

Abstract
The ability to accurately model evolutionary dynamics in cancer would allow for prediction of progression and response to therapy. As a prelude to quantitative understanding of evolutionary dynamics, researchers must gather observations of in vivo tumor evolution. High-throughput genome sequencing now provides the means to profile the mutational content of evolving tumor clones from patient biopsies. Together with the development of models of tumor evolution, reconstructing evolutionary histories of individual tumors generates hypotheses about the dynamics of evolution that produced the observed clones. In this review, we provide a brief overview of the concepts involved in predicting evolutionary histories, and provide a workflow based on bulk and targeted-genome sequencing. We then describe the application of this workflow to time series data obtained for transformed and progressed follicular lymphomas (FL), and contrast the observed evolutionary dynamics between these two subtypes. We next describe results from a spatial sampling study of high-grade serous (HGS) ovarian cancer, propose mechanisms of disease spread based on the observed clonal mixtures, and provide examples of diversification through subclonal acquisition of driver mutations and convergent evolution. Finally, we state implications of the techniques discussed in this review as a necessary but insufficient step on the path to predictive modelling of disease dynamics.
Cancer develops through evolutionary processes (Nowell 1976; Greaves and Maley 2012). Extrinsic and intrinsic factors combined with aberrant DNA-repair result in increased mutation rate in many cancers, leading to cell-specific genomic diversity. Tumor microenvironments, immune surveillance, therapeutic interventions, and other factors impose selective pressures on the substrate of diverse tumor clones, resulting in differential proliferation as a function of tumor clone fitness. We suggest that the desired end-goal of predicting fitness of tumor clones remains elusive in the field. However, it is well accepted that clonal dynamics (defined as the relative abundance of tumor clones over time or in different microenvironments in anatomic space) associate with major events in tumor progression including oncogenesis, treatment resistance, and metastasis. Thus, observing clonal dynamics and understanding their nature in different patient contexts represent a powerful first step on the path toward predictive fitness modelling in cancer.
We begin with the notion of generating in vivo observations in the form of evolutionary histories. A tumor’s evolutionary history comprises: (1) the genotypes of ancestral and extant clonal populations; (2) the evolutionary tree relating those genotypes; and (3) the distribution of clonal populations through time and anatomic space. Tumor biopsies collected for diagnostic purposes or during surgical resection provide tissues from which observations of tumor evolution can be made. We will focus on high-throughput sequencing methods applied to cancer tissue samples as a means of profiling the mutational content of tumor genomes, and inferring tumor genotypes present in tumor samples. Because each sample represents a population of only extant tumor cells, the evolutionary history of each tumor must be inferred computationally through reconstruction approaches. Herein, we will review principles of inferring the clonal genotypes, clone phylogenies, and the distribution of clones in tumor samples from high-throughput sequencing data. We will then present the application of these methods in two separate studies on patient tumor samples designed to observe clonal dynamics through time within follicular lymphoma (FL) patients’ clinical histories (Kridel et al. 2016 ) and over anatomic space in the peritoneal cavity of high-grade serous (HGS) ovarian cancer patients (McPherson et al. 2016).
FEATURES OF EVOLVING CANCER GENOMES
Genomic features distinguishing a cancer’s genome from its germline progenitor are broadly categorized into small variants, copy number (CN) changes, and rearrangements. Small variants include somatic point mutations, or single nucleotide variants (SNVs), and small insertions/deletions (InDels). Rearrangements involve the reordering of chromosomal segments and include translocations, inversions, tandem duplications, and deletions. CN changes include deletion or amplification of whole chromosomes or smaller chromosomal segments, and are often associated with rearrangements.
Of these three classes of genomic features, SNVs are particularly useful as markers of clones when studying tumor evolution. After acquisition of an SNV, a tumor cell’s progeny will, in general, all harbor the same SNV. Most SNVs can be assumed to be selectively neutral, and are thus passengers of a relatively small set of coincident driver mutations. The large size of the genome and the relatively small per nucleotide mutation rate leads to the “infinite sites assumption”: A specific nucleotide in the genome will be affected by mutation, at most, once throughout the life history of a tumor. Under this assumption, an SNV becomes an identifying feature of a clade of the tumor cell phylogeny, with its detection an indication of the presence of that clade. However, as is often the case in tumors harboring deficient DNA repair, deletion of chromosomal segments will remove concomitant SNVs. Thus, CN changes must be accounted for when assigning an SNV as an identifying feature of a tumor clade.
CLONAL EVOLUTION IN CANCER
Mutational processes produce genomically heterogeneous populations of tumor cells, providing the necessary substrate for evolution.3 Selective pressures acting on tumor cell populations will result in the expansion of tumor cells with higher fitness at the expense of less fit tumor cells. Over time, populations with higher fitness will out-compete ancestral populations, resulting in higher representation of fit genotypes in the mixture of tumor cells. Additionally, metastasis, invasion, and localized immune surveillance properties result in regional differences in clonal abundance across anatomic sites.
We define a tumor “clone” as a maximal set of tumor cells with shared ancestry.4 Modelling tumor evolution, in terms of clones, is a useful abstraction that greatly simplifies analysis over explicit modelling of evolutionary relationships between individual tumor cells (see Fig. 1). More specifically, modelling evolutionary relationships between ancestral tumor clones allows researchers to focus on the important selection events that produced a tumor genotype. In reality, an ancestral genotype may have arisen through successive stages of mutation until acquisition of a driver mutation, imposition of therapy, or some other selection event resulted in extinction of cells with intermediate genotypes and proliferation of cells harboring multiple, serially acquired mutations. However, when observing only the population of extant cells, loss of the intermediate genotypes makes it infeasible to order sets of mutations selected together, and it becomes convenient to consider those mutations as part of one ancestral clonal genotype.
Figure 1.
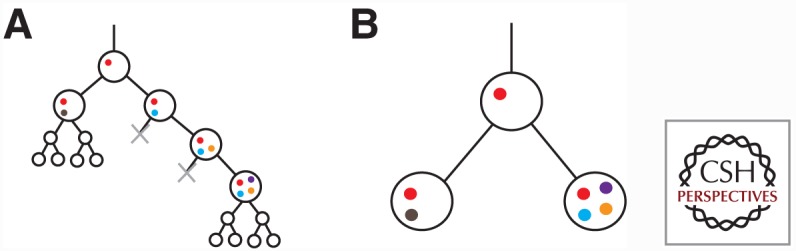
Phylogeny of individual cells (A) and a corresponding clone phylogeny (B). The cell phylogeny shows acquisition of mutations (colored circles) serially, although intermediate genotypes are extinguished (gray x) during selection for a clonal genotype. Neutrally evolving populations produce, in approximation, a full binary tree structure in the cell phylogeny. The clone phylogeny is a simplification of the cell phylogeny showing only the genotypes surviving selection events, and a common genotype for neutrally evolving extant populations.
Genomic heterogeneity may thus arise from the presence of divergent clones produced during ancestral selection events. Additionally, mutation without selection, or neutral evolution, will result in acquisition of many predominantly low-cellular prevalence mutations (Williams et al. 2016). Estimated mutation rates range in the 10s to 1000s per cell-division (Williams et al. 2016), implying that each cell likely harbors a unique genotype, and could be considered to be its own genomically distinct clone. In practice, it is often preferable to model only extant clones that have undergone a selection event and resulted in a significantly divergent genotype. Neutral evolution may also result in fixation of selectively neutral mutations caused by the stochastic nature by which future genotypes are sampled from existing genotypes in a birth death process. Such mutations will be indiscernible from the passenger mutations that were swept to fixation by the most recent selection event affecting the clonal population. Note that one of the selective mechanisms is related to the experiment itself: Sectioning of biopsied material for sequencing will sample a subset of the tumor cells, resulting in differential representation of specific genotypes in the sequence data, particularly if tumor clones are nonrandomly distributed throughout the tumor mass.
SEQUENCING OF HETEROGENEOUS TUMOR SAMPLES
Whole-genome sequencing (WGS) is particularly powerful when applied to the study of cancer evolution as it allows for the identification of all classes of genomic features mentioned above, and profiles the full length of the genome. However, the information gained from application of bulk WGS to a tumor biopsy is incomplete. Tumor cells are lysed, chromosomes are fragmented, and the resulting pool of fragments are sequenced en masse, without preserving the cell-specific origin of each fragment. Genomic features identified from sequenced fragments can therefore only be assigned to populations of cells using computational inference. Successful assignments of SNVs to cell populations lead to the ability to quantify population abundance within the original sample. This ultimately enables observations of cellular dynamics over time, or anatomic space.
The prevalence of each SNV in the cellular population can be approximately assessed by the fraction of reads with the variant (termed variant allele fraction [VAF]). The high breadth of WGS often necessitates sequencing to a lower depth to allow experiments to remain economically feasible. VAF accuracy is directly related to sequencing depth, and thus WGS is not ideally suited to quantification of low-prevalence clones. Targeted high- depth sequencing of a subset of phylogenetically informative SNVs is a useful method for augmenting low-depth WGS datasets, allowing for quantification of low-prevalence clones. As a result of CN change, VAF is not equal to the fraction of cells harboring an SNV, the mutation’s “cellular prevalence,” but is confounded by the SNV loci’s CN (Roth et al. 2014). As a basic example, an SNV with VAF of 0.5 could be heterozygous in 100% of the cells or homozygous in 50% of the cells. Accounting for CN is fundamentally important for accurate inference of evolutionary histories, especially for tumors that show even a moderate amount of deviation from diploid owing to CN change.
CLUSTERING MUTATIONS BY CELLULAR PREVALENCE
Clonal evolution will result in increased representation of specific fit genotypes, and increased cellular prevalence of mutations present in those genotypes. In the extreme case, all mutations present in the clone that survived the last selective sweep will be present with cellular prevalence equal to 1 (notwithstanding relevant CN changes deleting those mutations). Thus, within the context of a tumor evolving through nonneutral clonal dynamics, considerable benefit can be gained by clustering mutations with similar inferred cellular prevalence, as it allows for the discovery of sets of mutations with shared clonal ancestry. As cellular prevalence is not directly observable, in practice, mutations are clustered by VAF, while modelling, correcting for, or filtering by SNV loci CN. The resulting mutation clusters represent sets of mutations (termed clonal clusters) putatively sharing a common evolutionary history: They originate at the same point between selective sweeps, and are subject to the same CN changes that removed the SNVs from descendant subpopulations. An implicit assumption is that sets of mutations with no common history will not share the same cellular prevalence by chance. This assumption (termed weak parsimony in Deshwar et al. 2015) becomes less problematic when sequencing multiple samples from the same tumor biopsy. With an increasing number of samples, sets of mutations are less likely to have similar cellular prevalence across all samples by chance, with the corollary being that shared prevalences across multiple samples increases the likelihood that such sets were generated by a common evolutionary history.
Importantly, a mutation cluster does not, in general, correspond to the genotype of any clone. Instead, a tumor clone’s genotype consists of the cluster of mutations acquired immediately before the most recent selective sweep, in addition to any mutations acquired since the germline progenitor.
CLONAL GENOTYPES AND CLONE PHYLOGENIES
To understand mutation order, convergence, clonal interference, and other evolutionary properties of a tumor requires a level of inference beyond mutation clustering to clonal genotypes and clone phylogenies. In the context of tumor evolution, a clone phylogeny refers to the tree structured graph that represents the evolutionary relationships between extant and ancestral tumor clones. Given direct observations of extant clonal genotypes and an appropriate evolutionary model, it is possible to reconstruct both the evolutionary tree and ancestral clonal genotypes using standard phylogenetic approaches such as maximum parsimony. Sequencing multiple tumor samples is a powerful method for measuring the genotypes of tumor clones and reconstructing clonal phylogenies. If each sequenced sample is populated by a single tumor clone, the set of mutations identified in each sample may be used as a proxy for extant genotypes, allowing for straightforward phylogenetic reconstruction.
In general, however, bulk sequencing results are confounded by the unknown mixture of clones in each tumor sample, and the mutations identified in a sample represent a superposition of multiple clonal genotypes. From a biological perspective, observing a mixture of clones generates interesting hypotheses. Early proliferation of an emergent clone caused by a recently acquired driver mutation, invasion of one clone’s anatomic space by another clone, or interference or cooperation between clones, are all plausible interpretations. Thus, methods that are able to infer clonal genotypes, clone phylogenies, and sample clone mixtures (evolutionary histories) are important for accurate measurement of clonal dynamics.
Inference of evolutionary histories from tumor sequencing data is an ongoing topic of research. CITUP (Malikic et al. 2015), LICHeE (Popic et al. 2015), and SPRUCE (El-Kebir et al. 2016) take a combinatorial optimization approach, attempting to build evolutionary histories from a set of mutation clusters in a two-step process. PhyloSub (Jiao et al. 2014), PhyloWGS (Deshwar et al. 2015), and Canopy (Jiang et al. 2016) use Markov chain Monte Carlo (MCMC) to sample from the full posterior of evolutionary histories. The fundamental difficulty of the problem lies in the size of the search space over which methods optimize or generate MCMC samples. The space of trees is combinatorially large in the number of tumor clones, as is the space of assignments of mutations to tumor clones. Identifiability is also an issue. For many datasets, multiple evolutionary histories provide an equivalent fit to the data, especially when sequencing a smaller number of samples (Jiao et al. 2014).
WORKFLOW FOR RECONSTRUCTING EVOLUTIONARY HISTORIES
We synthesized the methods above into a workflow for reconstructing an evolutionary history by sequencing multiple tumor samples (Fig. 2). WGS is applied to each sample to discover patient-specific SNVs, estimate VAF of each SNV in each sample, and infer CN changes in each sample. Phylogenetically informative SNVs are selected uniformly from clusters of SNVs in the k-dimensional space of VAFs across the k samples. Deep sequencing is applied to the selected SNVs to provide accurate VAF measurements. PyClone (Roth et al. 2014) is applied to the deep sequenced SNVs to accurately cluster SNVs and estimate the cellular prevalence of each clonal cluster.5 Finally, CITUP (Malikic et al. 2015) is applied to the PyClone clusters to simultaneously infer the sets of clonal clusters comprising each clonal genotype, the clone phylogeny, and the sample clone prevalences.
Figure 2.

An experimental and analytical workflow for observing clonal dynamics. (A) Identify variant allele fraction (VAFs) of mutations in more than one sample with whole-genome sequencing (WGS). (B) Deep targeted sequencing of mutations from (A) provide more precise VAFs. (C) Input VAF into PyClone (Roth et al. 2014) for projection of VAF onto cellular prevalence estimates. (D) Estimate the mean cellular prevalence of coclustered mutations. (E) Using principles of parsimony under tree-additivity constraints, infer clonal phylogenies using CITUP (Malikic et al. 2015). (F) Estimate the clonal prevalences in each sample of interest.
Inference of clonal genotypes, and measurement of their abundance in bulk tumor samples have been used to great effect in observing clonal dynamics in human cancers. Here, we describe two recent studies, utilizing the workflow above, which show the type of inference that can be performed to interpret distributions of clones over time and across anatomic space.
CLONAL DYNAMICS THROUGH TIME: TUMOR EVOLUTION UNDERLYING HISTOLOGICAL TRANSFORMATION AND EARLY PROGRESSION OF FOLLICULAR LYMPHOMA
In a study of follicular lymphoma (FL), our group recently inferred the clonal dynamics through time of 21 patients with FL who experienced disease progression (Kridel et al. 2016). Although the majority of FL patients eventually experience disease progression (Kridel et al. 2012), a subset of patients are marked by early treatment resistance (progressed follicular lymphoma [PFL] patients) (Casulo et al. 2015a) and/or histological transformation (transformed follicular lymphoma [TFL]) of their tumors to a more aggressive form (Casulo et al. 2015b). Both of these clinical inflection points are associated with poor survival. As such, profiling and comparing the clonal composition between FL diagnostic (T1) and relapse/transformed (T2) specimens presents an opportunity to study the evolutionary dynamics underpinning FL disease progression.
By using WGS and deep sequencing data, in conjunction with bioinformatic methods such as PyClone (Roth et al. 2014) and Citup (Malikic et al. 2015), we reconstructed the evolutionary history of each tumor and characterized the patterns of clonal dynamics associated with transformation and early progression (Kridel et al. 2016). For TFL patients (n = 15), our results showed that the aggressive phenotype was associated with a vast change in clonal composition between the diagnostic and transformed specimen (Fig. 3A,B). Specifically, the majority of these patients (13/15, 87%) showed a pattern of a rare clone at diagnosis (<1% clonal prevalence) arising to dominant the clonal composition at transformation. This striking pattern was independent of both treatment regimen and time to transformation. Importantly, the dominant clone often harbored known driver mutations (e.g., CCND3, B2M, EZH2) suggesting that these punctuated expansions are under positive selection.
Figure 3.

Modes of evolution in follicular lymphoma. (A, C) Clonal dynamics of a selected transformed follicular lymphoma (TFL) patient (FL1004) and progressed follicular lymphoma (PFL) patient (FL2008). Progression from SNV cellular prevalences to clonal phylogenies and prevalences. (B, D) Schematic models of evolutionary progression in TFL and PFL. (From Kridel et al. 2016, reproduced, with permission from the author.)
In contrast, PFL patients (n = 6) showed a different pattern of clonal dynamics between diagnostic and early progressed specimens (Fig. 3). These patient’s tumors typically harbored readily detectable clones at diagnosis, which expanded to full clonal prevalence during treatment with immunochemotherapy. These patterns suggest that clones harboring treatment resistance properties were already present at diagnosis, and that symptomatic disease progression may be attributed to selection of clones that were major constituents of the diagnostic sample.
Collectively, our study reveals divergent modes of tumor evolution that underlie histological transformation and early progression. For TFL patients, the massive expansion of clones in T2 samples that were rare or detectably absent in T1 samples indicates that diagnostic samples are not likely to possess reliable predictors of transformation in the majority of cases. Moreover, the results suggest that clonal dynamics occurring after diagnosis likely underpin the histological change. As such, to determine of exact timing of transformation likely requires more frequent monitoring using techniques such as circulating tumor DNA (Scherer et al. 2016).
For PFL patients, there was stability in the clonal architecture between diagnosis and relapsed specimens. This suggests that the fitness could be attributed to nongenetic factors or that these tumors acquire resistance properties very early in their evolutionary histories and in the absence of therapeutic selective pressure. Importantly, the abundance of these clones at diagnosis substantiates the feasibility of predicting early treatment resistance. Additional studies that comprehensively profiling the clonal architecture and associated gene mutations at diagnosis in PFL patients will be needed for this.
CLONAL DYNAMICS ACROSS ANATOMIC SPACE: HIGH-GRADE SEROUS OVARIAN CANCER PERITONEAL SPREAD
The distribution of tumor clones resulting from disease spread as measured by multisample genome sequencing provides a window into the evolutionary dynamics that shaped a patient’s cancer. In a study of HGS ovarian cancer, our group reconstructed the evolutionary histories of the pretreatment cancers of seven patients (McPherson et al. 2016). HGS is characterized by widespread and early intraperitoneal spread to organs, including the omentum and contralateral ovary. The genomes of HGS show pervasive instability because of TP53 loss in nearly all cases (Ahmed et al. 2010), accompanied by homologous recombination and other DNA repair deficiencies in the majority of cases (Patch et al. 2015). Thus, combined dysregulation of apoptosis and DNA repair processes provide a mechanism for generating a substrate of genomically diverse clones (Bashashati et al. 2013; Ha et al. 2014; Schwarz et al. 2015) from monoclonal aetiology (Jacobs et al. 1992; Khalique et al. 2009).
We applied WGS to multiple samples collected from the seven HGS patients, and targeted deep sequencing of phylogenetically informative SNVs identified from WGS for accurate quantification of VAF. We inferred clonal genotypes and clone phylogenies using a combination of PyClone and maximum parsimony phylogenetic inference within the context of a stochastic Dollo model of evolution (Alekseyenko et al. 2008). The stochastic Dollo model assumes SNVs originate once within the evolutionary history of a tumor (infinite sites assumption), and also models the possibility that an SNV may be lost as a result of the CN change in descendant lineages, thus accounting for the pervasive chromosomal instability that is typical of HGS. Clone sample prevalence was estimated using a post hoc MCMC step. The output was a series of clonal “maps,” illustrating the spreading patterns of each patient (Fig. 4).
Figure 4.

Spatial distribution of high-grade serous (HGS) clones in seven patients. The anatomy of each patient is depicted in the center of each diagram, with a presentation of the clonal mixture by color within the inner circle, and the clones present in the sample overlaid on the clone phylogeny on the outer circle. The full clone phylogeny is shown at the top right for each patient. (From McPherson et al. 2016, reproduced, with permission of the author.)
We categorized samples as clonally pure or polyclonal; polyclonal samples were further classified into samples in which multiple clones were from within the same phylogenetic clade (monophyletic) or from different clades (polyphyletic). The majority of samples were monophyletic or clonally pure; however, each patient showed at least one polyphyletic sample. Polyphyletic samples may be indicative of an ancestral population having undergone branched clonal evolution. In patient 9, for example, acquisition of a ERBB2 amplification by a subclone of left ovary site 2 was followed by expansion of that clone into right ovary and omental sites (Fig. 4). In an alternative scenario, a polyphyletic sample may have been seeded either serially or simultaneously (polyclonal migration) by multiple clones. In patient 1, for example, the contralateral left ovary site was populated by both omentum and right ovary-specific clones. The most parsimonious explanation for this observation is remetastasis of the left ovary.
Accurately inferred clonal phylogenies allow for the investigation of parallel or convergent evolution of genomic features, an indication of positive selection toward shared genomic traits. We focused on convergent evolution of the structural genome toward amplification or loss of specific genes. Patient 7 showed convergent evolution toward loss of heterozygosity (LOH) of chromosome 14, with two distinct events independently deleting the reference allele of a single-nucleotide polymorphism (SNP) affecting a splice site of RAD51B (Fig. 5). Both RAD51B LOH clones coexist in the RUtD3 sample of timepoint 1, with clone B eventually dominating the brain metastasis of timepoint 2, clone D the right pelvic samples of timepoint 3, and RAD51B heterozygous clones extinguished in the second and third timepoints. Clone B also harbored an NF1 deletion that may have driven metastasis and expansion of that clone in the patient’s brain, providing of an example of clonal interference between the RAD51B LOH event of clone D and the NF1 deletion/RAD51B LOH genotype of clone B.
Figure 5.

Examples of convergent evolution of copy number (CN). (A) Major (red line) and minor (blue line) allele-specific CNs with experimentally validated breakpoints (colored lines/arrows) for patient 7, chromosomes 14, 8, 19, and 16. Both the brain metastasis (BrM) and right pelvic mass (RPvM) show loss of heterozygosity (LOH) of distinct segments of chromosome 14, corroborated by the presence of distinct sets of breakpoints. (B) The top plot shows total CN for omentum (Om1, Om2) and right ovary samples (ROv1, ROv2) in patient 2. The bottom plot shows the presence (blue) or absence (white) of rearrangement breakpoints, with break-ends in the CCNE1 amplified region. Green lines show correspondence between rearrangement breakpoints in the heatmap and positions of break-ends, highlighting the association between breakpoints and CN transitions. (C) Break-ends are associated with significantly higher CN change for samples in which they are predicted as present compared with absent. Shown is the distribution of the difference in CN between segments on the right and left of each break-end, correcting for break-end orientation. Higher CN difference indicates higher CN change induced by the breakpoint. For each break-end in each sample, we predicted whether the break-end exists in that sample. We then compare the CN difference in each sample for break-ends that are predicted as absent (left) versus present (right) in those samples. (From McPherson et al. 2016, reproduced, with permission from the author.)
In patient 2, presence of breakpoints within the CCNE1-amplified region recapitulated the clone phylogeny identified from SNVs. All 28 breakpoints within the CCNE1-amplified region were determined to be specific to either the right ovary or omentum. As evidence of the association between the breakpoints and CN changes, break-ends within the CCNE1-amplified region coincided with significantly higher CN change in samples in which they were predicted as present, compared with samples in which they were predicted as absent (Fig. 5). The combined effects of these breakpoints in both the right ovary and omentum was high level amplification of CCNE1, a known driver event in HGS.
CONCLUSIONS
The methods and examples we illustrate here exemplify progress in enumerating clones in cancer tissues and measuring their distribution across spatial and temporal axes. Interpretation of results obtained through analysis of our modestly sized cohorts is facilitated by leveraging prior information gained from studies in larger cohorts. Specifically, known driver mutations associated with emergence of more aggressive tumor clones in TFL, and convergent evolution in HGS, are suggestive of common patterns of evolution in these cancers. Nevertheless, we view the primary objective of our initial work as observational, necessary for providing an initial window into the evolutionary complexity of the diseases studies and generating additional important and unresolved questions to be addressed in future studies. For example, can we predict which clones might expand in different microenvironments? How do clonal dynamics relate to immunodynamics? Which clones might expand over a patient’s clinical trajectory? Under what selective pressures are some clones more fit than others? We suggest that the methods and examples presented here offer a prelude to future work that might elucidate progress addressing these questions.
As part of future efforts to build predictive models of clonal dynamics, it will be necessary to tackle both the complexity of fitness landscapes and the heterogeneity between cancers and patients. The extensive clinical heterogeneity in many cancers weakens our ability to make predictive statements from clonal phylogenies of a small number of patients. We propose that future studies involving orders of magnitude more patients for which clonal analysis is standard may allow researchers to build predictive models robust to the many factors that currently confound such analyses. Furthermore, the complex effects of genetic backgrounds, tumor microenvironments, and immune surveillance, all impact the observed clonal mixtures in each patient. We can envision future datasets that attempt to capture and provide an integrated analysis of each of these factors, and model their effects on fitness landscapes, providing additional power for the creation of accurate predictive models.
Finally, high-fidelity physical models of cancer will allow for controlled replicate experiments, providing the reproducibility that is a perquisite for predictive science. A recent study by our group provides a useful illustrative example. Patient derived immunodeficient mouse xenograft (PDX) systems offer the opportunity to study cancer cell populations in laboratory settings, where repeated experiments can be performed. Using the methods described in this review, we reported that clonal dynamics in serially passaged breast cancer PDX models are reproducible in parallel PDX lines (Eirew et al. 2014). This important result establishes a strong likelihood of determinism, setting the stage for measuring reproducible dynamics in interventional PDX studies using pharmacologic or genetic perturbation. We suggest that reproducible dynamics in the context of controlled interventional studies will provide a substrate for quantitative fitness models of cancer. If successful, these models will ultimately permit prediction of clonal response profiles of treatment interventions, significantly augmenting quantitative understanding on the path from observations of dynamics toward mechanistic insight.
Other mechanisms of variation, such as epigenetic modification, are likely contributing to evolution as well, however, we focus only on genomic aberrations here.
Maximality is required, as a clone should include “all” descendants of a common ancestor.
Although other methods may be used in place of PyClone, one advantage of PyClone is its ability to correct for the CN of each SNV loci.
Editors: Charles Swanton, Alberto Bardelli, Kornelia Polyak, Sohrab Shah, and Trevor A. Graham
Additional Perspectives on Cancer Evolution available at www.perspectivesinmedicine.org
REFERENCES
- Ahmed AA, Etemadmoghadam D, Temple J, Lynch AG, Riad M, Sharma R, Stewart C, Fereday S, Caldas C, deFazio A, et al. 2010. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J Pathol 221: 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyenko AV, Lee CJ, Suchard MA. 2008. Wagner and Dollo: A stochastic duet by composing two parsimonious solos. Syst Biol 57: 772–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashashati A, Ha G, Tone A, Ding J, Prentice LM, Roth A, Rosner J, Shumansky K, Kalloger S, Senz J, et al. 2013. Distinct evolutionary trajectories of primary high-grade serous ovarian cancers revealed through spatial mutational profiling. J Pathol 231: 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casulo C, Byrtek M, Dawson KL, Zhou X, Farber CM, Flowers CR, Hainsworth JD, Maurer MJ, Cerhan JR, Link BK, et al. 2015a. Early relapse of follicular lymphoma after rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone defines patients at high risk for death: An analysis from the National LymphoCare Study. J Clin Oncol 33: 2516–2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casulo C, Burack WR, Friedberg JW. 2015b. Transformed follicular non-Hodgkin lymphoma. Blood 125: 40–47. [DOI] [PubMed] [Google Scholar]
- Deshwar AG, Vembu S, Yung CK, Jang GH, Stein L, Morris Q. 2015. Phylowgs: Reconstructing subclonal composition and evolution from whole-genome sequencing of tumors. Genome Biol 16: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eirew P, Steif A, Khattra J, Ha G, Yap D, Farahani H, Gelmon K, Chia S, Mar C, Wan A, et al. 2014. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature 518: 422–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Kebir M, Satas G, Oesper L, Raphael BJ. 2016. Inferring the mutational history of a tumor using multi-state perfect phylogeny mixtures. Cell Syst 3: 43–53. [DOI] [PubMed] [Google Scholar]
- Greaves M, Maley CC. 2012. Clonal evolution in cancer. Nature 481: 306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha G, Roth A, Khattra J, Ho J, Yap D, Pentice LM, Melnyk N, McPherson A, Bashashati A, Laks E, et al. 2014. TITAN: Inference of copy number architectures in clonal cell populations from tumor whole-genome sequence data. Genome Res 24: 1881–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs IJ, Kohler MF, Wiseman RW, Marks JR, Whitaker R, Kerns BA, Humphrey P, Berchuck A, Ponder BA, Bast RC. 1992. Clonal origin of epithelial ovarian carcinoma: Analysis by loss of heterozygosity, p53 mutation, and X-chromosome inactivation. J Natl Cancer Inst 84: 1793–1798. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Qiu Y, Minn AJ, Zhang NR. 2016. Assessing intratumor heterogeneity and tracking longitudinal and spatial clonal evolutionary history by next-generation sequencing. Proc Natl Acad Sci 113: E5528–E5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao W, Vembu S, Deshwar AG, Stein L, Morris Q. 2014. Inferring clonal evolution of tumors from single nucleotide somatic mutations. BMC Bioinformatics 15: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalique L, Ayhan A, Whittaker JC, Singh N, Jacobs IJ, Gayther SA, Ramus SJ. 2009. The clonal evolution of metastases from primary serous epithelial ovarian cancers. Int J Cancer 124: 1579–1586. [DOI] [PubMed] [Google Scholar]
- Kridel RR, Sehn LH, Gascoyne RD. 2012. Pathogenesis of follicular lymphoma. J Clin Invest 122: 3424–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kridel R, Chan FC, Mottok A, Boyle M, Farinha P, Tan K, Meissner B, Bashashati A, McPherson A, Roth A, et al. 2016. Histological transformation and progression in follicular lymphoma: A clonal evolution study. PLoS Med 13: e1002197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malikic S, McPherson AW, Donmez N, Sahinalp CS. 2015. Clonality inference in multiple tumor samples using phylogeny. Bioinformatics 31: 1349–1356. [DOI] [PubMed] [Google Scholar]
- McPherson A, Roth A, Laks E, Masud T, Bashashati A, Zhang AW, Ha G, Biele J, Yap D, et al. 2016. Divergent modes of clonal spread and intraperitoneal mixing in high-grade serous ovarian cancer. Nat Genet 48: 758–767. [DOI] [PubMed] [Google Scholar]
- Nowell P. 1976. The clonal evolution of tumor cell populations. Science 194: 23–28. [DOI] [PubMed] [Google Scholar]
- Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, Nones K, Cowin P, Alsop K, Bailey PJ, et al. 2015. Whole-genome characterization of chemoresistant ovarian cancer. Nature 521: 489–494. [DOI] [PubMed] [Google Scholar]
- Popic V, Salari R, Hajirasouliha I, Kashef-Haghighi D, West RB, Batzoglou S. 2015. Fast and scalable inference of multi-sample cancer lineages. Genome Biol 16: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth A, Khattra J, Yap D, Wan A, Laks E, Biele J, Ha G, Aparicio S, Bouchard-Côté A, Shah SP. 2014. PyClone: Statistical inference of clonal population structure in cancer. Nat Methods 11: 396–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer F, Kurtz DM, Newman AM, Stehr H, Craig AFM, Shahrokh Esfahani M, Lovejoy AF, Chabon JJ, Klass DM, Liu CL, et al. 2016. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci Transl Med 8: 364ra155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz RF, Ng CKY, Cooke SL, Newman S, Temple J, Piskorz AM, Gale D, Sayal K, Murtaza M, Baldwin PJ, et al. 2015. Spatial and temporal heterogeneity in high-grade serous ovarian cancer: A phylogenetic analysis. PLoS Med 12: e1001789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MJ, Werner B, Barnes CP, Graham TA, Sottoriva A. 2016. Identification of neutral tumor evolution across cancer types. Nat Genet 48: 238–244. [DOI] [PMC free article] [PubMed] [Google Scholar]