


Abstract
The COFACTOR web server is a unified platform for structure-based multiple-level protein function predictions. By structurally threading low-resolution structural models through the BioLiP library, the COFACTOR server infers three categories of protein functions including gene ontology, enzyme commission and ligand-binding sites from various analogous and homologous function templates. Here, we report recent improvements of the COFACTOR server in the development of new pipelines to infer functional insights from sequence profile alignments and protein–protein interaction networks. Large-scale benchmark tests show that the new hybrid COFACTOR approach significantly improves the function annotation accuracy of the former structure-based pipeline and other state-of-the-art functional annotation methods, particularly for targets that have no close homology templates. The updated COFACTOR server and the template libraries are available at http://zhanglab.ccmb.med.umich.edu/COFACTOR/.
INTRODUCTION
Due to recent advances in high-throughput sequencing technology, the gap between the number of known protein sequences and number of those with experimentally characterized functions is quickly growing. As of 2017, for example, there are more than 60 million protein sequences deposited in the UniProt database (1), but fewer than 0.8% of these sequences have the functions manually annotated in SwissProt (2). Automated and yet accurate in silico protein function prediction thus becomes crucial for making use of the recent explosion of genomic sequencing data. Most of the current function prediction approaches are based on sequence homologous transfer (3), which may not be able to accomplish the remarkable task since more than 80% of unannotated protein sequences lack close functional homologs (i.e. sharing >60% sequence identity) and 25% of unannotated proteins lack any homologs sharing a sequence identity above 30% in the current databases. Given that the function of a protein is ultimately defined by its structure, COFACTOR (4,5) has been previously proposed to transfer functional insights to the unknown proteins from structural homologies, providing an alternative approach to annotating non-homologous targets that sequence-homology based methods cannot model effectively (3).
Function annotation using structural homology alone, however, suffers several deficiencies. First, global structural similarity does not always lead to functional similarity. For example, the TIM barrel fold (6) is adopted by many proteins covering 60 distinct EC classification (7) as well as many non-enzyme proteins. Even for proteins with similar functions, global fold based comparisons may fail because the proteins often share only the local binding or active sites with completely different folds (8). Second, the current structure-function database is far from complete. For around 88% of proteins with known functions from the UniProt-GOA (9), for example, there are no experimentally solved structures in the PDB database (10), seriously limiting the power of structure-based detection of functional homologies. Finally, although structure is essential to protein function, the structure of proteins in cells is far from static and many functions are associated with the cellular environment of the molecules and the molecular motion of disordered regions that do not have a structure on their own (11). Therefore, composite approaches combining multiple and complementary information from different resources of sequence homologs and interaction networks should help increase the accuracy and coverage of structure-based function annotations.
In this note, we report our recent enhancement of the COFACTOR web server (4) to make use of hybrid models combining information from structure and sequence homologies, as well as protein–protein interaction (PPI) networks, for optimal protein function predictions. In addition, considerable effort has been made to improve user's experience and facility in analyzing and visualizing the modeling results, which include the introduction of new animation tools to display structural templates and ligand-protein interactions and directed acyclic graphs (DAG) to visualize the Gene Ontology (GO) annotation hierarchy. The new COFACTOR server and the functional libraries are freely available at http://zhanglab.ccmb.med.umich.edu/COFACTOR/.
MATERIALS AND METHODS
Gene Ontology (GO) term prediction
The approach of GO prediction in the COFACTOR web server consists of three pipelines for structure-, sequence- and PPI-based predictions (Figure 1). While the previous COFACTOR web server (4) only implemented the structure-based pipeline, the major new developments in the current version of the GO prediction algorithm are the introductions of two new sequence- and PPI-based pipelines and a consensus based approach to combining information from the complementary pipelines. For completeness of description, here we briefly describe all three pipelines that are currently used in the COFACTOR server, including the pipeline developed previously (4,5).
Figure 1.

The workflow of COFACTOR for template-based function predictions. The method consists of three pipelines for functional template identifications. The GO models are derived from a consensus of the structure-, sequence- and PPI-based pipelines, while the enzyme commission (EC) and ligand-binding predictions are obtained from structure-based template transfers.
Structure-based pipeline
The structure-homology based GO prediction method by COFACTOR was described previously (4). Briefly, the query structure is compared to a non-redundant set of known proteins in the BioLiP library (12) through two sets of local and global structural alignments based on the TM-align algorithm (13), for functional homology detections. Here, BioLiP is a semi-manually curated structure-function database containing known associations of experimentally solved structures and biological functions of proteins in terms of GO terms, enzyme commission (EC) number and ligand-binding sites. The current version of BioLiP contains 35 238 entries annotated with 465 838 GO terms, which are used in benchmarking the methods described in this study. The local structure similarity between query and template is defined by
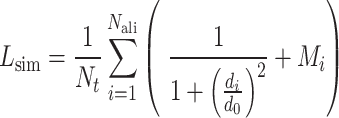 |
(1) |
where 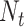 is the number of residues in the active/binding sites,
is the number of residues in the active/binding sites, 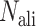 is the number of aligned residue pairs,
is the number of aligned residue pairs, 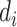 is the
is the 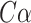 distance between ith aligned residue pair,
distance between ith aligned residue pair, 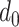 = 3Å is the distance cut-off and
= 3Å is the distance cut-off and 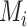 is the BLOSUM62 substitution matrix score (14) between ith pair of residues that has been normalized to the interval [0, 1]. The confidence score of a template hit is defined by
is the BLOSUM62 substitution matrix score (14) between ith pair of residues that has been normalized to the interval [0, 1]. The confidence score of a template hit is defined by
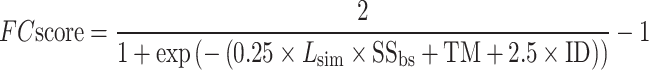 |
(2) |
where 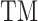 is the global structure similarity in terms of TM-score (15) between query and template,
is the global structure similarity in terms of TM-score (15) between query and template, 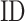 is the sequence identity between query and template in the aligned region and
is the sequence identity between query and template in the aligned region and 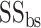 is the sequence identity at the binding site. The overall confidence score for a particular GO term λ is then calculated by
is the sequence identity at the binding site. The overall confidence score for a particular GO term λ is then calculated by
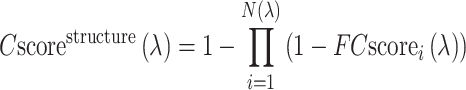 |
(3) |
where N(λ) is the number of templates associated with the GO term λ and 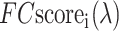 is the confidence score of the ith hit associated with λ as defined in Equation (2). The predicted GO terms are reconciled using the PIPA algorithm (16).
is the confidence score of the ith hit associated with λ as defined in Equation (2). The predicted GO terms are reconciled using the PIPA algorithm (16).
Sequence-based pipeline
In the second pipeline, the query sequence is searched against the UniProt-GOA database through both sequence and sequence-profile alignments by BLAST (17) and PSI-BLAST (18), respectively. Only manually reviewed GO terms of sequence templates are considered, with GO terms annotated with Inferred from Electronic Annotation (IEA) or No biological Data available (ND) evidence codes excluded. For BLAST, the query is directly searched against sequence template library with an E-value cut-off 0.01. The confidence score for a particular GO term λ resulting from a BLAST search is defined by
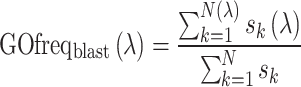 |
(4) |
where N is the number of templates identified, 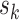 is the sequence identity between the query and the kth template and N(λ) and
is the sequence identity between the query and the kth template and N(λ) and 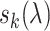 are those associated with a specific GO term λ. For PSI-BLAST, a sequence profile is obtained by searching with the query sequence through the Uniref90 sequence library (19) by three iterations under an E-value cut-off 0.01. The sequence profile is used to jump-start a PSI-BLAST profile-sequence search against the UniProt-GOA sequences. The confidence score for GO term λ is defined in the same way as in BLAST (Equation 4).
are those associated with a specific GO term λ. For PSI-BLAST, a sequence profile is obtained by searching with the query sequence through the Uniref90 sequence library (19) by three iterations under an E-value cut-off 0.01. The sequence profile is used to jump-start a PSI-BLAST profile-sequence search against the UniProt-GOA sequences. The confidence score for GO term λ is defined in the same way as in BLAST (Equation 4).
The final weighted average confidence score of the sequence-based pipeline is calculated as
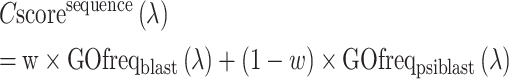 |
(5) |
where w equals the maximum sequence identity of the query to all the template hits. In this way, BLAST hits have a stronger weight if close homologs are found, while the weight of the PSI-BLAST hits is increased for the non-homologous cases for which PSI-BLAST profile alignments are usually more efficient than the sequence-based alignments.
PPI-based pipeline
In this pipeline, the query is first mapped to the STRING (20) PPI database by BLAST; only the BLAST hit with the most significant E-values is subsequently considered. GO terms of the interaction partners, as annotated in the STRING database, are then collected and assigned to the query protein (Inset of Figure 1). The underlying assumption is that interacting protein partners tend to participate in the same biological pathway at the same sub-cellular location and therefore may have similar GO terms.
Finally, the confidence score for GO term 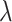 mapped by PPI is calculated by
mapped by PPI is calculated by
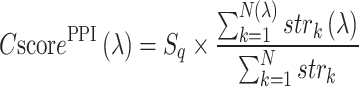 |
(6) |
where N is the number of interacting partners, 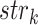 is the confidence score of interaction between query and the kth interaction partner as assigned by the STRING database and
is the confidence score of interaction between query and the kth interaction partner as assigned by the STRING database and 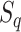 is the sequence identity in the first step of BLAST alignment between the query sequence and the STRING entry it is mapped to N(λ) and
is the sequence identity in the first step of BLAST alignment between the query sequence and the STRING entry it is mapped to N(λ) and 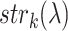 are those associated to the specific GO term λ.
are those associated to the specific GO term λ.
Consensus GO prediction
The final GO prediction is obtained by combining the GO terms from the structure-, sequence- and PPI-based pipelines, with the confidence score calculated by
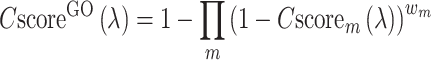 |
(7) |
where 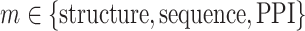 .
. 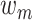 is the relative weight for each of the three methods, with
is the relative weight for each of the three methods, with 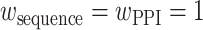 and
and 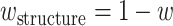 , where w equals to the maximum sequence identity among identified function templates. Hence, the weight of the structure-based model becomes stronger for the cases that have no homologous templates.
, where w equals to the maximum sequence identity among identified function templates. Hence, the weight of the structure-based model becomes stronger for the cases that have no homologous templates.
Enzyme Commission (EC) number prediction
The pipeline of EC number prediction is similar to the structure-homology based method used in GO prediction (Figure 1), as reported previously (4). Enzymatic homologs are identified by aligning the target structure, using TM-align (13), to a library of 8392 enzyme structures from the BioLiP library (12), with the active site residues mapped from the Catalytic Site Atlas database (21). The confidence score for each predicted EC number is estimated based on the global and local similarity between the target and top template hit:
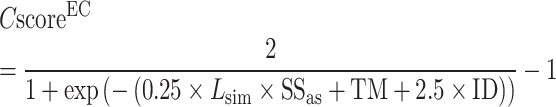 |
(8) |
where 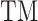 is the TM-score between query and template,
is the TM-score between query and template, 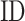 is the sequence identity,
is the sequence identity, 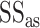 is the sequence identity at the active sites and
is the sequence identity at the active sites and 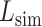 is local structure similarity as defined in Equation (1).
is local structure similarity as defined in Equation (1).
Ligand-binding site prediction
Following the previous implementation (4), ligand-binding prediction in COFACTOR consists of three steps (Figure 1). First, functional homologies are identified by matching the query structure through a non-redundant set of the BioLiP library (12), which currently contains 58 416 structure templates harboring in total 76 679 ligand-binding sites for interaction between receptor proteins and small molecule compounds, short peptides and nucleic acids. The initial binding sites are then mapped to the query from the individual templates based on the structural alignments.
Next, the ligands from each individual template are superposed to the predicted binding sites on the query structure using superposition matrices from a local alignment of the query and template binding sites. To resolve atomic clashes, the ligand poses are refined by a short Metropolis Monte Carlo simulation under rigid-body rotation and translation, guided by an empirical energy function of
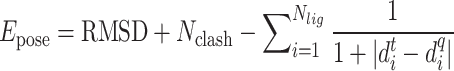 |
(9) |
where 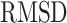 is the RMSD of current ligand pose and the origin ligand pose,
is the RMSD of current ligand pose and the origin ligand pose, 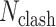 is the number of atomic clashes between ligand and protein,
is the number of atomic clashes between ligand and protein, 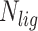 is the number of ligand atoms,
is the number of ligand atoms, 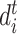 is the distance between ith ligand atom and the
is the distance between ith ligand atom and the 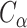 atom of the template residue in contact with the ligand atom and
atom of the template residue in contact with the ligand atom and 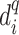 is the distance between the same ligand atom and the closest query
is the distance between the same ligand atom and the closest query 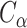 atom.
atom.
Finally, the consensus binding sites are obtained by clustering of all ligands that are superposed to the query structure, based on distances of the centers of mass of the ligands using a cut-off of 8 Å. Different ligands within the same binding pocket are further grouped by the average linkage clustering with chemical similarity, using the Tanimoto coefficient (22) with a cut-off of 0.7. The model with the highest ligand-binding confidence score (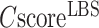 ) among all the clusters is selected, defined by
) among all the clusters is selected, defined by
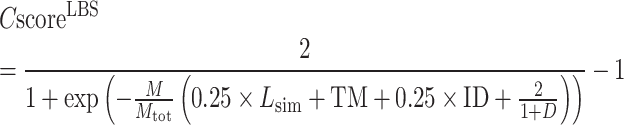 |
(10) |
where M is the number of ligands in the ligand cluster, 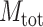 is the total number of ligands collected from all homologous templates,
is the total number of ligands collected from all homologous templates, 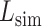 is the local similarity at the binding site defined in Equation (1),
is the local similarity at the binding site defined in Equation (1), 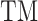 is TM-score between query and template,
is TM-score between query and template, 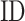 is the sequence identity between query and template in the structurally aligned region and
is the sequence identity between query and template in the structurally aligned region and 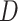 is the average distance between ligands within the cluster.
is the average distance between ligands within the cluster.
RESULTS
Benchmark results on GO predictions
The COFACTOR GO pipelines have been benchmarked on a non-redundant set of 1224 Escherichia coli proteins from UniProt database, with lengths ranging from 38 to 968 residues and pairwise sequence identity <40%. The input structures for COFACTOR were predicted by I-TASSER (23) with all homologous structural templates with a sequence identity >30% to the query excluded, thus simulating predictions for a target without any close homologs. Similar to the Critical Assessment of Function Annotation (CAFA) experiments (3,24), the GO performance is mainly assessed by the F-measure, which is defined as the harmonic average between precision and recall:
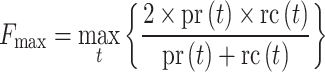 |
(11) |
where t is the confidence score threshold (ranging between 0 and 1) and pr(t) and rc(t) are the precision and recall at a threshold t, respectively.
Supplementary Figure S1 in the Supplementary Data shows the performance of the COFACTOR server on the three aspects of GO terms: molecular function (MF), biological process (BP) and cellular component (CC); results are shown in comparison with those of the GoFDR program (25), one of the top performing methods in CAFA2 (3), and three baselines methods: Naïve Baseline, BLAST and PSI-BLAST, as implemented in CAFA (3,24). To examine the effect of the combination of complementary pipelines, we also show the results from individual COFACTOR components from structure, sequence and PPI pipelines. To test the dependence of the pipelines on the homologies from known proteins, four levels of sequence identity cut-offs at 20, 30, 50 and 90% were used separately to filter out homologous templates.
Several interesting observations arise from Supplementary Figure S1. First, whereas the performance of sequence-based methods (GoFDR, BLAST/PSIBLAST and the sequence module of COFACTOR) declines rapidly below 50% sequence identity, the structure module of COFACTOR shows almost no loss of performance even down to 20% sequence identity, and at that point it outperforms all sequence-based methods. For example, 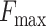 for MF is 0.538 at the 20% sequence identity cut-off, very close to 0.541 obtained at 50% cut-off (Supplementary Table S1). Second, the new sequence component of COFACTOR is a strong performer on its own, with performance exceeding all other sequence-based methods including GoFDR (except for the cases at a very low homology cut-off) and thus provides a useful complement to the structure-based module in the high sequence homology region. Finally, the new hybrid COFACTOR model not only outperforms the old structure-only COFACTOR model, but also outperforms all other methods used in our comparison (including, interestingly enough, the Naïve method for CC term predictions, which was not beaten by any prediction set in the CAFA2 competition (3)), at all levels of sequence identity cut-offs.
for MF is 0.538 at the 20% sequence identity cut-off, very close to 0.541 obtained at 50% cut-off (Supplementary Table S1). Second, the new sequence component of COFACTOR is a strong performer on its own, with performance exceeding all other sequence-based methods including GoFDR (except for the cases at a very low homology cut-off) and thus provides a useful complement to the structure-based module in the high sequence homology region. Finally, the new hybrid COFACTOR model not only outperforms the old structure-only COFACTOR model, but also outperforms all other methods used in our comparison (including, interestingly enough, the Naïve method for CC term predictions, which was not beaten by any prediction set in the CAFA2 competition (3)), at all levels of sequence identity cut-offs.
Here, since the structure-based pipeline is inherited from the former studies (4,5), the difference between ‘COFACTOR’ and ‘COFACTOR structure’ essentially calibrates the quantitative improvement of the COFACTOR server in GO prediction since the last release in 2012 (4), which is based on the same function library. At the sequence identity cut-off of 30%, for example, the Fmax of GO prediction on MF, BP and CC has been improved from 0.541, 0.495, 0.513 in ‘COFACTOR structure’ to 0.611, 0.579, 0.582 in ‘COFACTOR’, respectively. The difference becomes even larger with the sequence identity cut-off increasing, since the sequence-based pipeline can detect more accurate templates when close homologous templates are available (see Supplementary Table S1).
It is of interest to note that the sequence-based methods (GoFDR, PSI-BLAST, BLAST and the sequence pipeline of COFACTOR) do not converge in Supplementary Figure S1 with the sequence identity cut-off increasing, although the single-template based methods, BLAST and PSI-BLAST, do converge when sequence identity is >50%. The main reason is that both GoFDR and sequence-based COFACTOR combine multiple sequence templates, where templates of a low sequence identity can still affect their scoring function even at a very high sequence identity cut-off. Our benchmark data shows that such an approach using consensus information of high- and low-sequence identity templates almost always improves the prediction accuracy, due to the fact that the function similarity between proteins is not always proportional to the sequence identity and many protein pairs with a lower sequence identity can have a closer functional relation than those with a higher sequence identity (26).
To examine the specificity of the COFACTOR predictions, we present in Figure 2A a histogram of precision versus the confidence score by COFACTOR for the GO predictions, where a strong correlation is found for all aspects of GO terms, with the Pearson correlation coefficient (PCC) being 0.96, 0.94 and 0.86 for MF, BP and CC terms, respectively. Consistent with Supplementary Figure S1, at the same 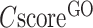 cut-off the precision of MF and BP is generally higher than that of CC. For example, the precision for both MF and BP will be >0.3 when
cut-off the precision of MF and BP is generally higher than that of CC. For example, the precision for both MF and BP will be >0.3 when 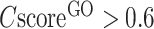 , while the precision of CC is only marginally close to 0.3 when
, while the precision of CC is only marginally close to 0.3 when 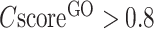 .
.
Figure 2.

Calibration curves showing the precision of COFACTOR models versus the confidence score in each category of function annotation. (A) GO, (B) EC and (C) Ligand-binding sites.
Structure-based approach for EC number prediction
COFACTOR's ability to predict EC numbers was tested on a set of 318 non-homologous enzymes, with the benchmark EC numbers extracted from the PDB entries. The structural models were again predicted by I-TASSER, which were used for the EC template detection as in Equation (8). As with the GO term predictions above, to simulate a challenging case with no close sequence homologs available, both structural and function templates homologous to the query (with a sequence identity >30%) were excluded from the I-TASSER and COFACTOR template libraries. Supplementary Figure S2 presents the benchmark results of COFACTOR on EC number prediction compared with the BLAST and PSIBLAST baseline predictors at the same homology cut-off. The data shows a significant advantage of COFACTOR's use of structural homology transfers over the sequence-homology approach of BLAST and PSIBLAST. For example, the F-measure for the first three digits of EC number for the first template of COFACTOR is 0.702, while those for the BLAST and PSIBLAST baseline predictors are just 0.243 and 0.450, respectively (Supplementary Figure S2).
Figure 2B shows the precision data of the EC models versus the confidence score (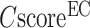 ), while a strong correlation with a PCC = 0.95 is obtained between
), while a strong correlation with a PCC = 0.95 is obtained between 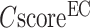 and the precision for the first enzyme homolog identified for each target. Generally, the precision of the prediction goes above 0.5 for any models with a
and the precision for the first enzyme homolog identified for each target. Generally, the precision of the prediction goes above 0.5 for any models with a 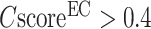 (Figure 2B).
(Figure 2B).
Ligand-binding site prediction
The performance of COFACTOR in ligand-binding site prediction was benchmarked on 814 ligand-binding sites from 500 non-homologous proteins from the PDB. Following the criterion used in the CASP experiment (27), a residue is defined as a binding site if it has at least one atom whose distance from the closest ligand atom is within 0.5 Å plus the sum of the van der Waals radii of the two atoms. As in the tests above, both structural and functional templates with a sequence identity >30% have been excluded from the I-TASSER structure prediction and COFACTOR binding site template recognitions, to avoid homologous contamination and simulate the case of a difficult target with no close annotated homologs.
The overall Matthews correlation coefficient (MCC, as defined in Supplementary Figure S3) between the actual and predicted binding sites by COFACTOR is 0.465. This compares favorably to other state of the art binding site predictors including Concavity (28) and Findsite (29) which have overall MCCs of 0.378 and 0.454, respectively, for the same set of proteins. The average precision and recall of the COFACTOR prediction are 0.501 and 0.485, respectively. Despite of the relatively low precision and recall, COFACTOR identifies at least one binding residue correctly in 88% of the test proteins. One reason for the low precision and recall on average is due to the alignment error in the query and template comparisons, which is most significant in comparing distantly homologous proteins; this alignment error can result in imprecise mapping of the binding residues from templates to query (although many of them are located near the center of the binding pocket). Nevertheless, considering that most binding site residues in natural proteins are spatially proximate to each other and all of them are located in the same binding pocket, correctly predicting one or more binding site residues is sufficient to locate the binding pocket that can be used to guide small molecule docking and/or to assist wet-lab experimentalists in designing mutagenesis experiment (30,31). In Supplementary Figure S3, we show an illustrative example from the C-chain of the GDPRan–NTF2 complex (PDB ID: 1a2k), where five residues were predicted by COFACTOR as ligand-binding sites and four of them were correct, resulting in an MCC = 0.723 for this case; while the fifth predicted residue is not correct, it is nevertheless located near the binding pocket.
Figure 2C displays the precision values of COFACTOR binding predictions versus the confidence score (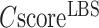 ), which demonstrates a strong correlation with PCC = 0.99. This correlation data should help provide users a quantitative estimation of their LBS predictions based on CscoreLBS. For example, 62.6% of the binding sites are predicted correctly for the models with a
), which demonstrates a strong correlation with PCC = 0.99. This correlation data should help provide users a quantitative estimation of their LBS predictions based on CscoreLBS. For example, 62.6% of the binding sites are predicted correctly for the models with a 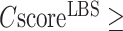 0.5; if the
0.5; if the 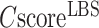 cut-off is increased to
cut-off is increased to 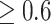 , the average precision will increase to 72.9%. In our benchmark test, 232 out of the 500 targets have a
, the average precision will increase to 72.9%. In our benchmark test, 232 out of the 500 targets have a 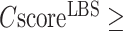 0.5 and 100 targets have a
0.5 and 100 targets have a 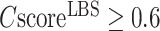 .
.
To further assess the significance of the ligand-binding site predictions, we present in Supplementary Figure S4 the enrichment factor of binding site prediction by COFACTOR at different 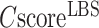 over a ‘naïve pocket’ approach in which ligand-binding sites are simply assigned by the largest cavities in the protein structure, as identified using Fpocket (32). The data shows a significantly higher accuracy of COFACTOR than the naïve pocket detection approach. At
over a ‘naïve pocket’ approach in which ligand-binding sites are simply assigned by the largest cavities in the protein structure, as identified using Fpocket (32). The data shows a significantly higher accuracy of COFACTOR than the naïve pocket detection approach. At 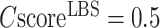 , for example, the precision of COFACTOR is 8.45 times higher than the naïve pocketing approach (Supplementary Figure S4).
, for example, the precision of COFACTOR is 8.45 times higher than the naïve pocketing approach (Supplementary Figure S4).
Web server
Server input
The mandatory input for the web server is a single-chain protein structure file for the query protein in PDB format. If the input structure contains multiple chains or multiple models, only the first chain of the first model will be parsed. In the absence of an experimentally solved structure, the user can use models generated by the online structure prediction tools, such as I-TASSER (33,34), QUARK (35), Rosetta (36), HHpred (37) or Phyre2 (38). The sequence of query will be extracted from ‘SEQRES’ records of the PDB file or ‘ATOM’ records if ‘SEQRES’ is absent. If the structure contains missing regions, users are encouraged to upload the full-length sequence separately using the ‘Advanced Options’ section, which may help the sequence- and PPI-based pipelines to generate more complete function predictions.
Server output
Upon job completion, the user will be notified by email with a link to the result page on the COFACTOR server website. The result page has been substantially updated since the original version of the COFACTOR server and consists of four major panels, including structural analogies, GO terms, EC numbers and ligand-binding sites; an example is shown in Figure 3.
Figure 3.

An illustration of the COFACTOR web server output consisting of four annotation panels. The example is from the Escherichia coli protein ysgA (UniProt accession: P56262) with a structural model generated by I-TASSER (23). (A) Top 10 analogous structures that are structurally closest to the query structure, displaying the structural similarity between ysgA and known hydrolases. (B) GO prediction results in three aspects of molecular function (MF), biological process (BP) and cellular component (CC), which are consistent with UniProt annotation of ysgA as a putative carboxymethylene butenolidase and EcoCyc (41) annotation as a predicted hydrolase. (C) EC prediction results from top-five enzyme homologous templates, suggesting carboxymethylene butenolide hydrolase activity (EC 3.1.1.45) and directly predicting the enzyme's active site. (D) Ligand-binding site prediction results from the top 10 homologous templates, including residues surrounding putative active sites that are in proximity to the ligand. The images are screen copied from the COFACTOR example webpage (http://zhanglab.ccmb.med.umich.edu/COFACTOR/example/). Larger size copies of the images with a higher resolution are listed in Supplementary Figures S5 and S6 in the Supplementary Data.
The first panel displays an ordered list of the top-ten analogous structures from the PDB library that are structurally closest to the query protein. The structural superimpositions are displayed in an interactive JSmol applet that allows users to rotate and annotate the pictures (39). The analogous template is shown together with the TM-score, RMSD of aligned region, sequence identity, and query coverage; and two links are given to the URL addresses for downloading the PDB template structure and the superposed query/template models from TM-align, respectively. By clicking on each of the radio buttons, the user can explore the JSmol applet of all different templates (Figure 3A).
The second panel shows the consensus GO prediction results, with models for the MF, BP and CC aspects listed separately (Figure 3B). The predicted GO terms are listed alongside the CscoreGO and their common name. For each of the three GO aspects, the predicted GO terms are plotted together with their parent terms as a DAG, in which the predicted GO terms are highlighted by a CscoreGO-specific color code, with blue to red representing the terms with 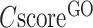 from [0.4–0.5] to [0.9–1.0]. Since there are usually multiple terms predicted for each target, only the confident predictions with
from [0.4–0.5] to [0.9–1.0]. Since there are usually multiple terms predicted for each target, only the confident predictions with 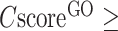 0.5 are displayed, although the full set of predictions is available for download. If none of the GO terms has a
0.5 are displayed, although the full set of predictions is available for download. If none of the GO terms has a 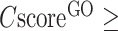 0.5, the GO terms with the highest
0.5, the GO terms with the highest 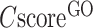 will be displayed.
will be displayed.
The third panel shows the top-five EC number predictions, each associated with the template structure and marked with predicted active sites that can be visualized in an accompanying JSmol applet (Figure 3C). In addition, the predicted EC number, the confidence score, TM-score between query and template, RMSD of aligned region, sequence identity, query coverage and predicted active sites are also listed for each model.
The last panel shows the ligand-binding site prediction results. For each set of binding sites, the structure templates are presented in order of descending confidence score, together with their TM-score, RMSD of aligned region, sequence identity, coverage and binding site residues. The positions of the ligand-binding site residues are highlighted in the target structure and can be viewed and interpreted using the JSmol applet (Figure 3D).
For every target protein, all prediction results are packed in a tarball file named ‘result.tar.bz2΄ that can be conveniently downloaded from the output page. Again, most of the animation applets and DAG tree images were newly developed in this version, which should provide useful facilities to help users better manipulate and interpret the results.
CONCLUSION
We report recent advancements made to the online COFACTOR server for hybrid protein function annotations. In general, the biological function of a protein can be intricate and often contains multiple levels of categorizations. The COFACTOR server focuses on the three most widely-used and computationally amenable categories of function: GO, EC number and ligand-binding sites. Compared with the previous version of COFACTOR, which generated function annotations purely based on structural homology transfer, the updated server introduced several new pipelines built on sequence profile and PPI network information to enhance the accuracy and coverage of the structure-based function predictions. Accordingly, new sources of function templates, including sequence homologs and PPI partners, have been incorporated into the default function library (BioLiP) of the COFACTOR server. Our large-scale benchmark tests have shown that the new composite pipelines can generate function predictions with accuracy outperforming the former version of COFACTOR, as well as many state-of-the-art methods in the literature.
To facilitate the use and interpretation of the prediction results, a confidence scoring system has been introduced (as calibrated in Figure 2), which can help users to quantitatively estimate the accuracy of the predictions. Meanwhile, new DAG combined with animation software are introduced to facilitate the viewing, analysis and manipulation of the prediction models. These developments and updates significantly enhance the accuracy and usability of an already widely applied structure function service system and will make it continue to be a powerful tool, powered by new state of the art algorithms, both for rapid annotation of uncharacterized proteins and for providing a starting point to understand and further characterize targets that may be identified in high-throughput experimental studies.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to Dr Ambrish Roy and Ms Fatima Zohra Smaili for helpful discussions and to Dr Wei Zheng for help in preparing the graphic abstract. Part of the method training and benchmarking work was done on the Extreme Science and Engineering Discovery Environment (XSEDE) (40) and the Open Science Grid.
Contributor Information
Chengxin Zhang, Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI 48109, USA.
Lydia Freddolino, Department of Biological Chemistry, University of Michigan, Ann Arbor, MI 48109, USA; Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI 48109, USA.
Yang Zhang, Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI 48109, USA; Department of Biological Chemistry, University of Michigan, Ann Arbor, MI 48109, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute of General Medical Sciences [GM083107, GM116960 to Y.Z., GM097033 to L.F.]; NSF [DBI1564756 to Y.Z.]. Funding for open access charge: National Institutes of Health [GM083107]; NSF [DBI1564756].
Conflict of interest statement. None declared.
REFERENCES
- 1. Bateman A., Martin M.J., O’Donovan C., Magrane M., Apweiler R., Alpi E., Antunes R., Ar-Ganiska J., Bely B., Bingley M.et al. UniProt: a hub for protein information. Nucleic Acids Res. 2015; 43:D204–D212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boutet E., Lieberherr D., Tognolli M., Schneider M., Bansal P., Bridge A.J., Poux S., Bougueleret L., Xenarios I. UniProtKB/Swiss-Prot, the manually annotated section of the UniProt knowledgebase: how to use the entry view. Methods Mol. Biol. 2016; 1374:23–54. [DOI] [PubMed] [Google Scholar]
- 3. Jiang Y.X., Oron T.R., Clark W.T., Bankapur A.R., D’Andrea D., Lepore R., Funk C.S., Kahanda I., Verspoor K.M., Ben-Hur A.et al. An expanded evaluation of protein function prediction methods shows an improvement in accuracy. Genome Biol. 2016; 17:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roy A., Yang J., Zhang Y. COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 2012; 40:W471–W477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roy A., Zhang Y. Recognizing protein-ligand binding sites by global structural alignment and local geometry refinement. Structure. 2012; 20:987–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nagano N., Orengo C.A., Thornton J.M. One fold with many functions: the evolutionary relationships between TIM barrel families based on their sequences, structures and functions. J.Mol. Biol. 2002; 321:741–765. [DOI] [PubMed] [Google Scholar]
- 7. Webb E.C. Enzyme Nomenclature 1992. Recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology on the Nomenclature and Classification of Enzymes. 1992; San Diego: Academic Press. [Google Scholar]
- 8. Zhang Y. Protein structure prediction: when is it useful. Curr. Opin. Struct. Biol. 2009; 19:145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barrell D., Dimmer E., Huntley R.P., Binns D., O’Donovan C., Apweiler R. The GOA database in 2009-an integrated Gene Ontology annotation resource. Nucleic Acids Res. 2009; 37:D396–D403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Berman H., Henrick K., Nakamura H. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 2003; 10:980–980. [DOI] [PubMed] [Google Scholar]
- 11. Dyson H.J., Wright P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Bio. 2005; 6:197–208. [DOI] [PubMed] [Google Scholar]
- 12. Yang J., Roy A., Zhang Y. BioLiP: a semi-manually curated database for biologically relevant ligand-protein interactions. Nucleic Acids Res. 2013; 41:D1096–D1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang Y., Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005; 33:2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Henikoff S., Henikoff J.G. Amino-acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang Y., Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins. 2004; 57:702–710. [DOI] [PubMed] [Google Scholar]
- 16. Yu C.G., Zavaljevski N., Desai V., Johnson S., Stevens F.J., Reifman J. The development of PIPA: an integrated and automated pipeline for genome-wide protein function annotation. BMC Bioinformatics. 2008; 9:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990; 215:403–410. [DOI] [PubMed] [Google Scholar]
- 18. Altschul S.F., Madden T.L., Schaffer A.A., Zhang J.H., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997; 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Suzek B.E., Wang Y.Q., Huang H.Z., McGarvey P.B., Wu C.H., Consortium U. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics. 2015; 31:926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K.P.et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015; 43:D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Furnham N., Holliday G.L., de Beer T.A.P., Jacobsen J.O.B., Pearson W.R., Thornton J.M. The Catalytic Site Atlas 2.0: cataloging catalytic sites and residues identified in enzymes. Nucleic Acids Res. 2014; 42:D485–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rogers D.J., Tanimoto T.T. A computer program for classifying plants. Science. 1960; 132:1115–1118. [DOI] [PubMed] [Google Scholar]
- 23. Yang J., Yan R., Roy A., Xu D., Poisson J., Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat. Methods. 2015; 12:7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Radivojac P., Clark W.T., Oron T.R., Schnoes A.M., Wittkop T., Sokolov A., Graim K., Funk C., Verspoor K., Ben-Hur A.et al. A large-scale evaluation of computational protein function prediction. Nat. Methods. 2013; 10:221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gong Q.T., Ning W., Tian W.D. GoFDR: a sequence alignment based method for predicting protein functions. Methods. 2016; 93:3–14. [DOI] [PubMed] [Google Scholar]
- 26. Sangar V., Blankenberg D.J., Altman N., Lesk A.M. Quantitative sequence-function relationships in proteins based on gene ontology. BMC Bioinformatics. 2007; 8:294–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schmidt T., Haas J., Gallo Cassarino T., Schwede T. Assessment of ligand-binding residue predictions in CASP9. Proteins. 2011; 79:126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Capra J.A., Laskowski R.A., Thornton J.M., Singh M., Funkhouser T.A. Predicting protein ligand binding sites by combining evolutionary sequence conservation and 3D Structure. PloS Comput. Biol. 2009; 5:e1000585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brylinski M., Skolnick J. A threading-based method (FINDSITE) for ligand-binding site prediction and functional annotation. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee H.S., Zhang Y. BSP-SLIM: a blind low-resolution ligand-protein docking approach using predicted protein structures. Proteins. 2012; 80:93–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang C.D., Buck M.A., Fraser C.M. Site-directed mutagenesis of alpha 2A-adrenergic receptors: identification of amino acids involved in ligand binding and receptor activation by agonists. Mol. Pharmacol. 1991; 40:168–179. [PubMed] [Google Scholar]
- 32. Le Guilloux V., Schmidtke P., Tuffery P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics. 2009; 10:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008; 9:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roy A., Kucukural A., Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 2010; 5:725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xu D., Zhang Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins. 2012; 80:1715–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim D.E., Chivian D., Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004; 32:W526–W531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hildebrand A., Remmert M., Biegert A., Soding J. Fast and accurate automatic structure prediction with HHpred. Proteins. 2009; 77:128–132. [DOI] [PubMed] [Google Scholar]
- 38. Kelley L.A., Mezulis S., Yates C.M., Wass M.N., Sternberg M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015; 10:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hanson R.M., Prilusky J., Renjian Z., Nakane T., Sussman J.L. JSmol and the next-generation web-based representation of 3D molecular structure as applied to Proteopedia. Isr. J. Chem. 2013; 53:207–216. [Google Scholar]
- 40. Towns J., Cockerill T., Dahan M., Foster I., Gaither K., Grimshaw A., Hazlewood V., Lathrop S., Lifka D., Peterson G.D.et al. XSEDE: accelerating scientific discovery. Comput. Sci. Eng. 2014; 16:62–74. [Google Scholar]
- 41. Keseler I.M., Mackie A., Peralta-Gil M., Santos-Zavaleta A., Gama-Castro S., Bonavides-Martinez C., Fulcher C., Huerta A.M., Kothari A., Krummenacker M.et al. EcoCyc: fusing model organism databases with systems biology. Nucleic Acids Res. 2013; 41:D605–D612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.