

Abstract
The RIG‐I‐like receptors (RLRs) are critical for protection against RNA virus infection, and their activities must be stringently controlled to maintain immune homeostasis. Here, we report that leucine‐rich repeat containing protein 25 (LRRC25) is a key negative regulator of RLR‐mediated type I interferon (IFN) signaling. Upon RNA virus infection, LRRC25 specifically binds to ISG15‐associated RIG‐I to promote interaction between RIG‐I and the autophagic cargo receptor p62 and to mediate RIG‐I degradation via selective autophagy. Depletion of either LRRC25 or ISG15 abrogates RIG‐I‐p62 interaction as well as the autophagic degradation of RIG‐I. Collectively, our findings identify a previously unrecognized role of LRRC25 in type I IFN signaling activation by which LRRC25 acts as a secondary receptor to assist RIG‐I delivery to autophagosomes for degradation in a p62‐dependent manner.
Keywords: ISG15, LRRC25, p62, RIG‐I, selective autophagy
Subject Categories: Autophagy & Cell Death, Immunology
Introduction
The innate immune responses, triggered by pathogen‐associated molecular patterns (PAMPs), are the first line of defense against invading viruses. In virus‐infected cells, viral RNAs can be detected by pattern recognition receptors (PRRs), including Toll‐like receptors (TLRs), RLRs, as well as several sensors of DNA (O'Neill, 2006; Takaoka et al, 2007; Unterholzner et al, 2010; Loo & Gale, 2011; Zhang et al, 2011; Sun et al, 2013). As the major members of RLRs family, RIG‐I and MDA5 (melanoma differentiation‐associated gene 5) play pivotal roles in sensing cytosolic viral RNAs (Loo & Gale, 2011). Both RIG‐I and MDA5 are composed of a conserved DEAD box helicase/ATPase domain, two caspase‐recruiting domains (CARDs), and a C‐terminal regulatory domain (CTD). Upon binding with viral RNAs via their CTD domains, RIG‐I and MDA5 use their CARDs to activate downstream adaptor MAVS (also known as IPS‐1, VISA, or CARDIF) (Kawai et al, 2005; Meylan et al, 2005; Seth et al, 2005; Xu et al, 2005). MAVS then triggers the signal cascades to initiate type I interferons (IFNs) production, as well as the downstream expression of multiple IFN stimulated genes (ISGs) and inflammation cytokines.
Due to its critical role in type I IFN activation, the activity of RIG‐I must be tightly regulated to protect the host from uncontrolled immune response such as autoimmune disease. Several negative regulators of RIG‐I have been reported to inhibit type I IFN signaling through the control of the RIG‐I degradation (Arimoto et al, 2007; Chen et al, 2013; Zhao et al, 2016). RNF125 is the first reported E3 ligase, which conjugates ubiquitin to RIG‐I to mediate the degradation of RIG‐I through proteasome pathway (Arimoto et al, 2007). Siglec‐G also promotes RIG‐I degradation through enhancing K48‐linked ubiquitination of RIG‐I by recruiting E3 ubiquitin ligase c‐Cbl (Chen et al, 2013). Recently, it has been shown that another E3 ligase CHIP associates with RIG‐I and promotes RIG‐I for proteasomal degradation (Zhao et al, 2016). Besides, it has been reported that RIG‐I can also be conjugated by ISG15, which results in the degradation of RIG‐I (Kim et al, 2008). However, the mechanisms underlying degradation of RIG‐I dependent on ISG15 remain to be explored. In particular, RIG‐I in general is degraded through ubiquitination‐mediated proteasome pathway, and it has not been reported that RIG‐I could be degraded through other degradation systems (such as lysosome and autolysosome).
Leucine‐rich repeat (LRR) domain is conserved in a variety of prokaryotic and eukaryotic proteins and displays significant functions in innate immunity (Ng et al, 2011). In mammals, the functions of LRR‐containing proteins, including NOD‐like receptors (NLRs) and TLRs in innate immune responses, are well characterized (Inohara et al, 2005; Takeuchi & Akira, 2010). Besides NLRs and TLRs, the functions of other LRR‐containing proteins in innate immunity remain to be determined. In this study, we identified LRRC25 as the negative regulator of RLR‐mediated type I IFN signaling. Ectopic expression of LRR‐containing protein 25 (LRRC25) inhibited the phosphorylation of endogenous IRF3 and antiviral response following RLR ligand stimulation, whereas knockdown or knockout of LRRC25 had the opposite effects. Upon RNA virus infection, LRRC25 specifically interacts with ISG15‐associated RIG‐I to mediate RIG‐I degradation via p62‐dependent selective autophagy. Depletion of ISG15 abrogates RIG‐I‐p62 interaction as well as the autophagic degradation of RIG‐I. Our findings provide new insights into the tight regulation of type I IFN signaling through its crosstalk with selective autophagy pathway and uncover a negative feedback loop to regulate RIG‐I‐mediated type I IFN signaling.
Results
LRRC25 inhibits RLR‐mediated type I IFN signaling pathway
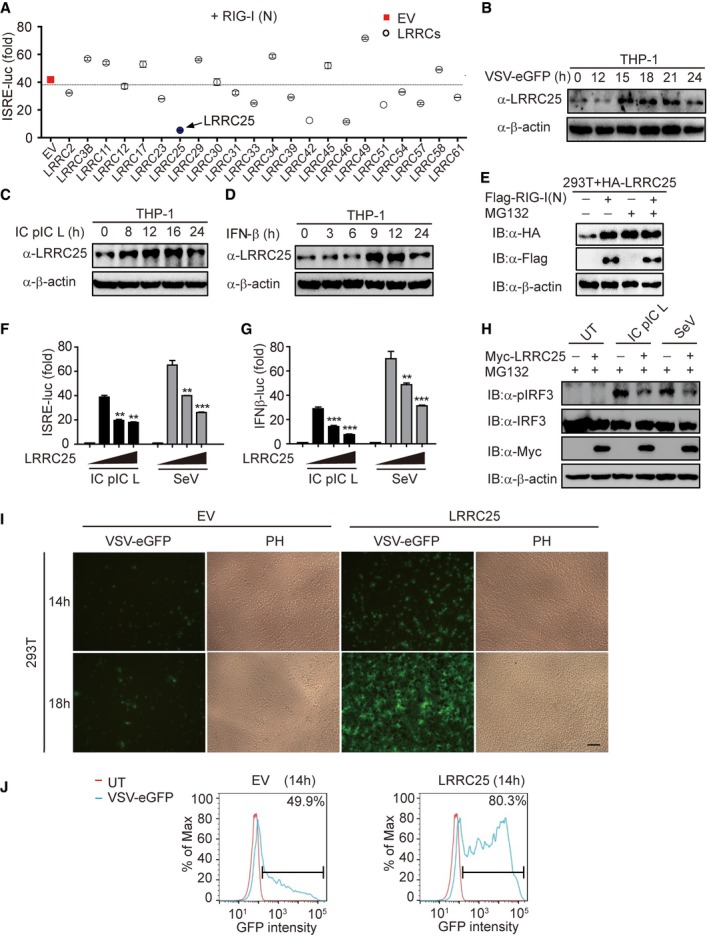
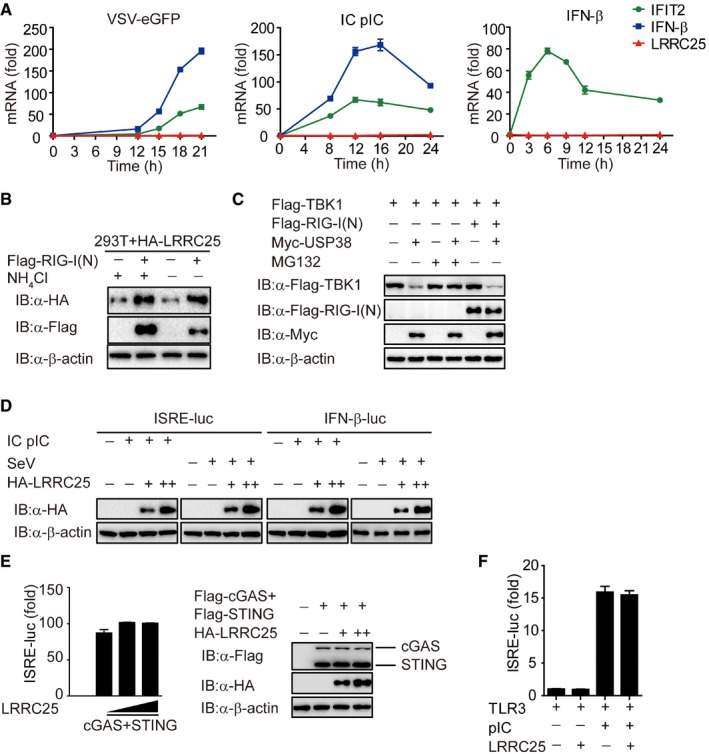
To identify the possible LRRC proteins that engage in type I IFN signaling pathway, we screened 22 candidate genes encoding LRRC proteins and found that LRRC25 significantly inhibited the ISRE activation induced by RIG‐I CARD domains (RIG‐I (N)) (Fig 1A). It has been reported that poly(I:C)‐low molecular weight (LMW) is a specific ligand for RIG‐I, but not for MDA5 (Kato et al, 2008; Takeuchi & Akira, 2010). We then analyzed the expression of LRRC25 in THP‐1 cells upon challenge with vesicular stomatitis virus with enhanced GFP (VSV‐eGFP), intracellular (IC) poly(I:C) (LMW), or IFN‐β and found that LRRC25 protein level was up‐regulated by all these treatments (Fig 1B–D). However, qPCR analysis showed that the mRNA level of LRRC25 was not changed by these treatments (Fig EV1A). These results suggest that LRRC25 protein can be stabilized by the activation of type I IFN signaling. Furthermore, we found that type I IFN signaling stabilized LRRC25 by blocking its proteasome‐dependent degradation, since the proteasome inhibitor MG132, but not the lysosome inhibitor NH4Cl, could stabilize LRRC25 and diminish the difference of LRRC25 protein level with or without RIG‐I (N) overexpression (Figs 1E and EV1B). In addition, we found that ectopic expression of RIG‐I (N) could not block the proteasome degradation of TBK1 mediated by USP38 (Lin et al, 2016), indicating the specific stabilization of LRRC25 mediated by RIG‐I‐type I IFN axis (Fig EV1C). To further test the role of LRRC25 in type I IFN signaling, we showed that LRRC25 inhibited ISRE‐luc and IFN‐β‐luc activities after treatment with IC poly (I:C) LMW or infection with Sendai virus (SeV) (Figs 1F and G, and EV1D). However, LRRC25 had no effect on TLR3‐ or cGAS‐mediated type I IFN activation (Fig EV1E and F), suggesting that LRRC25 specifically inhibits RLR‐induced type I IFN signaling pathway. Similarly, Myc‐LRRC25 significantly decreased the phosphorylation of endogenous IRF3 after treated with IC poly(I:C) LMW or infected with SeV (Fig 1H). To demonstrate a link between attenuated type I IFN response and antiviral immunity caused by LRRC25, we transfected empty vector and LRRC25 into 293T cells and subsequently infected the cells with VSV‐eGFP. LRRC25 rendered the cells more susceptible to viral infection and enhanced the replication of VSV‐eGFP at different time points (Fig 1I and J). Collectively, these data suggest that ectopic expression of LRRC25 markedly inhibits the type I IFN response and antiviral immunity.
Figure 1. LRRC25 inhibits RLR‐mediated type I IFN signaling pathway.
-
AHEK293T cells were transfected with a control plasmid or plasmids expressing 22 LRRCs along with RIG‐I (N) and a reporter plasmid carrying the ISRE promoter (ISRE‐Luc). 24 h after transfection, cells were analyzed for ISRE‐luc activity.
-
B–DTHP‐1 cells were treated with VSV‐eGFP (MOI = 0.1), intracellular (IC) poly(I:C) low molecular weight (5 μg/ml), or IFN‐β (10 ng/ml) for indicated time points. Cell lysates were used for immunoblot analysis with the indicated antibodies.
-
EHEK293T cells were transfected with plasmids for LRRC25, together with an empty vector or RIG‐I (N) for 24 h. Before harvesting, the cells were treated with DMSO or MG132 (5 μM) for 4 h. Cell lysates were used for immunoblot analysis with the indicated antibodies.
-
F, GHEK293T cells were transfected with plasmids for Myc‐LRRC25, plus an ISRE‐luc (F) or an IFN‐β‐luc (G) reporter plasmid. After 12 h, cells were treated with IC poly(I:C) LMW (5 μg/ml) or SeV (MOI = 0.1) for 24 h or 14 h, respectively, and analyzed for ISRE‐luc and IFN‐β‐luc activity.
-
HHEK293T cells were transfected with an empty vector or Myc‐LRRC25. After 12 h, cells were left untreated or treated with IC poly(I:C) LMW (5 μg/ml) or SeV (MOI = 0.1) for 24 h or 14 h, respectively. Before harvesting, the cells were treated with MG132 (5 μM) for 4 h. Protein extracts were analyzed by immunoblot using the indicated antibodies.
-
IHEK293T cells were transfected with an empty vector (EV) or Myc‐LRRC25. 24 h post‐transfection, cells were infected with VSV‐eGFP (MOI = 0.001) for the indicated time points and subjected to phase‐contrast (PH) and fluorescence microscopy analyses. Scale bar, 80 μm.
-
JFlow cytometry analyses of 293T cells in (I). Numbers at the top‐right corner indicate the percentage of cells expressing eGFP. Bar: population of GFP‐positive cells.
Figure EV1. LRRC25 has no effect on the type I IFN signaling pathway mediated by TLR3 or c‐GAS, related to Fig 1 .
- THP‐1 cells were treated with VSV‐eGFP (MOI = 0.1), intracellular (IC) poly(I:C) low molecular weight (5 μg/ml) or IFN‐β (10 ng/ml) for indicated time points. Total RNA was extracted for qPCR analysis for LRRC25, IFN‐β, and IFIT2.
- HEK293T cells were transfected with plasmids for LRRC25, together with an empty vector or RIG‐I (N) for 24 h. Before harvesting, the cells were treated with DMEM or NH4Cl (10 mM) for 6 h. Cell lysates were used for immunoblot analysis with the indicated antibodies.
- HEK293T cells were transfected with Flag‐TBK1, together with an empty vector, Flag‐RIG‐I (N), or Myc‐USP38 for 24 h. Before harvesting, the cells were treated with DMSO or MG132 (10 μM) for 6 h. Cell lysates were harvested and used to perform immunoblot analysis with the indicated antibodies.
- The expression of LRRC25 in Fig 1F and G was analyzed by IB analysis.
- HEK293T cells were transfected with an empty vector (no wedge) or increasing amounts (wedge) of vector for LRRC25, along with vectors for cGAS and STING. 24 h post‐transfection, the cells were analyzed for ISRE activity by a reporter gene assay, and the expressions of cGAS, STING, and LRRC25 were analyzed by IB analysis.
- HEK293T cells were transfected with an empty vector or plasmid for LRRC25, plus plasmids for TLR3 and ISRE‐luc reporter, followed by no treatment or treatment with poly (I:C) (10 μg/ml). After 24 h, cell lysates were analyzed for ISRE‐luc activity.
LRRC25 deficiency enhances antiviral responses
To determine the physiological function of LRRC25 during RNA virus infection, we designed two LRRC25‐specific small interfering RNA (siRNA) to knock down the expression of LRRC25. Both of them could efficiently knock down endogenous LRRC25 (Fig EV2A). To determine the effects of LRRC25 knockdown on ISRE‐luc activity, we showed that knockdown of endogenous LRRC25 increased the ISRE‐luc activity stimulated by IC poly(I:C) (Fig EV2B). Next, we tested the effect of LRRC25 knockdown on the replication of VSV‐eGFP and found that LRRC25 knockdown substantially inhibited viral infection compared to those of cells treated with scrambled siRNA (Fig EV2C and D).
Figure EV2. LRRC25 deficiency enhances antiviral immune responses, related to Fig 2 .
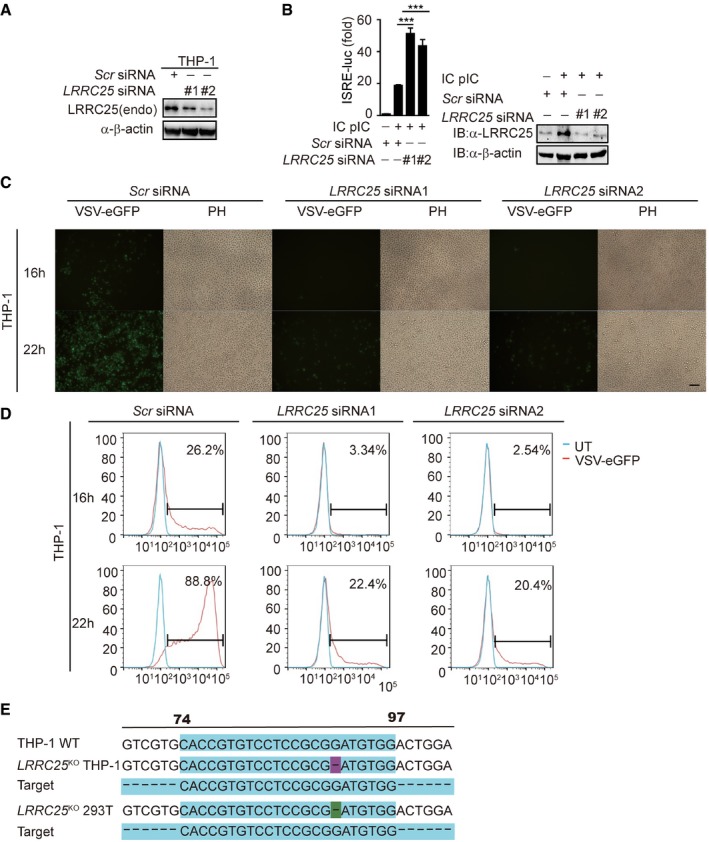
- THP‐1 cells were transfected with control or LRRC25‐specific siRNAs. Cell lysates were harvested and used to perform immunoblot analysis with the indicated antibodies.
- HEK293T cells were transfected with control or LRRC25‐specific siRNAs, together with an ISRE‐luc reporter plasmid. After 24 h, the cells were treated with IC poly(I:C) (5 μg/ml) for 24 h. The cells were analyzed for ISRE activity by a reporter assay, and the expression of LRRC25 was analyzed by IB analysis.
- THP‐1 cells were transfected with control or LRRC25‐specific siRNAs for 24 h, and then, the cells were infected with VSV‐eGFP (MOI = 0.01) for the indicated time points and subjected to phase‐contrast (PH) and fluorescence microscopy analyses. Scale bar, 80 μm.
- Flow cytometry analyses of THP‐1 cells in (C). Numbers at the top‐right corner indicate the percentage of cells expressing eGFP (infected cells).
- LRRC25 KO THP‐1 and LRRC25 KO HEK293T cells were generated by the CRISPR/Cas9 system. The sequences of target sgRNA are as indicated.
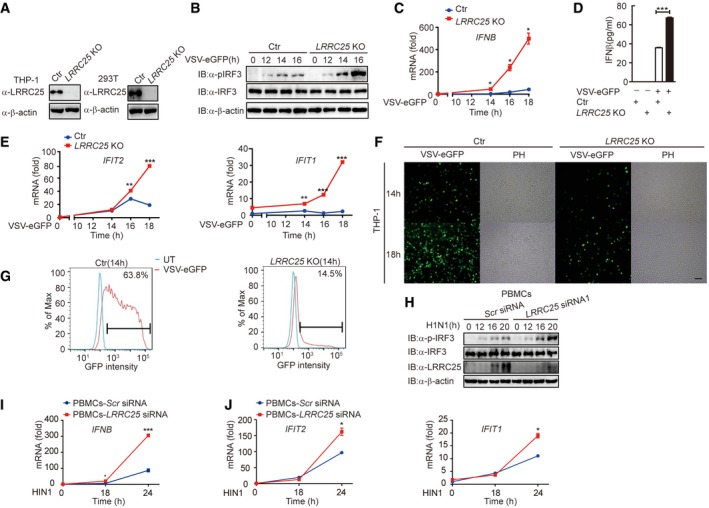
To further substantiate these findings, we used CRISPR/Cas9 system to generate LRRC25 knockout (KO) THP‐1 and 293T cells, respectively. The deletion of LRRC25 was confirmed at the DNA and protein levels (Figs 2A and EV2E). We found that the phosphorylation of IRF3 (p‐IRF3) in LRRC25 KO THP‐1 cells was higher than that in control cells after VSV‐eGFP infection (Fig 2B). We next sought to address whether the enhanced IRF3 phosphorylation by LRRC25 deficiency promotes type I IFN and ISG expressions. Using qPCR analysis, we showed that LRRC25 KO markedly increased mRNA abundance of IFN‐β following VSV infection (Fig 2C). Consistent with these observations, we found that VSV infection resulted in increased production of IFN‐β in LRRC25 KO THP‐1 cells compared to control cells (Fig 2D). Consistently, LRRC25 KO also resulted in higher expression of IFIT1 and IFIT2 after infection with VSV‐eGFP (Fig 2E). To further investigate whether the elevated IFN response is correlated with enhanced antiviral immunity, we infected LRRC25 KO and control THP‐1 cells with VSV‐eGFP. The percentage of GFP+ cells increased with the extended response time, but it was markedly inhibited by LRRC25 deficiency in LRRC25 KO THP‐1 cells (Fig 2F and G). We next isolated human peripheral blood mononuclear cells (PBMCs) and knocked down endogenous LRRC25 to evaluate the physiological importance of LRRC25 during influenza A (H1N1) infection. As expected, we found that knockdown of endogenous LRRC25 increased the phosphorylation of endogenous IRF3 after H1N1 infection in PBMCs (Fig 2H). Furthermore, qPCR analysis showed that the deficiency of LRRC25 highly enhanced the transcription of IFN‐β, IFIT1, and IFIT2 upon H1N1 infection in PBMCs (Fig 2I and J). Taken together, these results suggest that LRRC25 deficiency strongly potentiates the type I IFN activation and antiviral immunity.
Figure 2. LRRC25 deficiency enhances antiviral responses.
-
AProtein extracts of control, LRRC25 KO THP‐1, and LRRC25 KO HEK293T cells were used to perform immunoblot analysis with the indicated antibodies.
-
BControl or LRRC25 KO THP‐1 cells were infected with VSV‐eGFP (MOI = 0.1) for indicated time points. Cell lysates were harvested and analyzed by immunoblot.
-
C, DControl or LRRC25 KO THP‐1 cells were infected with VSV‐eGFP (MOI = 0.01) for indicated time points and subjected to qPCR analysis for IFN‐β (C) or ELISA analysis (D).
-
EControl or LRRC25 KO THP‐1 cells were infected with VSV‐eGFP (MOI = 0.01) for indicated time points and subjected to qPCR analysis for IFIT2 and IFIT1.
-
FControl or LRRC25 KO THP‐1 cells were infected with VSV‐eGFP (MOI = 0.01) for 0–18 h and subjected to phase‐contrast (PH) and fluorescence microscopy analyses. Scale bar, 40 μm.
-
GFlow cytometry analyses of THP‐1 cells in (F). Numbers at the top‐right corner indicate the percentage of cells expressing eGFP.
-
HPBMCs were transfected with control or LRRC25‐specific siRNAs for 24 h, and then, the cells were infected with influenza A/Puerto Rico/8/34 (H1N1) (PR8) (MOI = 5) for the indicated time points. Cell lysates were harvested and used to perform immunoblot analysis with the indicated antibodies.
-
I, JPBMCs were transfected with control or LRRC25‐specific siRNAs for 24 h, and then, the cells were infected with H1N1 (MOI = 5) for the indicated time points and subjected to qPCR analysis for IFNB (I), IFIT2, and IFIT1 (J).
LRRC25 interacts with RIG‐I
Since LRRC25 specifically inhibited RLR‐mediated type I IFN signaling, we next sought to determine the molecular targets for LRRC25. We co‐transfected 293T cells with RIG‐I (N), MAVS, TBK1, IKKi, and IRF3, together with increasing amounts of LRRC25 plus the ISRE luciferase reporter, and found that LRRC25 markedly inhibited activation of ISRE‐luc induced by RIG‐I (N), but had weak or no inhibition of ISRE‐luc reporter activity induced by MAVS, TBK1, IKKi, or IRF3 (Figs 3A and EV3A). Furthermore, we found that LRRC25 markedly inhibited RIG‐I‐mediated ISRE‐luc activation by IC poly(I:C) stimulation (Fig EV3B), suggesting that LRRC25 may block type I IFN signaling through active RIG‐I. In addition, we observed that LRRC25 also inhibited ISRE‐luc activation induced by MDA5, a RLR recognizing high molecular weight RNA fragments (Loo & Gale, 2011) (Fig EV3C).
Figure 3. LRRC25 interacts with RIG‐I.
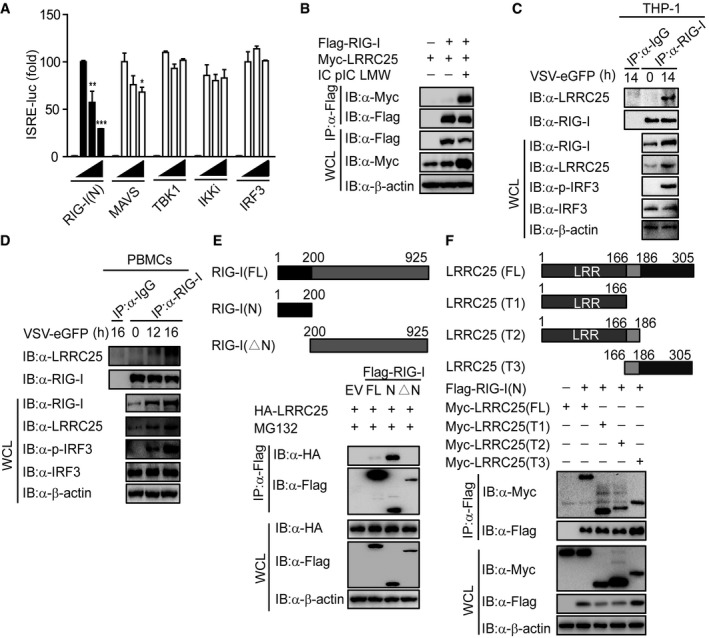
-
AHEK293T cells were transfected with an empty plasmid (no wedge) or increasing amounts (wedge) of plasmid for LRRC25, plus an ISRE‐luc reporter and plasmids for RIG‐I (N), MAVS, TBK1, IKKi or IRF3. 24 h post‐transfection, cell lysates were analyzed for ISRE‐luc activity.
-
BHEK293T cells were transfected with Flag‐RIG‐I and Myc‐LRRC25. After 12 h, cells were left untreated or treated with IC poly(I:C) LMW (5 μg/ml) for 24 h. Cell lysates were immunoprecipitated using anti‐Flag, followed by immunoblots using the indicated antibodies.
-
C, DTHP‐1 cells (C) or PBMCs (D) were infected with VSV‐eGFP (MOI = 0.1) for indicated time points, and cell lysates were immunoprecipitated using anti‐RIG‐I, followed by immunoblots using anti‐LRRC25.
-
EThe structure of RIG‐I and its mutants (top). HEK293T cells were transfected with deletion mutants of RIG‐I, along with HA‐LRRC25 for 24 h. Before harvesting, the cells were treated with MG132 (5 μM) for 4 h. Cell lysates were immunoprecipitated using anti‐Flag, followed by immunoblots using the indicated antibodies (bottom).
-
FThe structure of LRRC25 and its mutants (upper). HEK293T cells were transfected with deletion mutants of LRRC25, along with Flag‐RIG (N). 24 h post‐transfection, cell lysates were harvested and immunoprecipitated using anti‐Flag, followed by immunoblots using the indicated antibodies (lower).
Figure EV3. LRRC25 cannot interact with MAVS, related to Fig 3 .
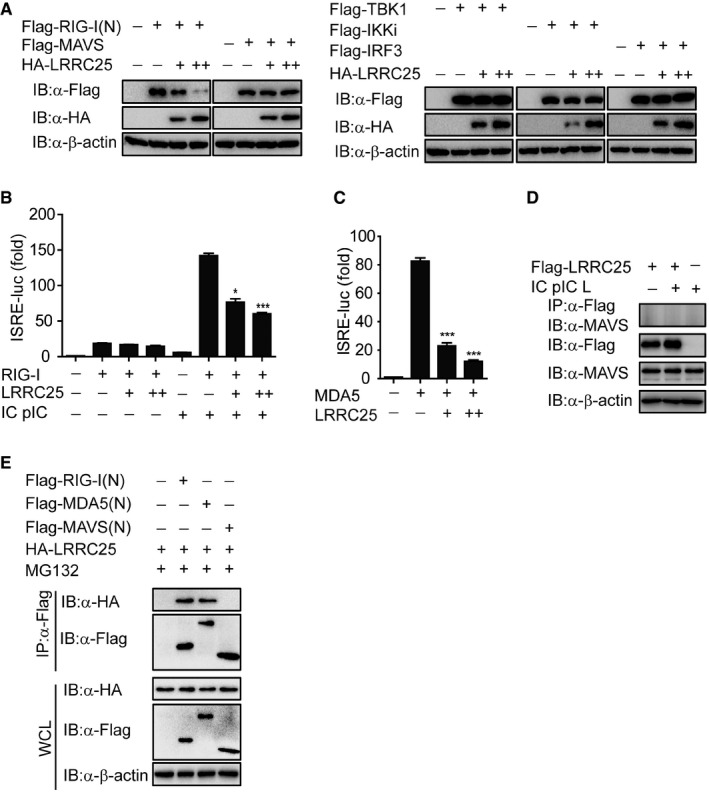
- The expressions of RIG‐I (N), MAVS, TBK1, IKKi, IRF3, and LRRC25 in Fig 3A were analyzed by IB analysis.
- HEK293T cells were transfected with an empty plasmid or increasing amounts of plasmid for LRRC25, plus an ISRE‐luc reporter and plasmids for RIG‐I. After 12 h, cells were left untreated or treated with IC poly(I:C) (5 μg/ml) for 24 h. Cell lysates were analyzed for ISRE‐luc activity.
- HEK293T cells were transfected with an empty plasmid (no wedge) or increasing amounts (wedge) of plasmid for LRRC25, plus an ISRE‐luc reporter and plasmid for MDA5. 24 h post‐transfection, cell lysates were analyzed for ISRE‐luc activity.
- HEK293T cells were transfected with Flag‐LRRC25. After 12 h, the cells were left untreated or treated with IC poly(I:C) LMW (5 μg/ml) for 24 h. Cell lysates were immunoprecipitated using anti‐Flag, followed by immunoblots using the indicated antibodies.
- HEK293T cells were transfected with Flag‐RIG‐I (N), Flag‐MDA5 (N), Flag‐MAVS (N), and HA‐LRRC25 for 24 h. Before harvesting, the cells were treated with DMSO or MG132 (5 μM) for 4 h. Cell lysates were immunoprecipitated using anti‐Flag, followed by immunoblot using the indicated antibodies.
Co‐immunoprecipitation (co‐IP) and immunoblot analyses further showed that LRRC25 interacted with RIG‐I after IC poly(I:C) treatment (Fig 3B). To examine the physiological relevance of these findings, we infected THP‐1 cells with VSV‐eGFP and found that LRRC25 strongly associated with RIG‐I after viral infection (Fig 3C). By contrast, LRRC25 did not associate with MAVS by IC poly(I:C) treatment (Fig EV3D). To further assess whether LRRC25 interacts with RIG‐I in primary cells, we isolated PBMCs and then challenged them with VSV‐eGFP and found that RIG‐I strongly interacted with LRRC25 after VSV‐eGFP infection in PBMCs (Fig 3D). Collectively, these data suggest that LRRC25 interacts with RIG‐I after viral infection. To distinguish which domain of RIG‐I was involved in the interaction with LRRC25, we constructed full‐length (FL) RIG‐I, truncated RIG‐I (N) and RIG‐I lacking CARDs (RIG‐I (ΔN)), and analyzed their ability to interact with LRRC25. We found that LRRC25 strongly interacted with RIG‐I (N), but not with RIG‐I (ΔN) (Fig 3E). In addition, we evaluated the interaction between LRRC25 and the CARDs of MDA5 and MAVS. We found that LRRC25 could interact with CARDs of RIG‐I and MDA5, but not with CARD domain of MAVS (Fig EV3E), suggesting that the interaction between LRRC25 and CARDs of RLRs is specific. In addition, using truncated LRRC25 fragments, we found that all of the LRRC25 domains can interact with RIG‐I (N) (Fig 3F). Taken together, these results suggest that LRRC25 strongly interacts with the CARD domain of RLRs.
LRRC25 targets RIG‐I for autophagic degradation
We next sought to determine how LRRC25 inhibits type I IFN signaling through its interaction with RIG‐I. When we transfected 293T cells with plasmids encoding RIG‐I (N) and LRRC25, we found that the protein levels of RIG‐I (N) were reduced with increasing LRRC25 protein expression (Fig 4A). However, LRRC25 had no effects on the mRNA level of RIG‐1 (N) (Fig 4B), indicating that LRRC25 destabilized RIG‐I (N) at the protein level. Consistently, we found that the concentration of RIG‐I protein reduced considerably with LRRC25 expression only after IC poly(I:C) stimulation, even in the presence of MG132 (Figs EV4A and 4C). Furthermore, domain deletions of LRRC25 containing LRR could attenuate the activation of ISRE induced by RIG‐I (N), as well as the abundance of RIG‐I (N) (Figs 4D and E, and EV4B). In addition, we found that MDA5 protein levels decreased considerably with increased LRRC25 expression (Fig EV4C). By contrast, LRRC25 had no effects on the protein level of MAVS (Fig EV4D). Collectively, these data indicate that LRRC25 promotes the degradation of RIG‐I and MDA5 after viral infection.
Figure 4. LRRC25 targets RIG‐I for autophagic degradation.
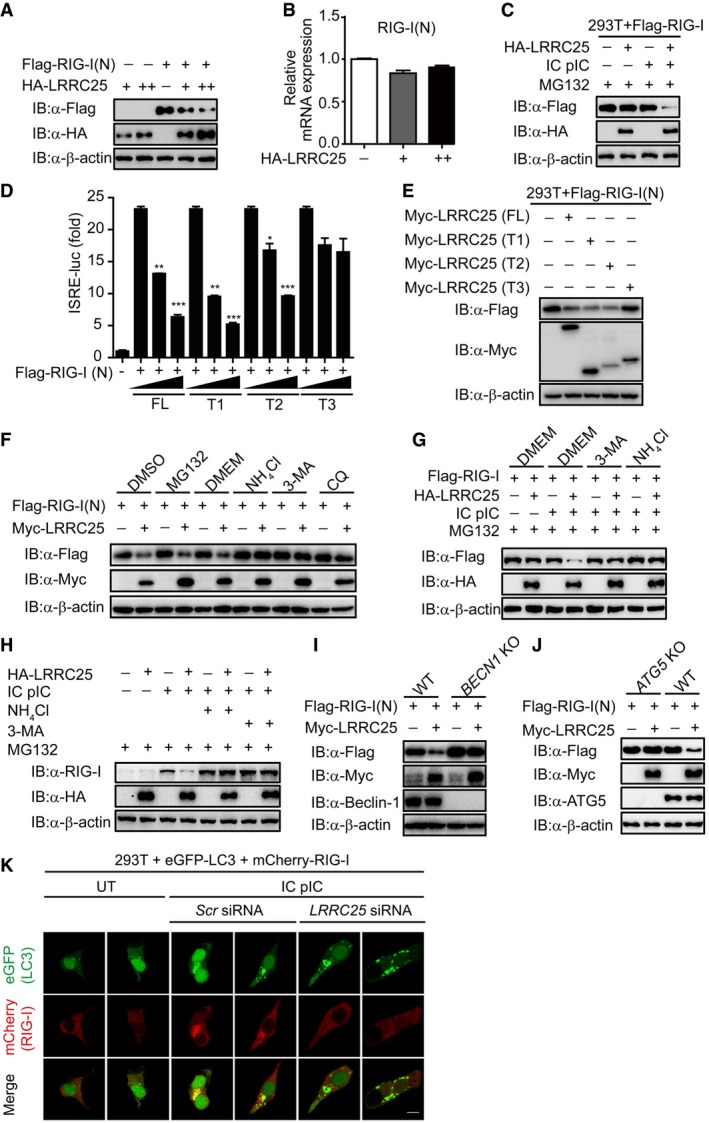
-
A, BHEK293T cells were transfected with Flag‐RIG‐I (N), together with an empty vector or HA‐LRRC25 (75 and 200 ng). 24 h post‐transfection, cells were harvested and used to perform immunoblot analysis with the indicated antibodies (A). Total RNA in was extracted for qPCR analysis for RIG‐I (N) (B).
-
CHEK293T cells were transfected with Flag‐RIG‐I together with an empty vector or HA‐LRRC25. After 12 h, the cells were left untreated or treated with IC poly(I:C) (5 μg/ml) for 24 h. Before harvesting, the cells were treated with MG132 (5 μM) for 4 h. Cell lysates were used for immunoblot analysis with the indicated antibodies.
-
DHEK293T cells were transfected with an empty vector (no wedge) or increasing amounts (wedge) of deletion mutants of LRRC25, along with RIG‐I (N) and ISRE‐luc. 24 h post‐transfection, cell lysates were analyzed for ISRE‐luc activity.
-
EHEK293T cells were transfected with Flag‐RIG‐I (N), together with an empty vector or deletion mutants of LRRC25. 24 h post‐transfection, cells were harvested and used to perform immunoblot analysis with the indicated antibodies.
-
FHEK293T cells were transfected with plasmids for Flag‐RIG‐I (N), together with an empty plasmid or Myc‐LRRC25. 12 h post‐transfection, the cells were treated with DMSO, MG132 (5 μM), DMEM, NH4Cl (10 mM), 3MA (2.5 mM), or CQ (20 μM) for 4 h, 4 h, 6 h, 6 h, 4 h, or 4 h, respectively. Cell lysates were used for immunoblot analysis with the indicated antibodies.
-
GHEK293T cells were transfected with Flag‐RIG‐I together with an empty vector or HA‐LRRC25. After 12 h, the cells were left untreated or treated with IC poly(I:C) (5 μg/ml) for 24 h. Before harvesting, the cells were treated with MG132 (5 μM) for 4 h, together with DMEM, 3MA (2.5 mM), NH4Cl (10 mM) for 6 h, 4 h, or 6 h, respectively. Cell lysates were used for immunoblot analysis with the indicated antibodies.
-
HHEK293T cells were transfected with empty vector or HA‐LRRC25. After 12 h, the cells were left untreated or treated with IC poly(I:C) (5 μg/ml) for 24 h. Before harvesting, the cells were treated with MG132 (5 μM) for 4 h, together with DMEM, 3MA (2.5 mM), NH4Cl (10 mM) for 6 h, 4 h, or 6 h respectively. Cell lysates were used for immunoblot analysis with the indicated antibodies.
-
I, JWild‐type (WT), BECN KO (I), or ATG5 KO (J) HEK293T cells were transfected with Flag‐RIG‐I (N), together with an empty vector or Myc‐LRRC25. Twenty‐four hours post‐transfection, cells lysates were harvested and analyzed by immunoblot.
-
KHEK293T cells were transfected with control or LRRC25‐specific siRNAs. 24 h post‐transfection, the cells were co‐transfected with mCherry‐RIG‐I and eGFP‐LC3, followed with IC poly(I:C) (5 μg/ml) treatment for 24 h. The cells were then subjected to confocal microscopy analysis. Scale bar, 10 μm.
Figure EV4. LRRC25 promotes the degradation of RIG‐I (N) and MDA5 but not MAVS, related to Figs 4 and 5 .
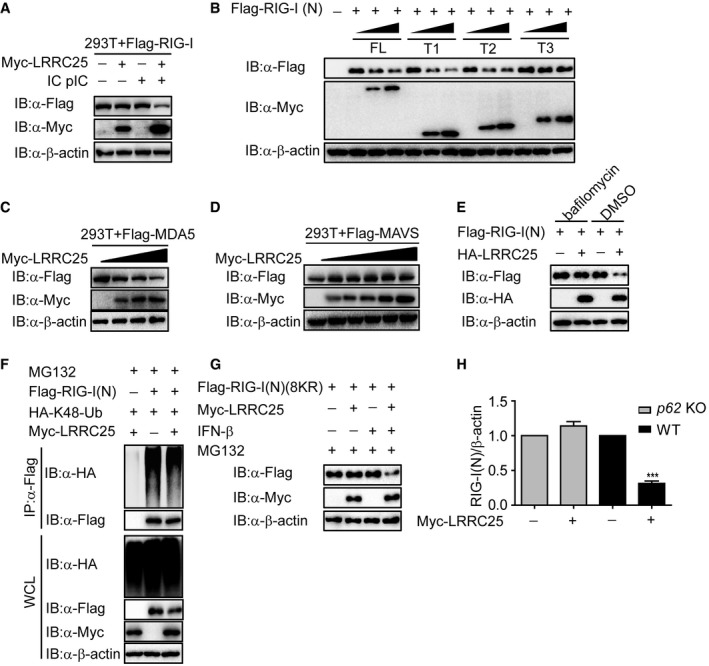
- HEK293T cells were transfected with Flag‐RIG‐I together with an empty vector or Myc‐LRRC25. After 12 h, the cells were left untreated or treated with IC poly(I:C) (5 μg/ml) for 24 h. Cell lysates were used for immunoblot analysis with the indicated antibodies.
- The expressions of RIG‐I (N) and deletion mutants of LRRC25 in Fig 4D were analyzed by IB analysis.
- HEK293T cells were transfected with Flag‐MDA5, together with an empty plasmid (no wedge) or increasing amounts (wedge) of plasmid for LRRC25. 24 h post‐transfection, cells were harvested and used to perform immunoblot analysis with the indicated antibodies.
- HEK293T cells were transfected with Flag‐MAVS together with an empty vector or Myc‐LRRC25. 24 h post‐transfection, cells were harvested and used to perform immunoblot analysis with the indicated antibodies.
- HEK293T cells were transfected with plasmids for Flag‐RIG‐I (N), together with an empty plasmid or HA‐LRRC25. 18 h post‐transfection, the cells were treated with DMSO or bafilomycin A1 (0.2 μM) for 6 h. Cell lysates were used for immunoblot analysis with the indicated antibodies.
- HEK293T cells were transfected with plasmids for HA‐K48‐Ub and Flag‐RIG‐I (N), together with an empty vector or Myc‐LRRC25. 24 h post‐transfection, the cells were treated with MG132 (5 μM) for 4 h. Cell lysates were immunoprecipitated using anti‐Flag, followed by immunoblots using the indicated antibodies.
- HEK293T cells were transfected with plasmids for Flag‐RIG‐I (N)(8KR) together with an empty vector, or Myc‐LRRC25 for 24 h. Before harvesting, the cells were treated with IFN‐β (10 ng/ml) and MG132 (5 μM) for 8 and 4 h, respectively. Cell lysates were harvested and used to perform immunoblot analysis with the indicated antibodies.
- Intensity analysis of the bands from the three independent experiments with the same setting in Fig 5E.
To determine how LRRC25 promotes the degradation of RIG‐I after stimulation, we found that LRRC25‐mediated degradation of RIG‐I (N) was completely inhibited by NH4Cl, 3‐methyladenine (3MA), chloroquine (CQ), and bafilomycin A1, which are inhibitors for lysosomal and autophagic degradation, respectively, but not MG132, which is the inhibitor for the proteasomal degradation pathway (Figs 4F and EV4E). In paralleled experiments, ectopically expressed LRRC25 could promote the degradation of FL RIG‐I after IC poly(I:C) stimulation, and such degradation could be rescued by NH4Cl and 3MA (Fig 4G). Consistently, NH4Cl and 3MA could block the degradation of endogenous RIG‐I promoted by LRRC25 upon viral infection (Fig 4H). In addition, LRRC25 did not enhance K48‐linked ubiquitination of RIG‐I (N) (Fig EV4F). To further validate these results, we examined the protein level of RIG‐I (N) in BECN1 and ATG5 knockout 293T cells and found that blockade of the autophagy process could impair the degradation of RIG‐I (N) promoted by LRRC25 (Fig 4I and J). LC3 is a well‐characterized marker of the autophagy process (Kuma et al, 2007). We next analyzed the effect of LRRC25 deficiency on RIG‐I‐LC3 co‐localization. As expected, we observed the puncta of RIG‐I‐LC3 upon IC poly(I:C) stimulation, and it was markedly inhibited by LRRC25 deficiency (Fig 4K). Taken together, these results suggest that LRRC25 targeted RIG‐I for autophagic degradation.
LRRC25 bridges RIG‐I to p62 for autophagic degradation
It is well documented that the removal of protein aggregates through autophagy is a highly selective process, which depends on cargo recognition by the cargo receptors (Stolz et al, 2014). To determine which cargo receptor was required in the degradation of RIG‐I through autophagosome pathway, we examined the interactions between RIG‐I and Tollip, NBR1, OPTN, p62, or Nix, respectively. We found that RIG‐I specifically interacted with p62, but not with other receptors after IC poly(I:C) stimulation (Fig 5A). It is well known that ubiquitination is a targeting signal on cargoes for the recognition by the ubiquitin‐associated (UBA) domain of p62. To test whether the ubiquitination of RIG‐I is required for the association between RIG‐I and p62, we performed co‐IP analysis and showed that RIG‐I could still interact with p62 ΔUBA mutant (Fig 5B), suggesting that the interaction between RIG‐I and p62 does not depend on the ubiquitination of RIG‐I. Furthermore, we observed that LRRC25 still reduced protein amount of RIG‐I (N) (Lys 48, 99, 154, 164, 169, 172, 181, 190 arginine) (8KR), a RIG‐I inactive mutant lacking all the known ubiquitination sites on RIG‐I (N) in the presence of IFN‐β (Fig EV4G). Together these data indicated that ubiquitination of RIG‐I is not required for its degradation mediated by p62. We reasoned that LRRC25 could act as a bridge for the interaction between RIG‐I and p62. As expected, the association between LRRC25 and p62, but not other receptors, was readily detected (Fig 5C). We next determined whether LRRC25 could enhance the interaction between RIG‐I and p62 after VSV stimulation. To test this possibility, we analyzed the association between RIG‐I and p62 in LRRC25 KO THP‐1 cells and found that the interaction between RIG‐I and p62 upon infection was abrogated in the absence of LRRC25 (Fig 5D). Furthermore, deletion of p62 in HEK293T cells completely impaired the degradation of RIG‐I (N) mediated by LRRC25 (Figs 5E and EV4H). These results suggest that LRRC25 bridges RIG‐I to p62 for autophagic degradation.
Figure 5. LRRC25 bridges RIG‐I to p62 for autophagic degradation.
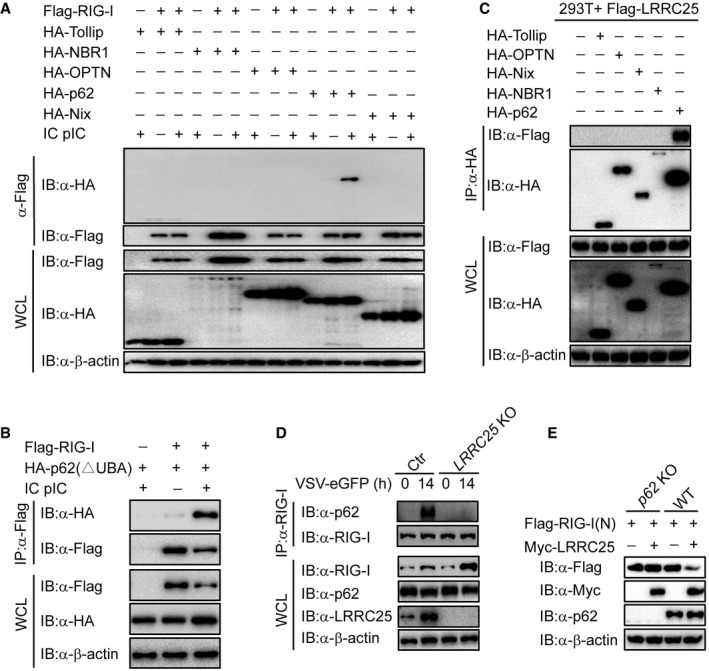
- HEK293T cells were transfected with Flag‐RIG‐I together with HA‐Tollip, HA‐NBR1, HA‐OPTN, HA‐p62, or HA‐Nix. After 12 h, the cells were left untreated or treated with IC poly(I:C) (5 μg/ml) for 24 h. Cell lysates were immunoprecipitated using anti‐Flag, followed by immunoblotting using the indicated antibodies.
- HEK293T cells were transfected with Flag‐RIG‐I and HA‐p62 (ΔUBA). After 12 h, the cells were left untreated or treated with IC poly(I:C) (5 μg/ml) for 24 h. Cell lysates were immunoprecipitated using anti‐Flag, followed by immunoblots using the indicated antibodies.
- HEK293T cells were transfected with Flag‐LRRC25, together with expression vector of HA‐Tollip, HA‐OPTN, HA‐Nix, HA‐NBR1, or HA‐p62. 24 h post‐transfection, cells lysates were immunoprecipitated using anti‐Flag, followed by immunoblot using the indicated antibodies.
- Control and LRRC25 KO THP‐1 cells were infected with VSV‐eGFP (MOI = 0.1) for 14 h. Cell lysates were immunoprecipitated using anti‐RIG‐I, followed by immunoblots using anti‐p62.
- WT and p62 KO HEK293T cells were transfected with plasmids for Flag‐RIG‐I (N), together with an empty vector or Myc‐LRRC25. 24 h post‐transfection, cell lysates were harvested and used to perform immunoblot analysis with the indicated antibodies.
ISG15 serves as an essential signal for LRRC25‐mediated RIG‐I degradation
Our results suggest that LRRC25 mediates the degradation of RIG‐I in a p62‐dependent manner, once RIG‐I is activated by viral infection. We next sought to determine whether the activation of type I IFN signaling or the exposure of RIG‐I CARD domain serves as an essential signal for autophagic degradation of RIG‐I. As MAVS is the key adaptor of RIG‐I to activate downstream TBK1 and IRF3 and induces the expression of multiple ISGs, we used MAVS KO HEK293T cells to block the activation of type I IFN signaling and re‐activated type I IFN signaling by introducing ectopic TBK1 expression to test our hypothesis (Fig 6A). As compared to wild‐type 293T cells, we observed no degradation of RIG‐I (N) and FL RIG‐I in MAVS KO 293T cells, indicating that simply exposure of RIG‐I CARD domain is not enough to initiate LRRC25‐mediated RIG‐I degradation (Fig 6B and C). Interestingly, the ability of LRRC25 to degrade RIG‐I (N) was restored when we introduced ectopic TBK1 to re‐activate type I IFN signaling in MAVS KO cells (Fig 6D). It is reported that TBK1 can also promote autophagosomal engulfment by phosphorylating p62 (Matsumoto et al, 2015). To exclude the possibility that TBK1 acts directly as a kinase on p62, we treated the cells with IFN‐β to activate downstream ISG genes and analyzed the effect of LRRC25 on the degradation of RIG‐I (N) and endogenous RIG‐I. We found that LRRC25 could still attenuate the abundance of RIG‐I (N) and endogenous RIG‐I in the presence of IFN‐β in MAVS KO cells (Fig 6E and F), which further validates that the activation of type I IFN signaling is an essential signal for autophagic degradation of RIG‐I. It has been reported that ISG15, an IFN‐inducible protein, negatively regulates RIG‐I level (Kim et al, 2008). However, its molecular mechanism is yet to be understood. We reasoned that ISG15 might contribute to the degradation of RIG‐I mediated by LRRC25. As expected, we observed that LRRC25 could destabilize RIG‐I (N) in the presence of ISG15 in MAVS KO HEK293T cells (Fig 6G). To further confirm these findings, we constructed ISG15 KO COS7 cells and THP‐1 cells using the CRISPR/Cas9 system (Fig EV5A and B). We found that the degradation of RIG‐I (N) and endogenous RIG‐I mediated by LRRC25 was abrogated in the absence of ISG15 (Fig 6H and I). In addition, we observed that endogenous RIG‐I was markedly reduced after treatment of cycloheximide (CHX), when ISG15 was ectopically expressed in control cells compared to that in LRRC25 knockout cells (Fig EV5C). These results suggest that LRRC25 degrades RIG‐I in an ISG15‐dependent manner.
Figure 6. ISG15 serves as an essential signal for LRRC25‐mediated RIG‐I degradation.
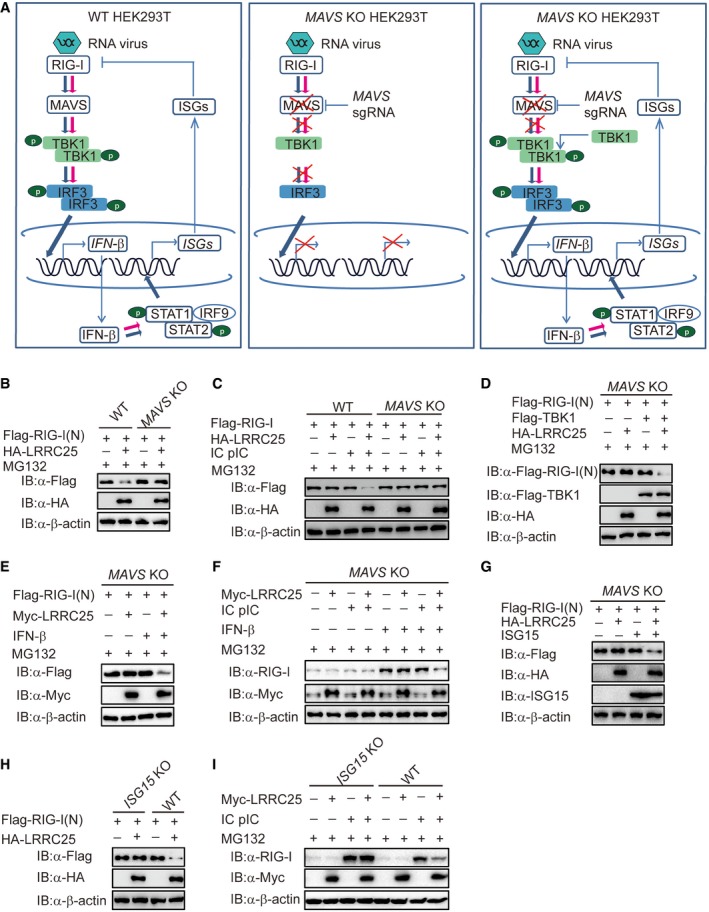
- The experimental strategy to investigate whether LRRC25 needs downstream signal to degrade RIG‐I.
- Wild‐type (WT) and MAVS KO HEK293T cells were transfected with plasmids for Flag‐RIG‐I (N), together with an empty vector or HA‐LRRC25 for 24 h. Before harvesting, the cells were treated with MG132 (5 μM) for 4 h. Cell lysates were harvested and used to perform immunoblot analysis with the indicated antibodies.
- WT and MAVS KO HEK293T cells were transfected with Flag‐RIG‐I, together with an empty vector or HA‐LRRC25. After 12 h, the cells were left untreated or treated with IC poly(I:C) (5 μg/ml) for 24 h. Before harvesting, the cells were treated with MG132 (5 μM) for 4 h. Cell lysates were used for immunoblot analysis with the indicated antibodies.
- MAVS KO HEK293T cells were transfected with Flag‐RIG‐I (N) together with an empty vector, Flag‐TBK1, or HA‐LRRC25 for 24 h. Before harvesting, the cells were treated with MG132 (5 μM) for 4 h. Cell lysates were harvested and used to perform immunoblot analysis with the indicated antibodies.
- MAVS KO HEK293T cells were transfected with Flag‐RIG‐I (N) together with an empty vector or Myc‐LRRC25 for 24 h. Before harvesting, the cells were treated with IFN‐β (10 ng/ml) and MG132 (5 μM) for 8 and 4 h, respectively. Cell lysates were harvested and used to perform immunoblot analysis with the indicated antibodies.
- MAVS KO HEK293T cells were transfected with an empty vector or Myc‐LRRC25. After 12 h, the cells were left untreated or treated with IC poly(I:C) (5 μg/ml) for 24 h. Before harvesting, the cells were treated with IFN‐β (10 ng/ml) and MG132 (5 μM) for 16 and 4 h, respectively. Cell lysates were used for immunoblot analysis with the indicated antibodies.
- MAVS KO HEK293T cells were transfected with Flag‐RIG‐I (N), together with an empty vector, ISG15, or HA‐LRRC25. 24 h post‐transfection, cell lysates were harvested and used to perform immunoblot analysis with the indicated antibodies.
- WT and ISG15 KO COS7 cells were transfected with Flag‐RIG‐I (N), together with an empty vector or HA‐LRRC25. 24 h post‐transfection, cell lysates were harvested and analyzed by immunoblot.
- WT and ISG15 KO COS7 cells were transfected with an empty vector or Myc‐LRRC25. After 12 h, the cells were left untreated or treated with IC poly(I:C) (5 μg/ml) for 24 h. Before harvesting, the cells were treated with MG132 (5 μM) for 4 h. Cell lysates were used for immunoblot analysis with the indicated antibodies.
Figure EV5. RIG‐I (N) and LRRC25 interact with ISG15 in an unconjugated manner, related to Figs 6 and 7 .
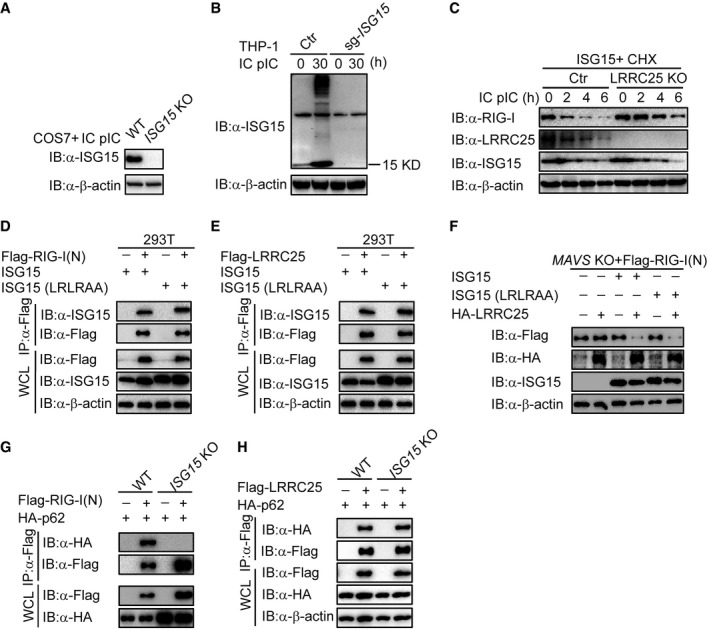
-
A, BControl, ISG15 KO COS7, and KO THP‐1 cells were left untreated or treated with IC poly(I:C) (5 μg/ml) for 24 or 30 h, respectively. Cell lysates were harvested and used to perform immunoblot analysis with the indicated antibodies.
-
CControl or LRRC25 KO HEK293T cells were transfected with ISG15. After 12 h, the cells were pre‐treated with CHX (100 μg/ml) for 2 h, followed by IC poly(I:C) (5 μg/ml) for indicated time points. Cell lysates were used for immunoblot analysis with the indicated antibodies.
-
D, EHEK293T cells were transfected with ISG15 or ISG15 (LRLRAA), together with Flag‐RIG‐I (N) (D) or Flag‐LRRC25 (E). 24 h post‐transfection, cell lysates were immunoprecipitated using anti‐Flag, followed by immunoblot using the indicated antibodies.
-
FMAVS KO HEK293T cells were transfected with Flag‐RIG‐I (N), together with an empty vector, ISG15, ISG15 (LRLRAA), or HA‐LRRC25. 24 h post‐transfection, cell lysates were harvested and used to perform immunoblot analysis with the indicated antibodies.
-
GWT and ISG15 KO COS7 cells were transfected with Flag‐RIG‐I (N) and HA‐p62 for 24 h. Cell lysates were immunoprecipitated using anti‐Flag, followed by immunoblots using the indicated antibodies.
-
HWT and ISG15 KO COS7 cells were transfected with Flag‐LRRC25 and HA‐p62 for 24 h. Cell lysates were immunoprecipitated using anti‐Flag, followed by immunoblots with the indicated antibodies.
The interaction between RIG‐I and LRRC25 is dependent on ISG15
Although previous reports showed that RIG‐I undergoes ISGylation upon viral infection (Kim et al, 2008), we observed that both RIG‐I (N) and LRRC25 could interact with ISG15 in an unconjugated manner in HEK293T cells (Fig 7A and B). Consistently, conjugation‐defect mutation (LRLRAA) of ISG15 still interacted with RIG‐I (N) (Fig EV5D) and with LRRC25 (Fig EV5E). Both wild‐type ISG15 and mutant ISG15 (LRLRAA) could mediate the degradation of RIG‐I (N) by LRRC25 in MAVS KO cells (Fig EV5F), suggesting that non‐covalent ISG15 promotes the degradation of RIG‐I mediated by LRRC25.
Figure 7. The interaction between RIG‐I and LRRC25 is dependent on ISG15.
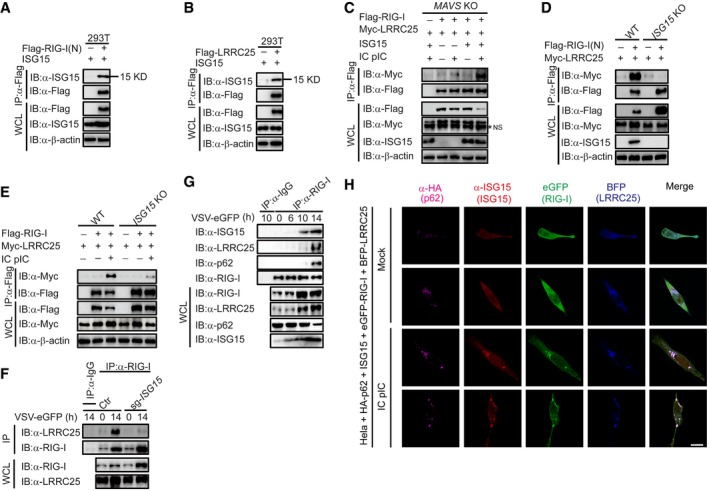
-
A, BHEK293T cells were transfected with ISG15, together with Flag‐RIG‐I (N) (A) or Flag‐LRRC25 (B). 24 h post‐transfection, cell lysates were immunoprecipitated using anti‐Flag, followed by immunoblot using the indicated antibodies.
-
CMAVS KO HEK293T cells were transfected with Flag‐RIG‐I and Myc‐LRRC25, together with an empty vector or ISG15. After 12 h, the cells were left untreated or treated with IC poly(I:C) (5 μg/ml) for 24 h. Cell lysates were immunoprecipitated using anti‐Flag, followed by immunoblots using the indicated antibodies. NS indicates non‐specific bands.
-
DWT and ISG15 KO COS7 cells were transfected with Flag‐RIG‐I (N) and Myc‐LRRC25. 24 h post‐transfection, cell lysates were immunoprecipitated using anti‐Flag, followed by immunoblot using the indicated antibodies.
-
EWT and ISG15 KO COS7 cells were transfected with Flag‐RIG‐I and Myc‐LRRC25. After 12 h, the cells were left untreated or treated with IC poly(I:C) (5 μg/ml) for 24 h. Cell lysates were immunoprecipitated using anti‐Flag, followed by immunoblots using the indicated antibodies.
-
FControl and ISG15 KO THP‐1 cells were infected with VSV‐eGFP (MOI = 0.1) for 14 h. Cell lysates were immunoprecipitated using anti‐RIG‐I, followed by immunoblots using anti‐LRRC25.
-
GTHP‐1 cells were infected with VSV‐eGFP (MOI = 0.2) for indicated time points, and cell lysates were immunoprecipitated using anti‐RIG‐I, followed by immunoblots using indicated antibodies.
-
HConfocal microscopic analysis of HeLa cells co‐transfected with HA‐p62, ISG15, eGFP‐RIG‐I, and BFP‐LRRC25, followed IC poly(I:C) (5 μg/ml) treatment for 24 h. Scale bar, 20 μm.
Based on these data, we hypothesized that the unconjugated interaction between ISG15 and RIG‐I as well as LRRC25 might affect the interaction between RIG‐I and LRRC25. As expected, co‐IP results showed that the impaired interaction between RIG‐I and LRRC25 in MAVS KO cells could be restored in the presence of ISG15 after IC poly(I:C) treatment (Fig 7C). However, the interactions between LRRC25 and RIG‐I or its CARDs domain were severely impaired in ISG15 KO cells (Fig 7D and E). Furthermore, the endogenous interaction between RIG‐I and LRRC25 was markedly inhibited in ISG15 KO THP‐1 cells after VSV infection (Fig 7F). Notably, the interaction between RIG‐I (N) and p62 was completely abolished in the absence of ISG15 (Fig EV5G). However, the deficiency of ISG15 had no effects on the association between LRRC25 and p62 (Fig EV5H). To further confirm the acting sequence of ISG15 after viral infection, we performed immunoprecipitation experiments to evaluate the endogenous interaction between RIG‐I, ISG15, LRRC25, and p62 at different time points during the viral infection. Consistent with our previous data, the immunoprecipitation results showed that after viral infection, RIG‐I firstly interacted with ISG15, and then, the ISG15‐associated RIG‐I interacted with LRRC25 and p62 (Fig 7G). In addition, confocal microscopy also showed that RIG‐I, ISG15, LRRC25, and p62 formed puncta upon IC poly(I:C) stimulation (Fig 7H). Taken together, these findings provide strong evidence that ISG15 plays a critical role in the RIG‐I‐LRRC25 interaction and subsequent autophagic degradation of RIG‐I through p62.
Discussion
In this study, we have shown that LRRC25 inhibits RIG‐I‐mediated type I IFN signaling through a p62‐dependent autophagy pathway. LRRC25 functions as a bridge for the interaction between RIG‐I and p62. Furthermore, we show that IFN‐inducible free ISG15 is required for LRRC25 to interact with RIG‐I. Our findings provide a previously unrecognized mechanism by which LRRC25 mediates inhibition of type I IFN signaling by targeting RIG‐I for degradation via a selective autophagy pathway. Because RLR‐mediated type I IFN signaling plays predominant roles in antiviral immunity, it is critically important to tightly control the activity of RIG‐I for avoiding excessive harmful immune response. Several studies have reported that the activity and stability of RIG‐I are regulated by multiple protein modifications. Proteasomal degradation of RIG‐I could be initiated through K48‐linked ubiquitination by several E3 ligases, including RNF125, CHIP, and Siglec‐G/c‐Cbl (Arimoto et al, 2007; Chen et al, 2013; Zhao et al, 2016). In addition, the phosphorylation and deamidation also play important roles in the regulation of RIG‐I activity. For instance, CKII and PKCα/β negatively regulate the activation of RIG‐I through phosphorylating the CTD and CARDs domain of RIG‐I, respectively, while PFAS could positively regulate RIG‐I via deamidation (Sun et al, 2011; Maharaj et al, 2012; He et al, 2015). Other and our own studies also show that several NLRs, including NLRC5 and NLRX1, could inhibit type I IFN signaling through physically blocking the interaction between RIG‐I and MAVS (Cui et al, 2010; Allen et al, 2011; Xia et al, 2011). However, whether RIG‐I stability is regulated by selective autophagy is not well investigated so far.
Autophagy is an evolutionarily conserved degradation pathway with diverse biological functions in immune response, including intracellular pathogen sensing, antigen presentation, lymphocyte development, and T‐cell selection (Starr et al, 2003; Dengjel et al, 2005; Lee et al, 2007; Shin et al, 2010; Arsov et al, 2011). Many autophagic proteins are involved in the regulation of type I IFN signaling. ATG5‐ATG12 has been reported to interfere the interaction between RIG‐I and MAVS to down‐regulate type I IFN signaling (Jounai et al, 2007). Beclin‐1 associates with cGAS and restricts cGAS enzymatic activity and subsequently attenuates cGAS‐induced antiviral immune response (Liang et al, 2014). More recently, we found that Beclin‐1 can also inhibit RLR‐mediated type I IFN signaling by blocking the interaction between RIG‐I and MAVS (Jin et al, 2016). p62 has been shown to recognize the poly‐ubiquitinated proteins and recruit them to autophagosome through its UBA domain as a cargo receptor for selective degradation (Pankiv et al, 2007). However, we found that p62 ΔUBA (lacking ubiquitin‐binding domain) still interacts with RIG‐I for autophagy degradation, suggesting that the interaction between RIG‐I and p62 is independent on poly‐ubiquitin signal. In addition, we showed that RIG‐I‐N (8KR), the RIG‐I mutant lacking all the known ubiquitination sites, could still be degraded by LRRC25 through the autophagy pathway. Further experiments show that LRRC25 acts as a secondary receptor to promote p62‐mediated autophagic degradation of RIG‐I in an ubiquitin signal‐independent manner.
To understand the molecular mechanisms by which LRRC25 promotes p62‐mediated autophagic degradation of RIG‐I, we show that ISG15 is critically required for the p62‐mediated degradation of RIG‐I. ISG15, an ubiquitin‐like protein, is highly induced by type I IFN signaling and exhibits antiviral activities (Skaug & Chen, 2010; Morales & Lenschow, 2013). Increasing evidence indicates that ISG15 mainly exerts its function by either conjugating to the lysine of its targeted proteins or free form (unconjugated ISG15) to regulate type I IFN signaling (Zhao et al, 2005; Tang et al, 2010; Morales & Lenschow, 2013). However, the functions of ISG15 in mice and humans are different. In ISG15 KO mice, antiviral immune response is reduced or mildly affected (Lenschow et al, 2007; Morales & Lenschow, 2013), but ISG15 deficiency in human increases viral resistance and type I IFN production (Zhang et al, 2015; Speer et al, 2016). It appears that human ISG15 functions as a key negative regulator to inhibit type I IFN signaling and IFN‐α/β production via USP18 stabilization (Zhang et al, 2015). Although ISG15 has been suggested to negatively regulate RIG‐I‐mediated antiviral signaling in a conjugated ISGylation‐dependent manner (Kim et al, 2008), its precise mechanisms, particularly free ISG15‐mediated mechanisms, are still not clear. Recent studies demonstrate that free ISG15 might play a role in antiviral responses. Free ISG15 has been reported to prevent the proteasome‐dependent degradation of USP18 to block JAK STAT signaling pathway (Zhang et al, 2015; Speer et al, 2016; Hermann & Bogunovic, 2017). However, the function of free ISG15 in autophagic degradation has not been reported. Our results presented in this study provide evidence that free ISG15 interacts with RIG‐I and acts as an essential recognition signal for LRRC25/p62‐mediated autophagic degradation of RIG‐I to dampen RIG‐I‐mediated signaling. We show that LRRC25 deficiency blocks RIG‐I degradation after viral infection; similarly, ISG15 KO impairs the interaction of LRRC25 with RIG‐I, thus reducing RIG‐I degradation. These results suggest that LRRC25 recognizes RIG‐I through free ISG15 after activation of RIG‐I signaling and targets it for degradation through p62‐mediated autophagy pathway. Thus, our results provide macular insights into the regulatory mechanisms of type I IFN signaling through the crosstalk with selective autophagy.
Based on these experimental data, we proposed a working model to illustrate how LRRC25 negatively regulates RLR‐mediated type I IFN signaling through facilitating the RIG‐I‐p62 interaction (Fig 8). Upon RNA virus infection, RIG‐I triggers the activation of type I IFN signaling and up‐regulates a variety of downstream ISGs, including ISG15. ISG15 then interacts with the CARD domains of RIG‐I and enhances the interaction between LRRC25 and RIG‐I. As a key secondary receptor, LRRC25 next bridges ISG15‐associated RIG‐I to cargo receptor p62 and assists RIG‐I delivery to autophagosomes for degradation. Thus, the RIG‐I/ISG15/LRRC25 axis forms a negative feedback loop to maintain the balance of type I IFN activation.
Figure 8.
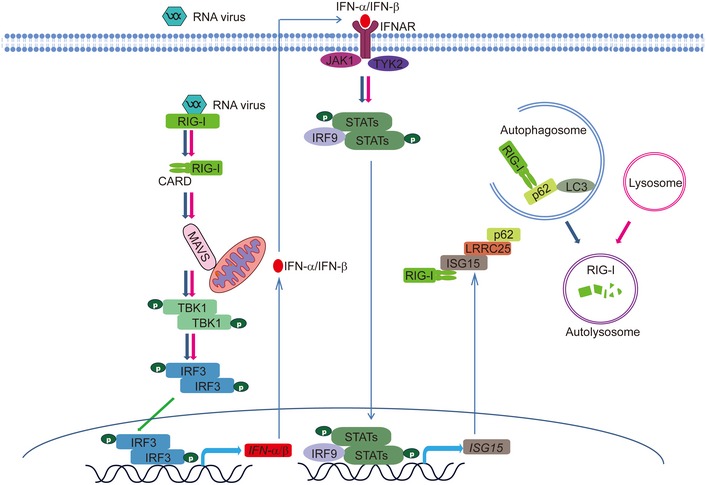
A proposed working model to illustrate how the ISG15‐LRRC25‐p62 axis negatively regulates type I IFN signaling.
In summary, our results shed new light on the function of ISG15 and LRRC25 in the regulation of RIG‐I mediated type I IFN signaling. More importantly, we have provided compelling evidence that innate immune signaling and selective autophagy are fully integrated. Furthermore, considering the pathological role of RIG‐I in many diseases, our findings have identified a novel therapeutic target for developing antiviral and cancer therapies.
Materials and Methods
Cell culture and antibodies
HEK293T (human embryonic kidney 293T) and COS7 cells were cultured in DMEM supplemented with 10% (vol/vol) FBS. Human THP‐1 cells and PBMCs were grown in RPMI 1640/glutamine medium (Gibco) containing 10% (vol/vol) fetal bovine serum (Gibco). The antibodies used in this study were purchased from indicated companies: Anti‐c‐Myc‐HRP (11814150001) and HRP‐anti‐hemagglutinin (12013819001) were purchased from Roche Applied Science; anti‐β‐actin (A1978) and horseradish peroxidase (HRP)‐anti‐Flag (M2) (A8592) were purchased from Sigma; anti‐IRF3 (sc‐9082), anti‐MAVS (E‐3) (sc‐166583), and anti‐GFP (sc‐8334) were purchased from Santa Cruz Biotechnology; anti‐RIG‐I (#3743), anti‐p‐IRF3 (#4947S), anti‐Beclin‐1 (#3738), anti‐ATG5 (#12994S), anti‐ISG15 (#2743), anti‐mouse IgG‐HRP (#7076), and anti‐rabbit IgG‐HRP (#7074) were purchased from Cell Signaling Technology; anti‐LRRC25 (#AB84954) was purchased from Abcam; and anti‐p62 (#18420‐1‐AP) was purchased from proteintech.
Transfection and reporter assays
HEK293T cells were plated in 96‐well plates and transfected, using Lipofectamine 2000 (Invitrogen), with plasmids encoding an ISRE or IFN‐β luciferase reporter (firefly luciferase; 25 ng) and pRL‐TK (renilla luciferase plasmid; 3 ng) together with 25 ng expression vector of Flag‐RIG‐I (N), Flag‐cGAS, Flag‐STING, Flag‐MAVS, Flag‐TBK1, Flag‐IRF3, or Flag‐MDA5, and increasing concentrations (0, 50, or 100 ng) of plasmid expressing LRRC25 or domain deletions of LRRC25. Empty pcDNA3.1 vector was used to maintain equal amounts of DNA among wells. Cells were collected at 24–36 h after transfection, and luciferase activity was measured with a Dual‐Luciferase Assay (Promega) with a Luminoskan Ascent luminometer (Thermo Scientific) according to the manufacturer's protocol. Reporter gene activity was determined by normalization of the firefly luciferase activity to renilla luciferase activity. Because poly(I:C)‐low molecular weight (LMW) is a specific ligand for RIG‐I but not for MDA5 (Kato et al, 2008; Takeuchi & Akira, 2010), we used poly(I:C)‐LMW as a RIG‐I ligand in this study (Invivogen (Catalog # tlrl‐picwlv).
Virus infection
VSV‐eGFP and Sendai virus were kindly provided by Dr. Xiaofeng Qin (Sun Yat‐sen University). Human influenza virus A/Puerto Rico/8/34 (H1N1) (PR8) was kindly provided by Dr. Hui Zhang (Zhongshan Medical School, Sun Yat‐sen University). Cells were infected at various MOI, as previously described by Cui et al (2012) and Shapira et al (2009).
siRNA transfection
siRNA targeting human LRRC25 was chemically synthesized from TranSheepBio and transfected using Lipofectamine® RNAiMAX (Invitrogen) according to the manufacturer's protocols. Oligonucleotide sequences are as follows:
LRRC25 siRNA #1: Sense: 5′‐GCACCAGUGGGAUGAACAA‐3′
Anti‐sense: 5′‐UUGUUCAUCCCACUGGUGC‐3′
LRRC25 siRNA #2: Sense: 5′‐CUCCCGACUAUGAGAACAU‐3′
Anti‐sense: 5′‐AUGUUCUCAUAGUCGGGAG‐3′
Real‐time PCR
Total RNA was isolated using TRIzol reagent (Invitrogen) and subjected to reverse transcription using PrimeScriptTM RT Master Mix (TAKARA). All gene transcripts were quantified by real‐time PCR with SYBR green qPCR Mix kit (Genstar). The following primers were used for real‐time PCR:
IFNβ: Forward 5′‐CCTACAAAGAAGCAGCAA‐3′
Reverse 5′‐TCCTCAGGGATGTCAAAG‐3′
IFIT2: Forward 5′‐GGAGGGAGAAAACTCCTTGGA‐3′
Reverse 5′‐GGCCAGTAGGTTGCACATTGT‐3′
IFIT1: Forward 5′‐TCAGGTCAAGGATAGTCTGGAG‐3′
Reverse 5′‐AGGTTGTGTATTCCCACACTGTA‐3′
GAPDH: Forward 5′‐TCAAGAAGGTGGTGAAGCAG‐3′
Reverse 5′‐GAGGGGAGATTCAGTGTGGT‐3′
LRRC25: Forward 5′‐TGCTACGCAACCCCTTGTC‐3′
Reverse 5′‐GGCCAGAGCAGTTGTCTCG‐3′
RIG‐I (N): Forward 5′‐CTGCAAGCCTTCCAGGATTATAT‐3′
Reverse 5′‐TCTGATCTGAGAAGGCATTCCA‐3′
ELISA
Control or LRRC25 KO THP‐1 cells were infected with VSV‐eGFP (MOI: 0.01) for 36 h. Cell culture supernatants were harvested and analyzed for IFN‐β production using a human IFN‐β ELISA kit (Cloud‐Clone Corp) according to the manufacturer's protocols.
Immunoprecipitation and immunoblot analysis
For immunoprecipitation, whole‐cell extracts were prepared after transfection or stimulation with appropriate ligands, followed by incubation overnight with the appropriate antibodies plus protein A/G beads (Pierce) or anti‐Flag agarose gels (Sigma) were used. Beads were then washed five times with low‐salt lysis buffer, and immunoprecipitates were eluted with 3× SDS loading buffer and resolved by SDS–PAGE. Proteins were transferred to PVDF or NC membranes (Bio‐Rad) and further incubated with the appropriate antibodies. Immobilon Western chemiluminescent HRP substrate (Millipore) was used for protein detection.
Generation of LRRC25 KO cells by the CRISPR/Cas9 technology
LRRC25 KO THP‐1 cells were generated by the CRISPR/Cas9 system, and the sequences of target sgRNA are as follows:
LRRC25‐sgRNA:
Forward 5′‐CACCGTGTCCTCCGCGGATGTGG‐3′
Reverse 5′‐CCACATCCGCGGAGGACACGGTG‐3′
Author contributions
YD, R‐FW, and JC designed the study; YD, TD, YF, QL and ML performed the experiments and analyzed the results. YD, JC, and R‐FW prepared the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Acknowledgements
This work was supported by National Natural Science Foundation of China (91629101, 31522018), Guangdong Natural Science Funds for Distinguished Young Scholar (S2013050014772), Guangdong Innovative Research Team Program (NO. 2011Y035 and 201001Y0104687244), Guangzhou Science and Technology Project (201605030012), and the Training Program for Outstanding Young Teachers in Higher Education institutions of Guangdong Province (YQ2015001). The National Basic Research Program of China (973 Program) (2014CB745203), R.‐F.W, was in part supported by grants (CA101795 and DA030338) from NCI and NIDA, NIH.
The EMBO Journal (2018) 37: 351–366
Contributor Information
Jun Cui, Email: cuij5@mail.sysu.edu.cn.
Rong‐Fu Wang, Email: rwang3@houstonmethodist.org.
References
- Allen IC, Moore CB, Schneider M, Lei Y, Davis BK, Scull MA, Gris D, Roney KE, Zimmermann AG, Bowzard JB, Ranjan P, Monroe KM, Pickles RJ, Sambhara S, Ting JP (2011) NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG‐I‐MAVS and TRAF6‐NF‐kappaB signaling pathways. Immunity 34: 854–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimoto K, Takahashi H, Hishiki T, Konishi H, Fujita T, Shimotohno K (2007) Negative regulation of the RIG‐I signaling by the ubiquitin ligase RNF125. Proc Natl Acad Sci USA 104: 7500–7505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsov I, Adebayo A, Kucerova‐Levisohn M, Haye J, MacNeil M, Papavasiliou FN, Yue Z, Ortiz BD (2011) A role for autophagic protein beclin 1 early in lymphocyte development. J Immunol 186: 2201–2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Han C, Xie B, Hu X, Yu Q, Shi L, Wang Q, Li D, Wang J, Zheng P, Liu Y, Cao X (2013) Induction of Siglec‐G by RNA viruses inhibits the innate immune response by promoting RIG‐I degradation. Cell 152: 467–478 [DOI] [PubMed] [Google Scholar]
- Cui J, Zhu L, Xia X, Wang HY, Legras X, Hong J, Ji J, Shen P, Zheng S, Chen ZJ, Wang RF (2010) NLRC5 negatively regulates the NF‐kappaB and type I interferon signaling pathways. Cell 141: 483–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Li Y, Zhu L, Liu D, Songyang Z, Wang HY, Wang RF (2012) NLRP4 negatively regulates type I interferon signaling by targeting the kinase TBK1 for degradation via the ubiquitin ligase DTX4. Nat Immunol 13: 387–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dengjel J, Schoor O, Fischer R, Reich M, Kraus M, Muller M, Kreymborg K, Altenberend F, Brandenburg J, Kalbacher H, Brock R, Driessen C, Rammensee HG, Stevanovic S (2005) Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci USA 102: 7922–7927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Zhao J, Song S, He X, Minassian A, Zhou Y, Zhang J, Brulois K, Wang Y, Cabo J, Zandi E, Liang C, Jung JU, Zhang X, Feng P (2015) Viral pseudo‐enzymes activate RIG‐I via deamidation to evade cytokine production. Mol Cell 58: 134–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann M, Bogunovic D (2017) ISG15. In sickness and in health. Trends Immunol 38: 79–93 [DOI] [PubMed] [Google Scholar]
- Inohara N, Chamaillard M, McDonald C, Nunez G (2005) NOD‐LRR proteins: role in host‐microbial interactions and inflammatory disease. Annu Rev Biochem 74: 355–383 [DOI] [PubMed] [Google Scholar]
- Jin S, Tian S, Chen Y, Zhang C, Xie W, Xia X, Cui J, Wang RF (2016) USP19 modulates autophagy and antiviral immune responses by deubiquitinating Beclin‐1. EMBO J 35: 866–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jounai N, Takeshita F, Kobiyama K, Sawano A, Miyawaki A, Xin KQ, Ishii KJ, Kawai T, Akira S, Suzuki K, Okuda K (2007) The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci USA 104: 14050–14055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Mikamo‐Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S (2008) Length‐dependent recognition of double‐stranded ribonucleic acids by retinoic acid–inducible gene‐I and melanoma differentiation–associated gene 5. J Exp Med 205: 1601–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S (2005) IPS‐1, an adaptor triggering RIG‐I‐ and Mda5‐mediated type I interferon induction. Nat Immunol 6: 981–988 [DOI] [PubMed] [Google Scholar]
- Kim MJ, Hwang SY, Imaizumi T, Yoo JY (2008) Negative feedback regulation of RIG‐I‐mediated antiviral signaling by interferon‐induced ISG15 conjugation. J Virol 82: 1474–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A, Matsui M, Mizushima N (2007) LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy 3: 323–328 [DOI] [PubMed] [Google Scholar]
- Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A (2007) Autophagy‐dependent viral recognition by plasmacytoid dendritic cells. Science 315: 1398–1401 [DOI] [PubMed] [Google Scholar]
- Lenschow DJ, Lai C, Frias‐Staheli N, Giannakopoulos NV, Lutz A, Wolff T, Osiak A, Levine B, Schmidt RE, Garcia‐Sastre A, Leib DA, Pekosz A, Knobeloch KP, Horak I, Virgin HW IV (2007) IFN‐stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc Natl Acad Sci USA 104: 1371–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q, Seo GJ, Choi YJ, Kwak MJ, Ge J, Rodgers MA, Shi M, Leslie BJ, Hopfner KP, Ha T, Oh BH, Jung JU (2014) Crosstalk between the cGAS DNA sensor and Beclin‐1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe 15: 228–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M, Zhao Z, Yang Z, Meng Q, Tan P, Xie W, Qin Y, Wang RF, Cui J (2016) USP38 inhibits type I interferon signaling by editing TBK1 ubiquitination through NLRP4 signalosome. Mol Cell 64: 267–281 [DOI] [PubMed] [Google Scholar]
- Loo YM, Gale M Jr (2011) Immune signaling by RIG‐I‐like receptors. Immunity 34: 680–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharaj NP, Wies E, Stoll A, Gack MU (2012) Conventional protein kinase C‐α (PKC‐α) and PKC‐β negatively regulate RIG‐I antiviral signal transduction. J Virol 86: 1358–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto G, Shimogori T, Hattori N, Nukina N (2015) TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum Mol Genet 24: 4429–4442 [DOI] [PubMed] [Google Scholar]
- Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J (2005) Cardif is an adaptor protein in the RIG‐I antiviral pathway and is targeted by hepatitis C virus. Nature 437: 1167–1172 [DOI] [PubMed] [Google Scholar]
- Morales DJ, Lenschow DJ (2013) The antiviral activities of ISG15. J Mol Biol 425: 4995–5008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng AC, Eisenberg JM, Heath RJ, Huett A, Robinson CM, Nau GJ, Xavier RJ (2011) Human leucine‐rich repeat proteins: a genome‐wide bioinformatic categorization and functional analysis in innate immunity. Proc Natl Acad Sci USA 108(Suppl 1): 4631–4638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill LAJ (2006) How Toll‐like receptors signal: what we know and what we don't know. Curr Opin Immunol 18: 3–9 [DOI] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282: 24131–24145 [DOI] [PubMed] [Google Scholar]
- Seth RB, Sun L, Ea CK, Chen ZJ (2005) Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF‐kappaB and IRF 3. Cell 122: 669–682 [DOI] [PubMed] [Google Scholar]
- Shapira SD, Gat‐Viks I, Shum BO, Dricot A, de Grace MM, Wu L, Gupta PB, Hao T, Silver SJ, Root DE, Hill DE, Regev A, Hacohen N (2009) A physical and regulatory map of host‐influenza interactions reveals pathways in H1N1 infection. Cell 139: 1255–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DM, Jeon BY, Lee HM, Jin HS, Yuk JM, Song CH, Lee SH, Lee ZW, Cho SN, Kim JM, Friedman RL, Jo EK (2010) Mycobacterium tuberculosis eis regulates autophagy, inflammation, and cell death through redox‐dependent signaling. PLoS Pathog 6: e1001230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaug B, Chen ZJ (2010) Emerging role of ISG15 in antiviral immunity. Cell 143: 187–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speer SD, Li Z, Buta S, Payelle‐Brogard B, Qian L, Vigant F, Rubino E, Gardner TJ, Wedeking T, Hermann M, Duehr J, Sanal O, Tezcan I, Mansouri N, Tabarsi P, Mansouri D, Francois‐Newton V, Daussy CF, Rodriguez MR, Lenschow DJ et al (2016) ISG15 deficiency and increased viral resistance in humans but not mice. Nat Commun 7: 11496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr TK, Jameson SC, Hogquist KA (2003) Positive and negative selection of T cells. Annu Rev Immunol 21: 139–176 [DOI] [PubMed] [Google Scholar]
- Stolz A, Ernst A, Dikic I (2014) Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 16: 495–501 [DOI] [PubMed] [Google Scholar]
- Sun Z, Ren H, Liu Y, Teeling JL, Gu J (2011) Phosphorylation of RIG‐I by casein kinase II inhibits its antiviral response. J Virol 85: 1036–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ (2013) Cyclic GMP‐AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339: 786–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, Ohba Y, Taniguchi T (2007) DAI (DLM‐1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448: 501–505 [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140: 805–820 [DOI] [PubMed] [Google Scholar]
- Tang Y, Zhong G, Zhu L, Liu X, Shan Y, Feng H, Bu Z, Chen H, Wang C (2010) Herc5 attenuates influenza A virus by catalyzing ISGylation of viral NS1 protein. J Immunol 184: 5777–5790 [DOI] [PubMed] [Google Scholar]
- Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, Fitzgerald KA, Paludan SR, Bowie AG (2010) IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol 11: 997–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X, Cui J, Wang HY, Zhu L, Matsueda S, Wang Q, Yang X, Hong J, Songyang Z, Chen Z, Wang R‐F (2011) NLRX1 negatively regulates TLR‐induced NF‐κB signaling by targeting TRAF6 and IKK. Immunity 34: 843–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB (2005) VISA is an adapter protein required for virus‐triggered IFN‐beta signaling. Mol Cell 19: 727–740 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu YJ (2011) The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol 12: 959–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Bogunovic D, Payelle‐Brogard B, Francois‐Newton V, Speer SD, Yuan C, Volpi S, Li Z, Sanal O, Mansouri D, Tezcan I, Rice GI, Chen C, Mansouri N, Mahdaviani SA, Itan Y, Boisson B, Okada S, Zeng L, Wang X et al (2015) Human intracellular ISG15 prevents interferon‐alpha/beta over‐amplification and auto‐inflammation. Nature 517: 89–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Denison C, Huibregtse JM, Gygi S, Krug RM (2005) Human ISG15 conjugation targets both IFN‐induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc Natl Acad Sci USA 102: 10200–10205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Zhang Q, Li X, Zhao D, Liu Y, Shen Q, Yang M, Wang C, Li N, Cao X (2016) Cytoplasmic STAT4 promotes antiviral type I IFN production by blocking CHIP‐mediated degradation of RIG‐I. J Immunol 196: 1209–1217 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2