


Abstract
DynOmics (dynomics.pitt.edu) is a portal developed to leverage rapidly growing structural proteomics data by efficiently and accurately evaluating the dynamics of structurally resolved systems, from individual molecules to large complexes and assemblies, in the context of their physiological environment. At the core of the portal is a newly developed server, ENM 1.0, which permits users to efficiently generate information on the collective dynamics of any structure in PDB format, user-uploaded or database-retrieved. ENM 1.0 integrates two widely used elastic network models (ENMs)—the Gaussian Network Model (GNM) and the Anisotropic Network Model (ANM), extended to take account of molecular environment. It enables users to assess potentially functional sites, signal transduction or allosteric communication mechanisms, and protein–protein and protein–DNA interaction poses, in addition to delivering ensembles of accessible conformers reconstructed at atomic details based on the global modes of motions predicted by the ANM. The ‘environment’ is defined in a flexible manner, from lipid bilayer and crystal contacts, to substrate or ligands bound to a protein, or surrounding subunits in a multimeric structure or assembly. User-friendly interactive features permit users to easily visualize how the environment alter the intrinsic dynamics of the query systems. ENM 1.0 can be accessed at http://enm.pitt.edu/ or http://dyn.life.nthu.edu.tw/oENM/.
INTRODUCTION
Proteins sample a spectrum of motions near their physiological conditions, which often assist in adapting to intermolecular interactions or accomplishing their biological functions (1). The native structure uniquely defines these motions, hence their description as intrinsic dynamics. They involve concerted subunit or domain rearrangements (global motions), as well as loop movements or side chain rotations (local motions).
While early studies focused on the intrinsic dynamics of individual proteins or domains, partly driven by available structural data, recent years have seen a significant increase in data, especially with the resolution of large structures and assemblies by cryo-EM, which also showed the significance of environment or intermolecular interactions in defining biological mechanisms of function. Functional motions are often carried out only in the right interaction context - as biological assemblies (BAs) or complexes, or in the proper cellular environment (e.g. lipid bilayer). Intermolecular interactions facilitate the catalysis of cognate substrates (2,3), trigger structural changes that enable biological activities (4,5) or stimulate allosteric responses (6–8) that selectively modulate different cellular pathways (for a review, see (9)).
In parallel with advances in structural proteomics, it became essential to build methodologies that can efficiently assess the dynamics of biomolecules not as single entities but in the presence of their environment. Coarse-grained (CG) approaches based on elastic network models (ENMs) and normal mode analysis (NMA) have proven successful in characterizing the dynamics of large systems, which led to the development of many web resources (10–23), but the need to examine the change in dynamics in the context of cellular interactions or localization remained unaddressed. Likewise, the full capabilities of network models, which, combined with spectral graph theoretical methods, yield insights into potential allosteric mechanisms of communication (see e.g. (6,24)), or those of linear response theories which permit to assess the response to perturbations at specific sites (25), are yet to be exploited.
We present here the DynOmics Portal that has been developed to meet these needs. The portal incorporates three essential components: (i) evaluation of collective motions of biomolecules not in isolation but in the presence of intermolecular interactions and/or their physiological environment, (ii) assessment of key sites potentially implicated in chemical, mechanical, binding or signaling properties of the biomolecular systems, as well as the response to perturbations based on structural dynamics and graph theoretical methods and (iii) resolution exchanges between full atomic and CG representations, so as to retrieve conformers and animations at full atomic details. The portal offers a highly elaborate, versatile and efficient server, ENM 1.0, for performing computations and analyses beyond the capabilities of our established resources—iGNM 2.0 database (21), ANM 2.0 server (19) or ProDy Application Programming Interface (API) (26). As will be illustrated in more detail below, as well as the Supplementary Data (SD) and the extensive Tutorial accessible on the web, ENM 1.0 has been designed to efficiently deliver both visual and quantitative outputs in a user-friendly platform that lends itself to intuitive interaction.
THEORY AND METHODS
Environment ANM/GNM (envANM/envGNM)
ANM and GNM, two ENMs at the core of ENM 1.0, have been introduced and reviewed earlier (27–29). Briefly, they represent the structure as a network of nodes and springs. GNM dynamics is exclusively based on inter-residue contact topology represented by an N × N connectivity (or Kirchhoff) matrix, Γ (also called Laplacian) for N nodes/residues. ANM uses harmonic potentials of uniform force constant γ, for all interacting residues; Γ is replaced by the 3N × 3N Hessian H of second derivatives of the potential. Mode spectra of N – 1 (or 3N – 6) non-zero modes are obtained upon eigenvalue decomposition of Γ (or H). Modes are rank-ordered by increasing frequency such that mode 1 is the slowest and softest (most easily accessible) mode. Soft modes are highly cooperative. Pseudoinverse of Γ (or H) scales with the cross-correlations between residue fluctuations, organized in N × N (or 3N × 3N) covariance matrix.
The ANM extension to evaluate the dynamics of a system (s) in the presence of an environment (e), called envANM, has been described earlier (30,31). The Hessian of the system is written as  = Hss – HseHee−1HseT where Hss, Hse and Hee refer to s–s, s–e and e–e submatrices of the Hessian constructed for the entire s + e. Comparison of the modes obtained using
= Hss – HseHee−1HseT where Hss, Hse and Hee refer to s–s, s–e and e–e submatrices of the Hessian constructed for the entire s + e. Comparison of the modes obtained using  with those deduced from
with those deduced from 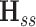 (for the isolated system) shows the effect of environment on the system dynamics. Likewise, envGNM compares the mode spectra for the isolated system (derived from
(for the isolated system) shows the effect of environment on the system dynamics. Likewise, envGNM compares the mode spectra for the isolated system (derived from 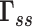 ) with that in the presence of environment (using
) with that in the presence of environment (using 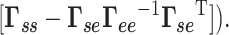 See the SD for more details. For membrane proteins, the lipid bilayer is constructed as a network model, and the dynamics of the system is evaluated using the methodology (membrANM) described earlier (32).
See the SD for more details. For membrane proteins, the lipid bilayer is constructed as a network model, and the dynamics of the system is evaluated using the methodology (membrANM) described earlier (32).
Potential functional sites, sensors, effectors, allosteric communication, IDDs
Potential functional residues (PFSs) are identified using the algorithm COMPACT, described in the SD (and Supplementary Figure S1). Mainly, hinge sites (residues that exhibit minimal displacements in the softest two modes) that are also surface-exposed are identified (2,3). These are rank-ordered based on their (i) proximity to mass center, (ii) level of solvent exposure relative to other sites equally distant from the mass center, and (iii) proximity to other hinges so as to form a possible pocket.
Sensors and effectors are residues distinguished by their responses to structural perturbations such as those induced by ligand binding, complexation, or any deformation originating from external applied force fields. Sensors are distinguished by their strong response to perturbations, often manifested by significant change in their local conformation. Effectors are distinguished by their ability to efficiently communicate perturbations or associated ‘information’ to other sites, often being located near sensors, but in a tightly packed environment so as to minimize the dissipation of structural change or the loss of information. They are evaluated using an extension of the Perturbation-Response-Scanning (PRS) method adapted to GNM (7,8). The analysis yields a response matrix, P, the ijth element of which provides a measure of the sensitivity of node i to perturbation at node j. Row- and column-averages of P, after normalizing each row with respect to the diagonal term, yield the sensitivity and effectiveness profiles of the nodes averaged with respect to all perturbations of the network. Peaks in these respective curves, shown on both sides of the response map, indicate sites likely to function as sensors and effectors, respectively.
Hitting times provide a metric of the efficiency of allosteric communication. While they are derived using graph theoretical concepts, based on a Markovian signal propagation model (6), our earlier work has also demonstrated their mathematical and physical equivalence to GNM-derived fluctuations in structural coordinates. As described in detail in the SD and Tutorial, the hitting time Hij scales with the average number of steps (network edges) that connect the origin (node j) and target (node i) of the signal. Smaller Hij means more efficient allosteric signaling between those sites, and the signal propagation is direction-dependent, i.e. Hij ≠ Hji. Thus, network nodes have distinctive ‘receiver’ and ‘broadcaster’ characteristics, provided in ENM 1.0 using the row- and column-averages of the hitting time matrix H. A useful quantity is the hitting rate, evaluated by dividing the distance Rij between nodes i and j, by Hij.
The method for identifying Intrinsic Dynamics Domains (IDDs), introduced earlier (5), is described in some detail in the SD (and Supplementary Figure S2) and in the Tutorial.
Building full-atomic structures for ANM-driven conformers
ANM-driven motions can result in unphysical stretching or compression of Cα–Cα pseudo-bonds that connect neighboring Cαs, especially during large conformational changes. The situation is particularly worsened at chain termini or exposed loop regions, termed as ‘tip effect’ (33). We designed a bond-regularization protocol of four steps (see SD) to correct for the unrealistic deformation of all Cα–Cα pseudo-bonds. The protocol restores the over-stretched or -compressed bond to their physical length (3.81 ± 0.02 Å). We then use these corrected CG coordinates to reconstruct the ANM-driven conformers (see Supplementary Figure S3): (i) first, unphysical Cα–Cα bonds are restored to their equilibrium distances while preserving the bond bending and torsion angles as driven by the ANM mode, after transforming the Cartesian coordinates into their internal space (generalized) counterparts; (ii) the Cα-coordinates of the new CG conformer are transformed back to the Cartesian system, and equilibrium atomic coordinates of backbone fragments of three sequential residues are superimposed onto the corresponding Cα sites of the CG conformer; (iii) side chains are built using the default rotamer library in Visual Molecular Dynamics (VMD); and (iv) possible steric clashes between newly added atoms are relieved by energy minimization. See details in SD and online tutorial.
DESCRIPTION OF WEB SERVER
DynOmics offers enhanced computing and data analysis capabilities commensurate with the current size of the structural proteome data. It is equipped with efficient algorithms and analytical tools to enable the generation of outputs on the fly for structures of 1000s of residues, and has access to stored pre-calculated GNM dynamics data (21) for all PDB structures. It uses as input any PDB structure or user-loaded coordinates in PDB format. A link to results page is provided for retrieval of results in the case of larger systems. It may take ∼15 min to generate output files for a complex of ∼104 residues, while those for N < 2000 are released within seconds.
The ENM 1.0 server enables users, for the first time, to evaluate and visualize the dynamics of biomolecular systems coupled to a fluctuating environment (see the flow chart in Supplementary Figure S4, also accessible in the Tutorial on the web). The definition of environment is broad; it may be the lipid environment, a substrate bound to the examined protein, the ‘other’ subunits in an oligomer or a complex, or neighboring molecules in the specific experimental setting (e.g. crystal contacts in X-ray structures). Users can select and view the effects of selected environment on the biomolecular system's dynamics.
ENM 1.0 further evaluates a broad range of properties organized in 10 output webpages. Each contains quantitative and qualitative data that can be visualized in a user-friendly interface, including molecular motions (animations) which permits users to reconstruct at full atomic resolution and download conformers visited during collective motions (Figure 1); mean-square fluctuation (MSF) profiles and comparison with B-factors; shapes, dispersion and degree of collectivity of individual modes of motions and cross-correlations between residue fluctuations; IDD-based domain separations; PFSs derived from mode shapes; residues acting as sensors and effectors based on PRS, or mediators of allosteric communication, based on signal propagation (hitting) rates and key residues that potentially mediate allosteric communication.
Figure 1.

A snapshot from Molecular Motions webpage, generated by ENM 1.0. A snapshot from the animation generated for 3-methyladenine DNA glycosylase (PDB: 1MPG) slowest mode (mode 1) is shown. The protein is in ENM representation, color-coded based on the size of motions (red: most mobile, blue: most rigid). Users download full atomic conformers (see orange box) after selecting the RMSD from the PDB structure (dotted-red box).
The interface is designed to allow efficient interrogation and intuitive mining by non-experts, using methods and default parameters that have been extensively tested and verified in the last decade. Further statistical data and illustrative examples can be found in the extensive tutorials and the SD.
EXAMPLES
Effect of environment
The X-ray crystallographic B-factors and the anisotropic displacement parameters (ADPs) provide experimental data on the spatial fluctuations of atoms in the crystal environment. The former provides a single value, MSF, per atom, implicitly assuming ‘isotropic’ fluctuations; the latter, reported for higher resolution structures, further provides information on the anisotropic character of the fluctuations. The MSF therein is replaced by a 3 × 3 matrix, composed of six distinct parameters (ADPs) representing the mean-square fluctuations along the three Cartesian coordinates, and their (symmetric) cross-correlations, for each individual atom (34). Figure 2A and B displays the results from our statistical analysis of ADPs and isotropic B-factors evaluated in the presence of crystal contacts, compared to those obtained in the absence. The computed data show marked improvement in correlations with observed crystallographic parameters when crystal contacts are included. The average correlation between envANM computations for ADPs and experiments is improved from 0.747 ± 0.013 to 0.838 ± 0.009, and that between envGNM-predicted B-factors and experimental counterparts increased from 0.578 ± 0.016 to 0.620 ± 0.014. To generate panels A and B, a dataset of 93 high-resolution (<1.5 Å) protein structures that share less than 15% sequence identity, previously used in ANM-predictions of ADPs (34), has been analyzed.
Figure 2.

Effect of environment on structural dynamics. (A and B) Distribution of correlations between computationally predicted and experimentally observed ADPs (A) and B-factors (B), computed in the presence (orange bars) and absence (blue bars) of crystal contacts using envANM (A) and envGNM (B). Average correlations are written in the inset. The difference between these values in A is supported by P-values of 1.74e–11 and 1.93e–07 from student's t-test and two-sample Kolmogorov–Smirnov (KS) test, respectively. Those in B yielded P-values of 1.35e–05 and 0.005 for the respective two tests. (C) Mode 1 sampled by γ-secretase in the presence (left) and absence (right) of lipid bilayer, color-coded as in Figure 1, based on an RMSD of 4 Å with respect to the PDB structure. Red arrows show residue movements larger than 7 Å. (D) Cross-correlations between residue motions in the presence (left) and absence (right) of lipid bilayer, based on 20 softest modes. Red blocks refer to correlated pairs and blue regions indicate the anticorrelated motions of ECD and TMD, enhanced in the presence of membrane.
Panels C and D illustrate the effect of lipid bilayer on the dynamics of a membrane protease complex, γ-secretase. Panel C compares the softest mode of motion accessible in the presence (left) and absence (right) of membrane. The closure of the ectodomain (ECD) over the transmembrane domain (TMD) is observed to be enhanced in the presence of membrane. The γ-secretase cleaves amyloid precursor proteins to release peptides that form amyloid-fibrils in Alzheimer's disease. The ECD motion directly modulates the opening/closure of the groove that has been reported to bind the substrate and/or modulators (35). Significantly, these movements are consolidated in the presence of the membrane, as also seen in the pronounced cross-correlations in panel D. Supplementary Figures S5 and S6 illustrate two other applications.
Identification of functional sites
Figure 3 illustrates the use of ENM 1.0 identifying functional sites and analyzing signal communication properties of studied structures. Computations for a set of 18 monomeric non-homologous enzymes from all the 6 enzyme classes (see SD) using the COMPACT algorithm with two slowest GNM modes (see above) show that catalytic sites can be predicted (among PFSs) with a high sensitivity and moderate specificity (Supplementary Table S1). Figure 3A shows, for example, the PFSs predicted alpha-lytic protease. All PFSs, including catalytic sites and others, are verified to be conserved (panel B), in support of their functional significance. Similar results for 18 non-homologous enzymes can be seen in the SD.
Figure 3.

Characterization of potentially functional sites by DynOmics. (A) PFSs predicted by COMPACT implemented in ENM 1.0, illustrated for alpha-lytic protease (PDB: 2LPR), shown in ball and stick (snapshot from interface). (B) Same enzyme, color-coded by sequence conservation (37) (purple: most conserved; cyan: most variable). Predicted hinges and PFSs are shown in van der Waals spheres, catalytic sites as ball-and-stick. (C) Signaling/communication properties. On the left is the hitting time matrix H as a function of signal communicating sites (abscissa) and signal receiving sites (ordinate), computed for phospholipase A2 (PDB: 1BK9). Red regions indicate efficiently communicating pairs. The (vertical) curve displays the average responses of all residues, with minima indicating the most efficient responders (colored red in the ribbon diagram). The three catalytic residues, labeled, lie among these most efficient receivers of signals. (D) Comparison of the hitting time distributions for catalytic (orange) and other (black) residues. Respective mean values are 1.679 ± 0.003 and 2.340 ± 0.023. (E) IDDs computed for oxidative DNA/RNA repair enzyme AlkB (PDB IDs: 2FDJ (apo) and 3BKZ (bound)), shown in red and blue, which move in opposite directions. Blue and green lines span the domain (D)-plane that cuts through the dsDNA.
Figure 3C shows the outputs for signaling/communication sites obtained for phospholipase A2. The left panel displays the hitting times (6), Hij, organized in a 2D map, color-coded from red to blue, in the order of increasing hitting time. The signaling rate is direction-dependent (the matrix is asymmetric). The profile on the right (average column-vector) shows of the propensity of individual residues to receive signals. Catalytic residues (labeled, and also shown in the ribbon) exhibit minimal hitting times (averaged over all signal communicating sites) consistent with earlier observations (6). Application to a set of 240 non-homologous enzymes from Catalytic Site Atlas (CSA) (36) (panel D) further demonstrates that catalytic residues exhibit shorter hitting times compared other sites. Using the slowest GNM mode, we have shown previously that a domain-plane (D-plane), which optimally divides the protein into two largest anti-correlated intrinsic dynamics domains (IDDs), cuts through protein-bound DNA molecules in 101 out of 104 studied cases (4). The probability of finding correct docking location and orientation for DNA is enhanced 2.5-fold upon usage of a filter that eliminates the unlikely protein–DNA docking decoys (4). Figure 3E illustrates the current implementation of IDDs/D-planes in ENM 1.0, which maps the structure into two domains based on its global dynamics and shows that the D-plane dissects through the bound DNA molecule. The D-plane was also reported to help locate ligand binding sites and correct protein–protein docking poses (5).
Supplementary Figure S7 further shows the sensors/effectors output from our server, for the bacterial chaperone DnaK. The color-coded diagrams indicate the sites that exhibit the highest propensity to serve as sensors (A) and effectors (B) of signals. Previous study has shown their relevance to substrate binding, and allosteric communication between the substrate-binding and ATPase domains of DnaK (8). The color-code map (in C) describes the strength of the response (by residue i, abscissa) to the perturbation (at residue j, ordinate). Peaks on the average profiles (shown in D for responding residues, and in E for perturbing residues) indicate the sites that are highly sensitive to perturbations and can serve as effectors or sensors, respectively.
CONCLUSION
The DynOmics portal has been designed with flexible and extensible features to address the need for learning about the dynamic and allosteric behavior of biomolecules, as modulated by their intermolecular interactions, and shaped by their different oligomerization states or assemblies. The resource provides an efficient means of harnessing the rapidly accumulating structural proteome data to provide users with a broad range of outputs that may guide in establishing the molecular basis of (dys)functional interactions.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [5R01GM099738-04, 5P41GM103712-03, P30DA035778, and P01DK096990]; Ministry of Science and Technology (MOST), Taiwan [104-2113-M-007-019]. Funding for open access charge: MOST [104-2113-M-007-019].
Conflict of interest statement. None declared.
REFERENCES
- 1. Haliloglu T., Bahar I.. Adaptability of protein structures to enable functional interactions and evolutionary implications. Curr. Opin. Struct. Biol. 2015; 35:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yang L.W., Bahar I.. Coupling between catalytic site and collective dynamics: a requirement for mechanochemical activity of enzymes. Structure. 2005; 13:893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yang L.W., Eyal E., Bahar I., Kitao A.. Principal component analysis of native ensembles of biomolecular structures (PCA_NEST): insights into functional dynamics. Bioinformatics. 2009; 25:606–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chandrasekaran A., Chan J., Lim C., Yang L.W.. Protein dynamics and contact topology reveal protein–DNA binding orientation. J. Chem. Theory Comput. 2016; 12:5269–5277. [DOI] [PubMed] [Google Scholar]
- 5. Li H., Sakuraba S., Chandrasekaran A., Yang L.W.. Molecular binding sites are located near the interface of intrinsic dynamics domains (IDDs). J. Chem. Inf. Model. 2014; 54:2275–2285. [DOI] [PubMed] [Google Scholar]
- 6. Chennubhotla C., Bahar I.. Signal propagation in proteins and relation to equilibrium fluctuations. PLoS Comput. Biol. 2007; 3:1716–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Atilgan C., Atilgan A.R.. Perturbation-response scanning reveals ligand entry-exit mechanisms of ferric binding protein. PLoS Comput. Biol. 2009; 5:e1000544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. General I.J., Liu Y., Blackburn M.E., Mao W., Gierasch L.M., Bahar I.. ATPase subdomain IA is a mediator of interdomain allostery in Hsp70 molecular chaperones. PLoS Comput. Biol. 2014; 10:e1003624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bahar I., Cheng M.H., Lee J.Y., Kaya C., Zhang S.. Structure-encoded global motions and their role in mediating protein-substrate interactions. Biophys. J. 2015; 109:1101–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Krebs W.G., Alexandrov V., Wilson C.A., Echols N., Yu H., Gerstein M.. Normal mode analysis of macromolecular motions in a database framework: developing mode concentration as a useful classifying statistic. Proteins. 2002; 48:682–695. [DOI] [PubMed] [Google Scholar]
- 11. Suhre K., Sanejouand Y.H.. ElNemo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004; 32:W610–W614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wako H., Kato M., Endo S.. ProMode: a database of normal mode analyses on protein molecules with a full-atom model. Bioinformatics. 2004; 20:2035–2043. [DOI] [PubMed] [Google Scholar]
- 13. Hollup S.M., Salensminde G., Reuter N.. WEBnm@: a web application for normal mode analyses of proteins. BMC Bioinformatics. 2005; 6:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lindahl E., Azuara C., Koehl P., Delarue M.. NOMAD-Ref: visualization, deformation and refinement of macromolecular structures based on all-atom normal mode analysis. Nucleic Acids Res. 2006; 34:W52–W56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lopez-Blanco J.R., Garzon J.I., Chacon P.. iMod: multipurpose normal mode analysis in internal coordinates. Bioinformatics. 2011; 27:2843–2850. [DOI] [PubMed] [Google Scholar]
- 16. Seo S., Kim M.K.. KOSMOS: a universal morph server for nucleic acids, proteins and their complexes. Nucleic Acids Res. 2012; 40:W531–W536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wako H., Endo S.. Normal mode analysis based on an elastic network model for biomolecules in the Protein Data Bank, which uses dihedral angles as independent variables. Comput. Biol. Chem. 2013; 44:22–30. [DOI] [PubMed] [Google Scholar]
- 18. Lopez-Blanco J.R., Aliaga J.I., Quintana-Orti E.S., Chacon P.. iMODS: internal coordinates normal mode analysis server. Nucleic Acids Res. 2014; 42:W271–W276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eyal E., Lum G., Bahar I.. The anisotropic network model web server at 2015 (ANM 2.0). Bioinformatics. 2015; 31:1487–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Frappier V., Chartier M., Najmanovich R.J.. ENCoM server: exploring protein conformational space and the effect of mutations on protein function and stability. Nucleic Acids Res. 2015; 43:W395–W400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li H., Chang Y.Y., Yang L.W., Bahar I.. iGNM 2.0: the Gaussian network model database for biomolecular structural dynamics. Nucleic Acids Res. 2016; 44:D415–D422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Emekli U., Schneidman-Duhovny D., Wolfson H.J., Nussinov R., Haliloglu T.. HingeProt: automated prediction of hinges in protein structures. Proteins. 2008; 70:1219–1227. [DOI] [PubMed] [Google Scholar]
- 23. Zimmermann M.T., Kloczkowski A., Jernigan R.L.. MAVENs: motion analysis and visualization of elastic networks and structural ensembles. BMC Bioinformatics. 2011; 12:264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dokholyan N.V. Controlling allosteric networks in proteins. Chem. Rev. 2016; 116:6463–6487. [DOI] [PubMed] [Google Scholar]
- 25. Yang L.W., Kitao A., Huang B.C., Go N.. Ligand-induced protein responses and mechanical signal propagation described by linear response theories. Biophys. J. 2014; 107:1415–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bakan A., Dutta A., Mao W, Liu Y., Chennubhotla C., Lezon T.R., Bahar I.. Evol and ProDy for bridging protein sequence evolution and structural dynamics. Bioinformatics. 2014; 30:2681–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Atilgan A.R., Durell S.R., Jernigan R.L., Demirel M.C., Keskin O., Bahar I.. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 2001; 80:505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bahar I., Lezon T.R., Yang L.W., Eyal E.. Global dynamics of proteins: bridging between structure and function. Annu. Rev. Biophys. 2010; 39:23–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang L.W. Models with energy penalty on interresidue rotation address insufficiencies of conventional elastic network models. Biophys. J. 2011; 100:1784–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ming D., Wall M.E.. Allostery in a coarse-grained model of protein dynamics. Phys. Rev. Lett. 2005; 95:198103. [DOI] [PubMed] [Google Scholar]
- 31. Bahar I., Lezon T.R., Bakan A., Shrivastava I.H.. Normal mode analysis of biomolecular structures: functional mechanisms of membrane proteins. Chem. Rev. 2010; 110:1463–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lezon T.R., Bahar I.. Constraints imposed by the membrane selectively guide the alternating access dynamics of the glutamate transporter GltPh. Biophys. J. 2012; 102:1331–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lu M., Poon B., Ma J.. A new method for coarse-grained elastic normal-mode analysis. J. Chem. Theory Comput. 2006; 2:464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Eyal E., Chennubhotla C., Yang L.W., Bahar I.. Anisotropic fluctuations of amino acids in protein structures: insights from X-ray crystallography and elastic network models. Bioinformatics. 2007; 23:i175–i184. [DOI] [PubMed] [Google Scholar]
- 35. Takeo K., Tanimura S., Shinoda T., Osawa S., Zahariev I.K., Takegami N., Ishizuka-Katsura Y., Shinya N., Takagi-Niidome S., Tominaga A. et al.. Allosteric regulation of gamma-secretase activity by a phenylimidazole-type gamma-secretase modulator. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:10544–10549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Porter C.T., Bartlett G.J., Thornton J.M.. The Catalytic Site Atlas: a resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Res. 2004; 32:D129–D133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Goldenberg O., Erez E., Nimrod G., Ben-Tal N.. The ConSurf-DB: pre-calculated evolutionary conservation profiles of protein structures. Nucleic Acids Res. 2009; 37:D323–D327. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.