


Abstract
The agriGO platform, which has been serving the scientific community for >10 years, specifically focuses on gene ontology (GO) enrichment analyses of plant and agricultural species. We continuously maintain and update the databases and accommodate the various requests of our global users. Here, we present our updated agriGO that has a largely expanded number of supporting species (394) and datatypes (865). In addition, a larger number of species have been classified into groups covering crops, vegetables, fish, birds and insects closely related to the agricultural community. We further improved the computational efficiency, including the batch analysis and P-value distribution (PVD), and the user-friendliness of the web pages. More visualization features were added to the platform, including SEACOMPARE (cross comparison of singular enrichment analysis), direct acyclic graph (DAG) and Scatter Plots, which can be merged by choosing any significant GO term. The updated platform agriGO v2.0 is now publicly accessible at http://systemsbiology.cau.edu.cn/agriGOv2/.
INTRODUCTION
An enrichment analysis is an efficient and fast method to determine the functions associated with large gene lists and to increase the likelihood of interpreting biological processes (1). Information on biological processes (BP), molecular functions (MF) and cell components (CC) is organized into structured vocabulary, known as gene ontology (GO), that could describe almost all organisms. The majority of emerging enrichment tools (2–7) employ GO as their annotation resource. However, these resources generally address human or model species but are limited when concerning agricultural species. For examples, the Database for Annotation, Visualization and Integrated Discovery (DAVID) provides a comprehensive platform of functional annotations and analyses based on any given gene list mainly focusing on humans and human diseases. The Gene Ontology enRIchment anaLysis and visuaLizAtion tool (GORILLA) is used for model species enrichment analyses. Our laboratory developed two GO-based tools for the agricultural community, easyGO (2) and the ensuing agriGO (8), and has continuously maintained them. With the rapid development of high-throughput technologies, these web servers cannot include the enormous amount of sequencing data in the agricultural field (9–14).
Additionally, the GO database is released monthly in several versions, while the definitions of terms and the parent–child relationship between terms changes towing to the integration of information from different sources (15). Many sources and methods induce irregular annotation levels and gene identities (IDs) (15–19). Relatively more comprehensive and accurate versions are being produced based on the evidence codes and/or comparative genomics to reduce the substantial number of false positives (20). For example, a rice gene or gene product's GO annotation contains versions 5.0, 6.1 and 7.0 from TIGR (21), Gramene release 50 (22), Phytozome v11 (18) and others. Thus, it is necessary to increase the GO annotation, as well as the corresponding GO terms under constantly expanding knowledge.
Up to January 2017, Google Scholar showed that agriGO was referenced 982 times with >80 000 analysis requests since it went online. At the same time, users have put forward a series of requirements for accurate analytical results with reliable backgrounds. To satisfy the ever-increasing number of sequenced species, maintain an up-to-date GO background (15) and meet user requirements, we have updated agriGO with new features and better support. The major updates are supporting more species and datatypes, and adding new visualization tools.
METHODS
AgriGO v2.0 mainly contains analysis tools for processing with the agriGO v2.0-provided background and custom analyses, including search, singular enrichment analysis (SEA), parametric analysis of gene set enrichment (PAGE), SEACOMPARE, Batch SEA, DAG drawer and Scatter Plots (Figure 1). The former four were derived from agriGO and updated with broad species and datatypes, while the DAG and Scatter Plots were newly developed for visualization. The difference between SEA and Batch SEA is that the latter can tackle multiple input datasets simultaneously based on one reference (Figure 2). SEACOMPARE is also convenient to compare the significant GO terms based on the false discovery rates (FDRs) from the Batch SEA results (Figure 3).
Figure 1.

The analysis framework for agriGO v2.0. AgriGO v2.0 provides three analysis entrances and six major tools. For all query lists, P-values are computed and adjusted by multiple testing corrections or permutation tests. Then, significant GO terms can be further analyzed for similarity or parent–child hierarchical structures. Four visualization methods can help users understand the results. The orange color represents the new function in agriGO v2.0.
Figure 2.

Batch SEA for up-regulated genes after cold treatments (0.5, 1, 3, 6, 12 and 24 h) in Arabidopsis thaliana. Input the query lists into the expanded text box with clicking on the ‘add’ button (A). All successful job IDs (B) corresponding to query lists can be selected for the SEACOMPARE analysis (C). A brief summary of one job for 24-h cold stress (D). Users can find the basic information on the reference ‘Affymetrix ATH1 Genome Array’ that contains 22,479 datatypes better matches the GO version 2010, and 34 significant GO terms were obtained with 165 annotated genes in the query list. The detailed information about the BP category was displayed in a tabular format, including significant GO terms with descriptions, gene numbers in input and background lists, P-values, FDR by multi-test adjustments, and P-value distribution (E). The DAG (F) and Scatter Plots (G) illustrates the BP category. A few GO terms were not in the top 1% and they were filtered out by multi-test adjustments, like the GO term ‘response to hormone stimulus’ (GO:0009725) (H).
Figure 3.

SEACOMPARE analysis based on multiple hypothesis tests (FDRs) and P-values of the up-regulated gene list after a 6-h cold treatment of Arabidopsis thaliana. The detailed information in each column, from left to right, represent the significant GO terms, GO category (BP, CC and MF), GO annotation, a heatmap (columns 4–10) for FDRs or P-values (columns 11–17) from different computational methods, and the last column is the top percent based on the P-value distribution as assessed by the permutation test. The colored blocks in the heatmap represent the level of significance in each term. The yellow-to-red indicates the level from low to high, and grey indicates not significant.
Statistical test method
The main default statistical method for Fisher's tests and z-scores in PAGE (23) can be calculated using the following formulae:
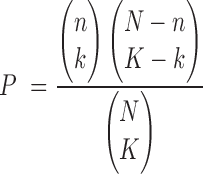 |
(1) |
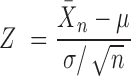 |
(2) |
where 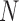 represents the total number of one organism's genes annotated with GO or of the user-provided background,
represents the total number of one organism's genes annotated with GO or of the user-provided background, 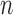 represents the number of genes mapped to the background in the query list,
represents the number of genes mapped to the background in the query list, 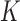 represents the total number of genes in one GO term and
represents the total number of genes in one GO term and 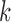 represents the counts of overlapped genes. Additionally,
represents the counts of overlapped genes. Additionally, 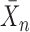 represents the mean of sample
represents the mean of sample 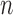 ,
, 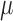 represents the average for all samples,
represents the average for all samples, 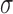 represents the standard deviation and
represents the standard deviation and 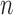 represents the sample counts. We also provided other statistical test methods, such as Hypergeometric and Chi-square tests. The Fisher's exact and Hypergeometric tests are more suitable for situation where the reference list covers the query list. The Chi-square is appropriate for fewer intersections between the target and reference lists.
represents the sample counts. We also provided other statistical test methods, such as Hypergeometric and Chi-square tests. The Fisher's exact and Hypergeometric tests are more suitable for situation where the reference list covers the query list. The Chi-square is appropriate for fewer intersections between the target and reference lists.
The P-value distribution (PVD) can display the distribution of P-values of significant GO terms. We also use the same formulas as in the SEA function to compute the P-values for another 99 lists of randomly selected genes of the same number as the observed query lists. Every GO term has a P-value distribution with 100 values, and the real P-value's position is recorded (Figure 2H).
Multi-test adjustment method
The multiple hypothesis adjustment can control the type I error effectively when there is an increase in the number of hypotheses tests. Several multiple hypotheses tests, including Holm (24), Hochberg (25), Hommel (26), Benjamini & Hochberg (BH) (27) and Benjamini & Yekutieli (BY) (28), are available. The first four methods have a greater control of the family-wise error rate. But, the ‘BH’ and ‘BY’ methods use FDRs under less stringent conditions than those of family-wise error rates, therefore, these methods are more powerful than the others.
Batch SEA and SEACOMPARE
With the development of high-throughput sequencing technologies, various platforms and the generation of multiple omics datasets across genomes, transcriptomes, epigenomes, proteomes and others, have been developed. Researchers usually focus on one key biology problem from different points of view (29). However, during transcriptome analysis, some studies on diurnal cycles (30), time-course (31), and the correlations of different abiotic or biotic stresses (31) often produce differentially expressed gene lists with intrinsic connections. Therefore, we developed the Batch SEA function to address requests to analyze multiple samples simultaneously. Batch SEA can cycle analyze the input gene lists with the same background, parameters and cut-offs, which accelerates the speed of processing and improves the efficiency.
SEACOMPARE was produced for the subsequent comparison of Batch SEA results. Two or more jobs can be selected for a comparative analysis with heatmaps to display the common or specific significant terms. The color in the tables indicates the P-values or FDRs.
The new developed visualization tools
The direct acyclic graph (DAG) drawer and Scatter Plots are two visualization tools with different formats to illustrate the significant GO terms. The DAG, based on the nature of the GO structure, can indicate submitted terms and the inter-relationships between terms, as in agriGO (8). Scatter Plots can display the significant GO terms after a multi-step filtering process. We first remove the GO terms above the P-value cut-off, then select one of the GO categories and filter the similar GO terms based on the semantic similarity measurement method (32). Scatter Plots remove similar GO vocabularies and can show the core GO terms.
The Semantic Similarity Measurement (32) estimates the functional similarities of gene products based on GO vocabularies (33) using the formula:
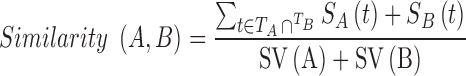 |
(3) |
Wang (33) defined the contribution of the GO term t (closer to term A) to the semantic GO term A as the S-value of GO term t related to term A, where 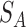 (t) is the S-value of GO term t related to term A and
(t) is the S-value of GO term t related to term A and 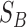 (t) is the S-value of GO term t related to term B. Multidimensional Scaling for Java (http://www.inf.uni-konstanz.de/algo/software/mdsj/) can translate the dissimilarity matrices to coordinate information.
(t) is the S-value of GO term t related to term B. Multidimensional Scaling for Java (http://www.inf.uni-konstanz.de/algo/software/mdsj/) can translate the dissimilarity matrices to coordinate information.
The processing flow for predicting the GO annotation
Glycyrrhiza uralensis was used as an example to introduce the comparative genomic approach to predict the GO annotation. We use BLAST-2.2.19 (-e 1e-3 -m 8) to compare the protein sequences of G. uralensis (34) and Arabidopsis thaliana (35). Then, the orthologous pairs with A. thaliana were determined, as was the corresponding GO annotation. Additionally, Blast2GO has a comprehensive bioinformatics platform that can automatically predict GO annotation based on the sequence information and a powerful remote annotation background (36). After the BLAST, interpro and mapping analyses steps in the Blast2GO application, we can export the GO annotation file and update agriGO v2.0. The InterPro (37) ID and Pfam (38) accessions have internal connections with the GO. Thus, we can map the GO terms to candidate genes using information on specific domains.
RESULTS
The updates in agriGO v2.0 are mainly embodied in three parts, adding a large number of species and datatypes, newly developed tools and more flexible visualization methods. These can increase the range, quality and strength of the statistical analyses, as well as present a convenient and intuitive graphical interface with new features.
Integrative collection of GO annotations
Reference files with higher whole genome coverage levels, as well as bigger ratios of GO terms (relative to all GO entries) are key points in producing reliable analysis results. We collected additional GO annotation files from different methods, including published databases (18,19,21,22,35,39), publications and those computed by comparative genomics (detailed in METHODS) (11,34,40–42).
Identifying the correct GO structure is another challenge (Supplementary Figure S1). We determined the structure for each GO annotation source by comparing its similarity with the GO files from the 2010 and 2016 versions. The background of version 2010 was retained to be consistent with agriGO v1, ensuring that former study results had a stable continuity. The 2016 version was corresponded to the new datatypes. We plan to update the older version and manually add into our web server later. Then, we ranked the datatype at the front based on their larger coverage of GO terms and genes, reliable data sources and newer annotated versions. The most recommended datatype was ranked first, and users are free to choose the appropriate datatype (Supplementary Table S1). In summary, a great deal of useful information regarding GO annotations was gained, including 394 species containing 865 datatypes.
Species classification
The previous display method for the organisms is not suitable for more species and datatypes become available. We categorized the species into plant, animal and fungi categories (Table 1). The plant category was further divided into Brassicaceae, Poaceae, Malvaceae, Fabaceae, Solanaceae, Medicinal plants, etc. Of those, the Brassicaceae family includes both vegetable and oilseed crops (43); the Poaceae (grass) family includes major cereal crops (Supplementary Table S1); the medicinal plants have attracted much attention, such as Cannabis sativa whose active components and their derivatives are Cannabinoids (44). In addition, Fish, Bird, Amphibia, Insecta and Mammalia were categorized under animal (19,45). Each category has its own group analysis webpage. Furthermore, to perform SEA for species that are being directly analyzed, we also created the single species analysis webpage that contain species that cannot be classified. The quick search for the Latin name or alias was also provided.
Table 1. Species classifications and statistics.
| Category | Classification | Species counts |
|---|---|---|
| Plant | Brassicaceae | 12 |
| Poaceae | 29 | |
| Malvaceae | 6 | |
| Fabaceae | 16 | |
| Solanaceae | 12 | |
| Rosaceae | 5 | |
| Medicinal plant | 12 | |
| Tree | 29 | |
| Algae | 20 | |
| Animal | Fish | 20 |
| Bird | 11 | |
| Amphibia | 3 | |
| Insecta | 56 | |
| Mammalia | 58 | |
| Fungi | Sordariomycetes | 5 |
Batch SEA
Several new functions have only been added to the single species analysis pages. We have now provided a new function, Batch SEA, to tackle multiple input datasets simultaneously based on one reference. The Batch SEA method often works best for time-course or continuous datasets, providing not only increased calculation speeds, but also benefits for the subsequent comparative analysis. SEACOMPARE is applied to compare the significant GO terms from the results of selected datasets to effectively identify GO terms.
We took six groups of up-regulated Arabidopsis genes under different cold treatment time intervals as inputs for a batch enrichment analysis at the A. thaliana SEA analysis webpage (8). There were 61, 64, 105, 150, 165 and 165 up-regulated genes corresponding to cold stress after 0.5, 1, 3, 6, 12 and 24 h (Supplementary Table S2), and these were inputted into the expanded text box after clicking on the ‘add’ button (Figure 2A). The detailed enrichment results, as well as the next visual analysis, with the significant GO terms are shown in Figure 2.
Job IDs are listed in the first analysis (Figure 2B), and the SEACOMPARE analysis can be performed using selected job IDs (Figure 2C). Thus, the GO terms were reproducible and a significant elevation occurred with the longer cold treatments. GO terms concerning ‘respond to cold’ (GO:0009409) were significantly represented throughout all of the stages, and ‘cold acclimation’ (GO:0009631) appeared after 24 h. Then, a brief summary about the job of 24-h cold stress is available. Based on the selected reference ‘Affymetrix ATH1 Genome Array’ (with GO version 2010), all 165 inputted genes were present in the 22 479 background genes and 34 significant GO terms were identified (Figure 2D). The detailed significant GO information in a tabular format ranked by FDR contains the GO term's description, P-value, FDR and PVD (Figure 2E). A DAG (Figure 2F) and Scatter Plots (Figure 2G) can be generated for each GO category (BP, CC and MF). The GO terms (GO:0009469, ‘response to cold’ and GO:0009414, ‘response to water deprivation’) were located at the lower level of the hierarchy but had higher significance levels (Figure 2F). Several GO terms (GO:0006950, ‘response to stress’ and GO:0009311, ‘oligosaccharide metabolic process’) that had too many or too few relationships with the rest of the GO terms were filtered out by Scatter Plot drawer (Figure 2G).
The PVD is represented by a line chart that can display the distribution of P-values of significant GO terms of observed query and random genes. It can decrease the false positive rate through the permutation test. The line chart can illustrate the location and percentage among 99 other random results to help users distinguish the real significant GO terms owing to their background bias or integrity. The PVD for each significant GO term can be illustrated when clicking on the line chart icon with a short description of the P-value position in the last column (Figure 2H). The P-value of the GO term concerning ‘response to hormone stimulus’ (GO:0009725) was not ranked first and was removed after a multi-test adjustment.
Additional tools for customized analyses
The accumulation of species in agriGO cannot catch up with the rate of genomic data generation or GO annotation updates, especially the de novo data that is user-defined. The custom tools independent of species were available in the previous version but were often overlooked by users. To facilitate user awareness, we highlighted these tools in the navigation bar and also added new tools to agriGO v2.0. The custom SEA and SEA share the same principle, but the custom tool can be used with any pair of queries and references. The SEACOMPARE tool can compare job IDs from SEA or a data matrix with GO terms as the first column and P-value as the leftover column (Figure 2C). Both the DAG drawer (Figure 2F) and Scatter Plots (Figure 2G) are graphics generation tools of different forms based on the significant GO terms and P-values in each of the categories, respectively. These can improve the usage range and compatibility with external data independent of the species and statistical methods. The DAG drawer can reproduce the structure with the significant GO terms based on the relationship between parent and child terms. The Scatter Plots can remove terms with similar definitions.
DISCUSSION
With the accumulation of sequencing data and various user requirements related to adding new species or datatypes, operating processes, downloads, GO versions and others (Supplementary Figure S2), we updated agriGO (v2.0) to contain more selected species and datatypes, a newly added species classification system and different GO structure versions. We maintained a similar interface and provide three analysis entrances: the group species analysis based on the classifications consistency and single species analyses with new Batch SEA and PVD options. Additionally, custom tools independent of species produced in the previous version (SEA and SEACOMPARE), as well as new developed tools (DAG drawer and Scatter Plots) were gathered and placed in a striking position (Figure 1).
The GO terms were updated monthly by changing the terms or relationship between terms. The current number of GO entries for BP, CC and MF are far greater than in the old version of agriGO (Supplementary Figure S1A). Additionally, the same GO term may correspond to different annotations, as well as a relationship between two parent–child terms (Supplementary Figure S1B). To have consistency between analyses from different periods, agriGO v2.0 must adapt to the changes in GO while maintaining uniformity between the GO annotations for each organism and the ontology structure (Supplementary Table S1).
Although the structure of GO and the coverage of GO annotations are continuously improving, the overall annotation level is low, except for those of some model species (46). Other means of gene functional annotations, like gene families and pathways, can be divided into gene sets for enrichment analyses (3). Furthermore, gene annotations can be more explicitly compared with the increasing number of relationships between genes and GO terms. Thus, we added gene annotations for some model species with high-quality gene annotations, including Arabidopsis, rice, maize, soybean and others.
The PVD option, which was embedded in the ‘optional advanced options’, provides a trend graph to display the P-values of query dataset and 99 random datasets (Figure 2H) (47). The line chart highlights the practical P-value with a red box and is displayed in two forms (normal and logarithmic scales). In SEA, we used Fisher's exact test and all of the multiple hypothesis tests (48) provided in agriGO v2.0 to analyze the up-regulated gene list under a 6-h cold stress treatment. The analysis results with six kinds of multiple hypothesis tests were compared by the SEACOMPARE tool displayed in Figure 3. We found that the GO terms selected by the multiple hypothesis test are always located at the top. The GO term concerning ‘calcium ion binding’ (GO:0005509) can be selected by the permutation test but is filtered out by the multi-test adjustment. Ca2+ plays an important role in the signal transduction pathways that lead to stress gene expression (49,50). Through the comparison between different statistical testing methods, the PVD can help users avoid missing important GO terms (47).
This was a practical demonstration that the distribution of P-values, to some extent, can distinguish false positive events (Figure 3). From our empirical point of view, lists of several hundred genes worked better (for A. thaliana as example). If there are too few genes on the list, the sensitivity is lost, but with too many, specificity is lacking. Some large input gene lists, encompassing >60–70% of the whole genome might be confused with the analysis results of other similar scale datasets. This is partially due to the incompleteness or preference of the GO terms in the reference background.
We have been constantly developing and enhancing our GO analysis tools and databases to support the agricultural community based on user experiences and feedback. The updated version agriGO v2.0 has an increased number of species and datatypes available, which have been classified into several groups. SEA, as the central tool, has been improved with a batch analysis to handle multiple input datasets simultaneously. Custom tools, including custom SEA and SEACOMPARE inherited from agriGO, and some new tools (DAG drawer and Scatter Plots) were placed together and highlighted in the navigation bar. These will provide users with more efficient and systematic analyses. We will continue to maintain our web server and welcome feedback on the need for new datatypes, the usability of existing tools, or suggestions for new features.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Professor Jia-Yang Li for his encouragement and critical suggestions. We are grateful to all of the agriGO users for their support and suggestions. The authors declare that they have no conflict of interest.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31371291, 31571360]. Funding for open access charge: National Natural Science Foundation of China [31371291, 31571360].
Conflict of interest statement. None declared.
REFERENCES
- 1. Huang da W., Sherman B.T., Lempicki R.A.. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009; 37:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhou X., Su Z.. EasyGO: Gene Ontology-based annotation and functional enrichment analysis tool for agronomical species. BMC Genomics. 2007; 8:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yi X., Du Z., Su Z.. PlantGSEA: a gene set enrichment analysis toolkit for plant community. Nucleic Acids Res. 2013; 41:W98–W103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Al-Shahrour F., Minguez P., Tarraga J., Medina I., Alloza E., Montaner D., Dopazo J.. FatiGO +: a functional profiling tool for genomic data. Integration of functional annotation, regulatory motifs and interaction data with microarray experiments. Nucleic Acids Res. 2007; 35:W91–W96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang da W., Sherman B.T., Lempicki R.A.. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009; 4:44–57. [DOI] [PubMed] [Google Scholar]
- 6. Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V., Clark N.R., Ma’ayan A.. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013; 14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Reimand J., Arak T., Adler P., Kolberg L., Reisberg S., Peterson H., Vilo J.. g:Profiler-a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 2016; 44:W83–W89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Du Z., Zhou X., Ling Y., Zhang Z., Su Z.. agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010; 38:W64–W70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang J., Liu D., Wang X., Ji C., Cheng F., Liu B., Hu Z., Chen S., Pental D., Ju Y. et al. The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat. Genet. 2016; 48:1225–1232. [DOI] [PubMed] [Google Scholar]
- 10. Iorizzo M., Ellison S., Senalik D., Zeng P., Satapoomin P., Huang J., Bowman M., Iovene M., Sanseverino W., Cavagnaro P. et al. A high-quality carrot genome assembly provides new insights into carotenoid accumulation and asterid genome evolution. Nat. Genet. 2016; 48:657–666. [DOI] [PubMed] [Google Scholar]
- 11. Qu Y., Zhao H., Han N., Zhou G., Song G., Gao B., Tian S., Zhang J., Zhang R., Meng X. et al. Ground tit genome reveals avian adaptation to living at high altitudes in the Tibetan plateau. Nat. Commun. 2013; 4:2071. [DOI] [PubMed] [Google Scholar]
- 12. Bombarely A., Moser M., Amrad A., Bao M., Bapaume L., Barry C.S., Bliek M., Boersma M.R., Borghi L., Bruggmann R. et al. Insight into the evolution of the Solanaceae from the parental genomes of Petunia hybrida. Nat. Plants. 2016; 2:16074. [DOI] [PubMed] [Google Scholar]
- 13. Chen X., Li H., Pandey M.K., Yang Q., Wang X., Garg V., Li H., Chi X., Doddamani D., Hong Y. et al. Draft genome of the peanut A-genome progenitor (Arachis duranensis) provides insights into geocarpy, oil biosynthesis, and allergens. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:6785–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. TÜRktaŞ M., YÜCebİLgİLİ KurtoĞLu K., Dorado G., Zhang B., Hernandez P., ÜNver T.. Sequencing of plant genomes – a review. Turkish J. Agric. Forestry. 2015; 39:361–376. [Google Scholar]
- 15. Harris M.A., Clark J., Ireland A., Lomax J., Ashburner M., Foulger R., Eilbeck K., Lewis S., Marshall B., Mungall C. et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004; 32:D258–D261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huntley R.P., Sawford T., Martin M.J., O’Donovan C.. Understanding how and why the Gene Ontology and its annotations evolve: the GO within UniProt. GigaScience. 2014; 3:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Proost S., Van Bel M., Vaneechoutte D., Van de Peer Y., Inze D., Mueller-Roeber B., Vandepoele K.. PLAZA 3.0: an access point for plant comparative genomics. Nucleic Acids Res. 2015; 43:D974–D981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goodstein D.M., Shu S., Howson R., Neupane R., Hayes R.D., Fazo J., Mitros T., Dirks W., Hellsten U., Putnam N. et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012; 40:D1178–D1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McCarthy F.M., Gresham C.R., Buza T.J., Chouvarine P., Pillai L.R., Kumar R., Ozkan S., Wang H., Manda P., Arick T. et al. AgBase: supporting functional modeling in agricultural organisms. Nucleic Acids Res. 2011; 39:D497–D506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rhee S.Y., Wood V., Dolinski K., Draghici S.. Use and misuse of the gene ontology annotations. Nat. Rev. Genet. 2008; 9:509–515. [DOI] [PubMed] [Google Scholar]
- 21. Kawahara Y., de la Bastide M., Hamilton J.P., Kanamori H., McCombie W.R., Ouyang S., Schwartz D.C., Tanaka T., Wu J., Zhou S. et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice. 2013; 6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tello-Ruiz M.K., Stein J., Wei S., Preece J., Olson A., Naithani S., Amarasinghe V., Dharmawardhana P., Jiao Y., Mulvaney J. et al. Gramene 2016: comparative plant genomics and pathway resources. Nucleic Acids Res. 2016; 44:D1133–D1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim S.Y., Volsky D.J.. PAGE: parametric analysis of gene set enrichment. BMC Bioinformatics. 2005; 6:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Holm S. A simple sequentially rejective multiple test procedure. Scand. J. Stat. 1979; 6:65–70. [Google Scholar]
- 25. Hochberg Y. A sharper Bonferroni procedure for multiple tests of significance. Biometrika Trust. 1988; 75:3. [Google Scholar]
- 26. Hommel G. A stagewise rejective multiple test procedure based on a modified Bonferroni test. Biometrika. 1988; 75:383–386. [Google Scholar]
- 27. Hochberg Y.B.A.Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995; 57:289–300. [Google Scholar]
- 28. Benjamini Y., Yekutieli D.. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001; 29:1165–1188. [Google Scholar]
- 29. He G., Zhu X., Elling A.A., Chen L., Wang X., Guo L., Liang M., He H., Zhang H., Chen F. et al. Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant cell. 2010; 22:17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kerr S.C., Gaiti F., Beveridge C.A., Tanurdzic M.. De novo transcriptome assembly reveals high transcriptional complexity in Pisum sativum axillary buds and shows rapid changes in expression of diurnally regulated genes. BMC Genomics. 2017; 18:221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Opitz N., Paschold A., Marcon C., Malik W.A., Lanz C., Piepho H.P., Hochholdinger F.. Transcriptomic complexity in young maize primary roots in response to low water potentials. BMC Genomics. 2014; 15:741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yu G., Li F., Qin Y., Bo X., Wu Y., Wang S.. GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics. 2010; 26:976–978. [DOI] [PubMed] [Google Scholar]
- 33. Wang J.Z., Du Z., Payattakool R., Yu P.S., Chen C.F.. A new method to measure the semantic similarity of GO terms. Bioinformatics. 2007; 23:1274–1281. [DOI] [PubMed] [Google Scholar]
- 34. Mochida K., Sakurai T., Seki H., Yoshida T., Takahagi K., Sawai S., Uchiyama H., Muranaka T., Saito K.. Draft genome assembly and annotation of Glycyrrhiza uralensis, a medicinal legume. Plant J. 2017; 89:181–194. [DOI] [PubMed] [Google Scholar]
- 35. Reiser L., Berardini T.Z., Li D., Muller R., Strait E.M., Li Q., Mezheritsky Y., Vetushko A., Huala E.. Sustainable funding for biocuration: The Arabidopsis Information Resource (TAIR) as a case study of a subscription-based funding model. Database. 2016; 2016:baw018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Conesa A., Gotz S., Garcia-Gomez J.M., Terol J., Talon M., Robles M.. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005; 21:3674–3676. [DOI] [PubMed] [Google Scholar]
- 37. Finn R.D., Attwood T.K., Babbitt P.C., Bateman A., Bork P., Bridge A.J., Chang H.Y., Dosztanyi Z., El-Gebali S., Fraser M. et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 2017; 45:D190–D199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Finn R.D., Coggill P., Eberhardt R.Y., Eddy S.R., Mistry J., Mitchell A.L., Potter S.C., Punta M., Qureshi M., Sangrador-Vegas A. et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016; 44:D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. The UniProt Consortium UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2016; 45:D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kellner F., Kim J., Clavijo B.J., Hamilton J.P., Childs K.L., Vaillancourt B., Cepela J., Habermann M., Steuernagel B., Clissold L. et al. Genome-guided investigation of plant natural product biosynthesis. Plant J. 2015; 82:680–692. [DOI] [PubMed] [Google Scholar]
- 41. Xu H., Song J., Luo H., Zhang Y., Li Q., Zhu Y., Xu J., Li Y., Song C., Wang B. et al. Analysis of the genome sequence of the medicinal plant Salvia miltiorrhiza. Mol. Plant. 2016; 9:949–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. VanBuren R., Bryant D., Edger P.P., Tang H., Burgess D., Challabathula D., Spittle K., Hall R., Gu J., Lyons E. et al. Single-molecule sequencing of the desiccation-tolerant grass Oropetium thomaeum. Nature. 2015; 527:508–511. [DOI] [PubMed] [Google Scholar]
- 43. Cheng F., Liu S., Wu J., Fang L., Sun S., Liu B., Li P., Hua W., Wang X.. BRAD, the genetics and genomics database for Brassica plants. BMC Plant Biol. 2011; 11:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guzman M. Cannabinoids: potential anticancer agents. Nat. Rev. Cancer. 2003; 3:745–755. [DOI] [PubMed] [Google Scholar]
- 45. Giraldo-Calderon G.I., Emrich S.J., MacCallum R.M., Maslen G., Dialynas E., Topalis P., Ho N., Gesing S., VectorBase C., Madey G. et al. VectorBase: an updated bioinformatics resource for invertebrate vectors and other organisms related with human diseases. Nucleic Acids Res. 2015; 43:D707–D713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rhee S.Y., Mutwil M.. Towards revealing the functions of all genes in plants. Trends Plant Sci. 2014; 19:212–221. [DOI] [PubMed] [Google Scholar]
- 47. Strasser H., Weber C.. On the asymptotic theory of permutation statistics. Math. Methods Statist. 1999; 8:220–250. [Google Scholar]
- 48. Noble W.S. How does multiple testing correction work. Nat. Biotechnol. 2009; 27:1135–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Solanke A.U., Sharma A.K.. Signal transduction during cold stress in plants. Physiol. Mol. Biol. Plants. 2008; 14:69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cheong Y.H., Kim K.N., Pandey G.K., Gupta R., Grant J.J., Luan S.. CBL1, a calcium sensor that differentially regulates salt, drought, and cold responses in Arabidopsis. Plant Cell. 2003; 15:1833–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.