genome Atlas Pan-cancer analysis project. Nat. Genet. 2013; 45:1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Forbes S.A., Beare D., Gunasekaran P., Leung K., Bindal N., Boutselakis H., Ding M., Bamford S., Cole C., Ward S. et al. . COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015; 43:D805–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yadav V.K., DeGregori J., De S.. The landscape of somatic mutations in protein coding genes in apparently benign human tissues carries signatures of relaxed purifying selection. Nucleic Acids Res. 2016; 44:2075–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Coordinators N.R. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2014; 42:D7–D17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Alexandrov L.B., Nik-Zainal S., Wedge D.C., Campbell P.J., Stratton M.R.. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013; 3:246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rosenthal R., McGranahan N., Herrero J., Taylor B.S., Swanton C.. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016; 17:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brunet J.P., Tamayo P., Golub T.R., Mesirov J.P.. Metagenes and molecular pattern discovery using matrix factorization. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:4164–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pascual-Montano A., Carazo J.M., Kochi K., Lehmann D., Pascual-Marqui R.D.. Nonsmooth nonnegative matrix factorization (nsNMF). IEEE Trans Pattern Anal. Mach. Intell. 2006; 28:403–415. [DOI] [PubMed] [Google Scholar]
- 44. Tomasetti C., Vogelstein B.. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015; 347:78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rogozin I.B., Pavlov Y.I.. Theoretical analysis of mutation hotspots and their DNA sequence context specificity. Mutat. Res. 2003; 544:65–85. [DOI] [PubMed] [Google Scholar]
- 46. Zhao Z., Jiang C.. Methylation-dependent transition rates are dependent on local sequence lengths and genomic regions. Mol. Biol. Evol. 2007; 24:23–25. [DOI] [PubMed] [Google Scholar]
- 47. Petitjean A., Mathe E., Kato S., Ishioka C., Tavtigian S.V., Hainaut P., Olivier M.. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007; 28:622–629. [DOI] [PubMed] [Google Scholar]
- 48. Petitjean A., Achatz M.I., Borresen-Dale A.L., Hainaut P., Olivier M.. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007; 26:2157–2165. [DOI] [PubMed] [Google Scholar]
- 49. Alexandrov L.B., Stratton M.R.. Mutational signatures: the patterns of somatic mutations hidden in cancer genomes. Curr. Opin. Genet. Dev. 2014; 24:52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rogozin I.B., Lada A.G., Goncearenco A., Green M.R., De S., Nudelman G., Panchenko A.R., Koonin E.V., Pavlov Y.I.. Activation induced deaminase mutational signature overlaps with CpG methylation sites in follicular lymphoma and other cancers. Sci. Rep. 2016; 6:38133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Greaves M. Evolutionary determinants of cancer. Cancer Discov. 2015; 5:806–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.