

Abstract
In the genome, most occurrences of transcription factor binding sites (TFBS) have no cis-regulatory activity, which suggests that flanking sequences contain information that distinguishes functional from nonfunctional TFBS. We interrogated the role of flanking sequences near Activator Protein 1 (AP-1) binding sites that reside in DNase I Hypersensitive Sites (DHS) and regions annotated as Enhancers. In these regions, we found that sequence features directly adjacent to the core motif distinguish high from low activity AP-1 sites. Some nearby features are motifs for other TFs that genetically interact with the AP-1 site. Other features are extensions of the AP-1 core motif, which cause the extended sites to match motifs of multiple AP-1 binding proteins. Computational models trained on these data distinguish between sequences with high and low activity AP-1 sites and also predict changes in cis-regulatory activity due to mutations in AP-1 core sites and their flanking sequences. Our results suggest that extended AP-1 binding sites, together with adjacent binding sites for additional TFs, encode part of the information that governs TFBS activity in the genome.
Local DNA sequence features influence the activity of transcription factor binding sites (TFBS) (White et al. 2013; Dror et al. 2015; Farley et al. 2015; Levo et al. 2015). The specific features in these local sequences that determine activity remain poorly characterized. Motifs for additional TFs are often implicated as determinants of activity, but whether additional motifs contribute additively or cooperatively is largely unknown. For example, both interacting and independent motifs contribute to the specificity of PPARG binding sites (Grossman et al. 2017). In most cases, however, these motifs remain unidentified because we do not know the typical distances over which other motifs contribute to the specificity of a TFBS.
In addition to motifs for TFs, other types of sequence features may specify high activity TFBS in the genome. Local DNA shape contributes to the specificity of homeodomain-containing TFs (Slattery et al. 2011; Gordân et al. 2013; Dror et al. 2014; Yang et al. 2017). The presence of particular dinucleotide repeats contributes to activity in other cases (Yáñez-Cuna et al. 2014; Farley et al. 2015), as does the presence or absence of nucleosome positioning signals (Lidor Nili et al. 2010). These types of sequence features are difficult to untangle as they depend on the local GC content of DNA, which is itself often correlated with activity (Landolin et al. 2010; Wang et al. 2012; White et al. 2013).
We have been using the Activator Protein 1 (AP-1) binding site as a model for studying the sequence determinants of local cis-regulatory activity. The AP-1 binding site is the most predictive sequence feature of cis-regulatory activity in K562 cells (Kwasnieski et al. 2014). Although the “core” AP-1 motif consists of seven high information nucleotides (5′-TGAG/CTCA-3′), the Position Weight Matrix (PWM) models of the full sites for AP-1 binding proteins each contain different low information positions directly flanking the core. Homodimers and heterodimers of JUN, FOS, ATF, and MAF protein families bind AP-1 sites and play key roles in proliferation, apoptosis, and differentiation (Ye et al. 2014). Active AP-1 sites make chromatin accessible for glucocorticoid receptor binding in a murine mammary epithelial cell line (Biddie et al. 2011). AP-1 binding sites are also enriched in ubiquitous and cell-type–specific clusters of DNase I Hypersensitive Sites (DHS) across 72 different human cell types (Sheffield et al. 2013). For these reasons, AP-1 is considered to be a pioneer, or chromatin accessibility, factor (Ng et al. 1997).
Although AP-1 is a pioneer factor in many cell types, the sequence features that specify AP-1 sites with high cis-regulatory activity from those with low activity remain unknown. Interactions with other DNA-binding proteins likely contribute some of the information that specifies active AP-1 sites in the genome (Chinenov and Kerppola 2001; Turpaev 2006; Gao et al. 2009). However, given a genomic sequence containing a high-scoring AP-1 binding site, we still cannot accurately predict which sequences will have AP-1 dependent cis-regulatory activity, even when that sequence is labeled as an Enhancer or DHS. In this study, we focused on understanding the contribution of local sequence context to AP-1 dependent cis-regulatory activity in DHS and Enhancer sequences.
Results
Sequence selection and expression quantification in K562 cells
Both local sequence features and regional chromosome properties contribute to the cis-regulatory activity of TFBS. Here, we focused on the local sequence contexts that help specify active AP-1 core motifs. Although the AP-1 binding site is predictive of enhancer activity in K562 cells (Kheradpour et al. 2013; Kwasnieski et al. 2014), only half the sequences that are annotated as “Enhancers” (The ENCODE Project Consortium 2012) and contain high-scoring AP-1 binding sites drive high levels of expression in K562 cells (Supplemental Fig. S1). Thus, the local context of AP-1-containing sequences must determine, at least in part, their functionality.
To identify local sequence features that control AP-1-dependent cis-regulatory activity, we selected two sets of previously assayed 130-bp AP-1-containing sequences, most of which are labeled as Enhancers based on epigenetic marks (Kwasnieski et al. 2014): 41 sequences that drive low reporter gene expression (LOW group) and 40 that drive high reporter gene expression (HIGH group) (Fig. 1A). All 81 sequences contain a high-scoring AP-1 binding site, and the distributions of motif scores for the two groups are nearly identical (Fig. 1B), suggesting important differences in their flanking sequences. We synthesized up to 130 bp of genome sequence surrounding each AP-1 binding site, centering the core motif to eliminate any effect of the position. We assayed the cis-regulatory activity of these sequences, and several variants of each sequence (discussed below) (Supplemental Data SD1), using a Massively Parallel Reporter Gene Assay (MPRA) (Kwasnieski et al. 2012; Patwardhan et al. 2012; Kheradpour et al. 2013; White 2015).
Figure 1.

Comparison of 41 LOW activity and 40 HIGH activity sequences. (A) Expression distribution for selected AP-1-containing sequences. HIGH activity sequences (orange) drive stronger expression than LOW sequences (blue). (B) Distributions of AP-1 motif scores for HIGH (orange) and LOW (blue) sequences. Scores were derived using FIMO (Grant et al. 2011) from the JUNB position weight matrix (PWM) in the JASPAR database (Bryne et al. 2008) (inset). (C) Dependency of expression on intact AP-1 binding sites. Expression driven by wild-type sequences (x-axis) plotted versus expression driven by sequences with inactivated AP-1 sites (y-axis). Most points are below the diagonal (solid black line), indicating the importance of an intact AP-1 binding site for cis-regulatory activity. (D) Schematic shows five different length variants created for each sequence. (E) HIGH and LOW sequences contain regulatory information near the AP-1 core motif. Box plots for the activities of 40 HIGH (orange) and 41 LOW (blue) sequences are shown for different length variants. (x-axis) Total length of sequence including the AP-1 core; (y-axis) expression measured in MPRA assay. HIGH and LOW sequences are significantly different at 50 bp (Wilcoxon test, P = 1.86 × 10−9).
We constructed libraries of barcoded AP-1-containing reporter genes in which four unique barcodes represented every sequence. After transfecting these libraries into K562 cells, we sequenced the barcodes from isolated mRNA and normalized those counts by the DNA counts of each barcode in the plasmid pool. The log ratio of the mRNA/DNA counts is a quantitative and reproducible measure of the cis-regulatory activity of each reporter gene in the library (Kwasnieski et al. 2012, 2014). The correlation between replicate experiments in this study ranged between 0.98 and 0.99 (Supplemental Fig. S2A). Expression of the newly synthesized, 130-bp AP-1-centered sequences was highly correlated with the previously measured activity of the noncentered sequences from Kwasnieski et al. (2014) (R2 = 0.68) (Supplemental Fig. S2B). We redefined new HIGH and LOW groups of sequences based on the median expression of the library. Only five sequences from each HIGH and LOW group switched groups after centering the AP-1 site (Supplemental Fig. S2B,C).
The activity of these elements on plasmids reflects the intrinsic activity of each sequence in the absence of regional chromosome effects. These intrinsic activities are highly correlated to the activities of genome integrated reporter genes and to functional genomic measures of activity in the genome (Kwasnieski et al. 2014; Inoue et al. 2017; Maricque et al. 2017).
Local sequence context specifies functional AP-1 sites
Most 130-bp genomic sequences containing an intact AP-1 binding site drove higher expression than their corresponding AP-1mut variants, in which the AP-1 site was inactivated by scrambling three bases of the binding site (Fig. 1C). The range of expression for AP-1mut sequences was threefold lower, suggesting that wild-type sequence activity is highly dependent on the AP-1 binding site. To narrow down the location of flanking information that controls AP-1-dependent activity, we assayed sequences with decreasing length of the genomic sequences flanking the AP-1 motifs, from 130 bp (∼62 bp per side) to 50 bp (∼21 bp per side) (Fig. 1D). Although activity did regress somewhat toward the mean as the sequences got shorter, HIGH and LOW sequences as short as 50 bp retained distinct activities (Wilcoxon test, P = 1.86 × 10−9) (Fig. 1E). This result suggests that most of the sequence features that are necessary for high activity of AP-1 sites reside close to the AP-1 core motifs.
To identify features within 50 bp that specify HIGH versus LOW activity AP-1 core motifs, we took two parallel approaches: We performed detailed saturation mutagenesis on a subset of HIGH and LOW sequences, and we performed a broad survey of 5000 additional genomic AP-1 sites located in DHS.
Saturation mutagenesis reveals interacting and independent features in the flanking sequence
We selected 20 of the 81 50-bp sequences described above for further analysis by saturation mutagenesis. We created AP-1mut versions of each sequence, in which the central AP-1 site was inactivated. In both the wild-type and AP-1mut sequences, we systematically mutated each base to every other base, one at a time. We measured library activity in K562 cells (Supplemental Data SD2, SD3) with high reproducibility between replicates (minimum R2 = 0.96) (Supplemental Fig. S3A,B). Based on their activity in this new library, we reassigned the wild-type sequences into the following categories: nine sequences as HIGH, nine as LOW, and two as MEDIUM (Supplemental Fig. S3C).
We assayed a total of 2565 single-base substitutions flanking the AP-1 sites across the 20 wild-type elements, of which 32% of these flanking substitutions caused a statistically significant change in activity (Wilcoxon test, P < 3.33 × 10−4). Of the substitutions that caused a significant change, 35% increased expression and 65% decreased expression, although only 11.3% (3.5% of the total) changed expression greater than twofold. These effect sizes are comparable to those observed previously (Melnikov et al. 2012; Patwardhan et al. 2012). When compared to sequences with HIGH activity, LOW sequences contained twofold more substitutions that increased expression, suggesting that LOW sequences may contain more sites for repressors compared to HIGH sequences, or that HIGH sequences contain more sites for activators compared to LOW sequences.
We next examined the results from the same 2565 substitutions made in the context of the AP-1mut sequences (Supplemental Table S1). By testing each substitution in the context of both wild-type and AP-1mut sequences, we could assess the interaction of the substitution with the AP-1 site. Substitutions that cause a similar expression change in both wild-type and AP-1mut sequences were designated as “independent” (Fig. 2A). Substitutions that have different effects on expression in wild-type and AP-1mut sequences were designated as “interacting” (Fig. 2B), because their effects depend on the presence of an intact AP-1 core motif (for assignment of independent and interacting substitutions, see Methods). Each substitution represents a disruption or creation of a sequence feature that interacts with AP-1 to determine AP-1 activity, or contributes to enhancer activity independent of AP-1, or has no effect on activity.
Figure 2.

Saturation mutagenesis of AP-1 dependent elements. (A,B) Results from saturation mutagenesis of an AP-1-dependent sequence: (x-axis) nucleotide position in the element; (vertical gray box) position of the AP-1 core motif; (y-axis) expression of point mutants relative to the parental sequence. Point mutations were created in the context of either the wild-type AP-1 site parent (blue) or the AP-1mut site parent (red). Substitutions that were not significantly different from wild-type are faded. The dashed box in A shows a cluster of mutants that affect expression only when the wild-type AP-1 binding site is present, and are thus designated as “interacting” substitutions. The dashed box in B shows a cluster of mutants in the first 10 bp that reduce expression in both the WT and AP-1mut background; thus they contribute to the activity of the sequence “independent” of AP-1. (C) The spatial distributions of independent (gray) and interacting (tan) features in the 20 sequences subjected to saturation mutagenesis. Interacting features tend to occur closer to the AP-1 core motif.
Independent and interacting features tended to occur in different locations relative to the central AP-1 core motifs (Fig. 2C). Interacting substitutions were more likely to be present within the 10-bp sequence flanking the AP-1 core, whereas independent features were enriched further away from the binding site.
When comparing the HIGH and LOW groups, we found that HIGH sequences had 1.6-fold more features in total than LOW sequences (P = 0.02, bootstrap analysis) (Table 1). Since sequences in the LOW group have low expression, we expected to find fewer substitutions that reduce expression further. However, HIGH sequences were more enriched for interacting features (twofold more than LOW) than independent features (1.3-fold more than LOW). Thus, a higher number of features, and interacting features in particular, might contribute to increased activity of HIGH sequences.
Table 1.
LOW sequences have disproportionately fewer interacting substitutions compared to HIGH sequences

Independent and interacting substitutions modify transcription factor motifs
Our results suggest that features that determine AP-1 binding site activity are located in proximity to the core binding site. These nearby features could represent matches to position weight matrices for various AP-1 binding proteins, other cooperating TFs (Chinenov and Kerppola 2001), composite sites (Jolma et al. 2015), or DNA shape features (Dror et al. 2015; Levo et al. 2015).
We mapped many of the features identified in the saturation mutagenesis experiment to motifs for other TFs. After filtering for positions outside of the AP-1 core motif at which substitutions caused a significant change in expression, we asked whether each substitution created or destroyed a motif in the JASPAR database (Mathelier et al. 2016). To control for spurious appearance of motifs based on the nucleotide composition of these elements, we shuffled the expression data and reassigned interacting and independent motifs. We performed the simulation 10,000 times and calculated the probability of each motif being assigned as independent/interacting by chance. Motifs that included the AP-1 core were discarded because of inactivation of the AP-1 site in AP-1mut sequences. Thirty-four independent and seven interacting motifs passed this test (P > 0.05) (Fig. 3; Supplemental Data SD5). In many cases, the evidence for independence or interaction of a motif came from multiple sequences.
Figure 3.

Identities and locations of potential independent and interacting TFs: (x-axis) position along regulatory sequence; (y-axis) transcription factor identity. Each row represents one TF, and points depict interacting (▴) or independent (•) substitutions under the TF motif at the indicated position on the x-axis. Colors represent the regulatory sequence in which the TF was present. When multiple TFs with similar motifs matched a single substitution pattern, one representative TF was chosen.
HIGH and LOW sequences showed differences in the numbers of interacting and independent motifs they contained. HIGH sequences had threefold more (P < 0.01, bootstrap analysis) substitutions in interacting or independent motifs than LOW sequences. Although 33% of the substitutions in HIGH sequences mutated motifs that showed a genetic interaction with AP-1, we observed no interacting motifs in the LOW sequences. LOW sequences are not refractory to interacting motifs because we identified several cases in which a substitution created an interacting motif in a LOW sequence (Table 2).
Table 2.
Wild-type LOW sequences are depleted for interacting motifs for TFs
Interacting and independent motifs also showed the same positional bias relative to the central AP-1 binding sites as individual features (Supplemental Fig. S4). Interacting motifs likely regulate AP-1 specificity through physical interactions with the AP-1 binding proteins, since they occur within 10 bp of the AP-1 binding site. Our results suggest that cis-regulatory activity of sequences with AP-1 sites is also regulated by TFs that function independently of AP-1 and occur further away from the binding site.
Selection and expression of 5000 genomic sequences with AP-1 binding sites
In a parallel set of experiments, we measured the cis-regulatory activity of 5000 additional genomic sequences containing AP-1 sites within DHS to identify sequence features that distinguish high from low activity sequences. We chose 5000 sequences that contain a perfect 7-bp AP-1 core motif: 5′-TGAG/CTCA-3′. Each sequence was 50 bp long and resided in a DHS region in the K562 cell line (Thurman et al. 2012). We mutated the AP-1 core in 250 randomly chosen sequences from this set. We assayed an MPRA library containing all these sequences tagged with five unique barcodes each in K562 cells (Supplemental Fig. S5A; Supplemental Data SD4).
Despite the fact that all 5000 sequences contained a perfect match to the AP-1 core motif and that all of the sequences were derived from DHS, we observed a wide range of cis-regulatory activities among these elements (Fig. 4A). Mutating the AP-1 site significantly reduced activity in 84% of the sequences, suggesting that most sequences in this collection have AP-1-dependent activity (Supplemental Fig. S5B). The median level of expression of the AP-1mut sequences was identical to that of basal controls (Supplemental Fig. S5C), demonstrating that activity in these elements depends on the AP-1 sites. We divided the expression data from the 5000 AP-1-containing sequences with DHS into HIGH (above the 80th percentile) and LOW (below the 20th percentile) groups and attempted to identify sequence features that distinguish the two groups (Fig. 4A).
Figure 4.
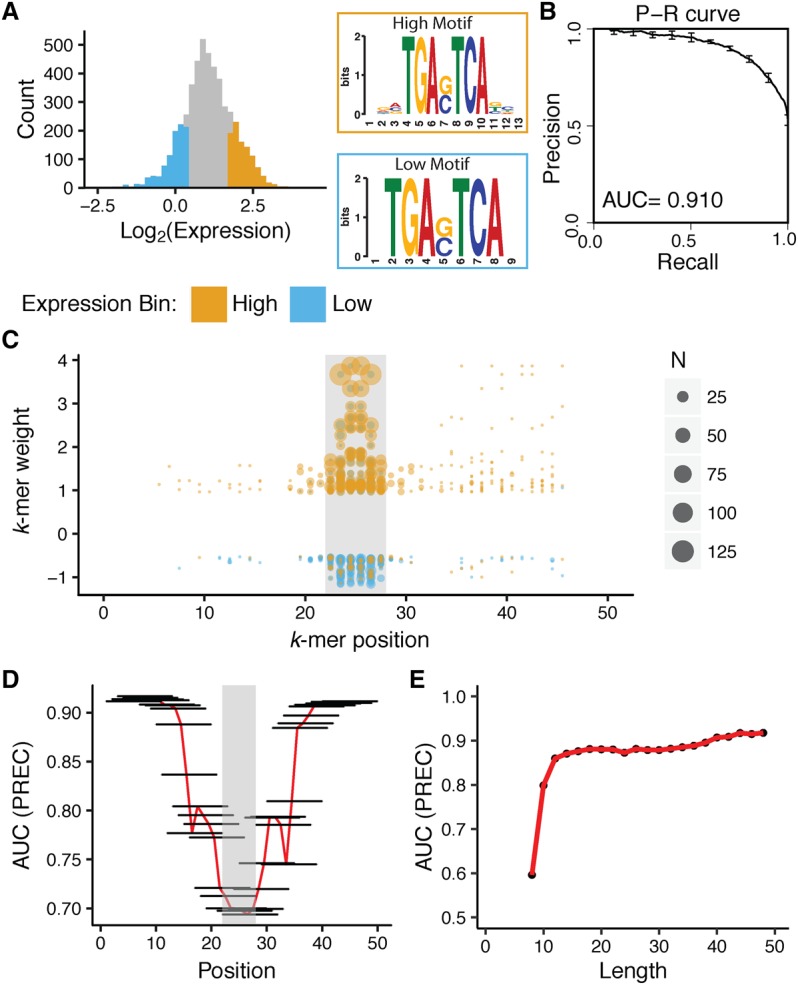
K-mers that distinguish HIGH and LOW DHS are enriched within 10 bp of the AP-1 core motif. (A) Expression distribution for 5000 sequences in DHS regions containing AP-1 sites. (x-axis) Log2(RNA/DNA) counts for barcodes representing particular cis-regulatory sequences (left). The top 1000 sequences are annotated as HIGH (orange), and the bottom 1000 sequences are annotated as LOW (blue). Motifs derived from HIGH and LOW sequences using MEME motif discovery tools (right). (B) gkm-SVM distinguishes between HIGH and LOW sequences within DHS. Precision-recall curve for a 10-mer gkm-SVM model trained on HIGH and LOW sequences (AUC = 0.91). Error bars show standard error from fivefold cross-validation. (C) K-mers that distinguish HIGH and LOW sequences overlap the AP-1 binding site. (x-axis) Position of the center of the k-mer along regulatory element in bp; (gray box) position of the AP-1 core motif; (y-axis) k-mer weight from gkm-SVM in B. Each point is an individual k-mer, and the size of the point denotes the number of sequences containing the k-mer. The color of the point indicates whether the k-mer was found in HIGH (orange) or LOW (blue) sequence. The top 400 k-mers, 200 with positive weights and 200 with negative weights are shown. (D) In silico deletion experiment also highlights that most informative k-mers overlap with the AP-1 core motif. A 10-bp region of every sequence was masked (horizontal black lines), and a 10-mer gkm-SVM model was refit. The x-axis shows the position of the masked segment along regulatory elements, and the y-axis shows the area under precision-recall curve from the resulting model. The gray box depicts the position of the AP-1 core motif, and the red line connecting the centers of the black bars highlights the trend of AUC values across the sequence. (E) Specification of HIGH and LOW groups lies within the central 12 bps. Sequences were shortened by removing one base from both ends and a 6-mer gkm-SVM model was refit: (x-axis) length of the shortened sequence; (y-axis) area under precision-recall curve. The red line connecting the centers the points highlights the trend of AUC values across the sequence.
Machine learning model identifies positions flanking the AP-1 core motif as contributing to activity
One hypothesis is that the AP-1 core by itself is not enough to drive expression, and specific flanking bases are necessary to recruit AP-1 proteins (Yáñez-Cuna et al. 2012; Gordân et al. 2013; Slattery et al. 2014; Dror et al. 2015; Farley et al. 2015; Levo et al. 2015). We ran the MEME motif discovery tool (Bailey et al. 2009) on the HIGH and LOW classes to determine if there were differences in the flanking bases surrounding these two types of sites. Like other motif finders, MEME identifies sequence motifs in which positions contribute independently to activity. The sequence logos (Schneider and Stephens 1990) derived from the HIGH and LOW groups are very similar (Fig. 4A) and do not distinguish between these two classes (Supplemental Fig. S6), suggesting that positions outside the core motif do not make independent contributions to cis-regulatory activity. It is, however, possible that correlated positions outside the core motif contribute to activity. Collections of k-mers can capture correlations between positions that might escape detection by motif finders that assume independence between positions. We trained a gapped 10-mer Support Vector Machine using gapped kmer-SVM (gkm-SVM) (Ghandi et al. 2014, 2016) (L = 10, K = 6, maxnmm = 1) to distinguish the 1000 HIGH and 1000 LOW sequences. The resulting model successfully classified HIGH and LOW sequences with a cross-validated area under the precision-recall curve of 0.91 (Fig. 4B). We obtained similar results with an ungapped 6-mer model and an ungapped 8-mer model (Supplemental Fig. S7).
Analysis of k-mers from gkm-SVM suggested that the information that distinguishes HIGH and LOW sequences resides directly adjacent to the AP-1 core motif. The highest weighted k-mers from gkm-SVM tended to flank, or even overlap the AP-1 core motif (Fig. 4C). We then performed an in silico deletion analysis by replacing 10 bp of sequence with Ns in a sliding window fashion and retraining the SVM for every in silico deletion. We found that when removed, the 10-mers that reduced predictive power the most overlapped the AP-1 core motif. The further a 10-mer was from the AP-1 core motif, the less it contributed to the predictive power of the model (Fig. 4D). We also trained 6-mer ungapped SVM models on data in which we systematically shortened the sequences by removing the positions at the two ends. Models trained on sequences as short as 12 bp, including the core motif, retained high power to discriminate between HIGH and LOW elements (Fig. 4E). These data suggest that much of the information that determines AP-1 activity resides in the 10 bp directly adjacent to the AP-1 site.
gkm-SVM predicts effect of mutations on regulatory potential
To assess the predictive power of the SVM trained on the DHS library, we tested whether the model could predict expression data from our previous libraries. The SVM successfully separated the original 81 sequences from their AP-1mut variants (Wilcoxon test, P = 3.5 × 10−9) (Supplemental Fig. S8A). Although the SVM was trained on categorical data, HIGH versus LOW, the output scores of the SVM were a reasonably quantitative measure of the activities of our original 81 sequences (R = 0.77) (Fig. 5A). The ability of the SVM trained on DHS data to predict activity in experiments performed on a different library measured in a different experiment is an important validation of the model.
Figure 5.

gkm-SVM scores quantitatively predict expression of wild-type sequences and effect of mutations. (A) gkm-SVM classifier trained on HIGH and LOW DHS sequences quantitatively predicted expression of sequences from the first library: (x-axis) expression of 81 cis-regulatory sequences; (y-axis) SVM score from the 10-mer gkm-SVM model in Figure 4B. (B) gkm-SVM classifier trained on HIGH and LOW DHS sequences accurately predicted the effect of substitutions in the AP-1 core motif and 2 bp flanking the AP-1 core tested in the saturation mutagenesis library. Data for substitutions in one sequence (Chr 3: 128734834–128734883) are shown: (x-axis) expression of sequences containing one substitution each; (y-axis) SVM score from 10-mer gkm-SVM model in Figure 4B. The model predicted loss in expression from wild-type sequence (black) when the substitutions are made in the AP-1 core (orange), and the effect of substitutions in 2 bp flanking the AP-1 site (red). Most substitutions outside of the core +2 bp flank (blue) have high expression and are not well predicted by the SVM. (C) Predictive power of the gkm-SVM model is inversely proportional to the absolute distance from AP-1 binding site. Substitutions from all 20 sequences in the saturation mutagenesis library were grouped by their distance from the AP-1 binding site: (x-axis) distance of the group of substitutions to the AP-1 core motif center; (y-axis) correlation coefficient between change in expression and change in SVM score compared to wild-type sequence for all substitutions in a group.
We also tested whether the trained SVM could predict the result from our saturation mutagenesis experiments. The model accurately separated sequences with intact AP-1 core motifs from those containing mutations in the AP-1 core (Supplemental Fig. S8B). The model also had reasonably good predictive power for mutations flanking the core site, in regions where the most predictive k-mers reside (Fig. 5B; Supplemental Fig. S9). Change in expression from wild type due to substitutions in the core motif and adjacent flanks was also well predicted by change in SVM score. The model was less predictive for substitutions further from the core motif, in regions where we detected fewer predictive k-mers (Fig. 5C). It is likely that the performance of the model drops because mutations in positions further from the AP-1 core cause smaller expression changes.
Flanking features represent other TF motifs and extensions of the AP-1 core motif
Our results from saturation mutagenesis (above) suggested that some of the information being captured by the SVM model might be motifs for other TFs adjacent to the AP-1 core motif. Alternatively, the SVM might be detecting extensions of the core motif, some of which might be present in the full motifs represented by different PWMs of AP-1 binding proteins. To address this question, we quantified the contribution of motifs for AP-1 binding TFs versus motifs for other TFs. Starting with the full collection of motifs in the JASPAR database, we converged on a logistic regression model using only 28 TF motifs that had nearly the same predictive power as the trained SVM models (AUC = 0.9) (Fig. 6A). This collection contained motifs for several TFs that directly bind the AP-1 site, as well as 16 additional motifs that overlapped motifs we identified in the saturation mutagenesis experiments (above) (Supplemental Data SD6; Supplemental Fig. S10). The identification of these additional motifs in two independent data sets lends support to the hypothesis that they contribute to AP-1 activity.
Figure 6.

AP-1 sites that bind multiple AP-1 binding proteins in forward and reverse orientations drive high activity. (A) A logistic regression model with additive terms for motifs matches for 28 TFs can distinguish between HIGH and LOW groups of DHS AP-1-containing sequences. Precision-recall curves for models with PWMs for 28 TFs (black, AUC = 0.9), six AP-1 PWMs (JUNB, MAF::NFE2, NFE2l2, FOSL1, MAFK, NFE2; blue, AUC = 0.86), and JUNB (red, AUC = 0.77) are shown. Error bars denote standard error from fivefold cross-validation. Motif matches in both orientations were included. (B) Ignoring orientation information of motifs reduced the predictive power of logistic regression models, especially for a model with JUNB alone. Precision-recall curves for models with PWMs for 28 TFs (black, AUC = 0.89), six AP-1 PWMs (blue, AUC = 0.84), and JUNB (red, AUC = 0.64) are shown again. (C) Expression of DHS sequences containing AP-1 sites in MPRA is correlated with genomic binding of AP-1 TFs to those sequences. (x-axis) Number of peaks observed in total in five ChIP-seq experiments (JUNB, MAFF, MAFK, NFE2 and FOSL1) (The ENCODE Project Consortium 2012); (y-axis) observed log2 (RNA/DNA) counts of cis-regulatory sequences. Expression distributions for sequences with three or more ChIP-seq peaks were not significantly different from each other (Wilcoxon test, Bonferroni-corrected P > 0.05); all other distributions were significantly different from each other.
We compared the performances of gkm-SVM and logistic regression models, which were trained on the DHS library, on the saturation mutagenesis data. SVM scores and probabilities from logistic regression were highly correlated (R2 = 0.69) (Supplemental Fig. S11). Motifs for TFs that directly bind the AP-1 core motif accounted for a large portion of the predictive power of the regression model. The motif for JUNB, a protein that directly binds the AP-1 motif, had reasonable predictive power on its own (AUC = 0.77) (Fig. 6A), suggesting that some of the flanking information outside the AP-1 core motif creates better matches to the JUNB motif. Although different AP-1 binding proteins share the same core motif, the preferences at positions flanking the core are different for different family members. A model that includes motifs for six different AP-1 binding proteins (JUNB, MAF::NFE2, NFE2l2, FOSL1, MAFK, NFE2) performs better than the model with only the JUNB motif (AUC = 0.86) (Fig. 6A). This result shows that specific dinucleotides flanking the core site make HIGH activity AP-1 sites better matches to PWMs for multiple AP-1 binding proteins. Although the core motif is symmetric, the flanking preferences for different family members are asymmetric. HIGH sites often scored as good matches to multiple AP-1 binding proteins in both the forward and reverse orientations, whereas LOW sites often scored well in only one orientation (Supplemental Fig. S12). As a result, the predictive power of the regression models drops if we ignore the contribution of AP-1 motifs in both orientations (Fig. 6B).
Sequences with higher cis-regulatory activities in our library were occupied by more AP-1 binding proteins (JUNB, MAFF, MAFK, NFE2, and FOSL1) (The ENCODE Project Consortium 2012) in their native chromosomal locations (Fig. 6C). Sequences containing ChIP-seq peaks for five different AP-1 family members had the highest cis-regulatory activity. In addition, there was no difference in the quality of JUNB motifs between sequences bound by different numbers of AP-1 binding proteins (Supplemental Fig. S13). This shows that sequences with higher numbers of ChIP-seq peaks do not necessarily contain a better site for any particular AP-1 binding protein, but rather that the information flanking the core AP-1 motif allows binding by multiple homologs.
Sequence features flanking the AP-1 core motif likely have effects in addition to their effects on the binding preferences for different AP-1 binding motifs. The full model containing motifs for TFs that do not bind the AP-1 core motif still outperformed the model with only AP-1 binding proteins. This suggests that HIGH sequences, in addition to having good sites for multiple AP-1 binding proteins, also contain motifs for additional interacting and independent factors. The overlap between motifs that predict activity of DHS sequences and motifs discovered using saturation mutagenesis, lends support to this hypothesis. In many cases, the effect on expression due to creation or disruption of a motif agrees with predictions from the logistic regression model (Supplemental Fig. S14).
Discussion
Transcription factor binding sites with high and low activities are specified, in part, by differences in their genomic contexts. Some of this contextual information resides near active binding sites and determines the intrinsic cis-regulatory activity of DNA elements (Wang et al. 2012). Other contextual information, which modifies the intrinsic activity of DNA elements, resides further from the binding sites and includes chromatin modifications, distally acting enhancers, and chromosome loops (Slattery et al. 2014). Plasmid-based reporter assays measure the local intrinsic activities of DNA elements in the absence of regional chromatin effects. In this study, we used MPRAs to identify local sequence features that distinguish highly active AP-1 binding sites from low activity sites. Because we were interested in identifying the flanking information that specifies high activity sites, we used an experimental design in which we compared groups of sequences with identical AP-1 core motifs, but with different intrinsic cis-regulatory activities.
Our study indicates that local sequence features within 50 bp often distinguish high from low activity AP-1 sites. In most cases, these sequence features reside within 10 bp of the AP-1 core motif. Elements with high activity AP-1 sites contain a high density of nearby TFBS, in particular, they contain motifs for TFs that depend on an intact AP-1 site for activity. These observations are consistent with a model in which extensive cooperativity between TFs underlies the specificity of cis-regulation in the genome. This cooperativity may result from direct protein–protein interactions between TFs, from indirect interactions mediated through contacts with the transcriptional machinery, or through cooperative displacement of nucleosomes (Mirny 2010; He et al. 2013; Jolma et al. 2015; Grossman et al. 2017). Many of the interacting TFs that we found in this study, were previously known to interact with AP-1 binding proteins (Chinenov and Kerppola 2001), highlighting the utility of MPRA methods for identifying interacting partners for TFs that are less well characterized.
Our results also suggest that nucleotides directly flanking the AP-1 core motif help specify high activity sites. PWMs for many AP-1 binding TFs include nucleotides flanking the AP-1 core motif. In highly active sequences, the flanking dinucleotides caused core motifs to score well for multiple AP-1 family members. Our observations suggest a model in which high cis-regulatory activity derives from the ability of multiple family members to bind a site in both orientations, which likely increases the overall occupancy of that site. It also remains a possibility that multiple imperfect PWMs are a better estimation of the true site for a single critical binding protein. The observation that elements with high intrinsic activity derive from genomic regions occupied by multiple AP-1 binding proteins supports the former hypothesis. These results are not unique to the AP-1 family of proteins. Regions bound by multiple members of the FOX family are more often functional than regions bound by a single member (Chen et al. 2016). Different members of the ETS family have been shown to bind the same consensus motifs in promoters of ubiquitously expressed housekeeping genes (Hollenhorst et al. 2007). We speculate that high occupancy by multiple family members may be a general signature of functional TFBS.
Computational models incorporating flanking sequence features distinguish between genome sequences with high and low AP-1 dependent cis-regulatory activity. In many cases these models also distinguish between point mutations with strong and weak effects on cis-regulatory activity. These models are especially useful for predicting changes in expression due to mutations in TFBS, which underlie many of the signals from human disease gene studies (Hindorff et al. 2009; Maurano et al. 2012; Schaub et al. 2012; Nishizaki and Boyle 2017). Our experimental approach for discovering interacting sequence features, combined with computational models of high-throughput measurements, will help build predictive models of TF specificity.
Methods
Design of MPRA libraries
Library 1
We screened sequences tested in a previous study (Kwasnieski et al. 2014) for AP-1 motifs obtained from JASPAR (Mathelier et al. 2016) and UniPROBE (Newburger and Bulyk 2009) databases using FIMO (Grant et al. 2011). From sequences with high-scoring AP-1 motifs (P < 1 × 10−4), we selected 40 sequences that drove high expression (2.47–9.41 units normalized to basal) and 41 that drove low expression (0.58–1.15 units normalized to basal). All 81 sequences were then centered on the AP-1 site, and flanking sequence was re-extracted from the genome, if necessary. All sequences were ∼130 bp long.
We mutated the AP-1 binding site in each sequence by scrambling three bases of the motif. Each binding site was mutated in two to three different ways to create AP-1mut variants. We scanned wild-type and AP-1mut sequences with all motifs from the JASPAR vertebrate database to ensure that no new binding sites for AP1 or other known factors were created in the process. For all the wild-type and AP-1mut sequences, we designed four versions with different lengths: 110, 90, 70, and 50 bp with the AP-1 site maintained in the center. We also included five sequences that are known AP-1-dependent enhancers as positive controls in this library (Ney et al. 1990; Zhang et al. 1993; Zutter et al. 1999; Iwasaki et al. 2006). Twenty sequences from a previous study (Kwasnieski et al. 2014) that drove expression at uniform intervals were included as experimental controls, and 25 empty constructs with no cis-regulatory elements were included as basal sequences. All 1625 sequences were designed with four unique barcodes resulting in a library with 6500 total number of elements. Library 1 composition is summarized in Supplemental Table S2.
Library 2
We picked 20 50-bp sequences from Library 1 for systematic dissection, 10 from HIGH and LOW groups each. Sequences were chosen to have maximal differences in expression and AP-1mut/wild-type ratio for HIGH and LOW categories, but minimal differences in GC content. We also picked one positive control sequence. For each of these 21 sequences, we chose an AP-1mut sequence from Library 1 that drove the least expression among all mutants of that sequence. We then generated the saturation mutagenesis library by changing every base to every other base for all selected wild-type sequences and their AP-1mut variants. About 150 variants were generated for every sequence (3 per bp × 50 bp). We filtered out any substitutions that created restriction sites important for cloning the library. The library was designed in two parts, each containing basal, positive, and experimental controls as described above. Additional sequences were included in common to compare the two parts of the library. All sequences were tagged with four unique barcodes, generating 13,500 sequences per part. The two parts were treated as independent libraries through all steps of library preparation and expression measurement. Library 2 composition is summarized in Supplemental Table S3.
Library 3
We downloaded DHS data for K562 cells generated by the ENCODE Analysis Working Group (Thurman et al. 2012; The ENCODE Project Consortium 2012) based on ENCODE Duke and UW DNase I hypersensitivity tracks. We screened the 150-bp DNase I hypersensitive regions for perfect matches to the AP-1 core (5′-TGAG/CTCA-3′). Of the 11,742 DHS hits with perfect AP-1 sites, we filtered out sequences in which AP-1 site was present within the first 25 bp or last 25 bp. For the remaining 9284 sequences, we extracted subsequences so as to generate 50-bp sequences with AP-1 in the center. These sequences were further filtered for restriction sites that would be used for cloning the library. We sampled the remaining sequences to achieve a wide distribution of GC contents and selected 5000 sequences. For 250 of these sequences, we mutated the AP-1 binding site by scrambling 3 bp of the AP-1 core (5′-TGAG/CTCA-3′→5′-TGAG/CATC-3′). We also included experimental, positive, and basal controls as described above. Each sequence was tagged with five unique barcodes to generate a total of 26,975 sequences. Library 3 composition is summarized in Supplemental Table S4.
Library construction
All three libraries were synthesized by Agilent Technologies through a limited licensing agreement. Each oligo was synthesized as follows: Left Primer/NheI site/Regulatory Sequence/HindIII site/Filler/XhoI site/SphI site/Barcode/SacI site/Right Primer. The total length of the oligos ordered was 200 bp for Library 1, 150 bp for Library 2, and 230 bp for Library 3. Random filler sequence, when necessary, was added between HindIII and XhoI sites to make all oligos the same length. All plasmid libraries were constructed as previously described (Kwasnieski et al. 2014). The synthesized oligos were amplified with left and right primers, and the annealing temperatures specified in Supplemental Table S5. Library 1 was amplified with 26 ng of template in a 50-µL reaction, whereas Libraries 2 and 3 were amplified with 9 and 15 ng of template, respectively. After amplification (multiple replicates) and PAGE purification, the oligos were cloned into a pGL backbone using NheI and SacI sites. Multiple ligations were pooled and purified with a PCR clean-up kit (NucleoSpin). We transformed the libraries into 5-alpha Electrocompetent E. coli (NEB) and collected colonies sufficient to cover the library. We then cloned hsp68 promoter driving dsRed reporter using HindIII and SphI sites. XhoI site was used to cut any constructs that did not receive the hsp68 promoter driving dsRed reporter before purification and transformation into E. coli.
Cell culture and library transfection
Cell culture and library transfections were performed as described in Kwasnieski et al. (2014). K562 cells were maintained in Iscove's Modified Dulbecco's Medium + 10% Fetal Bovine Serum + 1% non-essential amino acids (Gibco). Plasmid libraries were purified by phenol-chloroform extractions (2×) followed by ethanol precipitation. Then, 27 µg of the library was transfected into K562 cells by using Neon electroporation system (Life Technologies) in four replicates with 1.2 million cells each, and 3 µg of pmaxGFP plasmid (Ambion) was used as a transfection control.
Expression measurement of libraries
RNA extractions were performed 22 h after transfections using PureLink RNA Mini Kit (Life Technologies) and followed by DNase I treatment using TURBO DNA-free kit (Applied Biosystems). First strand cDNA was synthesized from RNA samples using SuperScript III Reverse Transcriptase (Life Technologies). Samples were prepared for RNA-seq as described previously (Kwasnieski et al. 2014). Barcodes were amplified from cDNA of transfected plasmids and DNA of the original plasmid pool. Amplified barcodes were digested with SphI and XhoI and ligated to indexed Illumina adapters. Ligation products were further amplified with enrichment PCR primers. A single pool was created from equal amounts of all four cDNA replicates and one DNA sample, which was then submitted for sequencing. All primers and adapter sequences are provided in Supplemental Table S5. We obtained greater than 1500× coverage across all libraries (Supplemental Table S6). We counted the number of reads per barcode and filtered out barcodes with fewer than 10 reads in the DNA or cDNA pool. Expression of a barcode was calculated as cDNA reads/DNA reads. For each replicate, we averaged the expression of all barcodes that tagged basal constructs to calculate basal expression. Expression of each barcode was then normalized by replicate-specific basal expression. Expression between barcodes was highly reproducible (average R2= 0.85 for sets of barcodes for sequences in Library 1). Lastly, to calculate expression driven by a particular sequence, we averaged expression across all barcodes for that sequence. Expression values for all libraries are provided in Supplemental Data SD1–SD4.
Assignment of independent and interacting substitutions
We calculated the effect of each substitution i on expression in wild-type and AP-1mut parental sequences by calculating the following ratios:
For each expression value, we had about 16 measurements (four barcodes in four replicates), which enabled us to perform statistical tests to determine if the expression change due to substitution was significantly different from the parental sequence. Substitutions that passed a Bonferroni-corrected P-value threshold (Wilcoxon test, P < 0.05/150) were assigned as significant.
The difference (Δi) between WTi and MUTi was calculated as
Each substitution i was assigned as independent or interacting as following:
Independent: Δi < 0.4; WTi and MUTi both significantly different from respective parent, or
Interacting: Δi > 0.5; WTi or MUTi significantly different from respective parent,
where respective parents are wild-type sequence or AP-1mut sequence.
Assignment of independent and interacting motifs
For each part of the library, we shuffled all WTi and MUTi ratios while preserving their significance assignment. Shuffled ratios were randomly assigned to all sequences. We then assigned motifs to 10,000 randomized data sets and the true data set as follows: We created a set of single-base substitution variants that caused a significant expression change in either the wild-type parent or the AP-1mut parent or both. We screened this set, wild-type parent, and AP-1mut parent sequences for matches to motifs from the JASPAR vertebrate database. We compared the motif matches between each variant and its parental sequence and kept the motifs that were present in only one of them, which were motifs that were created or disrupted because of the substitution. Next, for every single base variant, we filtered out any motifs unique to wild-type or AP-1mut background so as to eliminate any motifs contributed by mutations in the central AP-1 binding site. These motifs were then assigned as independent or interacting based on the underlying substitution. We created a list of independent and interacting motifs and their frequency of occurrence for the true and the simulated expression data sets. We then calculated the probability of a motif occurring as frequently or more in the simulated data set compared to the true data set.
To calculate the statistical significance of fold difference between HIGH and LOW groups for total number of interacting or independent substitutions and total number of motifs underlying those substitutions, we performed 10,000 randomizations for which we randomized the HIGH, MEDIUM, and LOW labels. For each randomized data set, we calculated the ratio of the total number of significant features in the HIGH and LOW groups. From the distribution of ratios, we calculated the probability of observing the true ratio or higher by chance.
Machine learning
We used the gkmSVM R package developed by the Beer laboratory (Ghandi et al. 2014, 2016) for learning features that distinguish HIGH activity AP-1 sites from the LOW activity AP-1 sites. We calculated a kernel matrix with HIGH sequences as the positive set, LOW sequences as the negative set, word length L = 10, nongapped positions K = 6, and maximum number of mismatches (maxnmm) = 1. We then performed SVM training with cross-validation. SVM kernel computed with 1000 HIGH and 1000 LOW sequences with DHS was used to classify sequences from Library 1 and 2. For Library 2, change in SVM score (ΔSVM score) for substitution i was calculated as SequenceWT score − Sequencei score.
Logistic regression
We used glm function in R (version 3.2.3) to perform logistic regression using transcription factor motifs from JASPAR (Bryne et al. 2008). We used FIMO (Grant et al. 2011) to scan HIGH and LOW sequences with all motifs from the JASPAR vertebrate database using default thresholds. Results from FIMO were then summarized in a Sequence by TF matrix, in which each cell represented the number of matches for a given TF in a given sequence. We first built a model with 424 TFs without any interaction terms and filtered out motifs with P > 0.1. For the remaining 54 motifs, we iteratively performed backward logistic regression using motifs with P ≤ 0.05 until all of the remaining motifs had significant coefficients. The final model with 28 TFs was fivefold cross-validated.
Data access
The tabulated barcode counts for every integrated reporter in the library are provided as Supplemental Material. The raw Illumina sequencing reads from this study, from which we tabulated the barcode counts, have been submitted to the NCBI BioProject database (http://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA389101 (SRA experiments: SRX2888431–SRX2888434).
Supplementary Material
Acknowledgments
We thank Gary Stormo and members of the Cohen laboratory for their feedback on the manuscript; Jessica Hoisington-Lopez for assistance with high-throughput sequencing; and Chris Fiore and Jamie Kwasnieski for assistance with CRE-seq protocols. This work was supported by grants from the National Institutes of Health, R01-GM092910 and R01-HG008687.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.226530.117.
References
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biddie SC, John S, Sabo PJ, Thurman RE, Johnson TA, Schiltz RL, Miranda TB, Sung M-H, Trump S, Lightman SL, et al. 2011. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol Cell 43: 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryne JC, Valen E, Tang MH, Marstrand T, Winther O, da Piedade I, Krogh A, Lenhard B, Sandelin A. 2008. JASPAR, the open access database of transcription factor-binding profiles: new content and tools in the 2008 update. Nucleic Acids Res 36: D102–D106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Ji Z, Webber A, Sharrocks AD. 2016. Genome-wide binding studies reveal DNA binding specificity mechanisms and functional interplay amongst Forkhead transcription factors. Nucleic Acids Res 44: 1566–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinenov Y, Kerppola TK. 2001. Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene 20: 2438–2452. [DOI] [PubMed] [Google Scholar]
- Dror I, Zhou T, Mandel-Gutfreund Y, Rohs R. 2014. Covariation between homeodomain transcription factors and the shape of their DNA binding sites. Nucleic Acids Res 42: 430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror I, Golan T, Levy C, Rohs R, Mandel-Gutfreund Y. 2015. A widespread role of the motif environment in transcription factor binding across diverse protein families. Genome Res 25: 1268–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley EK, Olson KM, Zhang W, Brandt AJ, Rokhsar DS, Levine MS. 2015. Suboptimization of developmental enhancers. Science 350: 325–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S-Y, Li E-M, Cui L, Lu X-F, Meng L-Y, Yuan H-M, Xie J-J, Du Z-P, Pang J-X, Xu L-Y. 2009. Sp1 and AP-1 regulate expression of the human gene VIL2 in esophageal carcinoma cells. J Biol Chem 284: 7995–8004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi M, Lee D, Mohammad-Noori M, Beer MA. 2014. Enhanced regulatory sequence prediction using gapped k-mer features. PLoS Comput Biol 10: e1003711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi M, Mohammad-Noori M, Ghareghani N, Lee D, Garraway L, Beer MA. 2016. gkmSVM: an R package for gapped-kmer SVM. Bioinformatics 32: 2205–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordân R, Shen N, Dror I, Zhou T, Horton J, Rohs R, Bulyk ML. 2013. Genomic regions flanking E-box binding sites influence DNA binding specificity of bHLH transcription factors through DNA shape. Cell Rep 3: 1093–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, Noble WS. 2011. FIMO: scanning for occurrences of a given motif. Bioinformatics 27: 1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman SR, Zhang X, Wang L. 2017. Systematic dissection of genomic features determining transcription factor binding and enhancer function. Proc Natl Acad Sci 114: E1291–E1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Chatterjee R, John S, Bravo H, Sathyanarayana BK, Biddie SC, FitzGerald PC, Stamatoyannopoulos JA, Hager GL, Vinson C. 2013. Contribution of nucleosome binding preferences and co-occurring DNA sequences to transcription factor binding. BMC Genomics 14: 428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. 2009. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci 106: 9362–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenhorst PC, Shah AA, Hopkins C, Graves BJ. 2007. Genome-wide analyses reveal properties of redundant and specific promoter occupancy within the ETS gene family. Genes Dev 21: 1882–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue F, Kircher M, Martin B, Cooper GM, Witten DM, McManus MT, Ahituv N, Shendure J. 2017. A systematic comparison reveals substantial differences in chromosomal versus episomal encoding of enhancer activity. Genome Res 27: 38–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki K, Mackenzie EL, Hailemariam K, Sakamoto K, Tsuji Y. 2006. Hemin-mediated regulation of an antioxidant-responsive element of the human ferritin H gene and role of Ref-1 during erythroid differentiation of K562 cells. Mol Cell Biol 26: 2845–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolma A, Yin Y, Nitta KR, Dave K, Popov A, Taipale M, Enge M, Kivioja T, Morgunova E, Taipale J. 2015. DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature 527: 384–388. [DOI] [PubMed] [Google Scholar]
- Kheradpour P, Ernst J, Melnikov A, Rogov P, Wang L, Zhang X, Alston J, Mikkelsen TS, Kellis M. 2013. Systematic dissection of regulatory motifs in 2000 predicted human enhancers using a massively parallel reporter assay. Genome Res 23: 800–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwasnieski JC, Mogno I, Myers CA, Corbo JC, Cohen BA. 2012. Complex effects of nucleotide variants in a mammalian cis-regulatory element. Proc Natl Acad Sci 109: 19498–19503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwasnieski JC, Fiore C, Chaudhari HG, Cohen BA. 2014. High-throughput functional testing of ENCODE segmentation predictions. Genome Res 24: 1595–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landolin JM, Johnson DS, Trinklein ND, Aldred SF, Medina C, Shulha H, Weng Z, Myers RM. 2010. Sequence features that drive human promoter function and tissue specificity. Genome Res 20: 890–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levo M, Zalckvar E, Sharon E, Dantas Machado AC, Kalma Y, Lotam-Pompan M, Weinberger A, Yakhini Z, Rohs R, Segal E. 2015. Unraveling determinants of transcription factor binding outside the core binding site. Genome Res 25: 1018–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidor Nili E, Field Y, Lubling Y, Widom J, Oren M, Segal E. 2010. p53 binds preferentially to genomic regions with high DNA-encoded nucleosome occupancy. Genome Res 20: 1361–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maricque BB, Dougherty JD, Cohen BA. 2017. A genome-integrated massively parallel reporter assay reveals DNA sequence determinants of cis-regulatory activity in neural cells. Nucleic Acids Res 45: e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathelier A, Fornes O, Arenillas DJ, Chen CY, Denay G, Lee J, Shi W, Shyr C, Tan G, Worsley-Hunt R, et al. 2016. JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 44: D110–D115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. 2012. Systematic localization of common disease-associated variation in regulatory DNA. Science 337: 1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnikov A, Murugan A, Zhang X, Tesileanu T, Wang L, Rogov P, Feizi S, Gnirke A, Callan CG Jr, Kinney JB, et al. 2012. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol 30: 271–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirny LA. 2010. Nucleosome-mediated cooperativity between transcription factors. Proc Natl Acad Sci 107: 22534–22539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newburger DE, Bulyk ML. 2009. UniPROBE: an online database of protein binding microarray data on protein–DNA interactions. Nucleic Acids Res 37: D77–D82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ney PA, Sorrentino BP, McDonagh KT, Nienhuis AW. 1990. Tandem AP-1-binding sites within the human β-globin dominant control region function as an inducible enhancer in erythroid cells. Genes Dev 4: 993–1006. [DOI] [PubMed] [Google Scholar]
- Ng KW, Ridgway P, Cohen DR, Tremethick DJ. 1997. The binding of a Fos/Jun heterodimer can completely disrupt the structure of a nucleosome. EMBO J 16: 2072–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizaki SS, Boyle AP. 2017. Mining the unknown: assigning function to noncoding single nucleotide polymorphisms. Trends Genet 33: 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan RP, Hiatt JB, Witten DM, Kim MJ, Smith RP, May D, Lee C, Andrie JM, Lee S-I, Cooper GM, et al. 2012. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol 30: 265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub MA, Boyle AP, Kundaje A, Batzoglou S, Snyder M. 2012. Linking disease associations with regulatory information in the human genome. Genome Res 22: 1748–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider TD, Stephens RM. 1990. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res 18: 6097–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield NC, Thurman RE, Song L, Safi A, Stamatoyannopoulos JA, Lenhard B, Crawford GE, Furey TS. 2013. Patterns of regulatory activity across diverse human cell types predict tissue identity, transcription factor binding, and long-range interactions. Genome Res 23: 777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery M, Riley T, Liu P, Abe N, Gomez-Alcala P, Dror I, Zhou T, Rohs R, Honig B, Bussemaker HJ, et al. 2011. Cofactor binding evokes latent differences in DNA binding specificity between Hox proteins. Cell 147: 1270–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery M, Zhou T, Yang L, Dantas Machado AC, Gordân R, Rohs R. 2014. Absence of a simple code: how transcription factors read the genome. Trends Biochem Sci 39: 381–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. 2012. The accessible chromatin landscape of the human genome. Nature 489: 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turpaev KT. 2006. Role of transcription factor AP-1 in integration of cell signaling systems. Mol Biol 40: 851–866. [DOI] [PubMed] [Google Scholar]
- Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, Pierce BG, Dong X, Kundaje A, Cheng Y, et al. 2012. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res 22: 1798–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MA. 2015. Understanding how cis-regulatory function is encoded in DNA sequence using massively parallel reporter assays and designed sequences. Genomics 106: 165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MA, Myers CA, Corbo JC, Cohen BA. 2013. Massively parallel in vivo enhancer assay reveals that highly local features determine the cis-regulatory function of ChIP-seq peaks. Proc Natl Acad Sci 110: 11952–11957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yáñez-Cuna JO, Dinh HQ, Kvon EZ, Shlyueva D, Stark A. 2012. Uncovering cis-regulatory sequence requirements for context-specific transcription factor binding. Genome Res 22: 2018–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yáñez-Cuna JO, Arnold CD, Stampfel G, Boryń LM, Gerlach D, Rath M, Stark A. 2014. Dissection of thousands of cell type-specific enhancers identifies dinucleotide repeat motifs as general enhancer features. Genome Res 24: 1147–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Orenstein Y, Jolma A, Yin Y, Taipale J, Shamir R, Rohs R. 2017. Transcription factor family-specific DNA shape readout revealed by quantitative specificity models. Mol Syst Biol 13: 910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye N, Ding Y, Wild C, Shen Q, Zhou J. 2014. Small molecule inhibitors targeting activator protein 1 (AP-1). J Med Chem 57: 6930–6948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Reddy PM, Yu CY, Bastiani C, Higgs D, Stamatoyannopoulos G, Papayannopoulou T, Shen CK. 1993. Transcriptional activation of human ζ 2 globin promoter by the α globin regulatory element (HS-40): functional role of specific nuclear factor-DNA complexes. Mol Cell Biol 13: 2298–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zutter MM, Painter AD, Yang X. 1999. The Megakaryocyte/Platelet-specific enhancer of the α2β1 integrin gene: two tandem AP1 sites and the mitogen-activated protein kinase signaling cascade. Blood 93: 1600–1611. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.