


Abstract
With advances in next-generation sequencing technologies, numerous novel transcripts in a large number of organisms have been identified. With the goal of fast, accurate assessment of the coding ability of RNA transcripts, we upgraded the coding potential calculator CPC1 to CPC2. CPC2 runs ∼1000 times faster than CPC1 and exhibits superior accuracy compared with CPC1, especially for long non-coding transcripts. Moreover, the model of CPC2 is species-neutral, making it feasible for ever-growing non-model organism transcriptomes. A mobile-friendly web server, as well as a downloadable standalone package, is freely available at http://cpc2.cbi.pku.edu.cn.
INTRODUCTION
Recent studies have well demonstrated that non-coding RNAs (ncRNAs) are pervasively transcribed from plant to animal genomes (1–4). Increasing evidences indicate that these ncRNAs play critical roles in numbers of important cellular processes, including transcriptional inhibition mediated by microRNAs (5), epigenetic inheritance by Piwi-interacting RNAs (6), cell-cycle regulation (7) or even acting as structural components in ribosomes (8).
With advances in next-generation sequencing technologies, numerous novel transcripts in a large number of diverse organisms, including several non-model ones, have been discovered in rapidly increasing RNA-seq data (9–12). Effective and efficient identification of ncRNAs in the massive dataset is an essential step for following-up function and evolution studies, and demands a fast, accurate and species-neutral assessment tool (13–19).
As a response to the challenge, we updated our Coding Potential Calculator (CPC) algorithm (20) to version 2. Employing a novel discriminative model based on four sequence intrinsic features, CPC2 not only runs ∼1000 times faster than CPC1 but is also more accurate. In addition, CPC2 is species-neutral, making it more useful for the ever-growing non-model organism transcriptomes. CPC2 is available freely at http://cpc2.cbi.pku.edu.cn as both a web server and a downloadable standalone package.
MATERIALS AND METHODS
To identify discriminative features, we first compiled a candidate list of sequence intrinsic features (i.e. features can be derived from transcript sequence directly) based on literature survey (see Supplementary Table S1). A hierarchical feature selection procedure was employed to identify effective features with recursive feature elimination method (random forest functions with 10-fold cross-validation, implemented with the caret R package (21)) adopted in each stage (see Supplementary Figure S1 for details). We identified a final set of four intrinsic features as Fickett TESTCODE score, open reading frame (ORF) length, ORF integrity and isoelectric point (pI). While the Fickett TESTCODE score is derived from the weighted nucleotide frequency of the inputted full length transcript (22), the rest of three features (ORF length, ORF integrity and isoelectric point) are calculated based on the longest putative ORF identified in silico (see http://cpc2.cbi.pku.edu.cn/help/feature_selection.php for the full candidate list as well as the script).
We then trained a support vector machine (SVM) model using these four intrinsic features. The LIBSVM (23) package was employed to train an SVM model using the standard radial basis function kernel (RBF kernel) with the training dataset containing 17 984 high-confident human protein-coding transcripts and 10 452 non-coding transcripts (18).
To evaluate the performance of CPC2 across species, we further built an independent testing set for human, mouse, zebrafish, fly, worm and the model plant Arabidopsis. We selected protein-coding and non-coding transcripts that met rigorous criteria to obtain a testing set of high quality: for the protein-coding testing set, we obtained all non-predicted mRNAs from the RefSeq database (24) with protein sequences annotated by Swiss-Prot (25) and redundant sequences (i.e identity ≥ 0.9) removed using CD-hit with default parameters. Non-coding transcripts were obtained from the Ensembl (v87) (26) and EnsemblPlants (v32) (26) databases with transcript status as ‘KNOWN’. All sequences in training set were further excluded (Table 1). The full training set and testing set are available for downloading as FASTA files at http://cpc2.cbi.pku.edu.cn/help/data_set.php.
Table 1. The independent testing set in human, mouse, zebrafish, fly, worm and the model plant Arabidopsis thaliana.
| Dataset type | Human | Mouse | Zebrafish | Fly | Worm | Arabidopsis |
|---|---|---|---|---|---|---|
| Coding | 6142 | 10 638 | 2344 | 3680 | 3551 | 13 986 |
| Non-coding | 12 019 | 12 251 | 1528 | 3556 | 9470 | 3853 |
All testing sets are available for downloading as FASTA file at http://cpc2.cbi.pku.edu.cn/help/data_set.php.
We employed standard performance measurements including sensitivity, specificity and accuracy, with protein-coding calls defined as ‘positive’ and non-coding calls as ‘negative’. The abbreviations in the equations below are as follows: FN, false negative; FP, false positive; TN, true negative; and TP, true positive.
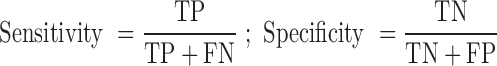 |
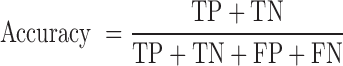 |
Back-end of the CPC2 web server is implemented in PHP running on Apache web server. The front-end interface is powered by JavaScript libraries Bootstrap (http://getbootstrap.com/), JQuery (http://jquery.com/), Tablecloth (http://cssglobe.com/lab/tablecloth/) as well as Highcharts (http://www.highcharts.com/).
RESULTS
CPC2 is fast, accurate and species-neutral
Given the large volume of transcriptome data generated by next generation sequencing, the efficiency is becoming vital for a useful tool in the real world. To measure the computational speed, we first randomly selected a sample of 200 sequences that consisted of 100 mRNAs and 100 lncRNAs from the human testing dataset. CPC2 completed its analysis in 1.8 s, whereas CPC1 required >1000-fold time (2815 s) on Intel Xeon E7-8830 2.13GHz CPU in single thread mode. To further evaluate the real world efficiency, we then measured the computational speed on all the coding and non-coding transcripts in Ensembl v87 (26) with gene and transcript status annotated as ‘KNOWN’. This dataset consists of 597 996 protein-coding transcripts and 55 277 non-coding transcripts from 69 organisms, which is more similar to the circumstances of users’ input. Similar to previous result, CPC2 showed a significant speedup (42 min) than CPC1 (4783 min).
In addition to being efficient, a sensible tool should pose high accuracy in a robust and species-neutral fashion across different organisms. Designed to use rather stringent criteria for non-coding calls, the CPC1 exhibits high sensitivity and relative poor specificity. As many important biological roles of long ncRNAs (lncRNAs) have been revealed by recent studies performed in this decade (7), CPC2 adopted a more balanced calling of protein-coding and non-coding transcripts, which is more suitable for current transcriptome studies. To evaluate the performance across various species, we ran both CPC1 and CPC2 against human, mouse, zebrafish, fly, worm and plant (Arabidopsis) testing set. The CPC2 showed better overall accuracy (0.961) than of CPC1 (0.932) with a much more improved specificity (0.970 versus 0.873) and a slightly lower sensitivity (0.952 versus 0.995). In particular, the CPC2 exhibited superior accuracy (0.942) for long non-coding transcripts, a newly discovered key regulators in several physiological and pathological processes (27–30), than of CPC1 (0.762, Figure 1A). Further comparison with other popular tools (14,17,19) also confirmed CPC2΄s superior performance (Supplementary Figure S2).
Figure 1.

Evaluation on accuracy of CPC1 and CPC2 in six species. (A) The overall accuracy (B) the detailed accuracy in six organisms. The Long ncRNAs were defined as non-coding RNAs longer than 200 nt.
Even the underlying model in CPC2 was trained based on transcript sequences from human only (the training set used in CPC1 is consist of sequences from multiple organisms), the CPC2 showed a more robust performance across species, with accuracy varied from 0.937 to 0.991 (from 0.826 to 0.997 for CPC1, Figure 1B), which may partly due to the fact that only sequence intrinsic features were employed in CPC2. In particular, while CPC1 shows higher accuracy than CPC2 in Arabidopsis, the inter-species variance of accuracy of CPC2 (0.04%) is one order of magnitude lower than CPC1 (0.4%) (Figure 1B), a property that we considered ‘species neutral’ (also see http://cpc2.cbi.pku.edu.cn/help/species_neutral.php for more details).
The web server of CPC2
For users to access CPC2 conveniently, we established a new web portal at http://cpc2.cbi.pku.edu.cn/. Briefly, the CPC2 web server accepts RNA transcripts as input and outputs its coding probability with detailed supporting features for the coding/non-coding call (Figure 2).
Figure 2.

Workflow of the CPC2 web server.
CPC2 web server currently supports both ‘interactive mode’, in which the nucleotide sequences in FASTA format can be directly copied and pasted into the input box at the home page, and ‘batch mode’ in which users can upload a local file in either FASTA format or BED/GTF/GFF format. When a new analysis task is submitted, a unique ‘Task ID’ (TID) will be assigned for tracking the analysis progress and retrieving results later.
As in CPC1, the results will be presented as an intuitive table online which can also be downloaded as a tabular file for further analysis (Figure 3A). In addition, detailed information of each transcript is provided in a separated ‘detailed’ page, including a summary paragraph, a graphic view of features’ distribution in known protein-coding and non-coding transcripts and additional functions (Figure 3B). More analysis such as querying against known databases, re-analyzing in alternative methods and annotating functions can also be run performed for given transcript (Figure 3C and D).
Figure 3.

Screenshot of the CPC2 web server. (A) Summary tabular output with coding probability; (B) graphical view of features’ distribution in the ‘Details’ page; (C) more analysis for querying against known databases, re-analyzing in alternative methods and annotating functions; (D) the rendered BLAST output including both ORF position and BLAST hits in queried databases.
The CPC2 web server implemented a responsive layout, enabling the optimal view for both desktop PCs and mobile devices. A standalone package of CPC2 can also be freely downloaded at http://cpc2.cbi.pku.edu.cn/download.php.
Example
We utilized online CPC2 on a human lncRNA MEG3 as an example. After inputting its sequence, CPC2 predicted it as a non-coding transcript (Figure 3A). By clicking the ‘View’ on the last column, more detailed information is shown.
The details page is divided into three parts. A description of MEG3 summarizing its coding probability and feature values is presented at the top (Figure 3B). In the middle of this page, an interactive visualization of three supporting features including Fickett score, peptide length (synonymous with ORF length) and pI are provided. Taking the graph of peptide length as an example, the black box indicates that MEG3 has a peptide length of 106 aa and was classified as non-coding. In addition, the position of MEG3 is noted in the background (Figure 3B). The blue area shows the feature's distribution in non-coding transcripts, whereas the orange one represents protein coding transcripts. Passing the mouse over the distribution curve, the feature value and transcripts frequency of the interval are displayed in a textbox. The static visualizations can be easily downloaded (Figure 3B).
At the bottom, CPC2 also provides additional functions to facilitate the coding/non-coding classification of input sequences (Figure 3C). The first function is querying the transcript against well-annotated databases, including Swiss-Prot (24), RNAdb (31) and lncRNAdb (32) by BLAST (33), to identify more evidence. By placing the mouse over the results, users can view details of predicted ORF and BLAST hits of MEG3 (Figure 3D). Moreover, the user can also send sequences to alternative tools like CPC1, CPAT and PORTRAIT for re-analysis through the ‘Re-analyze’ button.
SUMMARY
Employing a novel discriminative model, we upgraded our CPC to version 2. CPC2 runs ∼1000 times faster than CPC1. In addition, the CPC2 model is species-neutral, making it useful for ever-growing non-model organism transcriptomes and even transcriptomes of organisms that are poorly annotated or lack genome assembly. CPC2 is more accurate than CPC1, especially for long non-coding transcripts. In addition, the online CPC2 provides an informative graphic view of results and more integrated functions. The web server is mobile-friendly and more accessible on mobile devices such as the iPad.
Independent of external resources, CPC2 adopted four sequence intrinsic features that are easily comprehensible and biologically meaningful. At the DNA level, the Fickett score captures the position of each base favored in the sequence (18). At the RNA level, ORF length and integrity are powerful because the protein-coding transcript is more likely to have a long and high-quality ORF. Moreover, based on the assumption that the hypothetical peptide identified in a non-coding transcript should have different chemical properties than these real ones encoded by bona fide coding sequences, we also added several peptide level features into the candidate list, and eventually adopted pI in the final SVM model.
Since the first release of CPC1 at 2007, number of statistic-based tools have been developed to distinguish non-coding and protein-coding transcripts based on multiple lines of evidences. Many of them show high levels of accuracy (13–20). We hereby argue that the community should, in the coming years, shift from continuous improvement of discriminative performance to biological insights revealed by their statistical models which might further shed light onto the ultimate discriminative mechanism used by the Mother Nature.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Drs Cheng Li, Zemin Zhang and Jian Lu at Peking University for their helpful comments and suggestions during the study.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key Research and Development Program [2016YFC0901603]; China 863 Program [2015AA020108]; State Key Laboratory of Protein and Plant Gene Research; National Program for Support of Top-notch Young Professionals (to G.G.) (in part). Part of the analysis was performed on the Computing Platform of the Center for Life Sciences of Peking University. Funding for open access charge: National Key Research and Development Program [2016YFC0901603].
Conflict of interest statement. None declared.
REFERENCES
- 1. Eddy S.R. Non-coding RNA genes and the modern RNA world. Nat. Rev. Genet. 2001; 2:919–929. [DOI] [PubMed] [Google Scholar]
- 2. Fu X.D. Non-coding RNA: a new frontier in regulatory biology. Natl. Sci. Rev. 2014; 1:190–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. He S., Liu C., Skogerbo G., Zhao H., Wang J., Liu T., Bai B., Zhao Y., Chen R.. NONCODE v2.0: decoding the non-coding. Nucleic Acids Res. 2008; 36:D170–D172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mattick J.S., Makunin I.V.. Non-coding RNA. Hum. Mol. Genet. 2006; 15:R17–R29. [DOI] [PubMed] [Google Scholar]
- 5. Ambros V. microRNAs: tiny regulators with great potential. Cell. 2001; 107:823–826. [DOI] [PubMed] [Google Scholar]
- 6. Brennecke J., Malone C.D., Aravin A.A., Sachidanandam R., Stark A., Hannon G.J.. An epigenetic role for maternally inherited piRNAs in transposon silencing. Science. 2008; 322:1387–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rinn J.L., Chang H.Y.. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012; 81:145–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cole J.R., Chai B., Marsh T.L., Farris R.J., Wang Q., Kulam S.A., Chandra S., McGarrell D.M., Schmidt T.M., Garrity G.M. et al. . The Ribosomal Database Project (RDP-II): previewing a new autoaligner that allows regular updates and the new prokaryotic taxonomy. Nucleic Acids Res. 2003; 31:442–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cahais V., Gayral P., Tsagkogeorga G., Melo-Ferreira J., Ballenghien M., Weinert L., Chiari Y., Belkhir K., Ranwez V., Galtier N.. Reference-free transcriptome assembly in non-model animals from next-generation sequencing data. Mol. Ecol. Resour. 2012; 12:834–845. [DOI] [PubMed] [Google Scholar]
- 10. Ellegren H., Galtier N.. Determinants of genetic diversity. Nat. Rev. Genet. 2016; 17:422–433. [DOI] [PubMed] [Google Scholar]
- 11. Junttila S., Rudd S.. Characterization of a transcriptome from a non-model organism, Cladonia rangiferina, the grey reindeer lichen, using high-throughput next generation sequencing and EST sequence data. BMC Genomics. 2012; 13:575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schunter C., Vollmer S.V., Macpherson E., Pascual M.. Transcriptome analyses and differential gene expression in a non-model fish species with alternative mating tactics. BMC Genomics. 2014; 15:167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arrial R.T., Togawa R.C., Brigido M.M.. Screening non-coding RNAs in transcriptomes from neglected species using PORTRAIT: case study of the pathogenic fungus Paracoccidioides brasiliensis. BMC Bioinformatics. 2009; 10:239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hu L., Xu Z., Hu B., Lu Z.J.. COME: a robust coding potential calculation tool for lncRNA identification and characterization based on multiple features. Nucleic Acids Res. 2017; 45:e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li A., Zhang J., Zhou Z.. PLEK: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinformatics. 2014; 15:311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lin M.F., Jungreis I., Kellis M.. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 2011; 27:i275–i282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun L., Luo H., Bu D., Zhao G., Yu K., Zhang C., Liu Y., Chen R., Zhao Y.. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013; 41:e166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang L., Park H.J., Dasari S., Wang S., Kocher J.-P., Li W.. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013; 41:e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Washietl S., Findeiss S., Muller S.A., Kalkhof S., von Bergen M., Hofacker I.L., Stadler P.F., Goldman N.. RNAcode: robust discrimination of coding and noncoding regions in comparative sequence data. RNA. 2011; 17:578–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kong L., Zhang Y., Ye Z.-Q., Liu X.-Q., Zhao S.-Q., Wei L., Gao G.. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007; 35:W345–W349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kuhn M. Building Predictive Models in R Using the caret Package. Journal of Statistical Software. 2008; 28:https://www.jstatsoft.org/article/view/v028i05. [Google Scholar]
- 22. Fickett J.W. Recognition of protein coding regions in DNA sequences. Nucleic Acids Res. 1982; 10:5303–5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chang C.-C., Lin C.-J.. LIBSVM : a library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011; 2:27. [Google Scholar]
- 24. O’Leary N.A., Wright M.W., Brister J.R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D. et al. . Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016; 44:D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boutet E., Lieberherr D., Tognolli M., Schneider M., Bansal P., Bridge A.J., Poux S., Bougueleret L., Xenarios I.. UniProtKB/Swiss-Prot, the Manually Annotated Section of the UniProt KnowledgeBase: how to Use the Entry View. Methods Mol. Biol. 2016; 1374:23–54. [DOI] [PubMed] [Google Scholar]
- 26. Yates A., Akanni W., Amode M.R., Barrell D., Billis K., Carvalho-Silva D., Cummins C., Clapham P., Fitzgerald S., Gil L. et al. . Ensembl 2016. Nucleic Acids Res. 2016; 44:D710–D716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kitagawa M., Kitagawa K., Kotake Y., Niida H., Ohhata T.. Cell cycle regulation by long non-coding RNAs. Cell Mol. Life Sci. 2013; 70:4785–4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee J.T., Bartolomei M.S.. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell. 2013; 152:1308–1323. [DOI] [PubMed] [Google Scholar]
- 29. Ng S.Y., Johnson R., Stanton L.W.. Human long non-coding RNAs promote pluripotency and neuronal differentiation by association with chromatin modifiers and transcription factors. EMBO J. 2012; 31:522–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Park J.Y., Lee J.E., Park J.B., Yoo H., Lee S.H., Kim J.H.. Roles of long non-coding RNAs on tumorigenesis and glioma development. Brain Tumor Res. Treat. 2014; 2:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pang K.C., Stephen S., Dinger M.E., Engstrom P.G., Lenhard B., Mattick J.S.. RNAdb 2.0–an expanded database of mammalian non-coding RNAs. Nucleic Acids Res. 2007; 35:D178–D182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Quek X.C., Thomson D.W., Maag J.L., Bartonicek N., Signal B., Clark M.B., Gloss B.S., Dinger M.E.. lncRNAdb v2.0: expanding the reference database for functional long noncoding RNAs. Nucleic Acids Res. 2015; 43:D168–D173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J.. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997; 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.