

Abstract
Protein aggregation is associated with neurodegeneration and various other pathologies. How specific cellular environments modulate the aggregation of disease proteins is not well understood. Here, we investigated how the endoplasmic reticulum (ER) quality control system handles β‐sheet proteins that were designed de novo to form amyloid‐like fibrils. While these proteins undergo toxic aggregation in the cytosol, we find that targeting them to the ER (ER‐β) strongly reduces their toxicity. ER‐β is retained within the ER in a soluble, polymeric state, despite reaching very high concentrations exceeding those of ER‐resident molecular chaperones. ER‐β is not removed by ER‐associated degradation (ERAD) but interferes with ERAD of other proteins. These findings demonstrate a remarkable capacity of the ER to prevent the formation of insoluble β‐aggregates and the secretion of potentially toxic protein species. Our results also suggest a generic mechanism by which proteins with exposed β‐sheet structure in the ER interfere with proteostasis.
Keywords: endoplasmic reticulum, protein aggregation, proteostasis, quality control
Subject Categories: Membrane & Intracellular Transport; Post-translational Modifications, Proteolysis & Proteomics; Protein Biosynthesis & Quality Control
Introduction
The endoplasmic reticulum (ER) is the site of synthesis and processing of roughly one‐third of the eukaryotic proteome (Ghaemmaghami et al, 2003), including secretory and plasma membrane proteins, as well as proteins destined for the endomembrane system. To mediate the correct folding and assembly of secretory proteins, the ER contains a high concentration of quality control factors (Araki & Nagata, 2011). Multiple components, including a unique set of molecular chaperones, co‐chaperones, protein disulfide isomerases, glycosylating enzymes, and the ER‐associated degradation (ERAD) machinery, cooperate to maintain ER protein homeostasis (proteostasis; Balch et al, 2008; Araki & Nagata, 2011; Benyair et al, 2011; Brodsky & Skach, 2011; Walter & Ron, 2011; Vincenz‐Donnelly & Hipp, 2017). The proteostasis network (PN) of the ER ensures that misfolded or conformationally destabilized proteins are prevented from transport through the secretory pathway and are instead targeted for degradation. Failure of ER quality control may lead to either accumulation of misfolded proteins within the ER or to secretion of potentially toxic, aggregation‐prone proteins (Walter & Ron, 2011; Tipping et al, 2015). Accumulation of misfolded proteins in the ER induces the unfolded protein response (UPR), a complex stress response pathway that activates factors involved in re‐establishing proteostasis (Walter & Ron, 2011).
Protein misfolding and aggregation are hallmarks of numerous neurodegenerative diseases. Many of the proteins associated with neurodegeneration or systemic amyloidosis are co‐translationally targeted to the ER and processed within the secretory pathway, including the Alzheimer amyloid precursor, the prion protein PrPc, the neuroserpins, and the transthyretins (Davis et al, 1999; Soto, 2003; Sorgjerd et al, 2006). The aggregates of these proteins are rich in β‐sheet structure and often form amyloid‐like fibrils, although the aggregating proteins have no significant sequence similarity (Chiti & Dobson, 2006). How the ER quality control machinery recognizes and interacts with amyloidogenic proteins is not well understood. Interestingly, proteins with extended polyglutamine sequences, which readily aggregate in the cytosol, have been reported to remain soluble when targeted to the ER (Rousseau et al, 2004), suggesting important differences between cytosolic and ER quality control.
Here, we explored the capacity of the ER to retain proteins with a high propensity to form β‐sheet aggregates and to maintain them in a soluble state. We employed artificial proteins that were designed to aggregate into amyloid‐like fibrils based on an alternating pattern of polar and nonpolar amino acid residues that determines β‐strand secondary structure (West et al, 1999). When expressed in the cytosol of human cells, the propensity of these proteins to aggregate correlates with both their cytotoxicity and their ability to engage in aberrant interactions with key cellular factors (Olzscha et al, 2011; Woerner et al, 2016).
We find the ER‐targeted versions of these artificial β‐proteins (ER‐β) to be significantly less toxic than their non‐targeted counterparts. ER‐β is maintained in a soluble polymeric state within the ER, even though its concentration is estimated to be in the low millimolar range, exceeding that of available chaperones. Retention is independent of the specific sequence of the model β‐proteins analyzed. We further show that ER‐β interacts with ER‐resident chaperones and components of the ERAD pathway, such as the low abundance adaptor protein SEL1L. However, ER‐β is not effectively degraded and instead interferes with the degradation of other ERAD substrates. This inhibition of ERAD is reversed by overexpression of SEL1L, suggesting that ER‐β sequesters components that are limiting for ERAD. These results demonstrate a remarkable capacity of the ER to prevent both the secretion of toxic aggregation‐prone proteins and the formation of insoluble β‐aggregates. The three‐dimensional restriction of the aggregation process in the narrow ER lumen may be critical in this context. Furthermore, our results suggest a generic mechanism by which proteins with exposed β‐sheet structure interfere with ERAD of misfolded protein species.
Results
Targeting an aggregation‐prone β‐protein to the ER
To understand how the ER quality control machinery handles aggregation‐prone proteins, we determined the fate of an ER‐targeted version of the artificial β‐sheet protein β23. β23 is a member of a combinatorial library of proteins designed to fold into six β‐strands (West et al, 1999; Fig EV1, Appendix Table S1). It forms amyloid‐like fibrils with cross‐β structure in vitro and when expressed in the mammalian cytosol (West et al, 1999; Olzscha et al, 2011). We targeted β23 to the ER by addition of an N‐terminal signal peptide derived from the secretory pulmonary surfactant‐associated protein B (SP‐B; Dolfe et al, 2016; ER‐β, Fig EV1, Appendix Table S1). This signal peptide directs SP‐B toward co‐translational translocation into the ER, followed by processing and transport through the secretory pathway (Ueno et al, 2004). We also attached the same signal peptide to the control protein α‐S824, a de novo‐designed α‐helical protein similar in amino acid composition to β23 (ER‐α; Wei et al, 2003; Olzscha et al, 2011; Fig EV1, Appendix Table S1).
Figure EV1. Design of ER‐targeted model proteins.
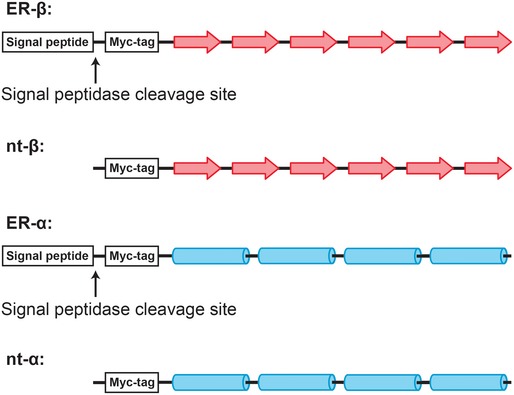
- ER‐targeted constructs of β‐protein (ER‐β) and α‐protein (ER‐α) were designed by adding an N‐terminal signal peptide upstream of the Myc‐tag of the non‐targeted constructs (nt‐β and nt‐α, respectively). The model β‐proteins were designed to form a β‐sheet consisting of six β‐strands (indicated by red arrows), and α‐proteins were designed to form four‐helical bundles (indicated by blue rods). The signal peptidase cleavage sites are indicated by black arrows.
The ER‐targeted proteins displayed the same apparent molecular weight on SDS–PAGE as their non‐targeted counterparts (nt‐β and nt‐α; Fig 1A), suggesting that the signal peptide was efficiently cleaved by the signal peptidase in the ER membrane. ER‐β accumulated to ~fourfold higher cellular levels than nt‐β. Analysis by confocal microscopy demonstrated that ER‐β and ER‐α co‐localized with ER marker proteins in HeLa and HEK293T cells (Fig 1B). In contrast, nt‐β and nt‐α did not co‐localize with ER markers, and nt‐β formed aggregate foci in the cytosol and nucleus (Fig 1B), as reported previously (Olzscha et al, 2011; Woerner et al, 2016).
Figure 1. ER‐β is efficiently targeted to the ER .
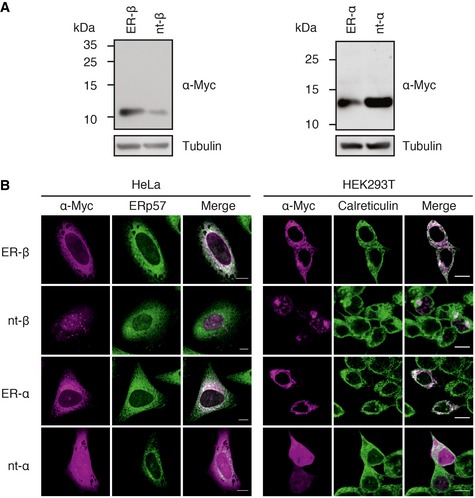
- Immunoblot analysis of ER‐targeted β‐protein (ER‐β) and α‐protein (ER‐α) constructs as well as the non‐targeted (nt) proteins after 48 h of expression in HEK293T cells revealed no shifts in mobility but differential expression levels.
- HeLa or HEK293T cells were seeded on poly‐L‐lysine (PLL)‐coated coverslips and transiently transfected with ER‐targeted and non‐targeted (nt) constructs. Cells were fixed 48 h post‐transfection and stained with anti‐Myc (magenta) and either anti‐ERp57 or anti‐calreticulin (green) antibodies, followed by fluorescently labeled secondary antibodies and analysis by confocal microscopy. Representative images from three independent experiments are shown. Scale bars represent 10 μm.
ER‐β is retained in the ER
Misfolded proteins are retained within the ER until they are either properly folded or degraded (Araki & Nagata, 2011). To determine whether ER‐β and ER‐α are secreted from cells, we analyzed their levels in conditioned medium. As expected for a protein without an ER retention signal, the majority of ER‐α was recovered in the medium (twofold higher levels than intracellular, P < 0.01), indicating efficient secretion (Fig 2A). Extracellular levels were strongly reduced when protein transport from the ER to the Golgi was blocked with brefeldin A (BFA; Klausner et al, 1992; Fig 2A). In contrast, ER‐β was not detectable in the medium, indicating that it was retained in the secretory pathway, consistent with its high intracellular concentration (Fig 2A).
Figure 2. ER‐β is retained in the ER .
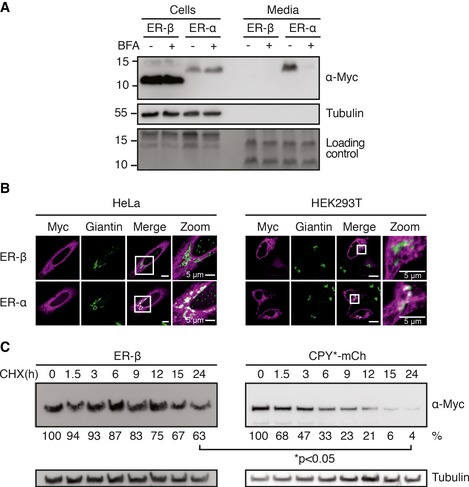
- HEK293T cells were transfected with ER‐β or ER‐α. After 24 h, media were exchanged for a minimal volume of growth medium containing 1 μg/ml brefeldin A (BFA) where indicated. After overnight treatment, cells and media were collected. Proteins from media samples were TCA‐precipitated and equal fractions of total protein from media and cell samples were analyzed by immunoblotting. After transfer, the PVDF membrane was stained to provide a loading control for media samples. Levels of ER‐β and ER‐α were analyzed by immunoblotting with anti‐Myc antibody. Tubulin served as a loading control and to demonstrate absence of cell leakage.
- HeLa cells or HEK293T cells were seeded on PLL‐coated coverslips and transfected with ER‐β or ER‐α. Cells were fixed 48 h post‐transfection and stained with anti‐Myc (magenta) and anti‐giantin (green) antibodies followed by fluorescently labeled secondary antibodies and analyzed by confocal microscopy. Representative images from three independent experiments are shown. Scale bars represent 10 μm unless otherwise indicated.
- HEK293T cells were transfected with either ER‐β or CPY*‐mCh, and 72 h later treated with 0.5 mM cycloheximide (CHX). Cells were harvested at the indicated time points and protein levels analyzed by immunoblotting against the Myc‐tag of ER‐β. For quantification, mean protein levels from untreated cells (0 h) were set to 100%. Mean values from three independent experiments are shown with P‐values determined using Student's t‐test (unpaired t‐test).
Addition of a C‐terminal mCherry‐tag allowed us to monitor ER‐β dynamics by live cell imaging (Movie EV1). The mCherry‐tag did not alter the retention of ER‐β within cells (Fig EV2A and B). Imaging of HEK293T cells over several minutes revealed that a large fraction of ER‐β‐mCh localized to network‐like structures that showed only little mobility (Movie EV1), whereas ER‐α‐mCh was mainly present in vesicles that rapidly moved laterally through the cell (Movie EV2).
Figure EV2. ER‐β‐mCh is retained in the ER and is less soluble than ER‐β.
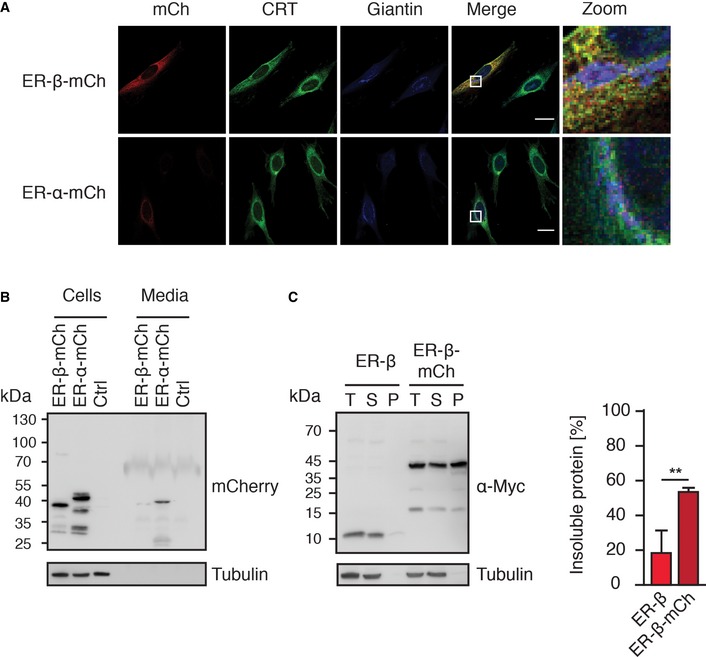
- HeLa cells transfected with ER‐β‐mCh or ER‐α‐mCh were fixed and stained with anti‐calreticulin (green) and anti‐giantin (blue) antibodies, followed by fluorescently labeled secondary antibodies and analysis by confocal microscopy. Scale bars represent 30 μm.
- HEK293T cells were transfected with ER‐β‐mCh or ER‐α‐mCh. After 48 h, cells and media were collected separately. Proteins from media samples were concentrated by TCA precipitation and equal fractions of total protein from media, and cell samples were analyzed by immunoblotting with anti‐mCherry antibody.
- Solubility of ER‐β and ER‐β‐mCh was analyzed 48 h after transfection by fractionation of lysates by centrifugation and immunoblotting with anti‐Myc antibody. T, total lysate; S, soluble fraction; P, pellet fraction. Right panel: Protein amounts in the pellet fraction were quantified. Error bars represent SD from three independent experiments. P‐values are based on Student's t‐test (unpaired t‐test). **P ≤ 0.01.
Having observed a defect in the secretion of ER‐β, we determined at what stage of the secretory pathway ER‐β trafficking was impaired. Some ER‐α co‐localized with the Golgi marker giantin, but ER‐β did not (Fig 2B), indicating that ER‐β was retained within the ER. Thus, in contrast to the artificial α‐helical protein, the aggregation‐prone ER‐β fails to pass ER quality control for secretion.
Degradation of misfolded proteins by ERAD involves retro‐translocation across the ER membrane and degradation in the cytoplasm by the ubiquitin–proteasome system (Olzmann et al, 2013). To investigate if ER‐β is a substrate of ERAD, we analyzed its turnover by inhibiting protein synthesis with cycloheximide (CHX). We found ER‐β to be a remarkably stable protein with a half‐life of more than 24 h (Fig 2C), considerably exceeding the half‐life of other ER proteins with folding defects (Finger et al, 1993). For example, the classic ERAD substrate mutant carboxypeptidase Y (CPY*), fused to mCherry (CPY*‐mCh), was degraded with a half‐life of less than 3 h (Fig 2C). Thus, although ER‐β is efficiently retained in the ER, it is not successfully targeted for degradation by ERAD.
Targeting β‐proteins to the ER reduces toxicity and aggregation propensity
Expression of nt‐β strongly reduces cell viability (Olzscha et al, 2011; Woerner et al, 2016). In contrast, we found that directing the β‐protein to the ER significantly attenuated its toxicity (Fig 3A). This reduction in toxicity was observed, despite the more than fourfold higher expression levels of ER‐β compared to nt‐β (Figs 1A and 3B). We have previously shown that directing these β‐sheet proteins to the nucleus results in a reduction in toxicity that is accompanied by the formation of insoluble inclusions (Woerner et al, 2016). We therefore tested whether the environment of the ER lumen affects the aggregation behavior of ER‐β in a similar manner. Strikingly, in contrast to the largely insoluble nt‐β, ER‐β was predominantly soluble in buffer containing 1% Triton X‐100 (Fig 3C), consistent with the absence of ER‐β inclusion bodies observable by light microscopy (Fig 1B). This behavior is even more remarkable when taking into consideration that the ER makes up only a fraction of the mammalian cell volume [~15% in hepatocytes (Alberts et al, 2014; Weibel et al, 1969), ~25% in COS‐7 cells (Valm et al, 2017)], and thus, the local concentration of ER‐β in the ER is at least 10 times higher than the concentration of nt‐β. We replicated these findings with two other β‐sheet proteins (β4 and β17) targeted to the ER (Appendix Fig S1). These proteins are of similar size as nt‐β but differ in their amino acid sequence and relative β‐aggregation propensity (Olzscha et al, 2011; Appendix Table S1). We found that the solubility of both ER‐β4 and ER‐β17 was comparable to the solubility of ER‐β (Appendix Fig S1A and B) and both were also retained in the ER (Appendix Fig S1C and D). This is in line with a previous report that ER‐targeting of proteins containing an expanded polyglutamine repeat prevented the formation of insoluble aggregates by these proteins (Rousseau et al, 2004).
Figure 3. ER‐targeting prevents β‐protein aggregation.
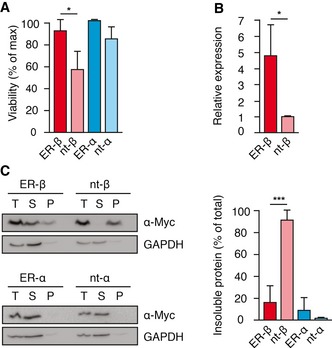
- Cell viability was measured by the MTT assay 72 h after transfection of HEK293T cells with the constructs indicated. Error bars represent SD from three independent experiments. P‐values are based on Student's t‐test (unpaired t‐test). *P < 0.05.
- Relative expression levels of ER‐β and nt‐β in HEK293T cells, based on data from Fig 1A. Total levels of ER‐β were quantified and plotted as fold increase over nt‐β. Mean values from four independent experiments are shown, error bars represent SD, and P‐values were determined using Student's t‐test (unpaired t‐test). *P < 0.05.
- Solubility of ER‐β, ER‐α, and their non‐targeted counterparts (nt) in HEK293T cells was analyzed 48 h after transfection by fractionation of lysates by centrifugation and immunoblotting with anti‐Myc antibody. T, total lysate; S, soluble fraction; P, pellet fraction. Right panel: Protein amounts in the pellet fraction were quantified. Error bars represent SD from three independent experiments, and P‐values are based on Student's t‐test (unpaired t‐test). ***P ≤ 0.001.
To test whether ER‐β is present as a monomeric protein in the ER, we analyzed complete cell extracts under non‐denaturing conditions by blue native PAGE (Fig 4A). Although some ER‐β migrated at the size of the monomer, the majority of ER‐β formed higher molecular weight species of 200 to more than 1,200 kDa, indicative of the presence of oligomers up to higher order polymers, possibly associating with protein quality control machinery. Note that ER‐β contains only a single cysteine and thus disulfide formation is not the cause of polymer formation. Only background levels of ER‐α were detected in regions of the gel were oligomers or complexes between ER‐α and the cellular quality control machinery would be expected and monomers could hardly be observed due to low intracellular levels.
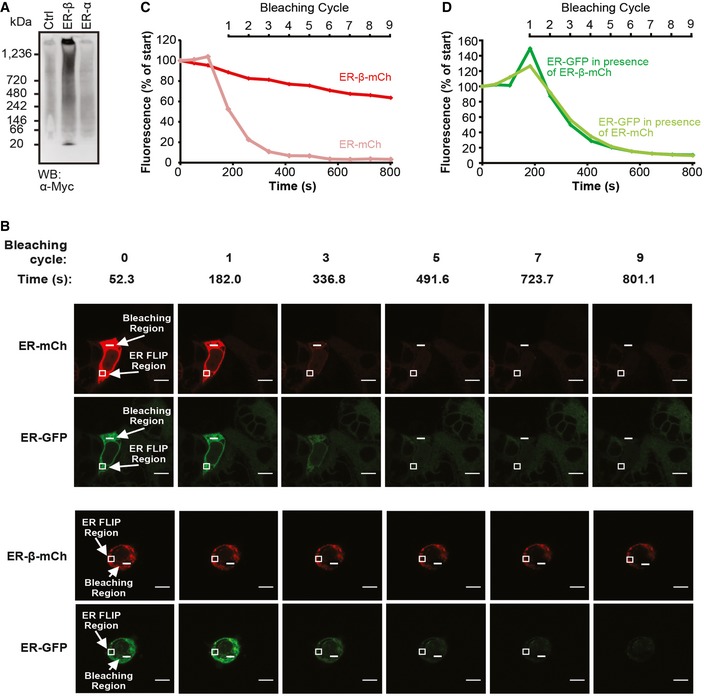
Figure 4. ER‐β forms oligomers with restricted mobility.
-
ALysates from HEK293T cells transfected with either pcDNA (Ctrl), ER‐β, or ER‐α were separated on blue native PAGE and analyzed by immunoblotting against the Myc‐epitope.
-
B–DFluorescence loss in photobleaching (FLIP) was performed in HEK293T cells 48 h after co‐transfection with ER‐β‐mCh and ER‐GFP‐KDEL (ER‐GFP) or ER‐mCherry‐KDEL (ER‐mCh) and ER‐GFP. After recording three images by confocal microscopy, small areas within the ER of cells (bleaching region) were repeatedly bleached using the 561 nm (for mCherry), 488 and 458 nm (for GFP) lasers and images recorded after each bleaching cycle (every 77.4 s) (B). Relative changes in mean fluorescence of areas within the ER of the same cells (ER FLIP region) were plotted for (C) mCherry and (D) GFP normalized to the fluorescence measured at the first data point. Representative data from three independent experiments are shown. Scale bars represent 10 μm.
The presence of oligomeric species of ER‐β was further supported by an analysis of the mobility of ER‐resident β‐proteins using fluorescence loss in photobleaching (FLIP; Fig 4B–D). By bleaching one region of the ER (bleaching region, Fig 4B) while measuring changes of fluorescence levels in a different region (ER FLIP region, Fig 4B), it is possible to draw conclusions about the mobility and size of the fluorescent particles. We compared the mobility of ER‐β‐mCh to a control mCherry protein (ER‐mCh) that contains the same signal sequence but is retained in the ER by a C‐terminal KDEL retention signal. As expected, the ER‐mCh control protein was rapidly bleached within the whole ER (Movie EV3, Fig 4B and C). In contrast, bleaching of ER‐β‐mCh resulted only in a slow fluorescence decrease in distant parts of the ER (Movie EV4, Fig 4B and C). This indicates that ER‐β‐mCh is substantially less mobile than a monomeric protein of comparable size. The increased viscosity is reminiscent of the liquid‐like behavior of phase transitions driven by intrinsically disordered protein regions (Brangwynne et al, 2009; Hyman et al, 2014). A reduction in fluorescence was observed in the immediate vicinity of the bleached ER‐β‐mCh, indicative of local dynamics (Movie EV4, Fig 4B). It is important to note, however, that the levels of the mCherry‐tagged β‐proteins were higher than those of ER‐β without a mCherry‐tag, consistent with an increased insolubility of ER‐β‐mCh, which might affect protein mobility (Fig EV2C). ER‐β mobility was slightly reduced in cells with the highest expression of the mCherry‐tagged protein compared to cells with very low expression, but this trend did not reach statistical significance (Appendix Fig S2). Thus, ER‐β without a mCherry‐tag, though efficiently retained in the ER, may be more dynamic than the more highly expressed ER‐β‐mCh.
The reduced mobility of ER‐β‐mCh measured by FLIP can be explained either by the presence of higher order polymers or by fragmentation of the ER into structures that are unable to exchange their components with the remainder of the ER network. To distinguish between these possibilities, we investigated whether ER‐β‐mCh affected the mobility of another fluorescent protein, ER‐targeted GFP (ER‐GFP). After continuous photobleaching of a small area of the ER, ER‐GFP fluorescence rapidly decreased within the whole ER both in the presence of ER‐mCh (Movie EV5, Fig 4B and D) and in the presence of ER‐β‐mCh (Movie EV6, Fig 4B and D). This indicates that the β‐protein did not impair the movement of monomeric proteins through the ER lumen, consistent with ER‐β‐mCh demonstrating liquid‐like behavior. We next tested if the unique ion composition of the ER (Vincenz‐Donnelly & Hipp, 2017) is responsible for the unusual aggregation behavior of ER‐β. To this end, we analyzed the mobility and detergent solubility of ER‐β in the presence of the SERCA inhibitor Thapsigargin, which causes a depletion of ER calcium (Thastrup et al, 1990). Thapsigargin treatment did not result in any alterations in ER‐β mobility (Fig EV3A) or changes of detergent solubility (Fig EV3B and C). It seems likely, therefore, that the reduced mobility of ER‐β‐mCh is due to the formation of a liquid‐like state and/or interactions with ER‐resident components. Formation of this physical state may be assisted by three‐dimensional constraints on the aggregation process in the narrow ER lumen, which is only ~50 nm wide (Shibata et al, 2010; Westrate et al, 2015).
Figure EV3. Thapsigargin does not alter ER‐β aggregation, and ER‐β expression is not increasing sensitivity toward external stress.
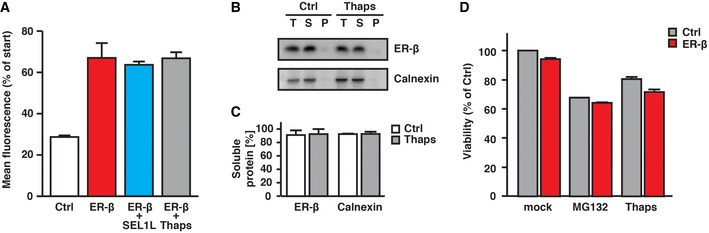
- HEK293T cells were co‐transfected with ER‐β‐mCh or ER‐mCh (Ctrl) and either empty pcDNA3.1 or SEL1L in a ratio of 1:1. After 48 h, cells were treated with 1 μM Thapsigargin (Thaps) for 5 h before analysis, where indicated. After recording three images by confocal microscopy, small areas within the ER of cells were repeatedly bleached using the 561 nm laser and images recorded after each bleaching cycle (˜22 s). Relative changes in mean fluorescence of total cells after seven bleaching cycles are plotted. Error bars represent SD from three independent experiments. At least five cells per condition per independent repeat were analyzed.
- Solubility of ER‐β and endogenous calnexin in HEK293T cells was analyzed 48 h after transfection by fractionation of lysates by centrifugation and immunoblotting with anti‐Myc and anti‐calnexin antibody. The culture media was replaced 5 h before analysis to DMEM without (Ctrl) or with 1 μM Thapsigargin (Thaps). T, total lysate; S, soluble fraction; P, pellet fraction.
- Quantification of (B). Error bars represent SD from three independent experiments.
- Viability of HEK293T cells expressing ER‐β or pcDNA3.1 (Ctrl), as measured by MTT [3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide] assay. Cells were treated for 16 h with 5 μM MG132 or 1 μM Thapsigargin (Thaps), where indicated. Error bars represent SD from three independent experiments.
ER‐β interacts with a distinct set of ER chaperones and ERAD components
To identify components of the ER quality control machinery that interact with ER‐β, we performed a quantitative proteomic analysis using stable isotope labeling with amino acids in cell culture (SILAC; Ong & Mann, 2006). ER‐β was isolated from cell lysates by immunoprecipitation against the N‐terminal Myc‐tag. Of a total of 3,317 proteins identified by mass spectrometry (MS), 83 proteins were enriched twofold or more over background (enrichment factors up to 8.7‐fold) and were thus defined as ER‐β interactors (Table EV1, Dataset EV1). Analysis of total protein levels showed that these proteins were not significantly upregulated in cells expressing ER‐β, indicating that the observed interactions are not due to alterations in total protein levels (Dataset EV2).
The ER‐β interactors were significantly enriched in ER luminal proteins and with lower significance in mitochondrial membrane proteins. ER‐β also interacted with a set of nuclear proteins, including proteins of the nuclear envelope, although nuclear proteins were not generally enriched among the interactors. We cannot exclude that the observed interactions between ER‐β and nuclear and mitochondrial proteins are due to interactions that occurred after cell lysis.
In contrast to ER luminal proteins, other components of the secretory pathway as well as the cytosol were not significantly enriched in the ER‐β interactome, consistent with the finding that ER‐β is retained in the ER. Indeed, less than 10% of the ER‐β interactors were previously found to interact with nt‐β (Olzscha et al, 2011; Fig EV4A). This confirms that the proteins have distinct cellular localizations and that import of ER‐β into the ER is efficient.
Figure EV4. ER‐β interacts with a distinct set of cellular proteins and is not glycosylated.
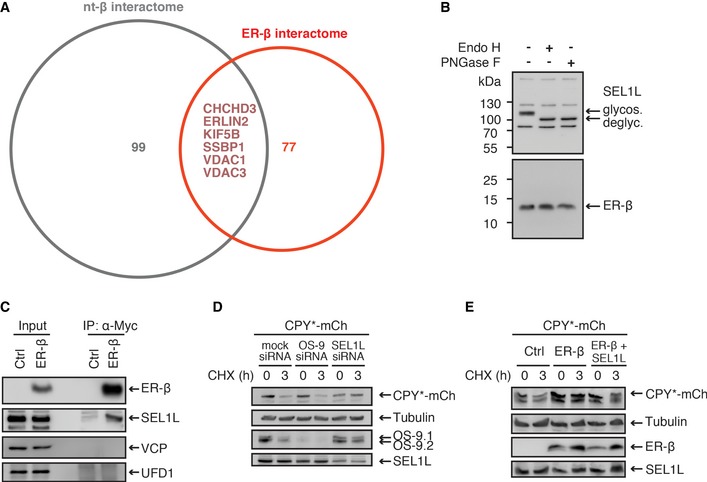
-
AComparison of the interactors of nt‐β (Olzscha et al, 2011; 105 proteins, represented by gray circle) and the identified interactors of ER‐β (83 proteins, represented by red circle) revealed six common interactors. The gene names of the common interactors are indicated.
-
BLysates from HEK293T cells transfected with ER‐β were treated with Endo H or PNGase F. Samples were then analyzed by immunoblotting with anti‐SEL1L and anti‐Myc antibodies, using 8% and 12% Bis–Tris gels for SEL1L and ER‐β, respectively.
-
CImmunoblotting against SEL1L, VCP, and UFD1 after immunoprecipitation of ER‐β as in Fig 5B.
-
D, EImmunoblots corresponding to Fig 5E and F.
Components of the ERAD machinery were among the most highly enriched interactors of ER‐β. These include the adaptor protein SEL1L, the substrate binding protein OS‐9, and the intramembrane adaptor protein Erlin‐2, which was also found to interact with nt‐β (Figs 5A and EV4A, and Table EV1). As expected, ER‐β also associated with several ER‐resident chaperones (Fig 5A, Table EV1). The HSP70 BiP (GRP78/HSPA5) made up ~20% of the total amount of all interacting proteins (Table EV2). We also identified as interactors the HSP90 family member GRP94 (HSP90B1), the lectin chaperone calnexin (CNX), the nucleotide exchange factor of the HSP110 family HYOU1 (GRP170/ORP150) and protein disulfide‐isomerase A6 (PDIA6), which has both isomerase and chaperone activities (Kikuchi et al, 2002), and is thought to play a role in protein folding and regulation of the UPR (Eletto et al, 2014). These findings are reminiscent of the interactions of nt‐β with cytosolic members of the HSP70 and HSP110 chaperone families (Olzscha et al, 2011). We confirmed the specific interaction of ER‐β with SEL1L, OS‐9, HYOU1, PDIA6, GRP94, Erlin‐2, CNX, and BiP by immunoblotting (Fig 5B). We found that only a small fraction of the total amount of the different chaperone proteins present in the cells was co‐immunoprecipitated (Fig 5B), arguing against a depletion of the free chaperone pool by ER‐β. In contrast, the ERAD components SEL1L and OS‐9 interacted more extensively (about 10% of total SEL1L and about 50% of total OS‐9), suggesting that their sequestration by ER‐β might have functional consequences. Notably, based on intensity‐based absolute quantification (iBAQ; Schwanhausser et al, 2011), the levels of SEL1L and OS‐9 in the total proteome were over 200‐fold lower than the levels of GRP94 and BiP (Table EV3, Dataset EV3).
Figure 5. ER‐β interacts with ER quality control factors.
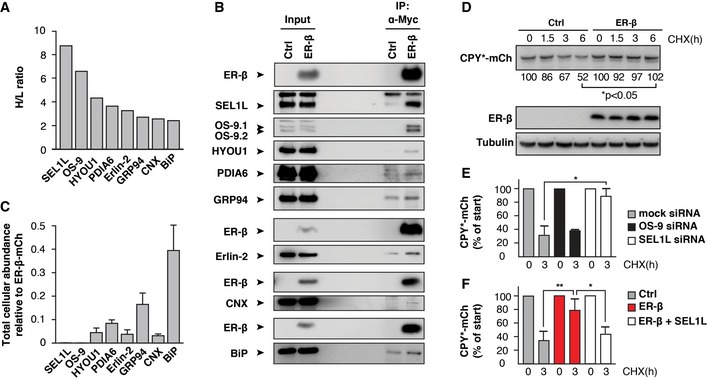
- Enrichment ratios (H/L) of ER‐β‐interacting ER quality control factors from three independent experiments [combined ratios calculated by MaxQuant (version 1.3.0.5)] are shown.
- Immunoblotting against SEL1L, OS‐9, HYOU1, PDIA6, GRP94, Erlin‐2, CNX, and BiP after immunoprecipitation of ER‐β confirmed specific interactions with ER‐β.
- Cellular abundance values of ER quality control factors relative to the abundance of ER‐β‐mCh. iBAQ values of proteins measured in ER‐β‐mCh‐expressing cells are plotted as fraction of the iBAQ value of ER‐β‐mCh after normalization for transfection efficiencies. Error bars represent SD from three independent experiments.
- HEK293T cells were co‐transfected with CPY*‐mCh and either empty pcDNA3.1 (Ctrl) or ER‐β. Degradation of CPY*‐mCh was analyzed by treatment with 0.5 mM CHX for the indicated time periods, and determination of CPY*‐mCh levels by immunoblotting with anti‐Myc antibody. For quantification, CPY*‐mCh levels from untreated cells (0 h) were set to 100%. Mean values from three independent experiments are shown. P‐values are based on Student's t‐test (unpaired t‐test).
- HEK293T cells were co‐transfected with CPY*‐mCh and either non‐targeting siRNA (mock) or siRNA targeting either OS‐9 or SEL1L. Degradation of CPY*‐mCh after 3 h was analyzed as in (D). *P < 0.05. Error bars represent SD from three independent experiments, and P‐values are based on Student's t‐test (unpaired t‐test).
- HEK293T cells were co‐transfected with CPY*‐mCh and either pcDNA3.1 (Ctrl), ER‐β and pcDNA or ER‐β and SEL1L. Degradation of CPY*‐mCh after 3 h was analyzed as in (D). *P < 0.05, **P ≤ 0.01. Error bars represent SD from three independent experiments, and P‐values are based on Student's t‐test (unpaired t‐test).
We hypothesized that binding of ER‐β to chaperones might contribute to its retention in the ER. It was therefore of interest to determine the abundance of ER chaperones relative to ER‐β. Tryptic digestion of nt‐β or ER‐β did not result in peptides that could be detected by MS, and thus, it was not possible to quantify ER‐β by proteomics. However, tryptic digestion of ER‐β‐mCh generated sufficient peptides to allow quantification using iBAQ. We observed that total ER‐β‐mCh levels were significantly higher than the levels of each individual chaperone found to interact with ER‐β, and even exceeded the total abundance of all interacting ER‐resident chaperones combined (Fig 5C). BiP (HSPA5) has been reported to reach low millimolar concentrations in the ER (up to 5 mM; Weitzmann et al, 2007), providing an estimate for the concentration of ER‐β. When we take into account that only a few percent of the total of each of the major chaperones was co‐immunoprecipitated with ER‐β (Fig 5B), a 1:1 molar interaction between a major fraction of ER‐β and any specific chaperone is unlikely, even when considering the difference in expression levels between ER‐β and ER‐β‐mCh (Fig EV2C). We conclude that chaperones either bind to only a small fraction of ER‐β at any given time (e.g., the monomeric form), or that a single chaperone molecule is able to interact with, and retain, larger ER‐β oligomers. Alternatively, ER‐β is not retained by chaperone binding but rather by the formation of a network of oligomers and higher order polymers that permeates the ER lumen.
Since several of the ER‐β interactors are involved in binding glycosylated substrates, we investigated whether ER‐β is subject to glycosylation. ER‐β contains several asparagine residues (Appendix Table S1) but no predicted N‐glycosylation sites (NetNGlyc 1.0 Server (Blom et al, 2004), http://www.cbs.dtu.dk/services/NetNGlyc/). ER‐β migrated at the same size as nt‐β (Fig 1A). Moreover, ER‐β did not show any shift in electrophoretic mobility after treatment of lysates with the deglycosylating enzymes endoglycosidase H (Endo H) or peptide‐N‐glycosidase F (PNGase F), in contrast to the glycosylated ER protein SEL1L (Fig EV4B). Thus, ER‐β either interacted with lectin chaperones despite the absence of glycosylation, or these proteins were co‐immunoprecipitated as parts of larger complexes.
ER‐β inhibits ERAD
Although ER‐β was only slowly degraded, it interacted strongly with factors that play a role in ERAD (Fig 5A and B, Table EV1). ERAD substrates are normally recruited to dislocons for transport from the ER to the cytosol (Olzmann et al, 2013). This recruitment to the ER membrane involves a variety of auxiliary factors, including the adaptor protein SEL1L, which is thought to link substrate recognition to retro‐translocation (Christianson et al, 2008; Olzmann et al, 2013). We found that both OS‐9 and SEL1L bind to ER‐β and also identified the intramembrane substrate adaptor Erlin‐2 as an interactor (Fig 5A and B, Table EV1). However, none of the downstream factors of the ERAD pathway involved in substrate dislocation or ubiquitination were identified as interactors of ER‐β (Fig EV4C, Dataset EV1). Together with our finding that ER‐β is rather stable, its exclusive interaction with luminal ERAD factors suggested that ER‐β is not translocated to the cytosol and thus apparently interacts with ERAD components in a non‐productive manner.
It seemed possible that these non‐productive interactions interfere with the degradation of other ERAD substrates. We hypothesized that the very low abundance proteins OS‐9 and SEL1L (4.8 and 4.9 ppm in the total proteome, respectively; Table EV3) may become limiting, especially when considering that a substantial fraction of these proteins interacted with ER‐β (Fig 5A and B). To test this possibility, we analyzed the effect of ER‐β on the degradation of the ERAD model substrate CPY*‐mCh. Co‐expression of ER‐β significantly stabilized CPY*‐mCh (Fig 5D), indicating inhibition of ERAD by ER‐β. It has previously been shown that Yos9p and Hrd3p, the yeast homologues of OS‐9 and SEL1L, are required for degradation of CPY* in yeast (Buschhorn et al, 2004; Bhamidipati et al, 2005; Gauss et al, 2006) and that SEL1L is required for ERAD in mammalian cells (Sun et al, 2014). The observed interactions of OS‐9 and/or SEL1L with ER‐β might therefore be sufficient to stabilize CPY*. To test whether degradation of CPY*‐mCh by the mammalian ERAD system depends on these factors, we performed siRNA knockdowns of OS‐9 and SEL1L. Simultaneous knockdown of both OS‐9 isoforms, OS‐9.1 and OS‐9.2, had no significant effect on CPY*‐mCh turnover (Figs 5E and EV4D). This finding may be explained by the redundant activities of OS‐9 and the ERAD lectin XTP3‐B in the degradation of luminal ERAD substrates (Bernasconi et al, 2010). Redundant activities in the ERAD pathway might also explain the low toxicity observed after ER‐β expression, even in the presence of additional forms of external proteostasis stress (Fig EV3D). In contrast, knockdown of SEL1L caused a significant stabilization of CPY*‐mCh with almost 90% of CPY*‐mCh remaining after 3 h of CHX chase (Figs 5E and EV4D). This strong dependence of CPY*‐mCh degradation on SEL1L suggests that sequestration of SEL1L by ER‐β is sufficient to block degradation. Indeed, mild overexpression of SEL1L rescued the degradation defect of CPY*‐mCh caused by ER‐β, without changing the mobility of ER‐β (Figs 5F, EV3A, and EV4E). Thus, the accumulation of ER‐β in the ER leads to a block of ERAD likely by titration of SEL1L and perhaps other ERAD machinery.
ER‐β inhibits a functional UPR
Accumulation of misfolded proteins in the ER normally leads to induction of the unfolded protein response (UPR) via activation of the transmembrane stress sensors ATF6, IRE1α, and PERK (Walter & Ron, 2011). Notably, despite its high levels, ER‐β expression did not increase the levels of ERQC proteins, as shown by MS analysis (Dataset EV2) and by immunoblotting of several UPR‐inducible proteins (Fig 5B). Similar results were previously obtained with cells expressing mutant α1‐Antitrypsin Z (Hidvegi et al, 2005) and mutant neuroserpin; however, in that case it is possible that the polymers represent a folded, near‐native conformation (Davies et al, 2009). For further analysis, we measured genomewide changes in the transcriptome by RNA sequencing (RNA‐seq) of three independent cell culture replicates. ER‐β expression led to significantly altered expression of 420 genes (Fig EV5A). To identify cellular pathways activated upon expression of ER‐β, we performed a Gene Ontology (GO) enrichment analysis of the 50 most upregulated genes using the EnrichR tool (Chen et al, 2013). As shown in Fig 6A, a gene expression signature of the UPR was clearly visible in the GO analysis, even though the magnitude of this response is only moderate in comparison with the response induced by tunicamycin (Raina et al, 2014). Furthermore, RNA‐seq analysis identified 12 genes previously shown to be associated with UPR as significantly upregulated upon ER‐β expression (adjusted P‐value < 0.05; Figs 6B and EV5). This discrepancy between UPR induction on the protein and mRNA levels indicates that although ER‐β expression is recognized as an ER stressor by the UPR sensors in the ER, this does not result in a functional UPR response. Interestingly, we previously found that expression of nt‐β‐proteins also failed to induce the cytosolic stress response or heat‐shock response, although the β‐proteins interact with several molecular chaperones (Olzscha et al, 2011).
Figure EV5. ER‐β expression leads to UPR transcriptional response.
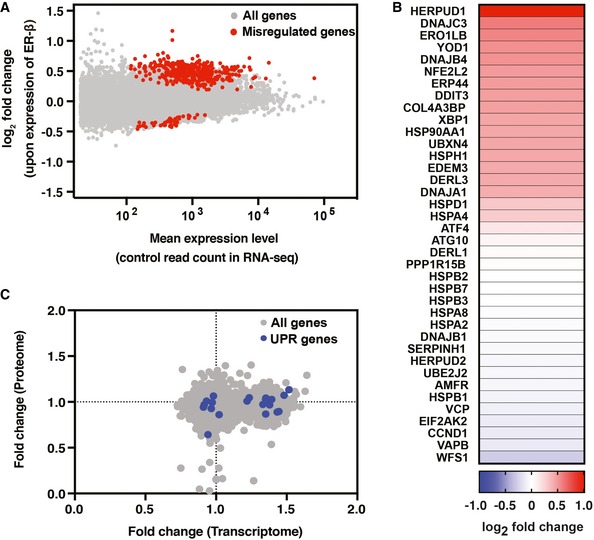
- Changes in gene expression of all annotated genes after ER‐β expression in HEK293T cells are plotted against mean expression levels under empty vector control conditions. Data obtained by RNA‐seq analysis are shown as means from three independent cell cultures. Genes with significant misregulation are shown in red (adjusted P‐values < 0.05, on the basis of DESeq2 analysis, as outlined in Materials and Methods).
- A heat map of UPR target genes upon ER‐β expression as identified by RNA‐seq analysis. The UPR target genes were defined in an earlier study (Raina et al, 2014).
- Scatter plot of the protein‐ and transcript‐level changes upon ER‐β expression. Misregulated UPR target genes are highlighted in blue.
Figure 6. ER‐β inhibits expression of UPR target proteins.
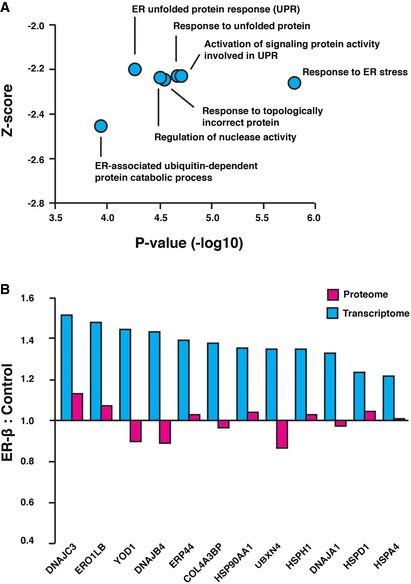
- The 50 most upregulated genes upon ER‐β expression were clustered on the basis of common Gene Ontology (GO) enrichment terms as indicated. The y‐axis indicates the Z‐score for each annotation, and the x‐axis depicts the significance of the respective enrichment.
- Induction of UPR target genes is not reflected in the proteome. Ratios of protein‐ and transcript‐level changes upon ER‐β expression of misregulated UPR target genes are plotted (n = 3 independent cell culture replicates).
Taken together, ER‐β is recognized by the ER quality control machinery as an aberrant protein and is retained in the ER, although it is maintained in a largely soluble, polymeric state. Despite its high concentration in the ER lumen, ER‐β fails to induce an appropriate cellular stress response and interferes with successful induction of a functional UPR and the degradation of other misfolded proteins by ERAD, thereby causing proteostasis imbalance.
Discussion
We have demonstrated in this study that the ER as a cellular compartment has a remarkable capacity to prevent the secretion of aggregation‐prone β‐proteins and maintain them in a more soluble and less toxic state than the cytosolic quality control system. Unlike the α‐helical protein ER‐α, which passes ER quality control and is secreted (Fig 7A), or the terminally misfolded protein CPY*, which is retained and efficiently degraded via ERAD (Fig 7B), the β‐proteins are neither secreted nor efficiently degraded, but rather accumulate in the ER to very high levels (Fig 7C). Since failure of retention would lead to secretion of potentially toxic protein species, ER retention is a protective mechanism that prevents extracellular and cytosolic protein aggregation. However, the retention of β‐proteins comes at a price: their accumulation leads to engagement and titration of critical components of the ERAD pathway (Fig 7C). The β‐proteins disturb proteostasis further by interfering with the induction of the ER stress response.
Figure 7. Proposed model of the fate of ER‐β in the ER .
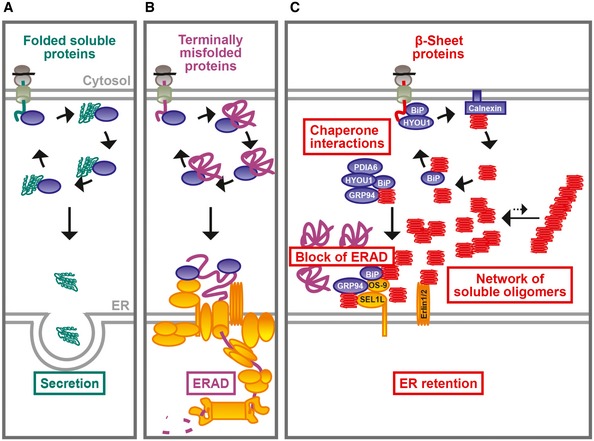
- After folding and assembly in the ER lumen, secretory proteins exit the ER via vesicular transport.
- Terminally misfolded proteins, such as CPY*, are recognized by the ERAD machinery and are retro‐translocated into the cytosol for ubiquitin–proteasome‐dependent degradation.
- ER‐β engages in substoichiometric interactions with ER‐resident chaperones and forms a “fluid” polymer network that permeates the ER lumen without interfering with the movement of other proteins, at least up to the size of GFP. ER‐β also interacts non‐productively with ERAD factors and inhibits ERAD of proteins such as CPY*. Chaperone proteins, blue; ERAD components, yellow.
ER‐targeting rendered the β‐proteins significantly more soluble than their cytosolic counterparts, similar to findings with polyglutamine expansion proteins (Rousseau et al, 2004). The capacity of the ER to maintain the β‐sheet proteins in a soluble state is particularly striking when considering that these proteins accumulate in the ER lumen to very high concentrations in the low millimolar range. In comparison, the β‐proteins β4, β17, and β23 form insoluble fibrillar aggregates in vitro already at low micromolar concentrations (Olzscha et al, 2011). Thus, in the environment of the ER these proteins reach levels ~1,000‐fold higher than their in vitro solubility, a state referred to as supersaturation (Ciryam et al, 2015). We suggest that three‐dimensional constraints exerted on the aggregation process in the ER are critical in generating this physical state. The width of the ER lumen is only ~50 nm (Shibata et al, 2010; Westrate et al, 2015), while the insoluble aggregates of cytosolic β‐proteins are 1‐2 μm in diameter (Olzscha et al, 2011). How long the ER can counterbalance the high intrinsic aggregation propensity of these proteins is not clear, however. Prolonged expression or a further increase in abundance may lead to formation of insoluble aggregates, as has been observed for the mCherry‐tagged proteins that are expressed at higher levels.
We identified a set of ER‐resident chaperone proteins that interact with ER‐β and may contribute to prevention of aggregation and ER retention. However, these chaperones were less abundant in cells than ER‐β itself, suggesting that they do not bind ER‐β at a 1:1 ratio. It is also possible that ER retention is not solely due to direct interactions with ER quality control machinery but rather occurs as a consequence of ER‐β forming a network of oligomers and higher order polymers. We can also not exclude the possibility that the β‐sheet proteins lack a specific signal that allows them to exit the ER. Transient membrane interactions of oligomers are likely to be enhanced by the two‐dimensional organization of the ER and may contribute to retention. In contrast to the ordered polymerization of members of the serine protease inhibitor superfamily (Ekeowa et al, 2010), ER‐β oligomers cannot be explained by the formation of linear chains due to insertion of loops from one molecule into β‐sheets of a second molecule. The decreased diffusion rate, when compared to a monomeric protein, as well as the finding that the mobility of a non‐interacting protein is not affected (Fig 4) would be consistent with ER‐β being kept in a state that bears similarity to the liquid‐like assemblies recently reported for various proteins with low sequence complexity (Patel et al, 2015). We also note that ER‐β does not appear to be retained due to failure of packaging into COPII vesicles, as ER‐β did not localize to ER exit sites (ERES; Fig 1B) and did not detectably interact with proteins known to be involved in COPII vesicle assembly (such as Sar1A, Sec16, Sec23, Sec24, Sec31, or ERGIC‐53; Budnik & Stephens, 2009; Table EV1).
Although ER‐β interacts with ER‐resident chaperones and engages ERAD machinery, it is not efficiently degraded, and apparently does not interact with the 26S proteasome. The failure of ER‐β to undergo ERAD may be due to its higher order structure preventing passage through the dislocon or due to the lack of specific post‐translational modifications that may be required for efficient export. It is also possible that, independent of its folding status, ER‐β cannot be recognized by the cytosolic components of the ERAD machinery due to features of its non‐evolved amino acid sequence, for example, the position of the lysine residues in the constructs.
We note that many of the identified ER‐β interactors have previously been shown to recognize disease‐related mutants of neuroserpins and α1‐antitrypsin (A1AT). These proteins accumulate in the ER as polymers and cause dementia in familial encephalopathy with neuroserpin inclusion bodies (FENIB) and lung and liver disease in A1AT deficiency, respectively. A1AT mutants that are retained in the ER interact with the chaperones BiP, GRP94, CNX, and HYOU1/GRP170 (Schmidt & Perlmutter, 2005), which are also interactors of ER‐β. Furthermore, OS‐9 and GRP94 have previously been shown to be involved in the delivery of both neuroserpin and A1AT mutants to SEL1L for ERAD (Christianson et al, 2008; Schipanski et al, 2014). OS‐9 is a lectin that has been suggested to have a dual function in ER quality control: the retention of misfolded proteins in the ER lumen and the delivery of misfolded proteins for ERAD (Bernasconi et al, 2008; Olzmann et al, 2013). In addition to recognizing misfolded glycoproteins, OS‐9 has been demonstrated to bind non‐glycosylated substrates (Bernasconi et al, 2008; Christianson et al, 2008; Hosokawa et al, 2009; Olzmann et al, 2013). However, both OS‐9.1 and OS‐9.2, while binding the non‐glycosylated mutant A1AT mutant NHKQQQ, failed to target NHKQQQ for degradation (Bernasconi et al, 2008; Hosokawa et al, 2009). We now show that the non‐glycosylated ER‐β also interacts with both isoforms of OS‐9, but is not efficiently targeted for ERAD. Thus, OS‐9 may recognize ER‐β through a mechanism comparable to its interaction with non‐glycosylated mutants of A1AT and neuroserpins, suggesting a specific affinity for β‐sheet conformations independent of glycosylation. However, it remains to be determined whether OS‐9 recognizes the non‐glycosylated β‐sheet proteins directly or interacts indirectly as part of a larger protein complex that includes BiP, SEL1L, Hrd1, and Grp94 (Christianson et al, 2008; Olzmann et al, 2013).
In summary, ER‐β‐proteins are retained in the ER where they interact with ER chaperones and ERAD factors, but are not efficiently degraded and instead inhibit the degradation of other ERAD substrates. This interference with ERAD causes proteostasis impairment and reveals a mechanism of proteotoxicity that may play a more general role in diseases associated with ER retention of aggregation‐prone proteins. However, considering the acute toxicity of the β‐proteins in the cytosol (Olzscha et al, 2011; Woerner et al, 2016), the inability of these proteins to undergo retro‐translocation from the ER may outweigh these negative effects. Indeed, retro‐translocation of the aggregation‐prone prion protein to the cytosol has been suggested to exert considerable toxicity (Ma & Lindquist, 2002), and ER‐targeted polyglutamine proteins were shown to aggregate only after retro‐translocation into the cytosol (Rousseau et al, 2004).
Materials and Methods
Plasmids
The signal peptide of human pulmonary surfactant‐associated protein (SP‐B; MAESHLLQWLLLLLPTLCGPGTA) was inserted upstream of the Myc‐tag of pcDNA3.1+ plasmids containing β23, αS‐824, β4, and β17 (Olzscha et al, 2011) to generate ER‐β, ER‐α, ER‐β4, and ER‐β17, respectively. For the mCherry‐tagged constructs, ER‐β and ER‐α were excised from the pcDNA3.1+ plasmids via KpnI and AgeI after generation of a downstream AgeI site via QuikChange (Agilent Technologies) and inserted into mCherry N1 vectors. The ER‐targeted GFP was encoded by the pShooter vector pCMV/Myc/ER/GFP (Invitrogen). To generate the ER‐mCherry constructs, the signal peptide of SP‐B followed by one alanine residue and a Myc‐tag was fused upstream to mCherry by PCR amplification. A C‐terminal KDEL sequence, as well as a 5′ BamH1 and a 3′ Not1 digestion site, was also added by PCR amplification. The purified PCR product was then inserted into the pcDNA3.1+ plasmid via BamH1/Not1. The ER‐targeted CPY*‐mCherry construct (CPY*‐mCh) was generated by insertion of the insert of CmCh* (Park et al, 2013) into the pShooter vector pCMV/Myc/ER (Invitrogen) via XhoI/NotI. The insert encoding human SEL1L flanked by BamH1 and Not1 digestion sites was purchased from Thermo Fisher Scientific and inserted into the pcDNA3.1+ plasmid via BamH1/Not1.
Cell culture and transfections
Human HEK293T and HeLa cell lines (ATCC) were cultured in Dulbecco's modified Eagle's medium (DMEM; Biochrom) supplemented with 10% FBS (Gibco), 1% penicillin–streptomycin (Gibco), and 1% L‐glutamine (Gibco) at 37°C in an atmosphere of 5% CO2. Cell lines were regularly tested for mycoplasma contamination. Cells were transfected with a total of 1 μg plasmid DNA in 6‐well plates using FuGene 6 transfection reagent (Promega) following the manufacturer's instructions.
Immunofluorescence imaging
Cells were seeded in μ‐Slide chambered coverslips (ibidi; for live cell imaging) or on poly‐L‐lysine‐coated glass coverslips (for immunofluorescence experiments) 24 h prior to transfection. Transfected cells were either directly analyzed in chambered coverslips or fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X‐100, blocked with 1% BSA, and stained with the following primary antibodies diluted 1:500 in 1% BSA: Myc (Santa Cruz #sc‐40), calreticulin (Abcam #ab14234), ERp57 (Abcam #ab10287), giantin (Abcam #ab24586). The following fluorescently labeled secondary antibodies were used at 1:200 dilutions in 1% BSA: anti‐mouse Cy3 (Jackson #115‐165‐062), anti‐rabbit FITC (Invitrogen #F2765), anti‐rabbit Alexa Fluor 405 (Life Technologies #A‐31556), anti‐chicken Alexa Fluor 488 (Life Technologies #A‐11039). For live cell imaging, cells were grown in μ‐Slide 8‐well plates (ibidi) in Dulbecco's modified Eagle's medium (DMEM; Biochrom) without phenol red supplemented with 10% FBS (Gibco), 1% penicillin–streptomycin (Gibco), and 1% L‐glutamine (Gibco). Throughout analysis, cells were kept in a chamber at 37°C, 5% CO2.
Images were recorded at the Imaging Facility of the Max Planck Institute of Biochemistry, Martinsried, on a ZEISS (Jena, Germany) LSM780 confocal laser scanning microscope equipped with a ZEISS Plan‐APO 63×/NA1.46 oil immersion objective and analyzed using the ImageJ software (ImageJ 1.49v or 2.0.0, http://imagej.nih.gov/ij/). Representative images or movies of at least three independent experiments are shown.
Solubility analysis
Cells were lysed in 1% Triton X‐100 in PBS supplemented with cOmplete Protease Inhibitor Cocktail (Roche) and benzonase for 1 h at 4°C. Protein concentrations were analyzed using a BCA assay (Pierce). A fraction of each lysate was put aside for analysis of total protein content, the remainder was centrifuged at 14,000 × g for 10 min at 4°C to separate soluble from insoluble proteins. The supernatant containing soluble protein was transferred to a fresh tube. Both soluble and total fractions were denatured by addition of 5× sample buffer. The pellet was resuspended in 1× sample buffer. All samples were heated to 70°C for 10 min and subsequently analyzed by immunoblotting. The fraction of insoluble protein was calculated by dividing the signal of the insoluble fraction through the sum of insoluble and soluble fraction.
Cell viability assay
Cell viability was analyzed by measuring the capacity of cells to reduce MTT [3‐(4,5‐dimethylthiazol‐2‐yl)‐2, 5‐diphenyltetrazolium bromide] to formazan after transfection with the indicated constructs. Cells were incubated with MTT at 37°C for 1 h before the reaction was stopped by the addition of 40% DMF and 20% SDS. Concentrations of formazan, the product of the reduction in MTT by cellular reductases, were analyzed by measuring absorbance at 570 nm.
Analysis of secreted proteins
Growth medium of HEK293T cells seeded and transfected in a 6‐well plate was exchanged 24 h after transfection for 600 μl full growth medium either with or without 1 μg/ml brefeldin A (BFA). After overnight incubation, media and cells were harvested separately; media samples were centrifuged at 5,000 × g for 5 min and transferred to fresh tubes to ensure elimination of any floating cells. Cell pellets were washed, lysed, quantified, and denatured as described above before addition of 6 μl 2% sodium deoxycholate and incubation at 4°C for 30 min followed by addition of 60 μl trichloric acid (TCA) and incubation at 4°C overnight. Precipitated proteins were collected by centrifugation at 16,000 × g for 14 min at 4°C. Pellets were dried and then resuspended in 1× sample loading buffer. Tris–Cl at pH 8.8 was added to neutralize the pH. Proteins were denatured at 70°C for 10 min. For immunoblotting, 20 μg of protein from cell lysates and equivalent volumes of concentrated protein from media samples were loaded per well. After transfer, PVDF membranes were stained using the MemCodeReversible Protein Stain Kit (Thermo Scientific).
Cycloheximide chase
Transfected cells were trypsinized and reseeded 24 h after transfection and after a recovery period of 24–48 h were treated with 0.5 mM cycloheximide (CHX). Cells were harvested at indicated time points for analysis by immunoblotting. Images were analyzed using the ImageJ software.
RNA isolation and processing
Twenty‐four hours after transfection, RNA was extracted using the RNA Clean & Concentrator kit (Zymo Research, cat. no. R1013) according to the manufacturer's protocol. On‐column DNAse I treatment was carried out to eliminate genomic DNA contamination. RNA‐seq libraries were prepared using the TruSeq Stranded mRNA library prep kit (Illumina cat. no. RS‐122‐2101) and analyzed by paired‐end poly(A) RNA sequencing with an Illumina HiSeq system.
Computational analyses of RNA‐seq data
RNA‐seq reads were trimmed with TrimGalore (v0.4.0) and mapped to GENCODE annotation release 19 with TopHat2 (v2.0.13). After mapping of RNA‐seq reads to the human genome build hg38, unique reads were counted with featureCounts2 (Liao et al, 2014). DESeq2 was used for differential gene expression analysis (Love et al, 2014). Genes with an adjusted P‐value < 0.05 were defined as significantly affected. Gene ontology (GO) analysis, Z‐scores, and P‐values for network enrichment in the RNA‐seq data were provided by Enrichr (Chen et al, 2013).
Data availability
All sequencing data generated in this study have been deposited in the Gene Expression Omnibus (GEO) database under accession number GSE98580.
siRNA knockdowns
HEK293T cells were transfected with 100 nM non‐targeting control siRNA 3 (mock), OS‐9 siRNA smart pool or SEL1L siRNA smart pool, and CPY*‐mCh using DharmafectDuo transfection reagent (Dharmacon) following the manufacturer's instructions. Protein levels were analyzed 48 h after transfection.
Deglycosylation
Transfected HEK293T cells were lysed in RIPA buffer supplemented with protease inhibitors (Roche) and benzonase. After removal of cell debris by centrifugation and quantification using the BCA assay (Pierce, Thermo Scientific), 40 μg protein was denatured and incubated with 2 μl Endo H (NEB) following the manufacturer's instructions.
Author contributions
LV‐D designed and performed the experiments, performed the bioinformatics analysis, and wrote the manuscript. HH designed and performed additional experiments during the revision. RK performed the MS analysis, performed bioinformatics analysis, and contributed to writing the manuscript. ECH and RS performed the RNA‐seq experiments and analyzed the RNA‐seq data. JP generated the ER‐targeted versions of the β‐protein constructs, contributed to the conception and design, and contributed reagents. JJ contributed to the conception and design and contributed reagents. FUH designed experiments, supervised the experimental design, and wrote the manuscript. MSH designed experiments, supervised the experimental design, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Dataset EV1
Dataset EV2
Dataset EV3
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Review Process File
Acknowledgements
We thank Ralf Zenke and Martin Spitaler from the MPIB Imaging Facility for assistance with confocal microscopy. The assistance of Louise Funke and Albert Ries is gratefully acknowledged. We acknowledge Rajat Gupta for generation of the CPY*‐mCh construct, and Daniel Hornburg and Barbara Hummel for their advice concerning the bioinformatics analysis. We are grateful to Courtney Klaips, David Balchin, and Neysan Donnelly for critically reading this manuscript. We thank Konstanze Winklhofer and Julia von Blume for sharing reagents and for fruitful discussions. The research leading to these results has received funding from the Boehringer Ingelheim Fonds (L.V.‐D.), the Max Planck Foundation (H.H.), the European Commission under Grant FP7 GA ERC‐2012‐SyG_318987–ToPAG (R.K., F.U.H. and M.S.H.), and the Deutsche Forschungsgemeinschaft (German Research Foundation) within the framework of the Munich Cluster for Systems Neurology (L.V.‐D., R.K., F.U.H., and M.S.H.). R.S. and E.C.H. acknowledge the financial support by the Max Planck Society, German Research Foundation (DFG) grant SA3190 and DFG collaborative research center “Medical Epigenetics”.
The EMBO Journal (2018) 37: 337–350
Contributor Information
F Ulrich Hartl, Email: uhartl@biochem.mpg.de.
Mark S Hipp, Email: hipp@biochem.mpg.de.
References
- Alberts B, Johnson A, Lewis J, Morgan D, Raff M, Roberts K, Walter P (2014) Molecular biology of the cell. New York, NY: Garland Sciences; [Google Scholar]
- Araki K, Nagata K (2011) Protein folding and quality control in the ER. Cold Spring Harb Perspect Biol 3: a007526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch WE, Morimoto RI, Dillin A, Kelly JW (2008) Adapting proteostasis for disease intervention. Science 319: 916–919 [DOI] [PubMed] [Google Scholar]
- Benyair R, Ron E, Lederkremer GZ (2011) Protein quality control, retention, and degradation at the endoplasmic reticulum. Int Rev Cell Mol Biol 292: 197–280 [DOI] [PubMed] [Google Scholar]
- Bernasconi R, Pertel T, Luban J, Molinari M (2008) A dual task for the Xbp1‐responsive OS‐9 variants in the mammalian endoplasmic reticulum: inhibiting secretion of misfolded protein conformers and enhancing their disposal. J Biol Chem 283: 16446–16454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernasconi R, Galli C, Calanca V, Nakajima T, Molinari M (2010) Stringent requirement for HRD1, SEL1L, and OS‐9/XTP3‐B for disposal of ERAD‐LS substrates. J Cell Biol 188: 223–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhamidipati A, Denic V, Quan EM, Weissman JS (2005) Exploration of the topological requirements of ERAD identifies Yos9p as a lectin sensor of misfolded glycoproteins in the ER lumen. Mol Cell 19: 741–751 [DOI] [PubMed] [Google Scholar]
- Blom N, Sicheritz‐Ponten T, Gupta R, Gammeltoft S, Brunak S (2004) Prediction of post‐translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4: 1633–1649 [DOI] [PubMed] [Google Scholar]
- Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, Julicher F, Hyman AA (2009) Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 324: 1729–1732 [DOI] [PubMed] [Google Scholar]
- Brodsky JL, Skach WR (2011) Protein folding and quality control in the endoplasmic reticulum: recent lessons from yeast and mammalian cell systems. Curr Opin Cell Biol 23: 464–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnik A, Stephens DJ (2009) ER exit sites–localization and control of COPII vesicle formation. FEBS Lett 583: 3796–3803 [DOI] [PubMed] [Google Scholar]
- Buschhorn BA, Kostova Z, Medicherla B, Wolf DH (2004) A genome‐wide screen identifies Yos9p as essential for ER‐associated degradation of glycoproteins. FEBS Lett 577: 422–426 [DOI] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma'ayan A (2013) Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14: 128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F, Dobson CM (2006) Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 75: 333–366 [DOI] [PubMed] [Google Scholar]
- Christianson JC, Shaler TA, Tyler RE, Kopito RR (2008) OS‐9 and GRP94 deliver mutant alpha1‐antitrypsin to the Hrd1‐SEL1L ubiquitin ligase complex for ERAD. Nat Cell Biol 10: 272–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciryam P, Kundra R, Morimoto RI, Dobson CM, Vendruscolo M (2015) Supersaturation is a major driving force for protein aggregation in neurodegenerative diseases. Trends Pharmacol Sci 36: 72–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies MJ, Miranda E, Roussel BD, Kaufman RJ, Marciniak SJ, Lomas DA (2009) Neuroserpin polymers activate NF‐kappaB by a calcium signaling pathway that is independent of the unfolded protein response. J Biol Chem 284: 18202–18209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL, Shrimpton AE, Holohan PD, Bradshaw C, Feiglin D, Collins GH, Sonderegger P, Kinter J, Becker LM, Lacbawan F, Krasnewich D, Muenke M, Lawrence DA, Yerby MS, Shaw CM, Gooptu B, Elliott PR, Finch JT, Carrell RW, Lomas DA (1999) Familial dementia caused by polymerization of mutant neuroserpin. Nature 401: 376–379 [DOI] [PubMed] [Google Scholar]
- Dolfe L, Winblad B, Johansson J, Presto J (2016) BRICHOS binds to a designed amyloid‐forming beta‐protein and reduces proteasomal inhibition and aggresome formation. Biochem J 473: 167–178 [DOI] [PubMed] [Google Scholar]
- Ekeowa UI, Freeke J, Miranda E, Gooptu B, Bush MF, Perez J, Teckman J, Robinson CV, Lomas DA (2010) Defining the mechanism of polymerization in the serpinopathies. Proc Natl Acad Sci USA 107: 17146–17151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eletto D, Eletto D, Dersh D, Gidalevitz T, Argon Y (2014) Protein disulfide isomerase A6 controls the decay of IRE1alpha signaling via disulfide‐dependent association. Mol Cell 53: 562–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger A, Knop M, Wolf DH (1993) Analysis of two mutated vacuolar proteins reveals a degradation pathway in the endoplasmic reticulum or a related compartment of yeast. Eur J Biochem 218: 565–574 [DOI] [PubMed] [Google Scholar]
- Gauss R, Jarosch E, Sommer T, Hirsch C (2006) A complex of Yos9p and the HRD ligase integrates endoplasmic reticulum quality control into the degradation machinery. Nat Cell Biol 8: 849–854 [DOI] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS (2003) Global analysis of protein expression in yeast. Nature 425: 737–741 [DOI] [PubMed] [Google Scholar]
- Hidvegi T, Schmidt BZ, Hale P, Perlmutter DH (2005) Accumulation of mutant alpha1‐antitrypsin Z in the endoplasmic reticulum activates caspases‐4 and ‐12, NFkappaB, and BAP31 but not the unfolded protein response. J Biol Chem 280: 39002–39015 [DOI] [PubMed] [Google Scholar]
- Hosokawa N, Kamiya Y, Kamiya D, Kato K, Nagata K (2009) Human OS‐9, a lectin required for glycoprotein endoplasmic reticulum‐associated degradation, recognizes mannose‐trimmed N‐glycans. J Biol Chem 284: 17061–17068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman AA, Weber CA, Julicher F (2014) Liquid‐liquid phase separation in biology. Annu Rev Cell Dev Biol 30: 39–58 [DOI] [PubMed] [Google Scholar]
- Kikuchi M, Doi E, Tsujimoto I, Horibe T, Tsujimoto Y (2002) Functional analysis of human P5, a protein disulfide isomerase homologue. J Biochem 132: 451–455 [DOI] [PubMed] [Google Scholar]
- Klausner RD, Donaldson JG, Lippincott‐Schwartz J (1992) Brefeldin A: insights into the control of membrane traffic and organelle structure. J Cell Biol 116: 1071–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930 [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Lindquist S (2002) Conversion of PrP to a self‐perpetuating PrPSc‐like conformation in the cytosol. Science 298: 1785–1788 [DOI] [PubMed] [Google Scholar]
- Olzmann JA, Kopito RR, Christianson JC (2013) The mammalian endoplasmic reticulum‐associated degradation system. Cold Spring Harb Perspect Biol 5: a013185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzscha H, Schermann SM, Woerner AC, Pinkert S, Hecht MH, Tartaglia GG, Vendruscolo M, Hayer‐Hartl M, Hartl FU, Vabulas RM (2011) Amyloid‐like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 144: 67–78 [DOI] [PubMed] [Google Scholar]
- Ong SE, Mann M (2006) A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC). Nat Protoc 1: 2650–2660 [DOI] [PubMed] [Google Scholar]
- Park SH, Kukushkin Y, Gupta R, Chen T, Konagai A, Hipp MS, Hayer‐Hartl M, Hartl FU (2013) PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell 154: 134–145 [DOI] [PubMed] [Google Scholar]
- Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, Pozniakovski A, Poser I, Maghelli N, Royer LA, Weigert M, Myers EW, Grill S, Drechsel D, Hyman AA, Alberti S (2015) A liquid‐to‐solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162: 1066–1077 [DOI] [PubMed] [Google Scholar]
- Raina K, Noblin DJ, Serebrenik YV, Adams A, Zhao C, Crews CM (2014) Targeted protein destabilization reveals an estrogen‐mediated ER stress response. Nat Chem Biol 10: 957–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau E, Dehay B, Ben‐Haiem L, Trottier Y, Morange M, Bertolotti A (2004) Targeting expression of expanded polyglutamine proteins to the endoplasmic reticulum or mitochondria prevents their aggregation. Proc Natl Acad Sci USA 101: 9648–9653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipanski A, Oberhauser F, Neumann M, Lange S, Szalay B, Krasemann S, van Leeuwen FW, Galliciotti G, Glatzel M (2014) Lectin OS‐9 delivers mutant neuroserpin to endoplasmic reticulum associated degradation in familial encephalopathy with neuroserpin inclusion bodies. Neurobiol Aging 35: 2394–2403 [DOI] [PubMed] [Google Scholar]
- Schmidt BZ, Perlmutter DH (2005) Grp78, Grp94, and Grp170 interact with alpha1‐antitrypsin mutants that are retained in the endoplasmic reticulum. Am J Physiol Gastrointest Liver Physiol 289: G444–G455 [DOI] [PubMed] [Google Scholar]
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M (2011) Global quantification of mammalian gene expression control. Nature 473: 337–342 [DOI] [PubMed] [Google Scholar]
- Shibata Y, Shemesh T, Prinz WA, Palazzo AF, Kozlov MM, Rapoport TA (2010) Mechanisms determining the morphology of the peripheral ER. Cell 143: 774–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorgjerd K, Ghafouri B, Jonsson BH, Kelly JW, Blond SY, Hammarstrom P (2006) Retention of misfolded mutant transthyretin by the chaperone BiP/GRP78 mitigates amyloidogenesis. J Mol Biol 356: 469–482 [DOI] [PubMed] [Google Scholar]
- Soto C (2003) Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci 4: 49–60 [DOI] [PubMed] [Google Scholar]
- Sun S, Shi G, Han X, Francisco AB, Ji Y, Mendonca N, Liu X, Locasale JW, Simpson KW, Duhamel GE, Kersten S, Yates JR III, Long Q, Qi L (2014) Sel1L is indispensable for mammalian endoplasmic reticulum‐associated degradation, endoplasmic reticulum homeostasis, and survival. Proc Natl Acad Sci USA 111: E582–E591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP (1990) Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)‐ATPase. Proc Natl Acad Sci USA 87: 2466–2470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipping KW, van Oosten‐Hawle P, Hewitt EW, Radford SE (2015) Amyloid fibres: inert end‐stage aggregates or key players in disease? Trends Biochem Sci 40: 719–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno T, Linder S, Na CL, Rice WR, Johansson J, Weaver TE (2004) Processing of pulmonary surfactant protein B by napsin and cathepsin H. J Biol Chem 279: 16178–16184 [DOI] [PubMed] [Google Scholar]
- Valm AM, Cohen S, Legant WR, Melunis J, Hershberg U, Wait E, Cohen AR, Davidson MW, Betzig E, Lippincott‐Schwartz J (2017) Applying systems‐level spectral imaging and analysis to reveal the organelle interactome. Nature 546: 162–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincenz‐Donnelly L, Hipp MS (2017) The endoplasmic reticulum: a hub of protein quality control in health and disease. Free Radic Biol Med 108: 383–393 [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334: 1081–1086 [DOI] [PubMed] [Google Scholar]
- Wei Y, Liu T, Sazinsky SL, Moffet DA, Pelczer I, Hecht MH (2003) Stably folded de novo proteins from a designed combinatorial library. Protein Sci 12: 92–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weibel ER, Staubli W, Gnagi HR, Hess FA (1969) Correlated morphometric and biochemical studies on the liver cell. I. Morphometric model, stereologic methods, and normal morphometric data for rat liver. J Cell Biol 42: 68–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzmann A, Baldes C, Dudek J, Zimmermann R (2007) The heat shock protein 70 molecular chaperone network in the pancreatic endoplasmic reticulum ‐ a quantitative approach. FEBS J 274: 5175–5187 [DOI] [PubMed] [Google Scholar]
- West MW, Wang W, Patterson J, Mancias JD, Beasley JR, Hecht MH (1999) De novo amyloid proteins from designed combinatorial libraries. Proc Natl Acad Sci USA 96: 11211–11216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westrate LM, Lee JE, Prinz WA, Voeltz GK (2015) Form follows function: the importance of endoplasmic reticulum shape. Annu Rev Biochem 84: 791–811 [DOI] [PubMed] [Google Scholar]
- Woerner AC, Frottin F, Hornburg D, Feng LR, Meissner F, Patra M, Tatzelt J, Mann M, Winklhofer KF, Hartl FU, Hipp MS (2016) Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 351: 173–176 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Dataset EV1
Dataset EV2
Dataset EV3
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Review Process File
Data Availability Statement
All sequencing data generated in this study have been deposited in the Gene Expression Omnibus (GEO) database under accession number GSE98580.