


Abstract
The accurate calculation of protein/nucleic acid–ligand interactions or condensed phase properties by force field-based methods require a precise description of the energetics of intermolecular interactions. Despite the progress made in force fields, small molecule parameterization remains an open problem due to the magnitude of the chemical space; the most critical issue is the estimation of a balanced set of atomic charges with the ability to reproduce experimental properties. The LigParGen web server provides an intuitive interface for generating OPLS-AA/1.14*CM1A(-LBCC) force field parameters for organic ligands, in the formats of commonly used molecular dynamics and Monte Carlo simulation packages. This server has high value for researchers interested in studying any phenomena based on intermolecular interactions with ligands via molecular mechanics simulations. It is free and open to all at jorgensenresearch.com/ligpargen, and has no login requirements.
INTRODUCTION
Recent advances in computer hardware, algorithm development and force field parameterization have contributed greatly to increased use of molecular simulations in many aspects of biomolecular studies (1,2). Nowadays, computational approaches have become widely popular to study the structure, mechanism of action and biomolecule–ligand interactions of proteins and nucleic acids. These methods, mostly based in molecular mechanics (MM), require force fields (FF) that provide an accurate description of the interactions to provide valid predictions. For instance, a 1.4 kcal/mol difference in binding free energy translates to an order of magnitude difference in binding. Most of the common MM force fields, such as CHARMM (3), OPLS-AA (4) or AMBER (5) provide complete parameters for proteins and nucleic acids, but the parameterization of the ligands remains an open problem due to the vast chemical space found in combinatorial and medicinal chemistry. Fortunately, bonded and van der Waals parameters can generally be ported from existing analogous atom types. Thus, the most critical issue in ligand parameterization is the estimation of a balanced set of partial atomic charges effective at reproducing experimental properties such as hydration free energies (HFE) or free energies of binding (ΔGbind).
The OPLS-AA force field was initially developed and parameterized to reproduce experimental heats of vaporization and densities of small organic molecules and was later extended to include proteins (6,7) and nucleic acids (8). As individual parameterization of sets of partial atomic charges for the countless possible organic ligands is not practical, several different quantum mechanics (QM) based charge models have been proposed. In particular, the CMx models developed by Cramer et al. (9–14) have been tested extensively in condensed phase simulations in combination with the OPLS-AA force field, usually by comparison between calculated and experimental hydration free energies, heats of vaporization and densities. The most accurate charge models to reproduce experimental HFEs with OPLS-AA have been found to be 1.20*CM5 (15) and 1.14*CM1A (16) charges. The accuracy of 1.14*CM1A charges is further improved by the use of localized bond charge corrections (LBCC), making 1.14*CM1A-LBCC (17) the best among existing QM charge models at the same computational cost of CM1A charges.
Once an appropriate set of parameters is obtained the problem is reduced to transferring them into the desired software. Each simulation program requires the specification of geometries, topologies and parameters in one or more files in specific formats. Although most of the packages provide utilities to prepare these files using their built-in parameter set and charge model, it is far from trivial to use a different set of parameters in any given program. The LigParGen web server was developed to address this problem by providing a simple, automatic procedure to assign the parameters and generate the files needed to do calculations in some of the most commonly used simulation packages.
There are other servers that provide force field parameters for MD simulations. Among them the CGenFF (18,19), MATCH (20), CHARMM-GUI (21) and CHARMMing (22) servers provide files formatted for CHARMM with parameters from the CHARMM36/CGenFF force field and charges assigned by analogy. The SwissParam (23) server produces CHARMM input files combining bonded parameters from MMFF and nonbonded ones from CHARMM22. Two servers that generate Amber-formatted files are H++ (24), which uses the ambertools (25) package to assign GAFF parameters with AM1-BCC charges, and R.E.D (26) that provides RESP and/or ESP charges fitted from an ab initio calculation. It must be noted that some of the simulation packages provide utilities to read files formatted for other packages. Lastly, the PRODRG (27) server provides GROMOS FF parameters and charges in a GROMACS formatted file.
The LigParGen server was designed to provide OPLS-AA parameters for neutral organic molecules using two CM1A charge models, 1.14*CM1A and 1.14*CM1A-LBCC, while unscaled CM1A charges are provided for charged molecules. The output files are formatted to be used directly in the popular molecular dynamics MD programs CHARMM (28), NAMD (29), GROMACS (30) and OpenMM (31) and the Monte Carlo (MC) software BOSS and MCPRO (32). The server can also generate a single file in PQR format, which are used for Poisson–Boltzmann calculations or docking. In addition, the input is very flexible. The input molecule can be specified with 3-dimensional coordinates in a MOL or PDB file, or a simplified 2D representation can be entered as a SMILES string, or the structure may be drawn on the web page through the use of the JSME (33) plugin, or it may be copied directly from a drawing program such as ChemDraw (34).
MATERIALS AND METHODS
The core of the LigParGen server is the internal use of the BOSS (32) software to assign the bonded and van der Waals parameters by analogy to the existing atom types in the latest OPLS-AA force field (4). Subsequently a semiempirical AM1 (9) calculation is performed to calculate and assign the charges. The server can, as directed by the user, utilize one of two CM1A-derived charge models as described briefly below. For further information about technical details and comparisons, please read the original papers (9,16).
In general, quantum mechanics population analysis methods distribute the total electron density of a molecule into partial charges centered on each atom of the molecule. As partial charges are not observables, there are different ways to partition the electron density. The CM1A method uses the Mulliken population analysis from the electron density obtained by the AM1 method from the ligand geometry. Mulliken charges for an atom A are computed using the following equation:
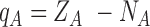 |
(1) |
where 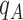 is the partial Mulliken charge,
is the partial Mulliken charge, 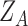 is the nuclear charge of the atom A and
is the nuclear charge of the atom A and 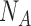 is the electron density assigned to atom A as described by the equation:
is the electron density assigned to atom A as described by the equation:
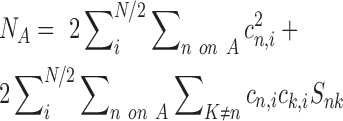 |
(2) |
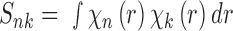 |
(3) |
where N is the total number of electrons in the molecule, 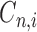 is the molecular orbital coefficient for the atomic orbital
is the molecular orbital coefficient for the atomic orbital 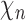 and
and 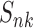 is the QM overlap integral. This electron density definition is based on the linear combination of atomic orbital–molecular orbital (LCAO–MO) method where the molecular electronic distribution per each molecular orbital is defined each as a linear combination of atomic orbitals (n).
is the QM overlap integral. This electron density definition is based on the linear combination of atomic orbital–molecular orbital (LCAO–MO) method where the molecular electronic distribution per each molecular orbital is defined each as a linear combination of atomic orbitals (n).
The CM1A charges are then computed using a multilinear transformation of the Mulliken charges based in the computed bond orders to improve the molecular dipole moment using empirical parameters. Then, for neutral molecules, the 1.14*CM1A model scales the charges by a factor 1.14, which was fitted to improve the agreement of the HFEs to the experimental values (16). If the total charge of the molecule is not zero, partial charges are not scaled. It should be noted that, as in all quantum mechanics based charges, the CM1A charges can have some variations due to the molecular geometry. The typical variations observed in our tests are in the 0.03–0.05 e range, with a few cases involving intramolecular hydrogen bonds reaching 0.1e.
A later evaluation of HFEs for a set of 426 organic molecules showed that some moieties such as phenyl rings, aldehydes or ketones are not well parameterized by the 1.14*CM1A charge model, leading to a mean unsigned error (MUE) of 1.5 kcal/mol with respect to experimental HFE data. The performance of CM1A charges was improved by adding Localized Bond Charge Corrections (LBCC), by which small charge adjustments are made to the partial charges for atoms in problematic bond types such as, CT-OH in aliphatic alcohols. Only 19 LBCCs were enough to reduce the errors with the 1.14*CM1A charges for the 426 HFE values to only 0.61 kcal/mol. These adjustments give rise to the 1.14*CM1A-LBCC charge method which can also be provided by the LigParGen server.
DESCRIPTION OF THE WEB SERVER
Overall description
The LigParGen web server is a free, robust and multi-platform tool to generate OPLS-AA parameters for organic molecules using the 1.14*CM1A and 1.14*CM1A-LBCC charge models. It has been implemented in CGI-python using the Apache HTTP server and the Bootstrap framework to allow access from any internet device (computers, tablets, cell phones, etc.) while keeping an optimal appearance. In order to facilitate its use, a sample button uses the benzene molecule to provide a quick exploration of the server features for novice users. It is important to mention that there is no login required for job submission (see Figure 1A). The server includes in its home web page the Clustrmap tracking system to analyze user relevant information such as location, operating system or internet browser for statistical purposes, future improvements, or for debugging in case specific failures are detected.
Figure 1.

LigParGen Screenshots. Panel A, main input web page including a sample button and the Clustrmap tracking system. Panel B, ‘Draw Molecule’ input page screenshot including a sample molecule visualization. Panel C, output web page including a 2D representation and the JSMOL 3D visualizer. Panel D, LigParGen server main scheme.
Additionally, a section of tutorials by example is included in the LigParGen web page to illustrate the preparation of ligand–water boxes or protein–ligand systems for molecular dynamics simulations using the output templates generated by the server. This tutorial section includes detailed explanations, comments, and snippets to understand and perform the equilibration and production phases of MD in GROMACS, NAMD and openMM. Furthermore, LigPargen server aims to be a dynamic tool with constant improvement in response to users’ feedback. For this reason, a contact section requests details for suggestions.
Input
The server currently accepts three different standard input formats for molecular structures: SMILES codes, PDB and MOL files. As the request will be processed by the BOSS package, which is used to calculate the CM1A charges, the maximum number of atoms is currently 200. A single-point calculation is done by default on the input structure, but users can request a structure optimization using the assigned OPLS-AA parameters and 1.14*CM1A or 1.14*CM1A-LBCC by checking the ‘Optimize Molecule’ radio button (see Figure 1A and B). The structure optimization is carried out by BOSS using the Broyden–Fletcher–Goldfarb–Shanno variable metric algorithm (35–38). The flow of the different operations in the server includes a robust error management system to detect possible problems in the submission process. For instance, if the proposed total charge of the ligand is not realistic an error page will be shown with an explanation (see Figure 1D).
Furthermore, the web server offers the possibility to interactively draw a 2D structure of the desired molecule using the JSME javascript plugin. JSME allows actions such as edit the molecule, export it in different formats or search in molecular databases, and it is compatible with iPhone, Android smartphones, iPad, and Android tablets. This option is located in the ‘Draw Molecule’ section and contains the same submission options than the default input section. Prior to the submission, any 2D structures generated by JSME, can be converted into SMILES code using the ‘make SMILES’ button, and the SMILES code can be copied for future submissions (See Figure 1B).
Output and representation of the results
The input molecule provided by the user in 2D (SMILES) or 3D (MOL or PDB) format is converted in several stages to a BOSS Z-matrix for processing by the BOSS program. If needed, the SMILES string is first transformed to a 3D MOL file using the RDKit chemoinformatics package. If no errors are detected in the input, the BOSS utility autozmat is used to create a Z-matrix. The latter is used by LigParGen to call the BOSS program to calculate the CM1A charges and assign parameters for bonded and van der Waals interactions. At this point the sum of the CM1A charges and the user defined charge are compared. If the two total charges match, the parameter and coordinate files are generated in all the different formats and displayed in the LigParGen output page (see Figure 1C).
The output page of LigParGen provides a publication quality image generated using Pymol (Schrodinger, LLC), as well as an interactive 3D visualization panel for the submitted molecule powered by the JSMOL (39) JavaScript application (see Figure 1C). The latter can be useful to identify possible mistakes in the submitted structures, particularly when the structure is generated from a SMILES code. The user can download each output template file needed to run an MD or MC simulation for CHARMM/NAMD, GROMACS, OpenMM, BOSS/MCPRO by clicking directly on the individual download buttons or a compressed zip file with all of the files can be downloaded by clicking the ‘ALL’ button. In addition to the MD/MC files, a PQR file format is generated with radii based on the OPLS-AA σ parameters and the selected charge model to perform Poisson-Boltzmann or docking calculations. It should be noted that all the files generated during a run are tagged by including in the name a unique six-character alphanumeric code used to manage different concurrent jobs and at the same time acting as a queue system. Also, it avoids vulnerabilities in the submissions. As an additional security measure, the server does not store any information about the users and the submitted structures, and all the job-related files are removed after an hour.
Processing time
The time required for processing depends on the ligand size and the number of optimization steps needed. The calculations of the charges are the most expensive compared to other parameters and are computed on the fly. Since these are based on semiempirical QM calculations, the total computational time for small or medium molecules is negligible, and output generation takes <5 s without optimization. If ligand optimization is requested in the submission, the running time can be increased up to 30–45 s depending on the system size and quality of the input structure. For SMILES input, the optimization time is decreased because structures are generated close to the equilibrium conformation.
RESULTS
The server capabilities and robustness are illustrated in two different sets of calculations. The first set present a direct comparison between single-point energy evaluations in vacuum from a representative test set of molecules to demonstrate the coherence between the different template outputs. The second case shows a comparison between experimental and computed HFE results, demonstrating a practical application of the output files generated by the server.
Comparison of gas phase energetics
Fifty two molecules from the SAMPL4 (40) dataset server were chosen to verify the coherence of parameters, topology and structure between the BOSS, OpenMM, NAMD and GROMACS programs using SMILES codes as input. BOSS single-point energies in vacuum were used as reference values to compute the mean unsigned error (MUE) for each energy term and package as shown in Table 1. A more detailed comparison of the energetics for all ligands can be found in the SI table. As seen in Table 1, the total single-point energies from GROMACS, NAMD and OpenMM are in very good agreement with the BOSS energies, showing a MUE of 0.03–0.02 kcal/mol for the entire test set. In most cases, the bond energy term produces the highest MUE, due to the sensitivity of the large stretching force constants to the loss of accuracy in the input coordinates (a BOSS Z-matrix uses five decimal figures while PDB/GRO uses just 3). On the other hand, the difference in non-bonded energies is due to the different values for the vacuum permittivity constants used by NAMD, OpenMM and GROMACS. Overall, the differences seen in the total energy is in the order of 10–20 cal/mol, and are not expected to have a significant effect on the outcome of MD simulations.
Table 1. Total energy MUE for the representative test set from SAMPL4 for different MD packages with respect to BOSS energies. All energies are shown in 10−2 kcal/mol.
| Package | Bond | Angle | Torsion | Nonbonded | Total |
|---|---|---|---|---|---|
| GROMACS | 1.5 | 0.8 | 0.1 | 0.7 | 1.9 |
| NAMD | 1.5 | 0.8 | 0.1 | 2.2 | 2.9 |
| OpenMM | 1.5 | 0.8 | 0.4 | 0.8 | 2.0 |
Evaluation of hydration free energies
Calculating HFE is an integral part of the estimation of binding free energy. To demonstrate the applicability of parameters and topology generated by the LigParGen server, MD/FEP calculations of the absolute HFE for 10 molecules which produce accurate results from the SAMPL4 data set were performed. The simulations were done both in water and gas phase using the NAMD package to calculate HFE. Water molecules were represented using the TIP3P water model while the ligands were represented using the OPLS-AA/1.14*CM1A-LBCC force field parameters generated by the server. Each ligand was solvated in a cubic box with a 13 Å padding, and a cutoff of 10 Å was used for calculating non-bonding interactions with PME for long range electrostatics. The Lennard–Jones interactions were smoothed off over last 0.5 Å and long-range corrections were included. All simulations were run at constant pressure and temperature, 1 atm and 300 K. A detailed description of the methodology is presented elsewhere (41). In brief, each ligand was annihilated in water and in the gas phase over 36 windows. Each window was equilibrated for 1 ns and the averaging was done over the next 1 ns. Following a decoupling scheme, the electrostatic interactions were switched off first, followed by LJ interactions. Soft-core potentials were used to avoid any end-point catastrophes. Both forward and backward simulations were performed to improve the accuracy of FEP calculations. The Bennett Acceptance Ratio estimator implemented in VMD was used to obtain the free energy changes (42). The free energies of hydration were obtained from the difference between the aqueous-phase and gas-phase free energies of annihilation, and are summarized in Table 2. From the comparison with the experimental data given in Table 2, it can be seen that the OPLS-AA/1.14*CM1A-BCC parameters produced by the LigParGen server perform notably well for hydration free energies with MUE and Mean Signed Error (MSE) of 0.6 kcal/mol and 0.0 kcal/mol, respectively.
Table 2. Comparison between experimental and computed free energies of hydration for 10 molecules selected from SAMPL4 data set.
| Molecule | Free energy of hydration (kcal/mol) | |||
|---|---|---|---|---|
| Experimental | Uncertainty | Calculated | Uncertainty | |
| 1-(2-Hydroxyethylamino)-9,10-anthraquinone | −14.21 | 1.10 | −12.82 | 0.48 |
| 1,1-Diphenylethene | −2.78 | 0.10 | −3.34 | 0.27 |
| 1-Amino-9,10-anthraquinone | −9.44 | 0.74 | −9.48 | 0.32 |
| 2,6-Dichlorosyringaldehyde | −8.68 | 0.76 | −8.18 | 0.36 |
| 2-Methylbenzaldehyde | −3.93 | 0.10 | −3.47 | 0.24 |
| 3,4-Dichlorophenol | −7.29 | 0.10 | −7.21 | 0.25 |
| 3,5-Dichlorosyringol | −6.44 | 0.38 | −6.45 | 0.33 |
| 5-Chloroguaiacol | −5.26 | 0.12 | −6.43 | 0.31 |
| Hexyl acetate | −2.29 | 0.12 | −3.72 | 0.32 |
| Nerol | −3.96 | 0.20 | −3.52 | 0.30 |
| MUE | 0.61 | |||
| MSE | 0.03 | |||
CONCLUSIONS AND FUTURE DEVELOPMENT
Here, we present the first server to generate, in an automated manner, OPLS-AA/CM1A parameter files for organic ligands starting from 3D MOL or PDB files, or even 2D chemical drawings or SMILES strings. The ligand parameters can be generated for common simulation software packages such as NAMD, GROMACS, OpenMM, BOSS and MCPRO. These capabilities allow the users to obtain high-quality parameters for MM simulations without extensive knowledge about MM force fields or QM methods. This server will be useful for any group interested in the study of biomolecule-ligand interactions, with direct applications to computational drug design, or condensed-phase ligand properties via MD or MC methods.
The LigParGen server is an ongoing project, and future improvements will reflect the feedback provided by the users. In the next update, we are planning to generate parameter files for the AMBER software.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are very grateful to the more than 30 researchers that helped us in the debugging process submitting their ligands and providing us constructive and helpful feedback about their experience with the LigParGen server. In particular, the authors would like to acknowledge to John Faver, Daniel Cole, Michael Robertson, Yue Qian and Andreas Mecklenfeld for their in-depth testing and detailed reports on bugs and suggestions that were crucial to increase robustness of the server.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [GM32136]; Yale University Faculty of Arts and Sciences High Performance Computing Center. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Hospital A., Goni J.R., Orozco M., Gelpi J.L.. Molecular dynamics simulations: advances and applications. Adv. Appl. Bioinform. Chem. 2015; 8:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sponer J., Banas P., Jurecka P., Zgarbova M., Kuhrova P., Havrila M., Krepl M., Stadlbauer P., Otyepka M.. Molecular dynamics simulations of nucleic acids. From tetranucleotides to the ribosome. J. Phys. Chem. Lett. 2014; 5:1771–1782. [DOI] [PubMed] [Google Scholar]
- 3. Best R.B., Zhu, Shim X., Lopes J., Mittal P.E., Feig J., Mackerell M., MacKerell A.D. Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi (1) and chi (2) dihedral angles. J. Chem. Theory Comput. 2012; 8:3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Robertson M.J., Tirado-Rives J., Jorgensen W.L.. Improved peptide and protein torsional energetics with the OPLS-AA force field. J. Chem. Theory. Comput. 2015; 11:3499–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., Simmerling C.. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006; 65:712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jorgensen W.L., Tirado-Rives J.. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988; 110:1657–1666. [DOI] [PubMed] [Google Scholar]
- 7. Jorgensen W.L., Maxwell D.S., Tirado-Rives J.. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996; 118:11225–11236. [Google Scholar]
- 8. Pranata J., Wierschke S.G., Jorgensen W.L.. OPLS potential functions for nucleotide bases. Relative association constants of hydrogen-bonded base pairs in chloroform. J. Am. Chem. Soc. 1991; 113:2810–2819. [Google Scholar]
- 9. Storer J.W., Giesen D.J., Cramer C.J., Truhlar D.G.. Class IV charge models: a new semiempirical approach in quantum chemistry. J. Comput. Aided Mol. Des. 1995; 9:87–110. [DOI] [PubMed] [Google Scholar]
- 10. Li J., Zhu T., Cramer C.J., Truhlar D.G.. New Class IV charge model for extracting accurate partial charges from wave functions. J. Phys. Chem. A. 1998; 102:1820–1831. [Google Scholar]
- 11. Kelly C.P., Cramer C.J., Truhlar D.G.. SM6: a density functional theory continuum solvation model for calculating aqueous solvation free energies of neutrals, ions, and solute-water clusters. J. Chem. Theory Comput. 2005; 1:1133–1152. [DOI] [PubMed] [Google Scholar]
- 12. Thompson J.D., Cramer C.J., Truhlar D.G.. Parameterization of charge model 3 for AM1, PM3, BLYP, and B3LYP. J. Comput. Chem. 2003; 24:1291–1304. [DOI] [PubMed] [Google Scholar]
- 13. Marenich A.V., Olson R.M., Kelly C.P., Cramer C.J., Truhlar D.G.. Self-consistent reaction field model for aqueous and nonaqueous solutions based on accurate polarized partial charges. J. Chem. Theory Comput. 2007; 3:2011–2033. [DOI] [PubMed] [Google Scholar]
- 14. Marenich A.V., Jerome S.V., Cramer C.J., Truhlar D.G.. Charge model 5: an extension of hirshfeld population analysis for the accurate description of molecular interactions in gaseous and condensed phases. J. Chem. Theory Comput. 2012; 8:527–541. [DOI] [PubMed] [Google Scholar]
- 15. Dodda L.S., Vilseck J.Z., Cutrona K.J., Jorgensen W.L.. Evaluation of CM5 charges for nonaqueous condensed-phase modeling. J. Chem. Theory Comput. 2015; 11:4273–4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Udier-Blagovic M., Morales De Tirado P., Pearlman S.A., Jorgensen W.L.. Accuracy of free energies of hydration using CM1 and CM3 atomic charges. J. Comput. Chem. 2004; 25:1322–1332. [DOI] [PubMed] [Google Scholar]
- 17. Dodda L.S., Vilseck J.Z., Tirado-Rives J., Jorgensen W.L.. 1.14*CM1A-LBCC: localized bond-charge corrected CM1A charges for condensed-phase simulations. J Phys Chem B. 2017; 121:3864–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vanommeslaeghe K., MacKerell A.D. Jr. Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model. 2012; 52:3144–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vanommeslaeghe K., Raman E.P., MacKerell A.D. Jr. Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012; 52:3155–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yesselman J.D., Price D.J., Knight J.L., Brooks C.L. 3rd. MATCH: an atom-typing toolset for molecular mechanics force fields. J. Comput. Chem. 2012; 33:189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jo S., Kim T., Iyer V.G., Im W.. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 2008; 29:1859–1865. [DOI] [PubMed] [Google Scholar]
- 22. Miller B.T., Singh R.P., Klauda J.B., Hodoscek M., Brooks B.R., Woodcock H.L. 3rd. CHARMMing: a new, flexible web portal for CHARMM. J. Chem. Inf. Model. 2008; 48:1920–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zoete V., Cuendet M.A., Grosdidier A., Michielin O.. SwissParam: a fast force field generation tool for small organic molecules. J. Comput. Chem. 2011; 32:2359–2368. [DOI] [PubMed] [Google Scholar]
- 24. Anandakrishnan R., Aguilar B., Onufriev A.V.. H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012; 40:W537–W541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Case D.A., Cheatham T.E. 3rd, Darden T., Gohlke H., Luo R., Merz K.M. Jr, Onufriev A., Simmerling C., Wang B., Woods R.J.. The Amber biomolecular simulation programs. J. Comput. Chem. 2005; 26:1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vanquelef E., Simon S., Marquant G., Garcia E., Klimerak G., Delepine J.C., Cieplak P., Dupradeau F.Y.. R.E.D. Server: a web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids Res. 2011; 39:W511–W517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schuttelkopf A.W., van Aalten D.M.. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D. Biol. Crystallogr. 2004; 60:1355–1363. [DOI] [PubMed] [Google Scholar]
- 28. Brooks B.R., Brooks C.L. 3rd, Mackerell A.D. Jr, Nilsson L., Petrella R.J., Roux B., Won Y., Archontis G., Bartels C., Boresch S. et al. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009; 30:1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kale L., Schulten K.. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005; 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindahl E.. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015; 1–2:19–25. [Google Scholar]
- 31. Eastman P., Friedrichs M.S., Chodera J.D., Radmer R.J., Bruns C.M., Ku J.P., Beauchamp K.A., Lane T.J., Wang L.P., Shukla D. et al. OpenMM 4: a reusable, extensible, hardware independent library for high performance molecular simulation. J. Chem. Theory Comput. 2013; 9:461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jorgensen W.L., Tirado-Rives J.. Molecular modeling of organic and biomolecular systems using BOSS and MCPRO. J. Comput. Chem. 2005; 26:1689–1700. [DOI] [PubMed] [Google Scholar]
- 33. Bienfait B., Ertl P.. JSME: a free molecule editor in JavaScript. J Cheminform. 2013; 5:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cousins K.R. Computer review of ChemDraw Ultra 12.0. J. Am. Chem. Soc. 2011; 133:8388. [DOI] [PubMed] [Google Scholar]
- 35. Broyden C.G. The convergence of a class of double-rank minimization algorithms 1. general considerations. J. Inst. Math. Its Appl. 1970; 6:76–90. [Google Scholar]
- 36. Fletcher R. A new approach to variable metric algorithms. Comput. J. 1970; 13:317–322. [Google Scholar]
- 37. Goldfarb D. A family of variable-metric methods derived by variational means. Math. Comp. 1970; 24:23–26. [Google Scholar]
- 38. Shanno D.F. Conditioning of quasi-Newton methods for function minimization. Math. Comp. 1970; 24:647–656. [Google Scholar]
- 39. Hanson R.M., Prilusky J., Renjian Z., Nakane T., Sussman J.L.. JSmol and the next‐generation web‐based representation of 3D molecular structure as applied to proteopedia. Isr. J. Chem. 2013; 53:207–216. [Google Scholar]
- 40. Guthrie J.P. SAMPL4, a blind challenge for computational solvation free energies: the compounds considered. J. Comput. Aided Mol. Des. 2014; 28:151–168. [DOI] [PubMed] [Google Scholar]
- 41. Chipot C., Pohorille A.. Free energy calculations. Springer Ser. Chem. Phys. 2007; 86: [Google Scholar]
- 42. Liu P., Dehez F., Cai W., Chipot C.. A toolkit for the analysis of free-energy perturbation calculations. J. Chem. Theory Comput. 2012; 8:2606–2616. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.