


Abstract
During the last decade, genome-wide association studies (GWAS) have represented a major approach to dissect complex human genetic diseases. Due in part to limited statistical power, most studies identify only small numbers of candidate genes that pass the conventional significance thresholds (e.g. P ≤ 5 × 10−8). This limitation can be partly overcome by increasing the sample size, but this comes at a higher cost. Alternatively, weak association signals can be boosted by incorporating independent data. Previously, we demonstrated the feasibility of boosting GWAS disease associations using gene networks. Here, we present a web server, GWAB (www.inetbio.org/gwab), for the network-based boosting of human GWAS data. Using GWAS summary statistics (P-values) for SNPs along with reference genes for a disease of interest, GWAB reprioritizes candidate disease genes by integrating the GWAS and network data. We found that GWAB could more effectively retrieve disease-associated reference genes than GWAS could alone. As an example, we describe GWAB-boosted candidate genes for coronary artery disease and supporting data in the literature. These results highlight the inherent value in sub-threshold GWAS associations, which are often not publicly released. GWAB offers a feasible general approach to boost such associations for human disease genetics.
INTRODUCTION
Genome-wide association studies (GWAS) are conducted to identify single nucleotide polymorphism (SNP) genetic variants that are associated with a disease population. Because candidate SNPs from GWAS can provide important clues regarding the genetics of complex traits, more than 2700 GWAS on human traits have been conducted and more than 31 000 unique trait-SNP associations have been reported to a central repository, the GWAS catalog (1), as of January 2017. Although they represent a major driving force in human genetics, GWAS have some challenges, including limited statistical power, which is due in part to the pathway nature of complex human diseases. Causal variants of most common diseases in humans are distributed across many genes in the disease-relevant pathway, resulting in genetic heterogeneity (2). Therefore, each causal variant occurs in only a subset of a disease population, reducing the test power of the association between a single variant and the disease. We can partly overcome the problems associated with the underpowered statistics of GWAS by increasing the population size, but this entails a higher cost.
The pathway nature of common diseases presents not only statistical challenges but also new opportunities for augmenting and interpreting GWAS data. Assuming that the genes in the same pathway are functionally coupled, co-functional gene networks can facilitate the analysis of GWAS data via mainly two approaches (3,4): (i) identifying disease-associated pathways by searching for subnetworks that are enriched for candidate genes identified in GWAS and (ii) reprioritizing disease-associated genes by integrating GWAS and network data. The network-assisted analysis of GWAS data requires gene-level scores for disease associations. Several methods have been developed to identify subnetworks enriched for candidate genes. Algorithms to search for high-scoring subnetworks in a node-weighted network such as jActiveModules (5) and dmGWAS (6) have been applied to identify disease-associated pathways in a network of genes weighted by P-values from GWAS (7,8). DAPPLE identifies disease-associated pathways through a permutation test for direct network connectivity among candidate genes identified in GWAS in a protein–protein interaction (PPI) network (9). NIMMI identifies disease-associated subnetworks using combined weights from GWAS P-values and network connectivity (10). The prix fixe strategy identifies disease-associated pathways by evaluating the significance of combinations of genes, with one gene from each GWAS candidate locus, in a gene network (11). The identified subnetworks are generally applied to a gene set enrichment analysis for pathway-centric interpretation, and the member genes that are not annotated for the disease can be investigated as novel candidate genes.
The second category of network-assisted analysis for GWAS data is reprioritizing genes for diseases based on the integration of GWAS and network data. For example, we previously identified disease genes of sub-threshold GWAS associations (e.g., P ≤ 5 × 10−8) by incorporating the GWAS P-values of neighbors in a co-functional network (12). The effectiveness of the network-based boosting of GWAS associations was validated in Crohn's disease (CD) and type 2 diabetes (T2D) using the updated GWAS candidates from a meta-analysis and literature review. Recently, another method for the network-based reprioritizing of GWAS associations, NetWAS (http://giant.princeton.edu/gwas/create_new), was developed and demonstrated to enhance the accuracy of disease gene predictions in hypertension, C-reactive protein levels, T2D, body mass index and advanced age-related macular degeneration (13).
Here, we present GWAB (genome-wide association boosting), a web server for the network-based boosting of GWAS data described in our previous work (12). We evaluated GWAB using seven disease GWAS for which summary statistics data are available for the whole set of SNPs and found that GWAB could more effectively identify disease-associated genes than GWAS could alone. We also validated many GWAB-boosted genes in coronary artery disease GWAS as new disease-associated candidates using supporting data in the literature.
WEB SERVER DESCRIPTION
Overview of the network-based boosting of GWAS data
Because the genes associated with a disease tend to be functionally coupled, the GWAS associations of a gene can also be a partial indication of the involvement of its co-functional partners in the disease. Thus, GWAS associations for individual genes can be propagated through co-functional network neighbors. If a gene shows sub-threshold associations in a disease GWAS but its network neighbors have strong GWAS associations, we may also consider the gene a considerable candidate based on the propagated significance score of the disease-association from its network neighbors (Figure 1A). To propagate GWAS data through the gene network, GWAB first assigns P-values of SNPs to genes based on chromosomal proximity. It assigns the best P-value within 10 kb from the beginning or end of the gene by default. Although there are more sophisticated methods for assigning SNP P-values to genes such as VEGAS (14), GWAB uses the simple distance-based assignment approach to achieve a favorable trade-off between prediction accuracy and calculation time. In supplemental analysis, we found that P-value assignment by VEGAS did not make significant improvement in boosting performance for the tested GWAS data in this study (data not shown).
Figure 1.

Overview of GWAB. (A) Schematic summary of the network-based boosting of GWAS data. (B) Components of the GWAS web service and their work flow. (C) A summary plot of GWAB results for the coronary artery disease GWAS data. The prediction performances for reference disease genes by GWAB, GWAS alone and 100 randomized networks are represented as AUC scores for FPR 5% (y-axis) for the given log(P) threshold (x-axis). The vertical dashed line indicates the optimal log(P) threshold where the best AUC score was achieved.
GWAB uses the scoring scheme described in our previous work (12). Let 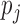 denotes the probability of disease involvement of a gene j. To make use of the information from the genes that are on the verge of being statistically significant, we implemented a ‘soft’ guilt-by-association (GBA) by
denotes the probability of disease involvement of a gene j. To make use of the information from the genes that are on the verge of being statistically significant, we implemented a ‘soft’ guilt-by-association (GBA) by 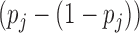 , where only genes that are very strongly associated with the disease are given full weight in the GBA. GWAB calculates the total contributions of the GWAS association scores from the network neighboring gene j of gene i as follows:
, where only genes that are very strongly associated with the disease are given full weight in the GBA. GWAB calculates the total contributions of the GWAS association scores from the network neighboring gene j of gene i as follows:
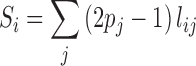 |
where 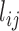 is the likelihood of the link between gene i and gene j in the co-functional network. The likelihood score of the co-functional links was calculated based on a Bayesian statistics framework, in which the ability to retrieve known pathway links are evaluated for the given evidence (15). When the data from GWAS and the data from the network are conditionally independent, we can integrate them in a naïve Bayes framework. The posterior log odds that gene i is involved in the disease (i.e. GWAB score) can be calculated using the following equation:
is the likelihood of the link between gene i and gene j in the co-functional network. The likelihood score of the co-functional links was calculated based on a Bayesian statistics framework, in which the ability to retrieve known pathway links are evaluated for the given evidence (15). When the data from GWAS and the data from the network are conditionally independent, we can integrate them in a naïve Bayes framework. The posterior log odds that gene i is involved in the disease (i.e. GWAB score) can be calculated using the following equation:
 |
where 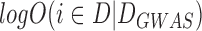 is the log odds of the association calculated from the GWAS data, which corresponds to the log Bayes factor for the disease-association plus the prior log odds for the association. Because the odds of the association are calculated from the P-value of GWAS, the GWAB will refer to the prior log odds as the P-value threshold (log(P) threshold).
is the log odds of the association calculated from the GWAS data, which corresponds to the log Bayes factor for the disease-association plus the prior log odds for the association. Because the odds of the association are calculated from the P-value of GWAS, the GWAB will refer to the prior log odds as the P-value threshold (log(P) threshold).
Web-based service design and implementation
The GWAB server consists of a front-end system that provides a user interface for submitting input data for the analysis and receiving of results and a back-end system that performs data preprocessing, boosting and optimizing procedures (Figure 1B). First, users need to submit a set of SNPs with P-value data from GWAS and a set of reference disease-associated genes for selecting an optimal P-value threshold for boosting and validating prediction performance. Using the given user-input data, GWAB sequentially performs the preprocess of assigning P-values to genes, boosting GWAS associations by incorporating GWAS association scores of network neighbors and optimizing boosting conditions by selecting a P-value threshold. In addition to the input data, several parameters need to be chosen from the job submission page. GWAS in humans have been conducted based on either an hg18 or hg19 genome build. Thus, users need to choose the correct version of the genome build for the given GWAS data. Users also need to choose a range for the chromosomal distance between SNPs and genes for use in the search for the SNP with the best P-value that will be assigned to each gene. Users may run an analysis with a different SNP-gene distance range, yet we found no significant changes in boosting by varying from 0 to 250 kb in our previous study (12). Next, GWAB reprioritizes genes by boosting GWAS associations using the scoring scheme described above. Then, the retrieval rate for reference disease genes is measured by receiver operating characteristic (ROC) analysis to assess the prediction performance. To determine how much of a boosting effect was gained by the given network, GWAB repeats the whole reprioritizing process for 100 randomized networks with the same parameters.
The effectiveness of the network boost could vary depending on the number of genes believed to be associated with the disease, in other words, the number of genes that can contribute their GWAS association scores to network neighbors. We can alter the number of genes that pass the ‘prior odds’ by using a different P-value threshold. Thus, only genes that pass the threshold can participate in network boosting. To identify the optimal P-value threshold for a given network boosting, GWAB repeats the analysis over various P-value thresholds within a given range (−6 < log(P) < −2 by default) with a set interval (0.3 by default). While the GWAB analysis is running, users can monitor the job status via a status page, which is refreshed every 10 s until it moves to the result page automatically.
The performance of each GWAB analysis is summarized as an area under the ROC curve (AUC) score for less than a 5% false positive rate (FPR). If the AUC score from GWAB is higher than that obtained with randomized networks and that obtained with GWAS alone, the high-ranked genes by GWAB are likely to be involved in the disease (Figure 1C). The final list of reprioritized genes for the disease is generated using the optimal log(P) threshold with which the highest AUC score was obtained (e.g. −3.6 for coronary artery disease).
All the backend programs in GWAB are implemented in Python and PHP, and all job information is saved as MYSQL to manage GWAB jobs. Moreover, the job controller, which acts as a daemon, is implemented as shell programs so that it can quickly respond to incoming work by calling a job list once per a second. The calculation time of GWAB depends on the number of SNPs and the availability of the CPU, but the process usually takes less than an hour.
EVALUATION OF GWAB USING DISEASE GWAS DATA
Network, GWAS data and reference disease genes
The quality of the co-functional network affects GWAB performance because GWAS data on disease involvement are propagated through the network. For the GWAB web server, we used an updated HumanNet constructed based on similar network inference and integration methods as in a previous study (12). We found that the updated HumanNet significantly improved GWAB performance for the disease GWAS data tested in this study. The manuscript for the updated HumanNet is currently under preparation.
The key advantage of GWAB is its ability to use sub-threshold GWAS association data in disease gene prediction. However, typically, GWAS deposit P-value data only for the subset of SNPs that pass the significance threshold. We were able to find public GWAS data that provide P-values for the whole set of SNPs in seven human diseases (Table 1): Alzheimer's disease (ALZ) GWAS (16) by the International Genomics of Alzheimer's Project (IGAP) Consortium, coronary artery disease (CAD) GWAS (17) by the Coronary Artery Disease Genome-Wide Replication and Meta-analysis plus the Coronary Artery Disease Genetics (CARDIoGRAMplusC4D) Consortium, CD GWAS (18) and ulcerative colitis (UC) GWAS (19) by the International Inflammatory Bowel Disease Genetics Consortium (IIBDGC), rheumatoid arthritis (RA) GWAS (20) by the Biologics in RA Control (BIRAC) Consortium, schizophrenia (SZ) GWAS (21) by the Psychiatric Genomics Consortium (PGC) and T2D GWAS (22) by the Diabetes Genetics Replication and Meta-analysis (DIAGRAM) Consortium. In practice, GWAB analysis ignores SNPs with a P-value > 0.01, which generally accounts for 95–98% of all SNPs. We found that this data filtration step substantially reduced the computation time with no significant changes in predictions.
Table 1. GWAS data and reference genes for the seven diseases tested in this study.
| Disease name | GWAS data sources* | # SNPs | # Cohorts | Sources of reference disease genes$ | # Disease genes |
|---|---|---|---|---|---|
| Alzheimer's disease (ALZ) | IGAP | 7 055 882 | 25 580 cases | OMIM, DO | 406 |
| 48 466 controls | |||||
| Coronary artery disease (CAD) | CARDIoGRAMplusC4D | 2 420 361 | 22 233 cases | OMIM, DO, CADgene | 678 |
| 64 762 controls | |||||
| Crohn's disease (CD) | IIBDGC | 12 255 197 | 22 027 cases | OMIM, DO | 182 |
| 29 082 controls | |||||
| Rheumatoid arthritis (RA) | BIRAC | 2 556 272 | 12 307 cases | OMIM, DO, RADB | 941 |
| 28 975 controls | |||||
| Schizophrenia (SZ) | PGC | 9 444 232 | 36 989 cases | OMIM, DO, SZGene | 1155 |
| 113 075 controls | |||||
| Type 2 diabetes (T2D) | DIAGRAM | 2 473 442 | 34 840 cases | OMIM, DO, T2DGADB | 615 |
| 114 981 controls | |||||
| Ulcerative colitis (UC) | IIBDGC | 12 276 506 | 16 315 cases | OMIM, DO | 204 |
| 32 635 controls |
*GWAS data sources:
IGAP (The International Genomics of Alzheimer's Project (IGAP): http://web.pasteur-lille.fr/en/recherche/u744/igap/igap_download.php
CARDIoGRAMplusC4D (Coronary ARtery DIsease Genome-wide Replication and Meta-analysis plus Coronary artery Disease Genetics): http://www.cardiogramplusc4d.org
IIBDGC (International Inflammatory Bowel Disease Genetics Consortium): https://www.ibdgenetics.org
BIRAC (Biologics in RA Control) Consortium: (http://www.broadinstitute.org/ftp/pub/rheumatoid_arthritis/Stahl_etal_2010NG)
PGC (Psychiatric Genomics Consortium): https://www.med.unc.edu/pgc
DIAGRAM (DIAbetes Genetics Replication And Meta-analysis): http://diagram-consortium.org
$Sources of reference disease genes
OMIM (Online Mendelian Inheritance in Man): http://omim.org
DO (Disease Ontology): http://disease-ontology.org)
CADgeneDB (Coronary Artery Disease Gene Database): http://www.bioguo.org/CADgene/
RADB (a database of rheumatoid arthritis-related polymorphisms): http://www.bioapp.org/RADB/
SZGene (SchizophreniaGene): http://www.szgene.org/
T2DGADB (type 2 diabetes genetic association database): http://www.diabetes.org
To benchmark the predictions for disease-associated genes, we constructed reference gene sets for the seven diseases based on several disease gene databases (Table 1). The two most popular disease gene databases, Online Mendelian Inheritance in Man (OMIM) as of May 2014 (23) and Disease Ontology (DO) as of March 2016 (24), were used to collect reference genes for all seven diseases. In addition, we employed several disease-specific databases: CADgeneDB (25) for CAD, RADB (26) for RA, SZGene database (27) for SZ and T2DGADB (28) for T2D. All GWAS data and reference disease genes used in this study are available from the GWAB server. In the web tool, we also provide pre-compiled disease gene sets derived from a database DISEASES (29), which enable users to provide reference gene sets easily for many other diseases in future analyses.
Benchmarking predictions for disease-associated reference genes
We first evaluated the ability of GWAB to retrieve disease-associated reference genes. We evaluated GWAB performance with 5-fold cross validation, in which the entire set of reference disease genes is divided into five subsets: four of them are used for optimal threshold search and one for testing predictions. By using the completely different data between searching parameters and testing prediction performance, over evaluation of GWAB was avoided. For the optimal log(P) threshold, GWAB more effectively retrieved the disease-associated reference genes than GWAS did alone for all tested diseases (Figure 2A–G). For all disease GWAS, GWAB found the optimal log(P) threshold that achieved the highest AUC: −2.4 for ALZ, −6 for CAD, −6 for CD, −1.8 for RA, −2.4 for SZ, −3.3 for T2D and −2.7 for UC. These results suggest that the GWAB approach can be generalized to many other disease GWAS to rescue false negatives and discover novel disease-associated genes.
Figure 2.

Evaluation of GWAB using seven disease GWAS. Summary plots of GWAB analyses for (A) Alzheimer's disease (ALZ), (B) coronary artery disease (CAD), (C) Crohn's disease (CD), (D) rheumatoid arthritis (RA), (E) schizophrenia (SZ), (F) Type 2 Diabetes (T2D) and (G) ulcerative colitis (UC). (H) Bar graphs representing the AUC scores for disease gene predictions by GWAB, NetWAS with all tissues and NetWAS with specific tissue (brain for ALZ, heart for CAD, intestine for CD, bone for RA, brain for SZ, liver for T2D, intestine for UC).
Next, we compared GWAB with NetWAS (13), another method for the network-based reprioritizing of GWAS associations. GWAB and NetWAS are distinct in several aspects. First, GWAB reprioritizes genes using the GWAS association scores of its network neighbors, whereas NetWAS reprioritizes genes using a support vector machine (SVM) classifier with training network features. Under the hypothesis that disease-relevant genes would be enriched among the nominally significant genes, NetWAS trains an SVM classifier using nominally significant (P-value < 0.01 by default) genes as positive examples and 10 000 randomly selected non-significant (P-value ≥ 0.01) genes as negatives. Second, GWAB uses only a single integrated co-functional network, whereas NetWAS can use 144 tissue-specific co-functional networks. In practice, users may conduct a NetWAS analysis with a specific tissue network (i.e. the most relevant tissue for the disease) or a network for all tissues. Third, GWAB provides the service of P-value assignment to genes, but NetWAS does not. Users need to assign P-values to genes using separate tools such as VEGAS (14). Fourth, GWAB runs many analyses using various P-value thresholds and finds the optimal threshold to output the final results automatically, whereas each job on NetWAS runs using a single parameter setting. Therefore, we used NetWAS results obtained with the default parameter setting in the comparison. We found that GWAB outperformed NetWAS in the seven diseases, with either a tissue-specific or an all-tissue network (Figure 2H). NetWAS predictions based on different tissue-networks or parameters did not result in significant changes in performance. Although we did not exhaustively test for different GWAS and reference genes, these results indicate that GWAB is one of the most useful tools for augmenting GWAS.
Validation of novel candidate genes by GWAB
In addition to rescuing known disease genes that could not be identified by GWAS alone, GWAB may predict new candidate genes for the disease. Figure 3 presents the largest component of the network of genes that are significantly associated with CAD after network boosting (GWAB score > 7.3). Genes with a larger node size are more likely to be involved in CAD based on the GWAB score. The intensity of the node color indicates the degree of the network boost. Genes with a darker color were raised to the higher ranks by more steps by a stronger effect of the network boost. The reference CAD genes are indicated by red gene names, and significant candidate genes retrieved by GWAS alone are indicated by red node borders. Four reference genes for CAD were retrieved by GWAS alone (Supplemental Table S1): CDKN2A, CDKN2B, LPA and ZPR1. The same GWAS identified seven new candidate genes for CAD, and we found that three of them, PSRC1 (30), SMARCA4 (31) and ZC3HC1 (17), were known to have CAD-associated genetic variants according to a literature review (Supplemental Table S2).
Figure 3.

The largest component of the network composed of significant candidate genes for CAD after GWAB network boosting. Edges were derived from HumanNet (an updated version). The node size represents the final GWAB score, and the intensity of the node color represents the degree of network boosting. Reference CAD genes are indicated by red names, and candidate CAD genes identified by GWAS alone are indicated by red node borders.
To evaluate effectiveness of the network boost, we focused on genes that were highly boosted by the network (dark nodes). Four reference genes for CAD were not retrieved by GWAS alone but were rescued by network boosting (Supplemental Table S3): EGFR, FN1, PECAM1 and PLG. The prediction ranks for these genes were raised by many steps after network boosting (e.g. EGFR was 2966th by GWAS alone but 44th by GWAB). Therefore, we could retrieve twice as many reference CAD genes using network boosting compared with by GWAS alone. Notably, we found 17 new candidate genes for CAD by network boosting that were not found by GWAS alone (Supplemental Table S4). Surprisingly, we validated that 11 of them (11/17 = 65%) were associated with CAD by a literature review. SMAD3-dependent regulation of COL4A1/COL4A2 was reported to have a functional significance in CAD pathogenesis (32), and SPARC was reported to be involved in CAD progression (33). COL5A2 was also reported as a novel candidate marker for the identification and treatment of ischemic cardiovascular disease (34). PTPN11 was reported to contain a CAD risk variant (35). SMAD3 was reported to be associated with CAD and suggested to be a useful biomarker for diagnosis and risk stratification (36). Recently, a cohort-based study reported that levels of amyloid-beta 1–40 peptides that are generated by proteolytic cleavage of the protein encoded by APP are significantly associated with arterial stiffness progression (37). CALD1 was reported to play an essential role in the regulation of smooth muscle (38), suggesting that its dysregulation causes cardiac disorders. A polymorphism of FBN1 has been suggested to be associated with aortic stiffness and disease severity in CAD patients (39). A recent GWAS analysis identified a genetic variant of FLT1 associated with increased risk of CAD in a Japanese population (40). GUCY1B3 encodes a key enzyme of the nitric oxide signaling pathway, and impaired nitric oxide signaling has been implicated in the pathogenesis of cardiovascular disease, including CAD (41).
We also examined the literatures for the top 20 GWAB candidates in other five diseases that are highly predictive for <5% FPR (see Figure 2A–G), and found that many of them are reference disease genes or validated by literature evidences: 17 candidates for AD, 11 for CD, 12 for SZ, 12 for T2D, 9 for UC (Supplemental Table S6). These results strongly suggest that GWAB can identify disease-associated genes through augmenting GWAS data by incorporating functional association between genes.
DISCUSSION
Network-based boosting of GWAS data has several advantages in discovery of disease-associated genes. First, it can effectively integrate complementary information from population-based approach and molecular profiling approach to disease gene identification. The complementarity between two approaches was demonstrated as many disease-associated genes (either known or validated by literatures) were retrieved not by statistical association from GWAS alone but by integration of GWAS with functional associations of the network. Second, it can utilize the inherent value of SNPs with sub-threshold significance. For example, CAD GWAS analyzed in the study used only 160 SNPs that pass the typical P-value threshold to identify disease gene candidates. However, 3079 SNPs that are below the typical threshold contributed to identify disease gene candidates with GWAB. Therefore, network-based boosting substantially increase the information usage of the given GWAS. Third, since neither GWAS nor the functional network makes any prior assumptions about the disease studies, this strategy is free from the study bias.
Since GWAB method uses the functional gene network, several limitations can come from network quality. First, only genes that are included in the network can be boosted. For example, the functional network used for GWAB covers approximately 93% of coding genes in human, thus the other 7% of coding genes cannot be boosted at all. Second, network topology can cause bias in predictions. For example, hub genes are more likely to be boosted due to the larger number of network neighbors. Third, bias in pathway information of the functional network may cause bias in disease predictions. Thus, we need to continue to expand the network coverage by incorporating new functional data regularly, and to reduce the functional bias of the network.
CONCLUSION
Previously, we demonstrated that GWAS data can be enhanced by incorporating co-functional network data, but we could not expand the application to a larger number of GWAS due to the lack of public data that provide summary statistics for whole sets of SNPs. In this study, we developed GWAB, a web-based service for GWAS boosting, and validated its effectiveness using seven public GWAS whose data were recently released by several consortia. Most GWAS generate trait-association probability data for more than a million SNPs. However, in general, only a subset of data including the most significant SNPs has been deposited in the public databases. Because the importance of data sharing has recently grown among the genomics community, we now expect to see more GWAS with complete summary statistics data. Our study clearly demonstrated the inherent value of sub-threshold GWAS associations. GWAB will offer a feasible tool to boost such associations for human disease genetics.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Research Foundation of Korea [2012M3A9B4028641, 2012M3A9C7050151, 2015R1A2A1A15055859 to I.L.]. Funding for open access charge: National Research Foundation of Korea [2015R1A2A1A15055859].
Conflict of interest statement. None declared.
REFERENCES
- 1. MacArthur J., Bowler E., Cerezo M., Gil L., Hall P., Hastings E., Junkins H., McMahon A., Milano A., Morales J. et al. . The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017; 45:D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. McClellan J., King M.C.. Genetic heterogeneity in human disease. Cell. 2010; 141:210–217. [DOI] [PubMed] [Google Scholar]
- 3. Leiserson M.D., Eldridge J.V., Ramachandran S., Raphael B.J.. Network analysis of GWAS data. Curr. Opin. Genet. Dev. 2013; 23:602–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jia P., Zhao Z.. Network.assisted analysis to prioritize GWAS results: principles, methods and perspectives. Hum. Genet. 2014; 133:125–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ideker T., Ozier O., Schwikowski B., Siegel A.F.. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics. 2002; 18(Suppl. 1):S233–S240. [DOI] [PubMed] [Google Scholar]
- 6. Jia P., Zheng S., Long J., Zheng W., Zhao Z.. dmGWAS: dense module searching for genome-wide association studies in protein-protein interaction networks. Bioinformatics. 2011; 27:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baranzini S.E., Galwey N.W., Wang J., Khankhanian P., Lindberg R., Pelletier D., Wu W., Uitdehaag B.M., Kappos L., Gene M.S.A.C. et al. . Pathway and network-based analysis of genome-wide association studies in multiple sclerosis. Hum. Mol. Gen. 2009; 18:2078–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bakir-Gungor B., Sezerman O.U.. A new methodology to associate SNPs with human diseases according to their pathway related context. PLoS One. 2011; 6:e26277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rossin E.J., Lage K., Raychaudhuri S., Xavier R.J., Tatar D., Benita Y., Cotsapas C., Daly M.J., Genetic I.I.B.D.. Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS Genet. 2011; 7:e1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Akula N., Baranova A., Seto D., Solka J., Nalls M.A., Singleton A., Ferrucci L., Tanaka T., Bandinelli S., Cho Y.S. et al. . A network-based approach to prioritize results from genome-wide association studies. PLoS One. 2011; 6:e24220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tasan M., Musso G., Hao T., Vidal M., MacRae C.A., Roth F.P.. Selecting causal genes from genome-wide association studies via functionally coherent subnetworks. Nat. Methods. 2015; 12:154–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee I., Blom U.M., Wang P.I., Shim J.E., Marcotte E.M.. Prioritizing candidate disease genes by network-based boosting of genome-wide association data. Genome Res. 2011; 21:1109–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Greene C.S., Krishnan A., Wong A.K., Ricciotti E., Zelaya R.A., Himmelstein D.S., Zhang R., Hartmann B.M., Zaslavsky E., Sealfon S.C. et al. . Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 2015; 47:569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu J.Z., McRae A.F., Nyholt D.R., Medland S.E., Wray N.R., Brown K.M., Investigators A., Hayward N.K., Montgomery G.W., Visscher P.M. et al. . A versatile gene-based test for genome-wide association studies. Am. J. Hum. Genet. 2010; 87:139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee I., Date S.V., Adai A.T., Marcotte E.M.. A probabilistic functional network of yeast genes. Science. 2004; 306:1555–1558. [DOI] [PubMed] [Google Scholar]
- 16. Lambert J.C., Ibrahim-Verbaas C.A., Harold D., Naj A.C., Sims R., Bellenguez C., Jun G., DeStefano A.L., Bis J.C., Beecham G.W. et al. . Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat. Genet. 2013; 45:1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schunkert H., Konig I.R., Kathiresan S., Reilly M.P., Assimes T.L., Holm H., Preuss M., Stewart A.F.R., Barbalic M., Gieger C. et al. . Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat. Genet. 2011; 43:333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Franke A., McGovern D.P.B., Barrett J.C., Wang K., Radford-Smith G.L., Ahmad T., Lees C.W., Balschun T., Lee J., Roberts R. et al. . Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat. Genet. 2010; 42:1118–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Anderson C.A., Boucher G., Lees C.W., Franke A., D’Amato M., Taylor K.D., Lee J.C., Goyette P., Imielinski M., Latiano A. et al. . Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 2011; 43:246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stahl E.A., Raychaudhuri S., Remmers E.F., Xie G., Eyre S., Thomson B.P., Li Y.H., Kurreeman F.A.S., Zhernakova A., Hinks A. et al. . Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet. 2010; 42:508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ripke S., Neale B.M., Corvin A., Walters J.T.R., Farh K.H., Holmans P.A., Lee P., Bulik-Sullivan B., Collier D.A., Huang H.L. et al. . Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014; 511:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morris A.P., Voight B.F., Teslovich T.M., Ferreira T., Segre A.V., Steinthorsdottir V., Strawbridge R.J., Khan H., Grallert H., Mahajan A. et al. . Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet. 2012; 44:981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Amberger J., Bocchini C.A., Scott A.F., Hamosh A.. McKusick's Online Mendelian Inheritance in Man (OMIM ®). Nucleic Acids Res. 2009; 37:D793–D796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kibbe W.A., Arze C., Felix V., Mitraka E., Bolton E., Fu G., Mungall C.J., Binder J.X., Malone J., Vasant D. et al. . Disease Ontology 2015 update: an expanded and updated database of human diseases for linking biomedical knowledge through disease data. Nucleic Acids Res. 2015; 43:D1071–D1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu H., Liu W., Liao Y.F., Cheng L., Liu Q.A., Ren X.A., Shi L.S., Tu X., Wang Q.K., Guo A.Y.. CADgene: a comprehensive database for coronary artery disease genes. Nucleic Acids Res. 2011; 39:D991–D996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang R.J., Luan M.W., Shang Z.W., Duan L., Tang G.P., Shi M., Lv W.H., Zhu H.J., Li J., Lv H.C. et al. . RADB: a database of rheumatoid arthritis-related polymorphisms. Database (Oxford). 2014; bau090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Allen N.C., Bagade S., McQueen M.B., Ioannidis J.P.A., Kavvoura F.K., Khoury M.J., Tanzi R.E., Bertram L.. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat. Genet. 2008; 40:827–834. [DOI] [PubMed] [Google Scholar]
- 28. Lim J.E., Hong K.W., Jin H.S., Kim Y.S., Park H.K., Oh B.. Type 2 diabetes genetic association database manually curated for the study design and odds ratio. BMC Med. Inform. Decis. Mak. 2010; 10:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pletscher-Frankild S., Palleja A., Tsafou K., Binder J.X., Jensen L.J.. DISEASES: text mining and data integration of disease-gene associations. Methods. 2015; 74:83–89. [DOI] [PubMed] [Google Scholar]
- 30. Samani N.J., Braund P.S., Erdmann J., Gotz A., Tomaszewski M., Linsel-Nitschke P., Hajat C., Mangino M., Hengstenberg C., Stark K. et al. . The novel genetic variant predisposing to coronary artery disease in the region of the PSRC1 and CELSR2 genes on chromosome 1 associates with serum cholesterol. J. Mol. Med. 2008; 86:1233–1241. [DOI] [PubMed] [Google Scholar]
- 31. Xiong X., Xu C., Zhang Y., Li X., Wang B., Wang F., Yang Q., Wang D., Wang X., Li S. et al. . BRG1 variant rs1122608 on chromosome 19p13.2 confers protection against stroke and regulates expression of pre-mRNA-splicing factor SFRS3. Hum. Gen. 2014; 133:499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Turner A.W., Nikpay M., Silva A., Lau P., Martinuk A., Linseman T.A., Soubeyrand S., McPherson R.. Functional interaction between COL4A1/COL4A2 and SMAD3 risk loci for coronary artery disease. Atherosclerosis. 2015; 242:543–552. [DOI] [PubMed] [Google Scholar]
- 33. Takahashi M., Nagaretani H., Funahashi T., Nishizawa H., Maeda N., Kishida K., Kuriyama H., Shimomura I., Maeda K., Hotta K. et al. . The expression of SPARC in adipose tissue and its increased plasma concentration in patients with coronary artery disease. Obes. Res. 2001; 9:388–393. [DOI] [PubMed] [Google Scholar]
- 34. Azuaje F., Zhang L., Jeanty C., Puhl S.L., Rodius S., Wagner D.R.. Analysis of a gene co-expression network establishes robust association between Col5a2 and ischemic heart disease. BMC Med. Genomics. 2013; 6:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Han X., Zhang L.J., Zhang Z.Q., Zhang Z.T., Wang J.C., Yang J., Niu J.M.. Association between phosphatase related gene variants and coronary artery disease: case-control study and meta-analysis. Int. J. Mol. Sci. 2014; 15:14058–14076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen C., Lei W., Chen W.J., Zhong J.F., Gao X.X., Li B., Wang H.L., Huang C.X.. Serum TGF-beta 1 and SMAD3 levels are closely associated with coronary artery disease. BMC Cardiovasc. Disord. 2014; 14:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stamatelopoulos K., Sibbing D., Rallidis L.S., Georgiopoulos G., Stakos D., Braun S., Gatsiou A., Sopova K., Kotakos C., Varounis C. et al. . Amyloid-beta (1-40) and the risk of death from cardiovascular causes in patients with coronary heart disease. J. Am. Coll. Cardiol. 2015; 65:904–916. [DOI] [PubMed] [Google Scholar]
- 38. Wang C.L. Caldesmon and smooth-muscle regulation. Cell Biochem. Biophys. 2001; 35:275–288. [DOI] [PubMed] [Google Scholar]
- 39. Medley T.L., Cole T.J., Gatzka C.D., Wang W.Y., Dart A.M., Kingwell B.A.. Fibrillin-1 genotype is associated with aortic stiffness and disease severity in patients with coronary artery disease. Circulation. 2002; 105:810–815. [DOI] [PubMed] [Google Scholar]
- 40. Konta A., Ozaki K., Sakata Y., Takahashi A., Morizono T., Suna S., Onouchi Y., Tsunoda T., Kubo M., Komuro I. et al. . A functional SNP in FLT1 increases risk of coronary artery disease in a Japanese population. J. Hum Genet. 2016; 61:435–441. [DOI] [PubMed] [Google Scholar]
- 41. Stasch J.P., Pacher P., Evgenov O.V.. Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation. 2011; 123:2263–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.