


Abstract
Molecular replacement (MR) is one of the most common techniques used for solving the phase problem in X-ray crystal diffraction. The success rate of MR however drops quickly when the sequence identity between query and templates is reduced, while the I-TASSER-MR server is designed to solve the phase problem for proteins that lack close homologous templates. Starting from a sequence, it first generates full-length models using I-TASSER by iterative structural fragment reassembly. A progressive sequence truncation procedure is then used for editing the models based on local variations of the structural assembly simulations. Next, the edited models are submitted to MR-REX to search for optimal placements in the crystal unit-cells through replica-exchange Monte Carlo simulations, with the phasing results used by CNS for final atomic model refinement and selection. The I-TASSER-MR algorithm was tested in large-scale benchmark datasets and solved 36% more targets compared to using the best threading templates. The server takes primary sequence and raw crystal diffraction data as input, with output containing annotated phase information and refined structure models. It also allows users to choose between different methods for setting B-factors and the number of models used for phasing. The online server is freely available at http://zhanglab.ccmb.med.umich.edu/I-TASSER-MR.
INTRODUCTION
Molecular replacement (MR) is a fast and cost-effective method for addressing the phase problem in X-ray diffraction, which is one of the most critical steps for determining the final crystal structure of proteins. MR estimates the phase of each diffraction amplitude by placing one or more homologous models in the unit cell of the crystal and searching for the best match between the calculated and experimental intensity data (1). With the expansion of the protein structure database and the advance of computational homology modeling, more and more proteins have had their structure determined by MR. In 2016, for example, MR has been used to solve around 78% of deposited macromolecular structures in the Protein Data Bank (PDB) (2).
Due to the efficiency of the MR, many efforts have been made to develop pipelines for automated phasing and protein structure determination. For example, Claude et al. combined T-COFFEE, MODELLER, AMoRe and CNS into a web server, CaspR, for MR using homology models (3). BALBES focused on designing a database which was chosen from the PDB with multimeric as well as domain organization (4). MrBUMP used FASTA and/or SSM to pick up templates and then used multiple methods to prepare search models for MR (5,6). Schmidberger et al. developed MrGrid, a portable grid based MR pipeline that allows multiple MR calculations across a grid of networked computers (7). Schwarzenbacher et al. designed the JCSG MR pipeline according to their finding that the more sophisticated profile–profile alignment methods (FFAS) can improve distant-homology template detection and increase the success rate of MR in cases of low target-template similarity (8,9). Recently, Bibby et al proposed the AMPLE pipeline which uses rapidly computed ab initio folding models to solve the crystal structures of small proteins based on a cluster-and-truncate approach (10).
Despite the significant effort and progress, the success rate of the MR models still relies essentially on the availability and quality of the homologous templates in the PDB, which limits the usefulness of the technique in a broader range of genome sequences. Recent years have witnessed considerable progress in distant homology-template detection and structure refinement (11). The I-TASSER method, for example, has shown to be able to generate correct fold for 3/4 of the sequences in the community-wide blind CASP experiment (12), where more than 80% of the templates were found to be drawn closer to their native structures (13). To examine the potential of using the cutting-edge structure prediction tools for improving MR, we recently developed an integrated pipeline, I-TASSER-MR (14), which starts with I-TASSER based protein structure prediction, followed by progressive model editing and then by the MR search and automated model refinement. By combining iterative fragmental structure assembly simulations and progressive trimming of poorly modeled regions, the pipeline increases the success rate of MR from 43 to 59% for the cases in which the target-template similarity is below 30%.
In this work, we propose to construct a new online MR server based on the I-TASSER-MR algorithm for automated phasing and structure determination, with focus on the proteins without closely homologous templates in the PDB. To enhance the efficiency and robustness of the server system, a new Monte Carlo based phasing program, MR-REX, is introduced to the I-TASSER-MR pipeline, which was shown to significantly improve the success rate of MR (J. Virtanen and Y. Zhang, submitted). Meanwhile, a variety of options are designed to enhance the facility and convenience of the system by providing optional parameter settings and intermediate structure and MR modeling data. The online system is freely available at http://zhanglab.ccmb.med.umich.edu/I-TASSER-MR/.
IMPLEMENTATION
The server is based on the I-TASSER-MR method (14) for MR using iterative fragmental structure assembly simulations followed by progressive sequence truncation. The flowchart of the server is depicted in Figure 1. In the first step, the query sequence is threaded through a representative PDB structure library to search for structural templates and super-secondary structure motifs by LOMETS (15). Continuous fragments are then excised from the threading aligned regions of the top-ranked templates, which are used to reassemble full-length models with the threading unaligned regions built by an on-and-off lattice-based folding procedure (16). The conformational space of the protein structure is searched by replica-exchange Monte Carlo (REMC) simulations (17). The structure trajectories are finally clustered by SPICKER (18), with cluster centroids obtained by averaging the coordinates of the decoys in the clusters.
Figure 1.

Flowchart of I-TASSER-MR. The target sequence is first threaded through a non-redundant PDB structure library to identify structural templates by LOMETS, with full-length 3D models constructed by iterative fragment reassembly simulations with I-TASSER. The structure models are then progressively edited based on AVS with the poorly-predicted regions truncated. The resulting models are submitted, together with the X-ray diffraction data, to MR-REX for Monte Carlo based MR search, with the output models finally refined by CNS.
Secondly, unreliably modeled residues in the I-TASSER simulations are identified and truncated based on the average variation score, AVS, defined as:
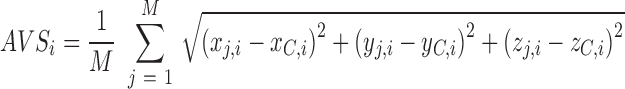 |
(1) |
where M is number of structure decoys in the SPICKER cluster. (xj,i, yj,i, zj,i) and (xC,i, yC,i, zC,i) are coordinates of the ith residue of the jth decoy and that of the cluster centroid model, respectively, after the TM-score structure superposition (19). Residues are first sorted by their respective AVSs, and those with the highest AVSs are progressively truncated to generate a series of search models at different level of AVS cut-offs. Up to 60 edited copies will be attempted for each I-TASSER model with the last copy having 40% of residues remaining.
In the third step, a newly developed MR method, MR-REX (Virtanen and Zhang, http://zhanglab.ccmb.med.umich.edu/MRREX/), is used to conduct an MR search in the unit cell of crystal. Briefly, MR-REX employs iterative REMC simulations to search for the correct placements of protein models into their unit cells in a 6n-dimensional space with n being the number of components in the asymmetric unit. One advantage of MR-REX is that the REMC simulations allow corporative rotation and translation searches and simultaneous clash and occupancy optimization. The large-scale benchmark tests have shown that MR-REX was able to generate more successful MR solutions than the state of the art MR program, Phaser (20), based on the same set of starting structure models, in particular for the cases that have a low quality (see ‘Results’ section below). However, since different MR programs are often complementary, the success rate of I-TASSER-MR can be further improved by combining MR-REX with other state of the art phasing tools such as Phaser (20), Molrep (21) and AMoRe (22). Since these tools have license limits that do not allow online uses, the server provides the link to download all the edited search models, so that users can use them as inputs to the other MR tools at their local computers in addition to MR-REX.
Finally, the solutions from MR-REX are quickly refined by the CNS program (23), and the Rfree factors, which measure the agreement between crystallographic model and experimental X-ray diffraction data, are used to rank the different MR solutions. Since the major purpose of the use of CNS here is for model ranking and evaluation, only part of the CNS procedure, i.e. the initial rigid-body refinement and simulated annealing, is implemented, whereas detailed refinement options, such as different protonation states and ligands and modified residues, have not been considered in the process.
The interface of the I-TASSER-MR server is handled on a Dell R620 server, with the MR modeling simulations implemented on an IBM NeXtScale nx360 super-computing cluster that can run 1200 simulation jobs in parallel. Part of the simulation jobs is also run on the community-wide resource from the XSEDE super-computing cluster (24).
USING THE WEB SERVER
Input
When using the I-TASSER-MR server, the user needs to give the amino acid sequence of the query protein, copy number (i.e. the number of monomers in asymmetric unit) and the crystallographic structure factors in MTZ format. To improve the efficiency, the users are allowed to choose whether the first or top five I-TASSER models are used for further MR processing and refinement. The first I-TASSER model is often the closest to the correct structure, but sometimes the lower ranked models from different SPICKER clusters represent better search models. Moreover, different B-factor schemes can be complementary and optimal MR results could be obtained from multiple B-factor predictors, so three different options for setting B-factors are provided, which are setting all B-factors to a constant value of 20 Å2, accessible surface-area based, or AVS-based methods. Given the time consumption and quality limitation of the I-TASSER-based structure prediction on large-size proteins, the sever is restricted to sequences with <1000 residues.
Output
The procedure for preparing the search models and phasing is fully automated, and the entire process of I-TASSER-MR, including I-TASSER modeling and the automated phasing and refinement, takes 15–24 h for a protein of 200 residues on a 2.8 GHz IBM NeXtScale machine. The actual waiting time may vary depending on the length of the target sequence, the space group, the number of molecules in the asymmetric unit, and sometime the server load although the jobs are submitted immediately to run with the highest priority in our cluster. An approximate time estimation, which is made by roughly combining these factors, is provided to users at the time when users submit a job. Meanwhile, a living status page is created which allows users to check the estimated job waiting time and the current status of their jobs, i.e. which of four steps (job waiting, I-TASSER running, MR and CNS running, and job finished) the job is on; the job status page reloads automatically every 10 min.
The server's outputs include: (i) a summary of the submission including query sequence and input parameters; (ii) the best MR and refined models with the lowest Rfree; (iii) the top five search models, MR models and refined models ranked by Rfree of the refined models; (iv) the I-TASSER full-length model and the histogram distribution of the AVSs of all the residues; (v) the top 10 templates used by I-TASEER for the query protein sequence. The results for each target are kept online for 3 months before they are removed from the server. A snapshot of the output page on an illustrative example is shown in Figure 2, with the major output results described briefly below.
Figure 2.

An illustration of I-TASSER-MR output page. (A) A summary of input reporting the query sequence and the crystal parameters. (B) Display of the best refined-model with lowest Rfree and the corresponding MR model. (C) A table listing the top five search models, MR models and refined models ranked by the Rfree of the refined models. (D) The histogram distribution of the AVS of the I-TASSER model and all the searching models edited by the progressive model truncation based on the AVS. (E) The top 10 threading templates used by I-TASSER for model construction. All the models are downloadable from the highlighted links.
The top five ranked models
In I-TASSER-MR, up to 60 edited copies of the I-TASSER models will be submitted to MR-REX for phasing and refinement. Only the top five MR models, as ranked by Rfree, are presented in this section, together with the seed models and the refined structure from CNS (see Figure 2C). The corresponding starting and final Rfree factors are also listed in the section when provided. The best refined-model and the corresponding MR model are displayed by JSmol to allow users to view and manipulate the structures (Figure 2B). All models can be downloaded to the local computer for further processing.
The search models produced according to the AVS
The average variability score is computed from the I-TASSER simulations to estimate the local accuracy of each residue. The higher the AVS is, the less reliable the residue model is. The unreliably modeled regions of the I-TASSER model can be easily viewed from the AVS plot as shown in Figure 2D. Users are allowed to download all of the search models edited according to the AVS profile in this section, which can be applied to other state of the art phasing tools. In addition, the section lists the confidence score (C-score) of the I-TASSER model, which is an estimation of the accuracy of the global fold and has been shown to strongly correlate with the actual quality of the I-TASSER model (25) and the success rate of MR (see ‘Results’ section below).
The top 10 templates used by I-TASSER
This section provides information about the top 10 threading templates collected by LOMETS, which were used by I-TASSER for the full-length structure generation. Although I-TASSER can generally draw the template structures closer to the native, the benchmark test has shown that the use of LOMETS template can sometime generate additional cases resulting in successful MR (see ‘Results’ section below). The template information of this section can be used by the users as input to other MR tools. The section includes: (i) the template PDB IDs, (ii) the length and coverage of the threading alignments, (iii) normalized threading Z-scores to assess the significance of the alignments; (iv) the threading program name and (v) alignments between the query and the templates (Figure 2E).
RESULTS
Benchmark results of I-TASSER-MR
The I-TASSER-MR pipeline was tested on two independent protein sets that consist of 61 targets from CASP8 and 100 non-redundant high-resolution proteins collected from the PDB.
To test whether the structure assembly simulation followed by AVS truncation performs better than the homologous templates that are identified from the PDB library, we compared the MR results using the first I-TASSER model and that using the best from the top 20 LOMETS templates that were used by I-TASSER for structure assembly. CHAINSAW (26) and Sculptor (27), two widely used programs for preparing homologous models for MR that prune non-conserved residues from the target-template alignments, were used to generate search models from the LOMETS templates. The default settings of CHAINSAW and 12 different predefined protocols of Sculptor (consisting of different combinations of methods for main-chain deletion, side-chain pruning and B-factor modification) were used to edit the LOMETS templates.
Results showed that I-TASSER-MR found correct MR solutions for 95 out of the 161 targets as judged by having a translation function Z-score (TFZ) of >8 or as having the final structure be closer to the native than the initial search models; this number is 36% higher than that obtained by the CHAINSAW and Sculptor programs (70 out of 161) based on the best threading template of the highest TM-score (19). There were four targets (PDB ID: 1NNX, 2O1Q, 3B79, 3DOJ) in the test cases for which the LOMETS-based MR succeeded but I-TASSER-MR failed. But there were 29 targets that were solved by I-TASSER-MR but not by LOMETS models.
Among these 29 targets, there were 15 targets for which the first successful truncated I-TASSER model had fewer residues than that of the corresponding LOMETS search models, due to the progressive truncation editing. Except for PDB ID: 1TU9, the I-TASSER models have a higher TM-score for all the targets than the best LOMETS template when considering the same threading aligned regions. The average TM-score of the I-TASSER models for these 29 targets was 0.76 that is 8.6% higher than that of the best LOMETS templates (0.70) calculated from the same threading aligned regions. These results confirm the advantage of using the I-TASSER structure assembly simulations and the progressive structural editing to improve the results of MR.
Comparison of phasing results by MR-REX and Phaser
One of the major updates of the current I-TASSER-MR server over the previous study (14), which uses Phaser (20), is the employment of MR-REX for the phasing search. To examine the impact of this update, we applied the Phaser and MR-REX programs to a large set of 1303 structural models generated by 3D-Robot (28), a structure decoy generator based on I-TASSER, from 38 non-redundant protein sequences. These decoy models have been selected with a nearly continuous range of quality, i.e. one decoy within each of 40 bins in the TM-score space from 0.59 to 0.99 for each sequence. Starting from random orientations, Phaser correctly places 542 models within <2 Å away from the best position, which is determined by directly superposing the decoy model and the target structure (called crystallography RMSD); while MR-REX successfully does so for 672 models using the same set of structural decoys and structural factor data. A detailed analysis reveals that the major difference between Phaser and MR-REX is on the lower resolution models. For high-quality decoys with a TM-score > 0.8, for example, both programs correctly places a substantial portion of models with a crystallography RMSD below 2 Å, i.e. Phaser succeeds in 76% of cases and MR-REX's success rate is only 16% higher. For the low-quality decoys with a TM-score < 0.8, however, Phaser succeeded in 53 cases while MR-REX's success rate is 94% higher than Phaser's. The ability to place low-resolution models is essential for I-TASSER-MR on modeling distant-homologous proteins, as many of the computational models have a low resolution for the distant-homologous proteins (29).
Given that the decoy models have been selected with a nearly continuous range of quality, we also counted the RMSD of the worst model that each program can successfully place for each target, since this can quantitatively assess the range of structural errors that the phasing programs can approximately tolerate. The average RMSD of the worst models that Phaser could success was 3.85 Å while that of MR-REX is 4.20 Å, suggesting that MR-REX could tolerate a slightly higher structural error from the target models in the MR phasing search.
Finally, we applied the phasing models to PHENIX.Autobuild (30) for model refinement and reconstruction. While Autobuild succeeds in creating models with a Rfree< 0.4 and meanwhile with TM-scores higher than the starting decoy for 382 cases using Phaser, it does so for 470 cases when using MR-REX. The average RMSD of the worst models in which Autobuild succeeds is 2.79 Å using Phaser, while that is 3.14 Å when using MR-REX. Although the success rate is generally lower when considering the more stringent criterion of Autobuild Rfree and TM-score cutoffs, the data confirm that MR-REX is able to generate better quality phasing models that can help increase the success rate of the MR solutions of I-TASSER-MR pipeline.
Correlation of MR results with I-TASSER C-score can be used to estimate the quality of the MR models
To quantitatively estimate the confidence of the MR solutions, we investigated the relationship between the quality of the I-TASSER models and their suitability as search models for MR. Here, the quality of I-TASSER models can be estimated by the C-score without knowing the native structure, which is calculated by a combination of the significance of threading alignments and the convergence of the structural assembly simulations i.e. 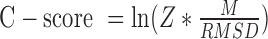 ,
, 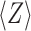 is the average Z-score of LOMET templates, M is the fraction of the decoys in the SPICKER cluster and
is the average Z-score of LOMET templates, M is the fraction of the decoys in the SPICKER cluster and 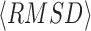 is the average RMSD of decoys to the centroid of the cluster (25). The C-score generally ranges from −5 to 2, with a higher value indicating better quality. Results showed that the success rate of MR increases almost linearly with the C-score (Figure 3). Nearly 91% of targets are solvable when the C-score is above 1.0, while I-TASSER-MR is unlikely to achieve a successful solution when the C-score drops below −1.5. Such data can be helpful for the users of I-TASSER-MR to estimate the confidence of the final MR solution.
is the average RMSD of decoys to the centroid of the cluster (25). The C-score generally ranges from −5 to 2, with a higher value indicating better quality. Results showed that the success rate of MR increases almost linearly with the C-score (Figure 3). Nearly 91% of targets are solvable when the C-score is above 1.0, while I-TASSER-MR is unlikely to achieve a successful solution when the C-score drops below −1.5. Such data can be helpful for the users of I-TASSER-MR to estimate the confidence of the final MR solution.
Figure 3.

Percentage of successful cases of MR by I-TASSER-MR versus of C-score of the I-TASSER models. The data were generated on a non-redundant set of 161 test proteins, where homologous templates with a sequence identity >30% to the query were excluded in the I-TASSER simulations.
CONCLUSION
We have developed a new online server pipeline, I-TASSER-MR, for automated MR from primary amino acid sequences. One of the major advantages of the I-TASSER-MR is the combination of the cutting-edge structure prediction method with the progressive modeling editing process, which allows for the successful MR for many proteins that do not have close homologous templates in the PDB. In addition, a new Monte Carlo-based phasing method MR-REX is incorporated to further improve the success rate of the MR calculations.
Much effort has been made to make the system convenient and easy to use. Options are provided to set the B-factors and the number of search models to be used. Detailed model annotations, including the global (C-score) and local (AVS) confidence scores of the I-TASSER models and the Rfree factors of refined structure models, have been provided for users to better interpret the structure modeling data and to quantitatively estimate the quality of the MR models.
It is important to note that the success rate of I-TASSER-MR essentially relies on the quality of the I-TASSER models, which can be assessed by the C-score of the structural assembly simulations. When a high-resolution model is predicted (e.g. with C-score > 0.5 or TM-score > 0.8), I-TASSER-MR can generate correct MR for the majority of the cases (with an average success rate = 82%). The challenging cases are, however, those with a correct fold (e.g. C-score in 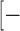 1.5, 0.5] or TM-score in [0.5, 0.8]) but with incorrect local structures (mostly in the loop and tail regions), for which the progressive model editing process often helps to select and truncate those incorrectly modeled regions to improve the MR solution. Nevertheless, a single phasing method such as MR-REX cannot solve all the challenging cases even given the correctly truncated models. At this point, the use of other state of the art phasing tools, including Phaser (20), Molrep (21) and AMoRe (22), may provide complementary results, where the intermediate modeling data (threading template, full-length and truncated I-TASSER models) provided by the I-TASSER-MR server are helpful for this purpose. Overall, with the continuous developments of new structure prediction and phasing methods, we expect that the I-TASSER-MR server will become an increasingly useful system for solving the phase problem of the X-ray crystallography, particularly for the non- and distantly-homologous protein targets.
1.5, 0.5] or TM-score in [0.5, 0.8]) but with incorrect local structures (mostly in the loop and tail regions), for which the progressive model editing process often helps to select and truncate those incorrectly modeled regions to improve the MR solution. Nevertheless, a single phasing method such as MR-REX cannot solve all the challenging cases even given the correctly truncated models. At this point, the use of other state of the art phasing tools, including Phaser (20), Molrep (21) and AMoRe (22), may provide complementary results, where the intermediate modeling data (threading template, full-length and truncated I-TASSER models) provided by the I-TASSER-MR server are helpful for this purpose. Overall, with the continuous developments of new structure prediction and phasing methods, we expect that the I-TASSER-MR server will become an increasingly useful system for solving the phase problem of the X-ray crystallography, particularly for the non- and distantly-homologous protein targets.
ACKNOWLEDGEMENTS
We are grateful to Dr Alex Brunger for the permit of using CNS in the online I-TASSER-MR server. Part of the method training and benchmarking work was done on the computing resource provided by the Extreme Science and Engineering Discovery Environment (XSEDE) (24).
FUNDING
National Institute of General Medical Sciences [GM083107, GM116960]; National Science Foundation [DBI1564756]; National Natural Science Foundation of China [30700162]; Fundamental Research Funds for the Central Universities of China [2014TS138]. Funding for open access charge: National Institute of General Medical Sciences [GM083107].
Conflict of interest statement. None declared.
REFERENCES
- 1. Rossmann M.G. The molecular replacement method. Acta Crystallogr. A. 1990; 46:73–82. [DOI] [PubMed] [Google Scholar]
- 2. Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E.. The Protein Data Bank. Nucleic Acids Res. 2000; 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Claude J.B., Suhre K., Notredame C., Claverie J.M., Abergel C.. CaspR: a web server for automated molecular replacement using homology modelling. Nucleic Acids Res. 2004; 32:W606–W609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Long F., Vagin A.A., Young P., Murshudov G.N.. BALBES: a molecular-replacement pipeline. Acta Crystallogr. D Biol. Crystallogr. 2008; 64:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Keegan R.M., Winn M.D.. Automated search-model discovery and preparation for structure solution by molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 2007; 63:447–457. [DOI] [PubMed] [Google Scholar]
- 6. Keegan R.M., Winn M.D.. MrBUMP: an automated pipeline for molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 2008; 64:119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schmidberger J.W., Bate M.A., Reboul C.F., Androulakis S.G., Phan J.M., Whisstock J.C., Goscinski W.J., Abramson D., Buckle A.M.. MrGrid: a portable grid based molecular replacement pipeline. PLoS One. 2010; 5:e10049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schwarzenbacher R., Godzik A., Grzechnik S.K., Jaroszewski L.. The importance of alignment accuracy for molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 2004; 60:1229–1236. [DOI] [PubMed] [Google Scholar]
- 9. Schwarzenbacher R., Godzik A., Jaroszewski L.. The JCSG MR pipeline: optimized alignments, multiple models and parallel searches. Acta Crystallogr. D Biol. Crystallogr. 2008; 64:133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bibby J., Keegan R.M., Mayans O., Winn M.D., Rigden D.J.. AMPLE: a cluster-and-truncate approach to solve the crystal structures of small proteins using rapidly computed ab initio models. Acta Crystallogr. D Biol. Crystallogr. 2012; 68:1622–1631. [DOI] [PubMed] [Google Scholar]
- 11. Zhang Y. Progress and challenges in protein structure prediction. Curr. Opin. Struct. Biol. 2008; 18:342–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moult J., Fidelis K., Kryshtafovych A., Schwede T., Tramontano A.. Critical assessment of methods of protein structure prediction: Progress and new directions in round XI. Proteins. 2016; 84:4–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang Y. Interplay of I-TASSER and QUARK for template-based and ab initio protein structure prediction in CASP10. Proteins. 2014; 82:175–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Y., Virtanen J., Xue Z., Tesmer J.J., Zhang Y.. Using iterative fragment assembly and progressive sequence truncation to facilitate phasing and crystal structure determination of distantly related proteins. Acta Crystallogr. D Struct. Biol. 2016; 72:616–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu S., Zhang Y.. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 2007; 35:3375–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang Y., Kolinski A., Skolnick J.. TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys. J. 2003; 85:1145–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang Y., Kihara D., Skolnick J.. Local energy landscape flattening: parallel hyperbolic Monte Carlo sampling of protein folding. Proteins. 2002; 48:192–201. [DOI] [PubMed] [Google Scholar]
- 18. Zhang Y., Skolnick J.. SPICKER: a clustering approach to identify near-native protein folds. J. Comput. Chem. 2004; 25:865–871. [DOI] [PubMed] [Google Scholar]
- 19. Zhang Y., Skolnick J.. Scoring function for automated assessment of protein structure template quality. Proteins. 2004; 57:702–710. [DOI] [PubMed] [Google Scholar]
- 20. McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J.. Phaser crystallographic software. J. Appl. Crystallogr. 2007; 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vagin A., Teplyakov A.. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:22–25. [DOI] [PubMed] [Google Scholar]
- 22. Navaza J. Implementation of molecular replacement in AMoRe. Acta Crystallogr. D Biol. Crystallogr. 2001; 57:1367–1372. [DOI] [PubMed] [Google Scholar]
- 23. Brunger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S. et al. . Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998; 54:905–921. [DOI] [PubMed] [Google Scholar]
- 24. Towns J., Cockerill T., Dahan M., Foster I., Gaither K., Grimshaw A., Hazlewood V., Lathrop S., Lifka D., Peterson G.D. et al. . XSEDE: accelerating scientific discovery. Comput. Sci. Eng. 2014; 16:62–74. [Google Scholar]
- 25. Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008; 9:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stein N. CHAINSAW: a program for mutating pdb files used as templates in molecular replacement. J. Appl. Cryst. 2008; 41:641–643. [Google Scholar]
- 27. Bunkoczi G., Read R.J.. Improvement of molecular-replacement models with Sculptor. Acta Crystallogr. D Biol. Crystallogr. 2011; 67:303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Deng H., Jia Y., Zhang Y.. 3DRobot: automated generation of diverse and well-packed protein structure decoys. Bioinformatics. 2016; 32:378–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang Y. Protein structure prediction: when is it useful. Curr. Opin. Struct. Biol. 2009; 19:145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. et al. . PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]