


Abstract
We develop a hierarchical pipeline, ThreaDomEx, for both continuous domain (CD) and discontinuous domain (DCD) structure predictions. Starting from a query sequence, ThreaDomEx first threads it through the PDB to identify multiple structure templates, where a profile of domain conservation score (DC-score) is derived for domain-segment assignment. To further detect DCDs that consist of separated segments along the sequence, a boundary-clustering algorithm is used to refine the DCD-linker locations. In case that the templates do not contain DCDs, a domain-segment assembly process, guided by symmetry comparison, is applied for further DCD detections. ThreaDomEx was tested a set of 1111 proteins and achieved a normalized domain overlap score of 89.3% compared to experimental data, which is significantly higher than other state-of-the-art methods. It also recalls 26.7% of DCDs with 72.7% precision on the proteins for which threading failed to detect any DCDs. The server provides facilities for users to interactively refine the domain models by adjusting DC-score threshold, deleting and adding domain linkers, and assembling domain segments, which are particularly helpful for the hard targets for which current methods have a low accuracy while human-expert knowledge and experimental insights can be used for refining models. ThreaDomEX server is available at http://zhanglab.ccmb.med.umich.edu/ThreaDomEx.
INTRODUCTION
Protein domains are subunits that can fold and evolve independently. The majority of eukaryotic proteins are found to consist of multiple domains for achieving various composite cellular functions (1). The identification of protein domains is thus the essential step for protein structure determination and functional annotations, where a variety of computational methods have been proposed to divide proteins into domains from the amino acid sequences (2).
One of the earliest and still widely used approaches is homologous modeling, which predicts protein domain boundaries through the identification of the evolutionarily conserved sequence families from multiple homologous sequence alignments. Examples that use such approach include Pfam (3,4), PASS (5,6), EVEREST (7,8), ADDA (9,10) and FIEFDom (11). Another popularly used approach obtains domain predictions through the statistical modeling or machine leaning trained from known domain structures in the PDB library. These include, for example, DGS (12), Armadillo (13), DPD (14), DomCut (15), CHOPnet (16), Dompro (17), DomNet (18), PPRODO (19), kemaDom (20), DLP-SVM (21), DROP (22), H-DROP (23), DOBO (24), and the methods proposed by Galzitskaya and Melnik (25) and Tanaka et al. (26). There are also methods, such as SnapDRAGON (27), RosettaDom (28) and OPUS-DOM (29), which first construct 3D models through ab initio folding and then extracted domain information directly from the structure prediction. Recently, we proposed a new method, ThreaDom (30), which deduces the domain boundary information from multiple query-to-template alignments derived by meta-server threading programs (31).
Most of the methods can only generate predictions for continuous domains (CDs) that consist of continuous residues along the query sequence, while a large number of proteins contain discontinuous domains (DCDs). For example, the current PDB library deposits about 18% proteins with at least one DCD which consists of two or more non-sequential segmental sequences (32). The detection and correct division of the DCDs represents a major challenging problem. The 3D-model based methods, such as SnapDRAGON (27), RosettaDom (28), OPUS-DOM (29), can in principle split the predicted 3D models into DCDs, but the success rate of ab initio structure predictions is very low (if not impossible) for the proteins of DCDs which usually have a medium to large size (33). ThreaDom (30) can break up the size limit due to the adoption of threading-based template recognition technique, but the success relies on the existence of similar DCD structure in the threading template library. DomEx (32) was recently proposed to predict DCDs by the reassembly of DCD segments guided with homology and symmetry alignments, which is able to detect DCDs beyond the structures in the template library and therefore significantly increases the accuracy of the DCD predictions.
Despite of the high number of method proposed, the number of on-line webservers available to biological community for automated domain prediction is quite low. Among the limit on-line servers, DOBO (24) generates domain boundary prediction by integrating evolutionary signals and machine learning. DomCut (15) collects linker preference profiles along the sequence with the domain boundary position predicted as the minima of the profiles. Scooby-domain (34) creates domain number and boundary predictions from the length and hydrophobicity analyses. The server by Galzitskaya and Melnik (25) generates fast domain boundary estimations based on chain entropy and amino acid composition. DLP-SVM (21), H-DROP (23) and DROP (22) provide domain linker positions and probability of the predictions based on SVM training. Finally, the ThreaDom server (30) uses multiple threading alignments and domain conservation score profiles to generate domain boundary assignments, which have witnessed successful applications in various 3D structure predictions (35–37) and structure-based function annotation studies (38–41).
Here, we propose a new domain prediction server, ThreaDomEx, which combines the ThreaDom and DomEx methods into a unified and user-friendly on-line server system for efficient structural domain predictions. Compared with many of the existing servers, one of the major novelties of ThreaDomEx is the enhanced ability to predict the DCD structures, mainly through the incorporation of the DomEx algorithm that assembles non-consecutive domain segments following multiple threading template alignments. Meanwhile, considerable efforts have been made to provide advanced visualization facilities, which, for the first time, allows users to conveniently integrate the human-expert knowledge and experimental insight to improve the domain prediction results of the sever pipelines.
IMPLEMENTATION
Method overview
The pipeline of ThreaDomEX for protein domain prediction consists of three consecutive steps (Figure 1). When a user uploads an amino acid sequence, the query is first threaded through a representative structure library of the PDB by LOMETS (31) to identify homologous and analogous structural templates. Meanwhile, PSSpred (42) and MUSTER (43) are employed to generate secondary structure and solvent accessibility predictions for further domain structure analyses. Secondly, a domain conservation score (DC-score) is calculated from the multiple threading template alignments to evaluate the conservativeness of each of the residues, where the initial boundaries of domain segments are decided on the distribution of DC-score along the sequence (30). Finally, a boundary-clustering based strategy is used to fine-tune the boundary positions, as well as to detect the DCDs from the templates. If no DCD is detected from the LOMETS templates, a segment assembly process guided by symmetric motif comparison, as proposed in DomEx (32), is employed for further detection of DCD structures.
Figure 1.

Flowchart of ThreaDomEx. The query sequence is first threaded through a representative PDB structure library to search for structural templates. Domain Conservation score (DC-score) is then calculated to evaluate the conservativeness of each amino acid in the query, with the domain boundaries assigned around the valley of the DC-score profile. Next, a boundary clustering-based strategy is used to optimize the boundary predictions and to detect discontinuous domains (DCDs). Finally, a symmetric alignment based segment assembly algorithm is employed for further DCD detection when no DCD was detected in the threading templates.
The on-line server system is constructed on a three-layer architecture of front-end, server-end and business logic layer. The front-end of server is implemented with HTML, CSS, JAVASCRIPT, bootstrap CSS and D3.js to preprocess the input data from the user submission and display the domain prediction results. The server-end is developed with PHP, Mysql and Perl for data persistence and constructing output pages. The business logic layer implements the backstage modeling calculations based on the ThreaDom and DomEx programs.
Domain conservation score and initial domain boundary assignment
The DC-score of the query protein is calculated based on a multiple sequence alignment matrix, which is created by matching all threading template sequences to query according to the individual query-template alignments derived by LOMETS (31). For ith query residue, the DC-score is calculated by
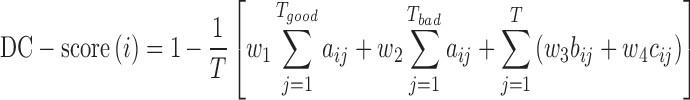 |
(1) |
where 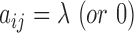 , if the ith residue on the query is (or is not) aligned with a linker region of the jth template;
, if the ith residue on the query is (or is not) aligned with a linker region of the jth template; 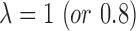 , if the domain of the template structure is defined by the CATH database (44) (or DomainParser program (45)).
, if the domain of the template structure is defined by the CATH database (44) (or DomainParser program (45)). 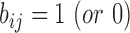 , if the ith residue is (or is not) aligned with a gap region of the jth template.
, if the ith residue is (or is not) aligned with a gap region of the jth template. 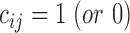 , if the ith residue is (or is not) aligned with a tail region of the jth template.
, if the ith residue is (or is not) aligned with a tail region of the jth template. 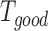 is the number of good (i.e. homologous) templates with the significance score (
is the number of good (i.e. homologous) templates with the significance score (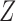 ) above the threshold (
) above the threshold (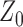 ) as specified by LOMETS, while
) as specified by LOMETS, while 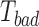 is that with
is that with 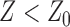 .
. 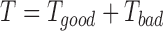 is the total number of templates identified by LOMETS.
is the total number of templates identified by LOMETS.
A residue is assigned as a potential domain linker residue if the DC-score is below the cutoff, i.e., 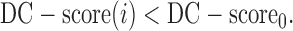 Here,
Here, 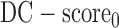 and the weight parameters (
and the weight parameters (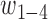 ) in Eq. (1) have been systematically trained by maximizing the Normalized Domain Overlap score (NDO-score) (46) on a non-redundant set of 800 proteins. The training dataset contains 400 single-domain, 314 two-domain, 57 three-domain and 29 four- or higher-order-domain proteins, which are defined according to the CATH 3.4 database (44). To increase the specificity, the parameters have been trained for Easy and Hard proteins, separately. Here, the protein type (easy or hard) is an estimation of the easiness or difficulty for the threading programs to detect homologous templates from the PDB library, i.e. a protein is considered as an Easy target if there are more than n homologous templates with
) in Eq. (1) have been systematically trained by maximizing the Normalized Domain Overlap score (NDO-score) (46) on a non-redundant set of 800 proteins. The training dataset contains 400 single-domain, 314 two-domain, 57 three-domain and 29 four- or higher-order-domain proteins, which are defined according to the CATH 3.4 database (44). To increase the specificity, the parameters have been trained for Easy and Hard proteins, separately. Here, the protein type (easy or hard) is an estimation of the easiness or difficulty for the threading programs to detect homologous templates from the PDB library, i.e. a protein is considered as an Easy target if there are more than n homologous templates with 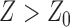 , where n = 7 is the number of threading programs in LOMETS (31); otherwise it is a hard target. The values of the optimized parameters summarized in Table 1.
, where n = 7 is the number of threading programs in LOMETS (31); otherwise it is a hard target. The values of the optimized parameters summarized in Table 1.
Table 1. Optimized parameters in ThreaDomEx.
| Parameters | Easy targets | Hard targets |
|---|---|---|
| w1 | 2.0 | 2.0 |
| w2 | 0.6 | 0.5 |
| w3 | 0.8 | 1.4 |
| w4 | 0.1 | 0.5 |
| DC-score0 | 0.6 | 0.76 |
Template-based prediction of DCD structures
A DCD consists of two or more segments that are from non-consecutive regions of the query sequence. If >30% of LOMETS templates contain DCDs, the query protein will be considered by ThreaDomEx as having DCDs.
To predict the boundary locations of DCDs, TheaDomEx first clusters all the templates that contain DCDs based on the similarity of their domain distributions, i.e. all templates, which have the same number of domains with each domain having similar boundaries, are grouped into the same cluster. Here, the ‘similar boundary’ means that the difference in boundary positions from different templates is within 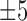 residues after the structure alignment of the two templates.
residues after the structure alignment of the two templates.
Following the clustering process, the domain structure from the largest template cluster will be used to guide the refinement of the DC-score based domain predictions. If the domain boundary difference between the domain structure of the largest template cluster and the assignments from the DC-score profile is within 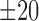 residues, for example, the separated domain segments in the DC-score models will be merged into a single DCD. Meanwhile, if the number of the domains from the DC-score assignment is >3 but less than the number of domains in the largest template cluster, ThreaDomEx will substitute the DC-score domain assignment by the domain structure from the largest template cluster, in case that the domain boundaries in >50% of all the templates are consistent (i.e. with average difference <20 residues).
residues, for example, the separated domain segments in the DC-score models will be merged into a single DCD. Meanwhile, if the number of the domains from the DC-score assignment is >3 but less than the number of domains in the largest template cluster, ThreaDomEx will substitute the DC-score domain assignment by the domain structure from the largest template cluster, in case that the domain boundaries in >50% of all the templates are consistent (i.e. with average difference <20 residues).
Further DCD prediction from symmetric segment reassembly
If there is no DCD found in the threading templates, a new symmetric segment assembly method is used to further detect DCDs. Here, when the number of domain segments assigned by the DC-score profile is 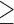 3, any pairs of two nonadjacent segments will be assembled from N-terminal to C-terminal into a putative DCD. The possibility of the putative domain to be a true DCD is assessed by three scores.
3, any pairs of two nonadjacent segments will be assembled from N-terminal to C-terminal into a putative DCD. The possibility of the putative domain to be a true DCD is assessed by three scores.
The putative domain sequence is searched by PSI-BLAST through a non-redundant domain library collected from SCOP (47), CATH (44) and Pfam (48) databases. The template similarity score (TS-score) is calculated by
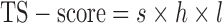 |
(2) |
where 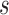 is the sequence identity between the putative domain and template sequences;
is the sequence identity between the putative domain and template sequences; 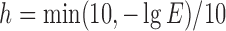 is the normalized E-value (
is the normalized E-value (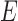 ) by PSI-BLAST; and
) by PSI-BLAST; and 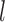 is a factor associated with the alignment coverage (32).
is a factor associated with the alignment coverage (32).
Second, a symmetry index score (SI-score) of the PSI-BLAST alignment is defined by
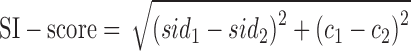 |
(3) |
where sid1,2 and c1,2 are, respectively, the sequence identity and alignment coverage of the two component segments compared to the PSI-BLAST templates.
Finally, a profile-profile alignment search, assisted with the secondary structure predictions, is performed by MUSTER (43) through the Pfam domain library, with the alignment score, 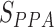 , returned.
, returned.
The putatively assembled domain sequence is predicted as a true DCD, if the scores calculated above satisfy the conditions of TS-score >TS-score0, 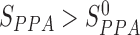 , and SI-score >SI-score0. The threshold parameters of TS-score0,
, and SI-score >SI-score0. The threshold parameters of TS-score0, 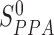 and SI-score0 have been determined by maximizing the Matthews correlation coefficient on an independent set of training proteins (32). This step of DCD detection and validation involves mainly the running of the PSI-BLAST search against a library of ∼500 millions single domain sequences and the profile-profile search through the Pfam database. It takes a much longer time (∼3–5 h) than the last step of template-based DCD detection through domain boundary clustering (<1 min).
and SI-score0 have been determined by maximizing the Matthews correlation coefficient on an independent set of training proteins (32). This step of DCD detection and validation involves mainly the running of the PSI-BLAST search against a library of ∼500 millions single domain sequences and the profile-profile search through the Pfam database. It takes a much longer time (∼3–5 h) than the last step of template-based DCD detection through domain boundary clustering (<1 min).
USING THE WEB SERVER
Input
To use the ThreaDomEx server, users need to upload the amino acid sequence of the query protein. Once the sequence is uploaded, a page containing the job ID and job status information will be displayed, which will be refreshed every 10 minutes. The users can retrieve the results in future by bookmarking this page or via the job ID from the on-line ThreaDomEx system. The server also allows the user to search the job with the submitted sequence. In addition, a URL link to the result page will be sent to the email address provided optionally by the user in the input page.
Output page and user-based interactive adjustment facilities
The procedure for multi-threading alignments, domain boundary prediction, boundary optimization and DCD detection is fully automated. The entire process of ThreaDomEx, from job submission to output generation, takes ∼4 h for a protein of 1000 residues. The data in the output page includes: (i) the DC-score profile; (ii) predicted secondary structure; (iii) histogram distribution of predicted solvent accessibility; (iv) top 50 LOMETS templates and alignments; (v) final domain model results. Most of the output data can be adjusted interactively by user based on the human-expert knowledge and experimental insights. A snapshot of the output page on an illustrative example from Chromodomain–helicase–DNA-binding protein (UniProt ID: Q86WJ1, 897 AA) is shown in Figure 2, which was taken from http://zhanglab.ccmb.med.umich.edu/ThreaDomEx/example.php. The output data for each target will be retained for three month before removed from the on-line server. The following paragraphs contain a brief description of the major items contained in the ThreaDomEx output results.
Figure 2.

An illustration of ThreaDomEx output page from chromodomain–helicase–DNA-binding protein (UniProt ID: Q86WJ1, 897 AA). (A) DC-score distribution along the query sequence; (B) predicted secondary structure; (C) predicted solvent accessibility; (D) top 50 threading templates by LOMETS; (E) the domain boundaries assigned by DC-score; (F) optimized boundaries and discontinuous domain (DCD) detection after the clustering process; (G) domain models editable by user; (H) DCD models after segment assembly and refinement.
DC-score profile
ThreaDomEx uses the DC-score profile to make initial domain segment assignments. As shown in Figure 2A, the predicted domain boundaries are marked by the orange vertical lines, which are determined by the DC-score cutoff that is marked by the green horizontal line. The default DC-score cutoff is shown in Table 1, but users are allowed to manually adjust the cutoff by dragging the horizontal line. Meanwhile, users are also allowed to modify (or add/delete) the domain boundaries by dragging (or right-clicking on) the vertical lines, where domain segment division results, as shown in color bar above the DC-score profile, are updated simultaneously according to the user's edit.
Predicted secondary structure and solvent accessibility
Domain boundaries of proteins often locate at the coils/loops and have a higher level of solvent accessibility than other regions (25). Figure 2B and C shows the distribution of predicted secondary structure (SS) and solvent accessibilities (SA), which may be used for users to fine-tune the domain boundary locations. To facilitate the referring of the SS and SA data, a thin vertical line will be displayed across the SS and SA figures when user moves the mouse on the DC-score profile figure. If a user moves the mouse along the SS figure, the detailed local secondary structure information is displayed in an enlarged pop-up window.
The top 50 LOMETS templates
This section provides information on the 50 highly ranked threading templates collected by LOMETS, which have been used by ThreaDomEx for calculated DC-score profile (Figure 2D). It includes: (i) the name of the threading programs creating the templates; (ii) the PDB ID and the link to the PDB entry for each template; (iii) visualization of query-template alignments. The domains along the alignment is marked by different colors based on the domain information of the template structure from CATH or DomainParser assignment, where segments in the same DCD are marked by the same color. The gray bars are the regions that are not within any domains defined by CATH or DomainParser. While the thin gray lines mark the regions of alignment gaps on the templates, the small red bulges above the domain bars are the insertions to query sequence. When user moves mouse along the alignment, an enlarged window will pop up to show the start and end residues of the segments or insertions for the query and template sequences.
The domain prediction results
The domain prediction results are summarized at right-top panel of the output page of the ThreaDomEx server. The data include the predicted domain boundaries according to the DC-score (Figure 2E), and the refined domain boundaries and DCDs detected by the template clustering and segment assembly process (Figure 2F). Both results are generated automatically by ThreaDomEx. This section also allows the user to edit the boundary by the ‘up to merge’ button; or to initialize, save and rollback the prediction results by the other corresponding buttons (Figure 2G), i.e. the ‘initiate’ button enabling the recovering of the original prediction results, the ‘rollback’ button helping user rollback the result to the last modification, and the ‘save’ button for result save. After the boundary modification and saving, users can re-run the DCD detection by clicking on the ‘DCD detect’ button in Figure 2H.
Despite that the server provide a variety of options for users to tweak and adjust the modeling process and results, it is worthy of noting that the ThreaDomEx server is fully automated and such manual refinement is not a required condition for successful domain model generation. In fact, the ThreaDomEx pipeline has been extensively trained and benchmarked on large-scale datasets, aiming to generate optimal domain models without using external information and human interventions. Thus, users are not encouraged to manually modify the automated modeling results, unless they have confident evidences, such as those from experimental data, biological functional analyses or reliable common sense, which are different from the automated models and deemed to be able to improve the model results.
RESULTS
Datasets and assessment criterions
Two independent datasets (Test-I and Test-II) were constructed to evaluate the performance of ThreaDomEX on the domain boundary prediction and DCD detection, respectively. The Test-I set contains 630 non-homologous proteins, which include 315 single-domain, 245 two-domain, 57 three-domain, and 13 four- or higher-order-domain proteins; the Test-II set has 481 non-redundant and multi-domain chains, which include 326 chains containing DCDs and 155 chains with only CDs. Each protein chain in Test-II has at least three segments, with segment length >40 residues. None of the testing proteins are homologous to any of the proteins that are used for training the ThreaDomEx program. To further rule out homology contamination, all the templates that have a sequence identity >30% to the query or are detectable by PSI-BLAST with an E-value <0.05 have been excluded in the threading template library.
The true domain boundaries of the test proteins are defined based on the CATH3.4 database (44). A predicted boundary is considered as correct if it is located with 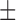 20 residues away from true domain boundary in the CATH3.4; this assessment criterion is the same as used in many previous studies (13,21). Moreover, a DCD detection is considered as correct only if the boundaries of all segments in the DCD are correct and the segment assembly is consistent to the DCD structure in the CATH3.4.
20 residues away from true domain boundary in the CATH3.4; this assessment criterion is the same as used in many previous studies (13,21). Moreover, a DCD detection is considered as correct only if the boundaries of all segments in the DCD are correct and the segment assembly is consistent to the DCD structure in the CATH3.4.
Domain boundary prediction on Test-I
The ability of domain boundary prediction is tested on the Test-I dataset, where ThreaDomEx correctly classifies the target sequences as being single- or multi-domain proteins in 81% of the cases. For the 522 ‘Easy’ proteins in which LOMETS has a higher confidence score, the average accuracy of classification is 84.7%, and for the rest of the 108 ‘Hard’ proteins, the accuracy is 68.5%. We also used the NDO-score, which is defined as the normalized overlap rate of all predicted domain and linker regions with the true assignments of the query structure (46), to evaluate the domain boundary prediction. The average NDO-score for the ‘All’, ‘Easy’ and ‘Hard’ targets is 0.893, 0.905 and 0.832, respectively. Figure 3A lists the NDO-score of ThreaDomEX, in control with that by the five publicly available domain predictor programs, including FIEFDom (11), DomPro (17), DROP (22) and PPRODO (19), on the same set of testing proteins. These programs represent a representative set of methods on homology and machine learning based approaches, where the data demonstrates the advantage and efficiency of the threading-based domain predictions on accurately assigning the protein domain locations.
Figure 3.

Benchmark results of ThreaDomEx in control with other methods. (A) Average NDO-score on the first test set of 630 proteins, with dark, gray and white histograms being for all, easy and hard proteins. (B) Average NDO-score on the second test set of 481 proteins. The dark or gray histograms mark the methods with or without ability for prediction discontinuous domains (DCD). X+DomEx refers to a hybrid method combining Method-X with DomEx to detect DCD.
We also control our method with a random domain predictor based on the regions that are allowable for domain boundary assignment along the sequence length of the testing proteins by cutting down 40 residues at each end (we have assumed that a domain is no <40 residues in size in this study), following the approach by Dovidchenko et al. (14). For the 245 two-domain proteins in the Test-I which have an average sequence length 272 residues, 61.8% (157 proteins) are correctly divided by ThreaDomEx into 2-domains within  20 residues to the true boundary, while the percentage of the random prediction is 32.6%. The probability of the random predictions to generate the same or a greater number of correct domain boundaries is very small (8.2E–72) based on the Gaussian error distribution (14), showing that the ThreaDomEx prediction is highly non-random.
20 residues to the true boundary, while the percentage of the random prediction is 32.6%. The probability of the random predictions to generate the same or a greater number of correct domain boundaries is very small (8.2E–72) based on the Gaussian error distribution (14), showing that the ThreaDomEx prediction is highly non-random.
DCD detection on Test-II
Figure 3B summarizes the NDO-score of ThreaDomEx with the other control programs for the 481 proteins from the Test-II dataset. Here, only ThreaDomEx and ThreaDom have the ability to detect DCDs, while the major difference between these two programs is that ThreaDomEx exploits DomEx to detect DCDs when ThreaDom does not detect any DCDs. Although the NDO-score of ThreaDomEx (0.759) is only slightly higher than ThreaDom (0.750), ThreaDomEx could recall 26.7% of the DCDs with 72.7% precision on the subset that ThreaDom failed to detect any DCDs, indicating that segment alignment and assembly process by DomEx can indeed help identify DCDs beyond template-based transferals.
To further examine the impact of the segment-assembly based DCD detection on the domain boundary prediction, we construct two hybrid methods that combine DomEx with the best two boundary predictors ThreaDom_Bdr and FIEFDom, denoted as ThrDm_Bdr+DomEx and FIEFDom+DomEx, respectively. ThrDm_Bdr represents a truncated program of ThreaDom that uses the DC-score profile to predict the domain boundaries but turns off the clustering-based boundary optimization and DCD detection. The results in Figure 3B shows that the inclusion of DomEx can improve the NDO-score of both programs, demonstrating a positive impact of the segment-assembly based DCD detection on the overall domain boundary prediction. But the ThreaDomEx pipeline, which includes the entire process of multiple threading, domain refinements, and segment-assembly based DCD detection, has a higher accuracy than all these testing programs.
ThreaDomEx on a public dataset
In addition to the tests on the two internal datasets, we applied ThreaDomEx on a publically available dataset at http://web.tuat.ac.jp/∼domserv/cgi-bin/LinkerList.txt, which was previously used by Ebina et al. (21). This dataset contains 206 proteins, with 174 two-domain, 24 three-domain and 8 four-domain proteins. Under a similar homologous template filter, i.e. excluding all templates with an identity identity >30% to the query or detectable by PSI-BLAST with an E-value <0.05 from the threading library, we obtained the domain boundary prediction with the average sensitivity and specificity being 0.795 and 0.601, respectively, compared to the true domain assignments. These values are significantly higher than the randomly domain assignments, which have the sensitivity and specificity being 0.221 and 0.262, respectively, calculated by Ebina et al. by randomly selecting a 11-residue window from the non-terminal region as the domain linker (21). The ThreaDomEx results also compare favorably to that by several other sequence-based predictors reported by Ebina et al., including DLP-SVM (sensitivity/specitificity = 0.597/0.436) (21) and Armadillo (0.486/0.342) (13). But it should be noted that the dataset that Ebina et al. used (containing 182 proteins) is slightly smaller than what we used here, which may account for part of the differences of the results between ThreaDomEx and these methods.
CONCLUSION
We developed a new on-line server system, ThreaDomEx, for efficient and user-friendly protein domain prediction, which was built on the multiple threading template alignments followed by domain boundary clustering and segment reassembly. Compared to traditional sequence homology and machine-learning based approaches, the threading based domain assignment, guided by a domain conservation scoring profile, achieves a significantly higher domain division accuracy, as shown in the large-scale benchmark tests. In particular, a segment assembly algorithm is introduced to enhance accuracy of both domain boundary prediction and DCD detection, which makes ThreaDomEx one of the very few on-line systems equipped with the ability to model DCD structures beyond template-based domain transferals.
The pipeline of the ThreaDomEx is fully automated. However, the overall accuracy of domain prediction can be low for the non-homologous hard proteins and those with DCDs, where the human-expert knowledge and insights from experimental data or biological function analyses will become valuable for further improving the automated domain prediction results. Considerable effort has been made to enable users to interactively edit and refine the domain predictions; these include the facilities to manually tune the DC-score threshold, delete and add domain linkers, and merge and assemble different domain segments. In addition to the final modeling results, various intermediate modeling data, including the DC-score profile, secondary structure and solvation prediction, and multiple threading template alignments, have been made available and visualizable, which not only help users to manually improve the domain predictions, but also provide valuable information to assist further structure prediction and function annotation studies for the submitted sequences.
FUNDING
National Institute of General Medical Sciences [GM083107, GM116960]; National Science Foundation [DBI1564756]; National Natural Science Foundation of China [30700162, 61073095]; Fundamental Research Funds for the Central Universities of China [HUST2014TS138, HUST2015QN101]; China Postdoctoral Science Foundation [2014M552043]. Funding for open access charge: Fundamental Research Funds for the Central Universities of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Han J.H., Batey S., Nickson A.A., Teichmann S.A., Clarke J.. The folding and evolution of multidomain proteins. Nat. Rev. Mol. Cell. Biol. 2007; 8:319–330. [DOI] [PubMed] [Google Scholar]
- 2. Kirillova S., Kumar S., Carugo O.. Protein domain boundary predictions: a structural biology perspective. Open Biochem. J. 2009; 3:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sonnhammer E.L., Eddy S.R., Durbin R.. Pfam: a comprehensive database of protein domain families based on seed alignments. Proteins. 1997; 28:405–420. [DOI] [PubMed] [Google Scholar]
- 4. Punta M., Coggill P.C., Eberhardt R.Y., Mistry J., Tate J., Boursnell C., Pang N., Forslund K., Ceric G., Clements J. et al. . The Pfam protein families database. Nucleic Acids Res. 2012; 40:D290–D301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kuroda Y., Tani K., Matsuo Y., Yokoyama S.. Automated search of natively folded protein fragments for high-throughput structure determination in structural genomics. Protein Sci. 2000; 9:2313–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hondoh T., Kato A., Yokoyama S., Kuroda Y.. Computer-aided NMR assay for detecting natively folded structural domains. Protein Sci. 2006; 15:871–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Portugaly E., Harel A., Linial N., Linial M.. EVEREST: automatic identification and classification of protein domains in all protein sequences. BMC Bioinformatics. 2006; 7:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Portugaly E., Linial N., Linial M.. EVEREST: a collection of evolutionary conserved protein domains. Nucleic Acids Res. 2007; 35:D241–D246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Heger A., Wilton C.A., Sivakumar A., Holm L.. ADDA: a domain database with global coverage of the protein universe. Nucleic Acids Res. 2005; 33:D188–D191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heger A., Holm L.. Exhaustive enumeration of protein domain families. J. Mol. Biol. 2003; 328:749–767. [DOI] [PubMed] [Google Scholar]
- 11. Bondugula R., Lee M.S., Wallqvist A.. FIEFDom: a transparent domain boundary recognition system using a fuzzy mean operator. Nucleic Acids Res. 2009; 37:452–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wheelan S.J., Marchler-Bauer A., Bryant S.H.. Domain size distributions can predict domain boundaries. Bioinformatics. 2000; 16:613–618. [DOI] [PubMed] [Google Scholar]
- 13. Dumontier M., Yao R., Feldman H.J., Hogue C.W.. Armadillo: domain boundary prediction by amino acid composition. J. Mol. Biol. 2005; 350:1061–1073. [DOI] [PubMed] [Google Scholar]
- 14. Dovidchenko N.V., Lobanov M.Y., Galzitskaya O.V.. Prediction of number and position of domain boundaries in multi-domain proteins by use of amino acid sequence alone. Curr. Protein Peptide Sci. 2007; 8:189–195. [DOI] [PubMed] [Google Scholar]
- 15. Suyama M., Ohara O.. DomCut: prediction of inter-domain linker regions in amino acid sequences. Bioinformatics. 2003; 19:673–674. [DOI] [PubMed] [Google Scholar]
- 16. Liu J., Rost B.. Sequence-based prediction of protein domains. Nucleic Acids Res. 2004; 32:3522–3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cheng J.L., Sweredoski M.J., Baldi P.. DOMpro: Protein domain prediction using profiles, secondary structure, relative solvent accessibility, and recursive neural networks. Data Mining Knowledge Discov. 2006; 13:1–10. [Google Scholar]
- 18. Yoo P.D., Sikder A.R., Taheri J., Zhou B.B., Zomaya A.Y.. DomNet: protein domain boundary prediction using enhanced general regression network and new profiles. IEEE Trans. Nanobiosci. 2008; 7:172–181. [DOI] [PubMed] [Google Scholar]
- 19. Sim J., Kim S.Y., Lee J.. PPRODO: prediction of protein domain boundaries using neural networks. Proteins. 2005; 59:627–632. [DOI] [PubMed] [Google Scholar]
- 20. Chen L.S., Wang W., Ling S.P., Jia C.Y., Wang F.. KemaDom: a web server for domain prediction using kernel machine with local context. Nucleic Acids Res. 2006; 34:W158–W163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ebina T., Toh H., Kuroda Y.. Loop-length-dependent SVM prediction of domain linkers for high-throughput structural proteomics. Biopolymers. 2009; 92:1–8. [DOI] [PubMed] [Google Scholar]
- 22. Ebina T., Toh H., Kuroda Y.. DROP: an SVM domain linker predictor trained with optimal features selected by random forest. Bioinformatics. 2011; 27:487–494. [DOI] [PubMed] [Google Scholar]
- 23. Ebina T., Suzuki R., Tsuji R., Kuroda Y.. H-DROP: an SVM based helical domain linker predictor trained with features optimized by combining random forest and stepwise selection. J. Comput.-Aided Mol. Des. 2014; 28:831–839. [DOI] [PubMed] [Google Scholar]
- 24. Eickholt J., Deng X., Cheng J.. DoBo: Protein domain boundary prediction by integrating evolutionary signals and machine learning. BMC Bioinformatics. 2011; 12:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Galzitskaya O.V., Melnik B.S.. Prediction of protein domain boundaries from sequence alone. Protein Sci. 2003; 12:696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tanaka T., Yokoyama S., Kuroda Y.. Improvement of domain linker prediction by incorporating loop-length-dependent characteristics. Biopolymers. 2006; 84:161–168. [DOI] [PubMed] [Google Scholar]
- 27. George R.A., Heringa J.. SnapDRAGON: a method to delineate protein structural domains from sequence data1. J. Mol. Biol. 2002; 316:839–851. [DOI] [PubMed] [Google Scholar]
- 28. Kim D.E., Chivian D., Malmstrom L., Baker D.. Automated prediction of domain boundaries in CASP6 targets using Ginzu and RosettaDOM. Proteins. 2005; 61:193–200. [DOI] [PubMed] [Google Scholar]
- 29. Wu Y., Dousis A.D., Chen M., Li J., Ma J.. OPUS-Dom: applying the folding-based method VECFOLD to determine protein domain boundaries. J. Mol. Biol. 2009; 385:1314–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xue Z., Xu D., Wang Y., Zhang Y.. ThreaDom: extracting protein domain boundary information from multiple threading alignments. Bioinformatics. 2013; 29:i247–i256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu S., Zhang Y.. LOMETS: A local meta-threading-server for protein structure prediction. Nucleic Acids Res. 2007; 35:3375–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xue Z., Jang R., Govindarajoo B., Huang Y., Wang Y.. Extending protein domain boundary predictors to detect discontinuous domains. PLoS One. 2015; 10:e0141541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Y. Progress and challenges in protein structure prediction. Curr. Opin. Struct. Biol. 2008; 18:342–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. George R.A., Lin K., Heringa J.. Scooby-domain: prediction of globular domains in protein sequence. Nucleic Acids Res. 2005; 33:W160–W163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang J., Yang J., Jang R., Zhang Y.. GPCR-I-TASSER: a hybrid approach to G protein-coupled receptor structure modeling and the application to the human genome. Structure. 2015; 23:1538–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Y. Interplay of I-TASSER and QUARK for template-based and ab initio protein structure prediction in CASP10. Proteins. 2014; 82:175–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Meng F.C., Kurgan L.. DFLpred: high-throughput prediction of disordered flexible linker regions in protein sequences. Bioinformatics. 2016; 32:341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Adam B., Michael S., Lutz F., Oliver B., Juri R.. Serum albumin domain structures in human blood serum by mass spectrometry and computational biology*. Mol. Cell. Proteomics MCP. 2015; 15:1105–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stojanoski V., Sankaran B., Prasad B.V., Poirel L., Nordmann P., Palzkill T.. Structure of the catalytic domain of the colistin resistance enzyme MCR-1. BMC Biol. 2016; 14:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Menon R., Panwar B., Eksi R., Kleer C., Guan Y., Omenn G.S.. Computational inferences of the functions of alternative/noncanonical splice isoforms specific to HER2+/ER−/PR− breast cancers, a chromosome 17 C-HPP study. J. Proteome Res. 2015; 14:3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ding Y.H., Gong Z., Dong X., Liu K., Liu Z., Liu C., He S.M., Dong M.Q., Tang C.. Modeling protein excited-state structures from ‘over-length’ chemical cross-links. J. Biol. Chem. 2017; 292:1187–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yan R., Xu D., Yang J., Walker S., Zhang Y.. A comparative assessment and analysis of 20 representative sequence alignment methods for protein structure prediction. Sci. Rep. 2013; 3:2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu S., Zhang Y.. MUSTER: improving protein sequence profile-profile alignments by using multiple sources of structure information. Proteins. 2008; 72:547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Orengo C.A., Michie A.D., Jones S., Jones D.T., Swindells M.B., Thornton J.M.. CATH—a hierarchic classification of protein domain structures. Structure. 1997; 5:1093–1108. [DOI] [PubMed] [Google Scholar]
- 45. Guo J.T., Xu D., Kim D., Xu Y.. Improving the performance of DomainParser for structural domain partition using neural network. Nucleic Acids Res. 2003; 31:944–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tai C.H., Lee W.J., Vincent J.J., Lee B.. Evaluation of domain prediction in CASP6. Proteins-Struct. Funct. Bioinformatics. 2005; 61:183–192. [DOI] [PubMed] [Google Scholar]
- 47. Murzin A.G., Brenner S.E., Hubbard T., Chothia C.. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995; 247:536–540. [DOI] [PubMed] [Google Scholar]
- 48. Bateman A., Coin L., Durbin R., Finn R.D., Hollich V., Griffiths-Jones S., Khanna A., Marshall M., Moxon S., Sonnhammer E.L. et al. . The Pfam protein families database. Nucleic Acids Res. 2004; 32:D138–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]