


Abstract
The PharmMapper online tool is a web server for potential drug target identification by reversed pharmacophore matching the query compound against an in-house pharmacophore model database. The original version of PharmMapper includes more than 7000 target pharmacophores derived from complex crystal structures with corresponding protein target annotations. In this article, we present a new version of the PharmMapper web server, of which the backend pharmacophore database is six times larger than the earlier one, with a total of 23 236 proteins covering 16 159 druggable pharmacophore models and 51 431 ligandable pharmacophore models. The expanded target data cover 450 indications and 4800 molecular functions compared to 110 indications and 349 molecular functions in our last update. In addition, the new web server is united with the statistically meaningful ranking of the identified drug targets, which is achieved through the use of standard scores. It also features an improved user interface. The proposed web server is freely available at http://lilab.ecust.edu.cn/pharmmapper/.
INTRODUCTION
Identification and confirmation of bioactive small-molecule targets is a crucial, often decisive step both in academic and pharmaceutical research (1). Through the development and availability of a variety of new experimental and computational techniques, the ‘one gene, one drug, one disease’ paradigm is, in principle, feasible and the number of successful examples steadily grows. As yet that era has not eventuated. However, what has become apparent is that compound promiscuity plays a key role in the efficacy of a significant number of approved drugs, which emphasizes the need to employ a new paradigm of polypharmacology. Polypharmacology focuses on searching for multi-target drugs to perturb disease-associated networks rather than designing selective ligands to target individual proteins (2,3). Web servers that allow the polypharmacology effects prediction have been developed and there is an increasing number of approaches that deal with targets prediction from different perspectives (4–7).
Pharmacophore modeling is a broadly used ligand-based method in drug target identification. It refers to a protein−ligand interaction pattern corresponding to a desired pharmacological effect and can be considered as the largest common denominator shared by a set of active molecules. PharmaGist (8) is a free web server that can identify a consensus pharmacophore of a set of ligands in a few minutes. Alternatively, structure-based methods require a ligand-binding structure and generate the corresponding pharmacophore model by analyzing the interaction site. ZINCPharmer (9) provides a mechanism for deriving a pharmacophore model directly from structures within the Protein Database Bank (PDB) and also supports searching the ZINC database using the Pharmer (10) pharmacophore search technology. Launched on the web in 2010, an open-source platform called PharmMapper has been designed to identify potential target candidates for the given small molecules using reverse pharmacophore mapping approach (11). Benefiting from the highly efficient and robust pharmacophore mapping approach, PharmMapper bears high-throughput ability and can identify the potential target candidates from the database within hours. Furthermore, a statistically meaningful score of the identified proteins has been proposed to improve the hits enrichment and enhance the liability of PharmMapper method (12). Collectively, PharmMapper is popular and has been widely used for targeting fishing. PharmMapper is backed up by a large, internal collection of pharmacophore database, namely PharmTargetDB annotated from all the targets information in BindingDB, TargetBank, DrugBank and PDTD. Over 7000 receptor-based pharmacophore models (covering 1627 drug target information, 459 of which are human protein targets) are stored and accessed by PharmMapper. However, the number of structures deposited in PDB (13) grows linearly yearly. As of December 2016, over 125 000 structures had already been deposited into PDB, which is much more than the number of target pharmacophore models in PharmTargetDB, indicating the inadequate coverage of target pharmacophores in PharmMapper.
In this update, we describe the new features and extensions of the PharmMapper web server. In the next section, we describe how we have substantially extended our collection of the targets pharmacophore databases. The following section reports on updates to the ranking of the identified target pharmacophores by utilizing a statistically meaningful score. Finally, we describe the updates to our user interface, in which we have improved the user experience, removed our dependency on Java applets in favor of embedded JavaScript components.
MATERIALS AND METHODS
Binding sites detection
A well-defined binding pocket is critical for ligand design and target identification. CAVITY is a newer method, which adopts a structural geometry-based ligand-binding sites detection and analysis strategy with the capability of predicting both the druggabilities and ligandabilities of the detected binding sites (14,15). This software uses CavityScore and CavityDrugScore to make quantitative predictions for the ligandability and druggability of a binding site, respectively. Druggability is defined as the possibility of being a good target for drug discovery, while ligandability is the possibility of finding a small molecule that binds to a certain target.
The value of CavityScore is related to the depth of the pocket, the size of the pocket lip, the physicochemical properties of hydrophobicity and the presence of hydrogen bonds. By using the training sets from PDBbind (16), the average experimental binding affinity pKd (Ave) showed linear relationship with CavityScore. Therefore, CavityScore is used to predict the average pKd of the binding site by a linear equation.
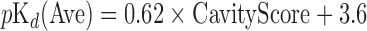 |
Protein druggability is a more complicated property that is related to higher level properties of ligands (ADME/T) as well as the role of the macromolecules acting in cellular pathways. Considering the complexity of druggability, many researchers have applied machine learning algorithms to make a qualitative prediction. CAVITY used the NRDLD dataset (17) to train and validate CavityDrugScore. It turned out that druggable and undruggable proteins were successfully separated by using CavityDrugScore.
Receptor-based pharmacophore modeling
We have also developed a standalone program, Pocket version 4.0, a new update of Pocket program (18,19), which is a software tool that allows rapid derivation of pharmacophores from the three-dimensional (3D) structure of a protein receptor. In Pocket version 4.0, five primary types of pharmacophore features were adopted in this process: hydrophobic center, positive-charged center, negative-charged center, hydrogen bond acceptor vector and hydrogen bond donor vector. Each cavity was automatically analyzed by scored-grids and the crucial features in the pharmacophore model were reduced to a reasonable number. Moreover, the shapes and boundaries of cavities were characterized by a number of excluded volumes centered at the grid clusters.
Enhanced pharmacophore-based target prediction method
In PharmMapper, target proteins with highest fit scores between corresponding pharmacophore models and query compound were predicted as potential targets, regardless of the distribution of fit scores for the given targets across different ligands, which could bias the prediction accuracy toward the protein targets with more pharmacophore features. Therefore, we have developed a novel method to improve the accuracy and discriminative ability of PharmMapper (12). A ligand–target pairwise fit score matrix reflecting the fit score distribution was generated by profiling all the pharmacophore models in PharmMapper target database with the corresponding ligands from the original protein–ligand complex structures. Based on the matrix, the probability of finding a given target pharmacophore by random can be estimated from the corresponding fit value with the query ligand. Two retrospective tests were carried out on DrugBank (20) and ChEMBL (21) databases. For the DrugBank dataset, about half of the targets were identified at 10% false positive rate (FPR) by both fit score and Z’-score, while 36.78% of targets were identified by Z’-score at 5% FPR versus 32.48% by fit score; while for the ChEMBL dataset, Z’-score achieves 15% more performance than fit score at top 1% rank stages and increases to 30% more at top 0.1% predictions. Those results demonstrated that the probability-based ranking score could enhance the hit rate or enrichment based on the previous pharmacophore database.
NEW FEATURES IN THE 2017 PharmMapper WEB SERVER
Pharmacophore databases extension
Originally, PharmMapper included PDB structures with co-crystallized ligand and extracted the pharmacophore models describing the binding modes at the ligand-binding sites (11). Thus, some potential targets of the query ligand could be missed due to the limited coverage of the database. Moreover, the presence of druggable, topographically distinct allosteric sites has offered new paradigm for small molecules. Ligands that target allosteric sites offer significant advantages over the corresponding orthosteric ligands in terms of selectivity (22).
To conduct an update, the non-redundant set of protein chains achieved with the BLASTClust (23) algorithm were downloaded from the RCSB PDB server (ftp://resources.rcsb.org/sequence/clusters/; January 1, 2016 snapshot), with the sequence similarity threshold set to 90%. The PDB structures with the best resolution in each cluster were chosen, leading to a total of 38 088 chains with 35 161 unique PDBs. We then used CAVITY to detect all the potential-binding sites for the above structures and finally a comprehensive, non-redundant dataset containing crystal structures of 23 236 PDBs with 16 159 druggable cavities and 51 431 ligandable cavities (a predicted pKd value higher than 6.0) was retained. This dataset was subsequently used to extract pharmacophore models using Pocket version 4.0. As a result, we generated 53 184 unique pharmacophore models, which is currently the largest collection of this kind. Table 1 gives a summary of the basic information of the two versions. One can see that the pharmacophore databases in PharmMapper have increased more than six times. Compared with version 2010, the number of proteins in the current release (version 2017) has almost tripled.
Table 1. A comparison of current and the previous version of PharmMapper.
| Category | Version 2010 | Version 2017 |
|---|---|---|
| Entries in PDB | 7302 | 23 236 |
| Number of pharmacophore models | 7302 | 53 184 |
| Number of unique indications | 110 | 450 |
| Number of unique molecular functions | 349 | 4800 |
The target annotations were extracted from UniProt (24) and were categorized as follows: UniProt ID, target name, target function and disease involved. The entire 640 indications were collected from KEGG (25) and DrugBank (20), and then the indication annotation was done automatically by running a string-match script between the indication term and the publication abstracts related to the target. Finally, the significantly expanded target data cover 450 indications and 4800 target functions, which are compared to 110 indications and 349 molecular functions in the initial version.
PharmMapper web services
Input
PharmMapper accepts a file with single drug-like molecule or natural product stored in MOL2 or SDF format. If the uploaded molecule does not have 3D structural information, the server will automatically convert it into a single 3D conformer. After uploading the file, the user is allowed to search either the target sets in version 2010 or the updated pharmacophore database (Figure 1A). Furthermore, the estimated time consumed by the complete screening and scoring protocol for each target set is also presented. Since PharmMapper adopts a semi-flexible alignment strategy, a conformer ensemble has to be generated prior to pharmacophore mapping. For the single 3D conformer, an in-house program Cyndi (26) is used by default to generate multiple conformations and the computational cost is proportional to the number of conformers.
Figure 1.

PharmMapper interface. (A) The target sets provided by PharmMapper. (B) The ranked list of hit target pharmacophore models from the newly updated database (Druggable pharmacophore database), which are sorted by fit score in descending order. The pharmacophore model candidate and the aligned pose of molecule are shown in the JavaScript 3D structure viewer JSmol.
Output
A typical run of PharmMapper task for the old version database takes 1–2 h, while it costs 4–6 h for the druggable target pharmacophore database and 20–24 h for the ligandable target pharmacophore database. Upon completion of the computations, the results of the hit target pharmacophore models are demonstrated in the form of a ranked list. Since a protein may have many PDB structures presenting identical or distinct pharmacophores in the binding site, we have regrouped those PDB structures and adjusted to list only the PDB file with the highest score.
An important aspect of the results table renovation was to replace the Java applets that were in use with appropriate JavaScript components and libraries. Due to the heavily publicized security failures, the Java install base is shrinking. Even when Java is installed, users are presented with multiple security prompts that must be correctly navigated before a Java applet can run. To address this concern, we have utilized a JavaScript-based JSmol web applet for the interactive ligand-pharmacophore alignment poses analysis and visualization (27). In addition, Z’-scores described in the Enhanced pharmacophore-based target prediction method section are added to the output of a specific PharmMapper run. Users can also re-rank the result list by Z’-scores in descending order via clicking the arrow icon in the corresponding column (Figure 1B). The above function currently only supported the old version database of PharmMapper.
TEST CASE
Identified targets ranking
Here we show a test case using kanamycin as the query structure to find its potential target proteins via a PharmMapper search. In this case the 16 159 druggable pharmacophore models were used as the searching database. Kanamycin is an antibiotic used to treat bacterial infections and tuberculosis. The top 1% (top 162) pharmacophore candidates identified are listed in Supplementary Table S1 and those corresponding to the proteins with experimental evidence are shown in Table 2. Two kinds of important protein targets of kanamycin, namely aminoglycoside phosphotransferases (Rank 26, 32, 39 and 80) (Figure 2) and nucleotidyltransferase (Rank 122) as validated in the DrugBank database (20) are identified by PharmMapper. Interestingly, the bacterial multidrug exporter AcrB (PDB ID: 3AOC, Rank 42) was also revealed by PharmMapper as a potential target of kanamycin. Only the structures of multidrug exporter AcrB bound to ifampicin and erythromycin were solved but AcrB was indeed reported to export kanamycin (28). In practice, users usually will only focus on the top 50–200 candidates for further experimental testing. In this test case six potential kanamycin targets were ranked in the top list, indicating the reliability of this server tool.
Table 2. Pharmacophore candidates of kanamycin identified by PharmMapper from the 16 159 druggable pharmacophore models.
| Rank | PDB ID | Target name |
|---|---|---|
| 26 | 3SG9 | Aminoglycoside-2΄-phosphotransferase type IVa |
| 32 | 4DFU | Aminoglycoside phosphotransferase APH (2΄)-ID/APH (2΄)-IVa |
| 39 | 4H05 | Aminoglycoside-3΄-phosphotransferase of type VIII |
| 42 | 3AOC | Multidrug exporter AcrB |
| 80 | 4GKI | Aminoglycoside phosphotransferase APH (3΄)-Ia |
| 122 | 4WQL | Aminoglycoside nucleotidylyltransferase ANT (2΄)-Ia |
Figure 2.

Kanamycin targeted on Aminoglycoside 2΄-phosphotransferase IVa (PDB ID: 3SG9). (A) Kanamycin (sticks), pharmacophores (balls) and surface (gray space) of the cavity kanamycin occupies, are presented. (B) Kanamycin, which is shown as sticks, as well as pharmacophores around the molecule, are presented. Among pharmacophores, purple balls indicate hydrogen bond acceptor centers, green balls indicate hydrogen bond donor centers and light blue balls indicate hydrophobic centers.
Performance comparison of old database version versus new database
The 7302 proteins in the initial version were used to extract pharmacophore models using Pocket 4.0. Tamoxifen was then used as a proof-of-concept compound to test the target identification accuracy between the newly constructed pharmacophore database and the old database version which was built up by using the macromolecule–ligand complexes-based 3D pharmacophore extraction tool LigandScout (29). The rankings of 11 known targets of 4H-tamoxifen from two databases retrieved by PharmMapper are shown in Table 3. Four known targets of tamoxifen were identified among the top 100 candidates for both two databases, while nine tamoxifen targets were covered for the new database in the top 1000 pharmacophore models versus 10 for the old version, indicating the reliability of the new version web server.
Table 3. Performance comparison of the target identification for 4H-tamoxifen against the two pharmacophore databases extracted using Pocket 4.0 and LigandScout, respectively.
| Rank | |||
|---|---|---|---|
| PDB ID | Target Name | Old database version | New database version |
| 1YA4 | Liver carboxylesterase | 130 | 2 |
| 2GPU | Estrogen-related receptor gamma | 1 | 5 |
| 1XA5 | Calmodulin | 297 | 30 |
| 1HJ1 | Microsomal antiestrogen binding site (AEBS) | >1000 | 69 |
| 1I5R | Estradiol 17-β-dehydrogenase | 18 | 188 |
| 1GSF | Glutathione S-transferase | 49 | 451 |
| 1XJD | Protein kinase C, theta type | 222 | 471 |
| 1HDC | 3-α, 20 β-hydroxysteriod dehydrogenase | 168 | 672 |
| 1PXX | Prostaglandin G/H synthase 2 | 124 | 957 |
| 1DG7 | Dihydrofolate reductase | 29 | >1000 |
| 3I7I | Collagenase 3 | 136 | >1000 |
DISCUSSION
PharmMapper is an open-source web server that identifies potential drug targets via large-scale reverse pharmacophore mapping strategy. Our previous platform provides pharmacophore databases for over 7000 protein–ligand complexes in PDB. The updated system included 23 236 proteins covering 16 159 druggable pharmacophore models and 51 431 ligandable pharmacophore models, which is six times larger than the previous version. The new pharmacophore database enables a more comprehensive target identification compared to the previous version. We have also reported new features and interface enhancements which have been added to PharmMapper. To our knowledge, there is no such freely available web server that would include such a large collection of target pharmacophore models. PharmMapper can provide useful insights for further bioassay in drug–target interaction research in pharmaceutical applications.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key Research and Development Program [2016YFA0502304, 2016YFA0502303, in part]; National Natural Science Foundation of China [81230090, 81273436]. Funding for open access charge: National Key Research and Development Program [2016YFA0502304, 2016YFA0502303, in part].
Conflict of interest statement. None declared.
REFERENCES
- 1. Ziegler S., Pries V., Hedberg C., Waldmann H.. Target identification for small bioactive molecules: finding the needle in the haystack. Angew. Chem. Int. Ed. Engl. 2013; 52:2744–2792. [DOI] [PubMed] [Google Scholar]
- 2. Hopkins A.L. Network pharmacology. Nat. Biotechnol. 2007; 25:1110–1111. [DOI] [PubMed] [Google Scholar]
- 3. Kitano H. A robustness-based approach to systems-oriented drug design. Nat. Rev. Drug Discov. 2007; 6:202–210. [DOI] [PubMed] [Google Scholar]
- 4. Li H., Gao Z., Kang L., Zhang H., Yang K., Yu K., Luo X., Zhu W., Chen K., Shen J. et al. TarFisDock: a web server for identifying drug targets with docking approach. Nucleic Acids Res. 2006; 34:W219–W224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang J.C., Chu P.Y., Chen C.M., Lin J.H.. idTarget: a web server for identifying protein targets of small chemical molecules with robust scoring functions and a divide-and-conquer docking approach. Nucleic Acids Res. 2012; 40:W393–W399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gfeller D., Grosdidier A., Wirth M., Daina A., Michielin O., Zoete V.. SwissTargetPrediction: a web server for target prediction of bioactive small molecules. Nucleic Acids Res. 2014; 42:W32–W38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gong J., Cai C., Liu X., Ku X., Jiang H., Gao D., Li H.. ChemMapper: a versatile web server for exploring pharmacology and chemical structure association based on molecular 3D similarity method. Bioinformatics. 2013; 29:1827–1829. [DOI] [PubMed] [Google Scholar]
- 8. Schneidman-Duhovny D., Dror O., Inbar Y., Nussinov R., Wolfson H.J.. PharmaGist: a webserver for ligand-based pharmacophore detection. Nucleic Acids Res. 2008; 36:W223–W228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Koes D.R., Camacho C.J.. ZINCPharmer: pharmacophore search of the ZINC database. Nucleic Acids Res. 2012; 40:W409–W414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Koes D.R., Camacho C.J.. Pharmer: efficient and exact pharmacophore search. J. Chem. Inf. Model. 2011; 51:1307–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu X., Ouyang S., Yu B., Liu Y., Huang K., Gong J., Zheng S., Li Z., Li H., Jiang H.. PharmMapper server: a web server for potential drug target identification using pharmacophore mapping approach. Nucleic Acids Res. 2010; 38:W609–W614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang X., Pan C., Gong J., Liu X., Li H.. Enhancing the enrichment of pharmacophore-based target prediction for the polypharmacological profiles of drugs. J. Chem. Inf. Model. 2016; 56:1175–1183. [DOI] [PubMed] [Google Scholar]
- 13. Rose P.W., Prlic A., Altunkaya A., Bi C., Bradley A.R., Christie C.H., Costanzo L.D., Duarte J.M., Dutta S., Feng Z. et al. The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017; 45:D271–D281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yuan Y., Pei J., Lai L.. Binding site detection and druggability prediction of protein targets for structure-based drug design. Curr. Pharm. Des. 2013; 19:2326–2333. [DOI] [PubMed] [Google Scholar]
- 15. Zhang W., Yuan Y., Pei J., Lai L.. Zhang W. Computer-Aided Drug Discovery. 2016; NY: Springer; 111–132. [Google Scholar]
- 16. Wang R., Fang X., Lu Y., Yang C.-Y., Wang S.. The PDBbind database: methodologies and updates. J. Med. Chem. 2005; 48:4111–4119. [DOI] [PubMed] [Google Scholar]
- 17. Krasowski A., Muthas D., Sarkar A., Schmitt S., Brenk R.. DrugPred: a structure-based approach to predict protein druggability developed using an extensive nonredundant data set. J. Chem. Inf. Model. 2011; 51:2829–2842. [DOI] [PubMed] [Google Scholar]
- 18. Chen J., Lai L.. Pocket v.2: further developments on receptor-based pharmacophore modeling. J. Chem. Inf. Model. 2006; 46:2684–2691. [DOI] [PubMed] [Google Scholar]
- 19. Chen J., Ma X., Yuan Y., Pei J., Lai L.. Protein-protein interface analysis and hot spots identification for chemical ligand design. Curr. Pharm. Design. 2014; 20:1192–1200. [DOI] [PubMed] [Google Scholar]
- 20. Law V., Knox C., Djoumbou Y., Jewison T., Guo A.C., Liu Y., Maciejewski A., Arndt D., Wilson M., Neveu V. et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014; 42:D1091–D1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gaulton A., Bellis L.J., Bento A.P., Chambers J., Davies M., Hersey A., Light Y., McGlinchey S., Michalovich D., Al-Lazikani B.. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012; 40:D1100–D1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Christopoulos A. Allosteric binding sites on cell-surface receptors: novel targets for drug discovery. Nat. Rev. Drug Discov. 2002; 1:198–210. [DOI] [PubMed] [Google Scholar]
- 23. Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J.. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997; 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Consortium U. UniProt: a hub for protein information. Nucleic Acids Res. 2014; 43:D204–D212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kanehisa M., Furumichi M., Tanabe M., Sato Y., Morishima K.. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017; 45:D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu X., Bai F., Ouyang S., Wang X., Li H., Jiang H.. Cyndi: a multi-objective evolution algorithm based method for bioactive molecular conformational generation. BMC Bioinformatics. 2009; 10:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hanson R.M., Prilusky J., Renjian Z., Nakane T., Sussman J.L.. JSmol and the next‐generation web‐based representation of 3D molecular structure as applied to proteopedia. Isr. J. Chem. 2013; 53:207–216. [Google Scholar]
- 28. Nakashima R., Sakurai K., Yamasaki S., Nishino K., Yamaguchi A.. Structures of the multidrug exporter AcrB reveal a proximal multisite drug-binding pocket. Nature. 2011; 480:565–569. [DOI] [PubMed] [Google Scholar]
- 29. Wolber G., Langer T.. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005; 45:160–169. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.