

SUMMARY
The BCL-2 family protein BAX is a central mediator of apoptosis. Overexpression of anti-apoptotic BCL-2 proteins contributes to tumor development and resistance to therapy by suppressing BAX and its activators. We report the discovery of BTSA1, a pharmacologically optimized BAX activator that binds with high affinity and specificity to the N-terminal activation site and induces conformational changes to BAX leading to BAX-mediated apoptosis. BTSA1-induced BAX activation effectively promotes apoptosis in leukemia cell lines and patient samples while sparing healthy cells. BAX expression levels and cytosolic conformation regulate sensitivity to BTSA1. BTSA1 potently suppressed human acute myeloid leukemia (AML) xenografts and increased host survival without toxicity. This study provides proof-of-concept for direct BAX activation as a treatment strategy in AML.
Keywords: apoptosis, BCL-2 family, BAX, BH3 domain, MOMP, BAX activator, BTSA1
Graphical Abstract
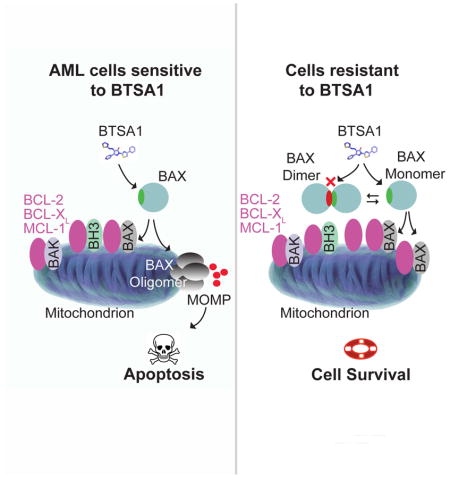
Reyna et al. develop BTSA1, a pharmacologically optimized BAX activator, and show that BTSA1-induced BAX activation effectively and selectively promotes apoptosis of acute myeloid leukemia cells. They further demonstrate that the BAX expression level and cytosolic conformation regulate the sensitivity to BTSA1.
INTRODUCTION
Cancer cells depend on their ability to enforce cell survival pathways and block cell death mechanisms (Hanahan and Weinberg, 2011). It is established that cancer cells reprogram the BCL-2 family interaction network that regulates mitochondrial apoptosis to ensure their growth, maintenance and resistance to current treatments (Hata et al., 2015; Llambi and Green, 2011). Most frequently, cancer cells overexpress anti-apoptotic BCL-2 family members such as BCL-2, BCL-XL and MCL-1 that bind and neutralize the BH3 death domains of the activated pro-apoptotic BCL-2 members: BAX, BAK and the BH3-only proteins such as BIM and BID (Letai, 2008; Youle and Strasser, 2008). Elucidation of the BCL-2 family interactions has led to the development of clinical inhibitors targeting anti-apoptotic BCL-2, BCL-XL and BCL-W Navitoclax (ABT-263) or BCL-2 only Venetoclax (ABT-199) that prevent neutralization of BH3 death domains and promote BAX/BAK-mediated apoptosis (Juin et al., 2013; Oltersdorf et al., 2005; Souers et al., 2013). Evaluation of Venetoclax and Navitoclax in preclinical models and clinical trials demonstrated promising efficacy in tumors that are highly dependent on BCL-2 and/or BCL-XL (Leverson et al., 2015; Roberts et al., 2015; Rudin et al., 2012). However, studies highlighted the limited efficacy and resistance in cancers that rely on or overexpress additional anti-apoptotic proteins, such as MCL-1, which are not inhibited by the aforementioned inhibitors (Beroukhim et al., 2010; Konopleva et al., 2006; van Delft et al., 2006). Efforts in developing drug-like MCL-1 inhibitors has been proven challenging although a recent study suggested that selective MCl-1 inhibitors would be effective for a range of tumors (Belmar et al., 2015; Kotchy et al., 2016; Perciavalle et al., 2013)
Pro-apoptotic BAX is the cardinal executioner BCL-2-family member that upon conformational activation and oligomerization at the mitochondrial outer membrane (MOM) causes permeabilization of the MOM and release of mitochondrial factors e.g. cytochrome c, and Smac/Diablo that activate the caspase cascade of apoptosis (Luna-Vargas and Chipuk, 2016; Walensky and Gavathiotis, 2011). Cells deficient for BAX become less sensitive to various apoptotic stimuli and become resistant when both BAX and BAK are deleted (Wei et al., 2001). Selective inhibitors of anti-apoptotic BCL-2 proteins including Venetoclax and Navitoclax are effective inducers of apoptosis because they release BH3-only proteins (e.g. BIM, BID) from the anti-apoptotic BCL-2 proteins to activate BAX and BAK (Juin et al., 2013; Oltersdorf et al., 2005; Souers et al., 2013). Moreover, induction of BAX-dependent apoptosis by BH3-only proteins can be induced by several classic chemotherapeutic agents (Zhang et al., 2000). However, in several tumors, BH3-only proteins can be suppressed or deleted at transcription, translation and post-translational levels, rendering these tumors less sensitive to current treatments (Hata et al., 2015; Juin et al., 2013). Interestingly, the vast majority of cancer cells contain functional BAX in an inactive conformation or suppressed by anti-apoptotic proteins; mutations or alterations in BAX that may cause its inactivation occur, albeit at a low frequency in different tumor types analyzed by the TCGA Research Network (<4%) (Gao et al., 2013). Thus, we hypothesized that induction of apoptosis by mimicking the BAX-activating interactions of select BH3 domains to deploy BAX offers the possibility of an anti-cancer strategy. Toward this end, the understanding of the precise regulatory mechanisms of pro-apoptotic BAX has dramatically evolved in recent years and details of the activation mechanism of BAX by BH3-only proteins such as BIM, BID and PUMA have been elucidated (Czabotar et al., 2013; Gavathiotis et al., 2008; Kim et al., 2009; Llambi et al., 2011; Lovell et al., 2008).
We set out to discover small molecules that activate BAX through interaction with the N-terminal activation site (trigger site) of BAX (Gavathiotis et al., 2008; Gavathiotis et al., 2010). We reasoned that this unique binding site compared to the canonical BH3 grooves of BAX, BAK and anti-apoptotic BCL-2 proteins may provide a BAX-selective targeting strategy. Previously, we used an in silico docking screen to target the trigger site of BAX and identified a BAX activator molecule 7 (BAM7), which induces activation of BAX in vitro, albeit, at moderate potency (Gavathiotis et al., 2012). The efficacy in cancer models in vitro and in vivo was undetermined and given that BAX is expressed in cancer cells as well as normal cells the specificity and therapeutic window for targeting BAX in cancer remains unknown. Moreover, to identify the therapeutic potential and utility for clinical application of BAX activators in cancer, compounds with potency, selectivity and drug-like properties need to be developed. Here, we sought to develop such a BAX activator to evaluate direct BAX activation through the BAX trigger site as a potential therapeutic strategy to promote apoptosis in cancer.
RESULTS
BTSA1 is a potent and selective BAX trigger site activator
We generated a pharmacophore model based on the structural information of previously reported models of BIM BH3 helix and BAM7 compound bound to the N-terminal activation site (trigger site) of BAX (Figure S1A, S1B). Synthesized compounds and chemical libraries were evaluated to fit the pharmacophore model and to have an increased interaction for the BAX trigger site. A competitive fluorescence polarization assay that evaluates the capacity of compounds to compete a fluorescein-labeled stapled peptide of the BIM BH3 helix, FITC-BIM SAHBA2, from the BAX trigger site, was used to determine the binding potency (Gavathiotis et al., 2008; Gavathiotis et al., 2012). A lead compound named BAX Trigger Site Activator 1 (BTSA1) (Figure 1A) competed FITC-BIM SAHBA2 with IC50 of 250 nM, and compared to the binding of BIM SAHBA2 helix (IC50 = 280 nM) and BAM7 (IC50 = 3.2 μM) (Figure 1B) demonstrated the most potent small-molecule binding to the BAX trigger site. Moreover, direct binding of fluorescein-labeled BTSA1 (Method S1) to BAX showed higher nanomolar affinity, EC50 = 144 nM (Figure 1C). BTSA1 has a pyrazolone group substituted with a phenyl, a thiazolhydrazone and a phenylthiazol. BTSA1 complies with the Lipinski’s rule of five for drug-likeness and is generated with a two-step synthetic protocol (Method S1). Because BIM BH3 helix binds the BH3 groove of anti-apoptotic BCL-2 proteins and BAX, we investigated whether BTSA1 binds selectively to BAX. Unlabeled BIM SAHBA2 helix effectively competed FITC-BIM SAHBA2 binding to the structurally diverse members of the anti-apoptotic BCL-2 proteins, BCL-XL, MCL-1 and BFL-1/A1 (Figure 1D). In contrast, BTSA1 had no capacity to compete FITC-BIM SAHBA2 from anti-apoptotic BCL-2 proteins at 50 μM, showing specificity for BAX and excluding nonspecific reactivity for BTSA1 (Figure 1D).
Figure 1. BTSA1 is a high affinity and selective BAX trigger site activator.

(A) Chemical structure of BTSA1. (B) Competitive fluorescence polarization binding assay of BTSA1, BAM7 and Ac-BIM SAHBA2 using FITC-BIM SAHBA2 bound to BAX. (C) Direct fluorescence polarization binding assay using fluorescent-labeled BTSA1 (F-BTSA1) and BAX. (D) Competitive fluorescence polarization binding assay of BTSA1 and Ac-BIM SAHBA2 using FITC-BIM SAHBA2 bound to BCL-XL, MCL-1 and BFL-1. (E) Measured chemical shift changes from comparative analysis of HSQCs using 15N-labelled BAX upon BTSA1 titration up to a ratio of 1:1 are plotted as a function of BAX residue number. (F) Mapping of the residues with significant backbone amide chemical shift change (orange) showing the co-localization of residues in the BAX trigger site (α1, α6) (G) Surface representation of BAX with BTSA1 (sticks) docked in the trigger site showing overlap with residues undergoing chemical shift changes (orange). (H) Docked structure of BTSA1 showing possible interactions formed predominantly with BAX sidechains of hydrophobic residues and a key hydrogen bond between the pyrazolone group and K21 residue (I) Structural overlay of the BAX:BIM BH3 structure and the BAX:BTSA1 docked structure suggest similar type of interactions between BIM BH3 helix and BTSA1 at the BAX trigger site. The phenylthiazol group of BTSA1 mimicks hydrophobic interactions of the I155 and L152 of the BIM BH3 helix. The thiazol ring and part of the hydrazono group of BTSA1 overlaps with the F155 of the BIM BH3 helix. The pyrazolone group of BTSA1 forms a hydrogen bond with the sidechain of the K21 of BAX and is aligned with the E158 residue of the BIM BH3 helix that also forms a hydrogen bond with the sidechain of the K21. Data represent mean ± SD (n=3) from three independent experiments or are representative of three independent experiments. See also Figure S1.
NMR analysis of 15N-labeled full-length BAX upon BTSA1 titration revealed backbone amide chemical-shift changes consistent with BTSA1 binding reversibly to residues of the BAX trigger site (Figure 1E, 1F). Molecular docking driven by the NMR data, determined a lowest-energy pose for BTSA1 that is also consistent with the observed chemical shifts upon BTSA1 binding to BAX (Figure 1G). BTSA1 is calculated to bind in the hydrophobic region at the juxtaposition of helices α1 and α6 formed by M20, K21, A24, L25, Q28, G138, L141, and R145 residues (Figure 1H). BTSA1 mimics features of the binding pose of the BIM BH3 helix (Figure 1I). Interestingly, the phenyl ring attached to the pyrazolone is in close proximity with the α1–α2 loop of BAX in its open conformation. This phenyl ring provides significant increase in potency as a BTSA1 analogue, BTSA2, that has the same structure but a methyl instead of a phenyl ring, displayed moderate binding affinity (IC50 = 3.7 μM) (Figure S1C, S1D). To gain further insights, we performed molecular dynamics (MD) simulations of the BTSA1-bound and unbound BAX (Figure S1E–S1G). BTSA1 retained its bound position during the MD trajectory and also the α1–α2 loop maintained an open conformation from interactions with the BTSA1; a feature that was shown to be essential for BAX activation and consistent with the exposure of the epitope (residues 12-24) that is recognized by the 6A7 antibody only upon BAX activation (Garner et al., 2016; Gavathiotis et al., 2010). In contrast, unbound BAX kept the α1–α2 loop in closed conformation during the MD trajectory (Figure S1E–S1G). Consistently, antibodies that either bind to or stabilize the closed conformation of α1–α2 loop of BAX were found to inhibit cytosolic BAX (Iyer et al., 2016, Uchime et al., 2016). Taken together, BTSA1 binds with high affinity to BAX at a critical region associated with BAX activation, which is consistent with the high binding selectivity for BAX.
Upon BH3-mediated activation, BAX undergoes conformational changes that transform the inactive cytosolic monomer into a mitochondrial oligomer capable of mitochondrial permeabilization (Figure 2A) (Bleicken et al., 2014; Czabotar et al., 2013; Gavathiotis et al., 2010; Zhang et al., 2015). Indeed, we found that binding of BTSA1 to inactive and soluble BAX progressively leads to BAX oligomerization as evidenced by the disappearance of the BAX monomer in Heteronuclear Single-Quantum Correlation (HSQC) spectra and size-exclusion chromatography (SEC) trace and appearance of the corresponding BAX oligomer HSQC spectra and SEC trace (Figure 2B, 2C and S2A). Using established liposomal assays we probed the capacity of BTSA1 to induce functional activation of BAX in an isolated system. BTSA1 effectively induced dose-dependent BAX membrane translocation and BAX-mediated membrane permeabilization (Figure 2D, 2E). To confirm that BTSA1 activated BAX by engaging the BAX trigger site, we mutated BAX K21 that is suggested to interact with BTSA1 from NMR studies (Figure 1E–1H). BTSA1 had significantly impaired capacity to induce BAX membrane translocation and BAX-mediated membrane permeabilization for BAX K21E mutant although the BAX mutant protein is functional upon tBID-mediated BAX activation (Figure 2D, 2E). Moreover, using liposomal assay we confirmed that BTSA1 had no capacity to directly activate the pro-apoptototic homologue BAK (Figure S2B). However combining BAX and BAK with BTSA1 resulted in a synergistic effect when compared to the permeabilization effect of BAX alone using the same concentrations (Figure S2B). To explain this effect, we hypothesized that BTSA1 induces exposure of the BAX BH3 domain, which can then activate BAK as previously demonstrated (Leshchiner et al., 2013). Indeed, using an antibody that specifically recognizes a BAX BH3 epitope, we found that BTSA1 induced dose-dependently exposure of the BAX BH3 domain (Figure S2C).
Figure 2. BTSA1 induces all steps of the BAX activation pathway.

(A) Cartoon representation of the different steps toward BTSA1-induced BAX activation leading to mitochondrial permeabilization. (B) 1H-15N HSQC spectra of unbound and inactive BAX. (C) Overlaid 1H-15N HSQC spectra of BAX activated by BTSA1 or BIM SAHBA2 shows the formation of the same BAX oligomers. The appearance of several amide correlation cross peaks originates from the significant increase of the large size of the BAX oligomers. (D, E) BAX translocation (D) and BAX-mediated membrane permeabilization (E) assay using ANTS/DPX liposomes and 200 nM BAX or BAX K21E, 100 – 400 nM BTSA1 and 60 nM tBID. (F) Immunoprecipitation assay of samples containing soluble BAX treated with increasing doses of BTSA1 using the 6A7 antibody. (G–I) BAX mitochondrial translocation assay (G), BAX mitochondrial oligomerization assay (H), and BAX-mediated cytochrome c release assay (I) upon BTSA1 titration with recombinant soluble BAX using isolated mitochondria from mouse livers. (J) Cytochrome c release assay upon BTSA1 titration in the absence of recombinant BAX. Data represent mean ± SD (n=3) from at least two independent experiments or are representative of three independent experiments respectively. See also Figure S2.
Using the conformation-specific antibody 6A7 that recognizes the exposure of the BAX activation epitope, we found that BTSA1 exposed the 6A7 epitope of recombinant BAX consistent with the functional activation of BAX and structural model of BTSA1-bound BAX (Figure 2F, 1H). We further investigated the activity of BTSA1 to BAX in the presence of isolated mitochondria from mouse liver that contain no BAX or BAK. BTSA1 treatment potently and dose-responsively induced membrane translocation of recombinant soluble BAX to mitochondrial membrane, which was followed by induction of BAX oligomerization (Figure 2G, 2H). Consistently, BTSA1-induced BAX activation resulted in mitochondrial outer membrane permeabilization and release of cytochrome c from mitochondria (Figure 2I). BTSA1 had no effect on cytochrome c release without addition of BAX (Figure 2J). Taken together, the biophysical, structural and biochemical data demonstrate that BTSA1’s high affinity and specific binding to the BAX trigger site leads to functional and effective BAX activation.
BTSA1-induced BAX activation promotes apoptosis in cancer cells
The BCL-2 protein family regulates apoptosis in hematologic malignancies, which are characterized by defective apoptosis and oncogenic alterations that promote cell growth (Reed et al., 2008). We evaluated the ability of BTSA1 to induce apoptosis in human AML cell lines that have p53 and/or NRAS mutations and a range of expression levels for BAX and anti-apoptotic BCL-2 proteins. BTSA1 reduced viability of all AML cell lines in a dose-dependent manner with half maximal inhibitory concentration (IC50) values ranged between 1–4 μM that leads to complete effect within 24 hr treatment (Figure 3A). Cellular viability measured for select cell lines at different time points displayed substantial cell death activity by BTSA1 within 6 hr of treatment (Figure 3B). The substantial and rapid loss of viability of leukemia cells is consistent with the biochemical effects that characterize BAX activation and apoptosis, which was observed promptly after BTSA1 treatment. Significant BAX mitochondrial translocation was induced in a BTSA1 dose-dependent manner (Figure 3C). BTSA1-induced BAX translocation coincided with the release of cytochrome c from the mitochondria to the cytosol (Figure 3C). BTSA1 induced dose-dependent caspase-3/7 activation in all five AML cell lines (Figure 3D). Caspase-3/7 activation was monitored within 4–24 hr and maximal caspase-3/7 activation was detected in 4 hr consistent with prompt BAX translocation and release of cytochrome c (Figure 3E). Moreover, loss of viability, apoptosis induction and cytotoxicity occurred at the same effective dose of BTSA1 (Figure S3A, S3B).
Figure 3. BTSA1 induced robust and rapid BAX-mediated apoptosis.

(A) Viability assay of human AML cell lines and mouse AML cell line WEHI exposed to BTSA1 for 24 hr. (B) Viability assay of human AML cell lines treated with 5 μM BTSA1 for 6, 12 and 24 hr. (C) Western blot analysis of BAX translocation and cytochrome c release from NB4 cells treated with BTSA1 for 6 hr. Actin and VDAC are loading controls for cytosolic and mitochondrial fractions respectively. (D) Caspase 3/7 assay in AML cells treated with BTSA1 for 6 hr. (E) Caspase 3/7 assay in OCI-AML3 cells treated with BTSA1 at 4, 6, 8, 12 and 24 hr. (F) TMRE mitochondrial potential assay in OCI-AML3 cells treated with serial dilution doses of BTSA1 at indicated time points. (G) Percent BAX and anti-apoptotics (BCL-2, BCL-XL and MCL-1) protein expression levels relative to Actin as measured by quantitative IR fluorescence western for five AML cell lines. (H) Correlation of percentage of BAX expression levels relative to Actin for AML cell lines with the amount of mitochondrial depolarization detected at 2 hr with 2.5 μM BTSA1. (I) Viability assay of BTSA1-treated NB4 cells and NB4 cells over-expressing BCL-2, BCL-XL and MCL-1 proteins treated with BTSA1 for 24 hr. (J) Viability assay of OCI-AML3 treated with serial dilution concentrations of BTSA1 alone or in combination with increasing concentrations of Venetoclax for 24 hr. Data represent mean ± SD (n=3) from three independent experiments or are representative of three independent experiments. See also Figure S3.
A more direct consequence of the BTSA1 activity is considered to be the loss of mitochondrial membrane potential that can be caused by BAX mitochondrial translocation. Consistently, significant mitochondrial potential loss was detected within 2 hr and in a dose-responsive manner upon BTSA1 treatment (Figure 3F). To investigate the correlation of the BTSA1 activity to BAX and mitochondrial apoptosis, we measured the protein levels of BAX, BCL-2, BCL-XL and MCL-1 by an infrared-based quantification method (Figure 3G, S3C–3E). Interestingly, we found a linear correlation with BAX protein levels and the degree of mitochondrial membrane potential loss induced by BTSA1, suggesting that mitochondrial damage induced by BTSA1 is proportional to BAX levels in leukemia cells (Figure 3H). In addition, there is no correlation of BTSA1 activity and levels of anti-apoptotic BCL-2 proteins. The protein levels for BCL-2, BCL-XL and MCL-1 were higher compared to BAX in the AML cell lines (Figure 3G, S3E). Consistent with the established mechanism of anti-apoptotic heterodimeric blockade of BAX, transient overexpression of BCL-XL, BCL-2 and MCL-1 decreased cell killing from BTSA1-induced BAX activation, (Figure 3I, S3F–S3H). Furthermore, addition of Venetoclax at non-lethal doses significantly enhanced BTSA1 activity consistent with the activity of Venetoclax to de-repress pro-apoptotic BCL-2 proteins (Figure 3J). Taken together, BTSA1 induces robust apoptosis in leukemia cells through rapid BAX activation. BTSA1’s activity directly correlates with increasing BAX protein levels, which can overcome blockade of the endogenous anti-apoptotic BCL-2 proteins.
BTSA1 cell killing is specific to BAX and is regulated by BAX cytosolic conformation
Since BAX is found at the nexus of several signaling pathways, cellular specificity of BTSA1 to directly induce BAX activation was investigated. Firstly, we used the weaker analogue BTSA2 and found that the reduction in binding capacity to BAX (~10 fold) (Figure S1C, S1D) resulted in a similar reduction in cell death induction in AML cell lines (Figure 4A). Moreover, BTSA1-mediated apoptosis was prevented with a pharmacological inhibitor of the BAX mitochondrial associated channel (Bombrun et al., 2003), which resulted in complete inhibition as measured by caspase-3/7 activation (Figure 4B). Likewise, knockdown of BAX using a siRNA for BAX markedly reduced cell death activity of BTSA1 (Figure S4A, S4B). Finally, we sought proof of direct engagement of BTSA1 with BAX in cells. Using the structural model of BTSA1 bound to BAX, we installed a biotin group to a BTSA1 analogue with a carboxylic acid at the phenylthiazol group and a hydrophilic linker of an appropriate size (Method S1). The biotin-labeled BTSA1 retained binding to recombinant BAX although binding affinity was reduced ten-fold (IC50 = 2 μM) (Figure S4C) We found that biotin-labeled BTSA1, at 2.5 μM dose, binds to cellular BAX and preferentially binds to cytosolic BAX but not to BAX associated with the mitochondrial outer membrane (Figure 4C, 4D), consistent with BTSA1 targeting the BAX trigger site that is only available in the cytosolic BAX conformation (Gavathiotis et al., 2008; Kim et al., 2009;). Moreover, biotin-labeled BTSA1 failed to engage other anti-apoptotic BCL-2 family proteins, confirming the selectivity to targeting BAX in cells (Figure S4D, S4E). Thus, these data provide strong evidence for the cellular specificity and on-target engagement of BTSA1.
Figure 4. Specificity of BTSA1 for cellular BAX, cytosolic BAX monomer and the BAX trigger site.

(A) Viability assay of human AML cells treated for 24 hr with increasing concentrations of BTSA2. (B) Caspase 3/7 assay of BTSA1-treated OCI-AML3 cells after co-treatment with a BAX channel inhibitor at indicated doses. (C, D) Western blot analysis of BAX from pulldown assay using biotin-labeled BTSA1 exposed to cytosolic (C) or mitochondrial (D) fractions from OCI-AML3. (E) Viability assay of non-cancerous cell lines treated with BTSA1 for 24 hr. (F) Plot of half maximal inhibitory concentration (IC50) values from viability assays of wild type, BAX KO, and BAK KO MEF treated with Navitoclax alone or in combination with 5 μM BTSA1. (G) Plot of IC50 values from viability assays of reconstituted human wild type and BAX mutants MEF cell lines treated with Navitoclax alone or in combination with 5 μM BTSA1. (H) Viability assay of OCI-AML3, HPB-ALL and MEFs cells treated with BTSA1 for 24 hr. (I) Size-exclusion chromatography (Superdex 200, HR 10/30) of cytosolic extracts from MEF, OCI-AML3 and HPB-ALL. Fractions corresponding to increasing elution volumes from left to right and indicated molecular weights were analyzed by anti-BAX western blot. HPB-ALL contained monomeric BAX, OCI-AML contained predominantly monomeric BAX and MEF have no detected monomeric BAX. (J) Structural model of BAX monomer showing BTSA1 docked to the trigger site (green). (K) Crystal structure of autoinhibited cytosolic BAX dimer showing the trigger site residues (green) blocked by residues of another BAX molecule within the dimer (magenta). Data represent mean ± SD (n=3) from at least two independent experiments or are representative of three independent experiments. See also Figure S4.
Non-leukemia cell lines that have no BAX expression such as mouse embryonic fibroblasts Bax−/− (BAX KO) MEF and doubly Bax−/−Bak−/− (DKO) MEF, were treated with BTSA1 and exhibit resistance to BTSA1 treatment (Figure 4E). The insensitivity of BAX KO and DKO MEF demonstrate the specificity of BTSA1 activity for BAX. However, additional cells that express BAX such as wild type MEF, H9c2 myocytes and 3T3 fibroblasts still exhibit resistance to BTSA1 treatment at similar doses that BTSA1 induces complete cell death in AML cells (Figure 4E). To further investigate the resistance to BTSA1 treatment, we tested whether BTSA1 can promote BAX activation in wild type MEF. BTSA1 had no capacity to induce BAX translocation, suggesting that the ability of BTSA1 to activate BAX is suppressed (Figure S4F). We reasoned that two mechanisms could operate in these cells to ensure insensitivity to specific pro-apoptotic agents. One mechanism is the sequestration of activated BAX from available anti-apoptotic BCL-2 proteins (Llambi et al., 2011) and a second is the formation of a cytosolic autoinhibited BAX dimer that regulates BAX-dependent apoptosis by sequestering the BAX trigger site of monomeric BAX (Garner et al., 2016). To dissect these two mechanisms, we tested established MEF cells with BAX KO, BAK KO, or BAX mutants that we have previously established to form either BAX monomers or dimers (Garner et al., 2016). MEF cell lines were insensitive to BTSA1 (Figure S4G), however Navitoclax showed activity in these cell lines with an IC50 between 1–5 μM consistent with its de-repression activity of pro-apoptotic proteins (Figure 4F, 4G, S4H). When Navitoclax was combined with BTSA1, cell-death inducing activity was observed in WT and BAK KO MEFs but not in BAX KO MEFs, consistent with the specificity of BTSA1 for BAX and not BAK (Figure 4F, S4H). The compounds combination had significant synergy in cell lines that express BAX monomer (BAX E75, BAX T172K) suggesting that BTSA1 can activate more effectively BAX monomers that have an available trigger site (Figure 4G, S4H). In contrast, the compound combination in cells that express autoinhibited BAX dimer (WT, DKO BAX, BAX P168G) had additive or no effect (Figure 4F, 4G, S4H). Importantly, cells expressing BAX monomers but with mutations (K21E, A24R) in residues of the trigger site that were found to interact with BTSA1 (Figure 1H) were insensitive to the compounds combination consistent with the loss of BTSA1’s binding to BAX and induced BAX activation (Figure 4G, S4H).
Next, using gel filtration analysis of cytosolic extracts, we found that leukemia cell lines that display increased sensitivity to BTSA1-induced killing compared to wild type MEF (Figure 4H) contain indeed cytosolic BAX monomers, whereas MEF contain cytosolic BAX dimers (Figure 4I). Therefore, leukemia cells’ sensitivity to BTSA1-induced BAX activation results from the ability of BTSA1 to bind the BAX trigger site, which is readily available in the BAX monomer (Figure 4J). In contrast, the autoinhibited dimeric BAX in cells such as MEF prevents prompt BAX activation by BTSA1 (Figure 4K). Thus, these data demonstrate the capacity of BTSA1 to activate BAX and induce BAX-mediated apoptosis is regulated by the availability of anti-apoptotic BCL-2 proteins to inhibit activated BAX and also by the cytosolic conformation of BAX.
BTSA1 kills primary AML samples and spares healthy cells
To evaluate the translational potential of BTSA1-induced BAX activation we tested BTSA1 in primary samples from patients with high-risk AML (Figure S5A). Strikingly, BTSA1 induced dose-dependent apoptosis in primary AML blast cells but not in healthy hematopoietic stem cells and progenitors (Figure 5A). Consistent with BTSA1 activity, we found that AML blasts from patients demonstrate higher expression of BAX compared to healthy controls by comparative expression analysis (Figure 5B) and by western blot of BAX protein levels (Figure S5B). Likewise, we found that BTSA1 has no pro-apoptotic activity on primary T lympocytes (CD4+) compared to AML blast cells (Figure S5C). BTSA1 is also effective to induce apoptosis in leukemic stem cell-enriched fractions (CD34+CD38−) from these high-risk AML patients (Figure 5C). In contrast, the same dosages of BTSA1 did not affect apoptosis of primary bone marrow CD34+CD38− cells from healthy donors (Figure 5C, S5D). Highly purified stem and progenitor cell populations from AML patients demonstrated higher expression of BAX when compared to healthy counterparts (Barreyro et al., 2003; Schinke et al., 2015) (Figure 5D). Interestingly, BCL-2, but no BCL-XL or MCL-1, has significantly higher expression in AML patient samples (Figure S5E). Expression analysis in pre-leukemic stem cells from AML patients showed higher expression for MCL-1, but not for BCL-2 or BCL-XL (Figure S5F). Taken together, the data demonstrate a significant killing selectivity for BTSA1-induced BAX activation in human AML blasts and pre-leukemic stem cells compared to healthy stem and progenitor cells, which is consistent with the observed elevation of BAX expression levels in human AML.
Figure 5. BTSA1 is effective against patient AML blasts and pre-leukemic stem cells without affecting normal hematopoietic progenitor cells and demonstrates significant synergy with Venetoclax.

(A) Apoptosis assay indicated by Annexin V binding in primary human AML blasts (n=4) and healthy control stem and progenitors cells (n=2) exposed to BTSA1. (B) Boxplot showing BAX mRNA expression in AML patients cells (n=542) and healthy control cells (n=74), p = 3.26 x 10−7. Line within box shows median expression, while edges of the box represent the 25th (bottom) and 75th percentile of expression (top). Whiskers indicate maximum and minimum values excluding outliers. (C) Apoptosis assay indicated by Annexin V binding in primary human CD34+CD38− (stem cell-enriched) AML cells (n=4), and healthy CD34+CD38− cells (n=2) exposed to BTSA1. (D) Boxplot showing BAX mRNA expression in highly purified stem cells (CD34+CD38−Lin−) and progenitor cell populations (Granulocytic Macrophage Progenitors, GMPs, (Lin−CD34+CD38+CD123+CD45RA+) from AML patients and healthy controls. Line within box shows median expression, while edges of the box represent the 25th (bottom) and 75th percentile of expression (top). Whiskers indicate maximum and minimum values excluding outliers. (E, F) Viability assay of THP-1 (E) and OCI-AML3 (F) cells treated with Venetoclax or BTSA1 alone or in combination for 24 hr. (G) Caspase 3/7 assay of OCI-AML3 cells treated with Venetoclax alone at indicated doses or in combination with 1.25 μM BTSA1 for 1 hr. (H) Caspase 3/7 assay of OCI-AML3 treated with Venetoclax or in combination with BTSA1 at the indicated doses and time points. (I) Immunoblot for BAX, BCL-2, BCL-XL and MCL-1 from immunoprecipitates of BAX from OCI-AML3 cells treated with Venetoclax (1.25 μM), BTSA1 (1.25 μM) or in combination after 2.5 hr and western blot detection for BAX, BCL-2, BCL-XL and MCL-1. Data represent mean ± SD (n=3) from three independent experiments or are representative of three independent experiments unless indicated otherwise. See also Figure S5.
Comparative expression analysis of AML blasts compared to healthy controls demonstrated upregulation of BAX and BCL-2 in AML (Figure 5B, S5E). We hypothesized that BTSA1 would potentiate the activity of Venetoclax (Konopleva et al., 2016). We tested, Venetoclax and BTSA1 in several AML cell lines that display similar killing potency. When both compounds are evaluated in combination, BTSA1 at ~IC10 dose displayed significant synergy with Venetoclax increasing its efficacy to low nM concentration from the low μM range (Figure 5E, 5F, S5G). Moreover, the synergistic killing activity of the compound combination is observed promptly (Figure S5H). To confirm that this synergistic activity is consistent with the kinetics of mitochondrial apoptosis induction, we found that the combination of the compounds promoted casapase-3/7 activation within min as opposed to hr when compounds are added alone (Figure 5G, 5H). Additionally, immunoprecipitation of BAX from vehicle or BTSA1 treated OCI-AML3 cells revealed that upon BTSA1 treatment some BAX is sequestered by BCL-2 but not by MCL-1 or BCL-XL (Figure 5I). Treatment of Venetoclax derepressed BCL-2/BAX complex (Figure 5I), which enabled BTSA1 to potentiate BAX activation and promote a more robust apoptosis (Figure 5E, 5F, S5G). Our results suggest that direct BAX activators such as BTSA1 can be effective therapeutics for AML as monotherapy by having also activity in pre-leukemic stem cells while sparing healthy counterparts. Combination of BTSA1 with Venetoclax could have more potent and broader activity in AML (Goff et al., 2013; Konopleva et al., 2016; Lagadinou et al., 2013; Pan et al., 2014).
BTSA1 suppresses human AML xenografts and is well tolerated
The in vivo activity and safety of BTSA1-induced BAX activation was next evaluated in human AML xenografts and in healthy cells and tissues. Pharmacokinetics studies in mice demonstrated that BTSA1 has substantial half-life in mouse plasma (T1/2 = 15 hr) and oral bioavailability (%F =51) while a 10 mg/kg dose reached sufficient levels (~15 μM) of BTSA1 to induce BAX activation and apoptosis in leukemia cells (Figure S6A–S6C). We generated human leukemia THP-1 xenografts in mice and after engraftment at 10 days, mice were randomly divided into two groups and treated with either vehicle or 10 mg/kg BTSA1 every 48 hr (Figure 6A). Mice treated with BTSA1 had significantly increased survival when compared to vehicle-treated mice (median survival 40 days in control vs 55 days in BTSA1-treated) and 43% of BTSA1-treated mice were alive at day 60 and had no signs of AML infiltrates (Figure 6B). To confirm anti-leukemic efficacy of BTSA1, the experiment was repeated and after 3 weeks of BTSA1 treatment, AML tumor burden from bone marrow and liver was evaluated by flow cytometry using anti-hCD45 and anti-hCD15 antibodies (Figure 6C). Human leukemia infiltration was detected in livers of vehicle treated mice while BTSA1 treatment induced significant suppression of leukemia growth (Figure 6D–6F).
Figure 6. BTSA1 demonstrates potent efficacy in killing human AML in vivo.

(A) Schematic of the THP-1 xenograft mouse model for survival experiment. (B) Kaplan-Meir survival curves of vehicle- and BTSA1-treated cohorts (n=7). (C) Schematic of THP-1 xenograft mouse model for analysis of human cell infiltrates experiment. (D, E) FACS analysis of tumor burden in liver using mouse and human CD45 (D) and human CD15 (E) antibodies from vehicle- and BTSA1-treated cohorts (n=4). (F) An example of liver infiltrates from a vehicle- and BTSA1-treated mice. (G) Schematic of MOLM-13 xenograft mouse model for analysis of human cell infiltrates and apoptosis markers. (H) FACS analysis of bone marrow infiltration using human and mouse CD45 and CD15 antibodies from vehicle- and BTSA1-treated cohorts (n=5). (I) FACS analysis of peripheral blood infiltration using mouse and human CD45 and human CD15 from vehicle- and BTSA1-treated cohorts (n=5). (J) Quantification of densitometric analysis for cleaved caspase-3 positive cells stained by IHC from bone marrow slides of vehicle- and BTSA1-treated cohorts (n=5). (K) Quantification of densitometric analysis for TUNEL positive cells stained by IHC in the bone morrow from vehicle- and BTSA1-treated cohorts (n=5). (L) Representative tissue sections from bone marrow using cleaved caspase-3 staining after treatment of vehicle and BTSA1. Scale bars, 100 μm. (M) Representative tissue sections from bone marrow using TUNEL staining after treatment of vehicle and BTSA1. Scale bars, 100 μm. (N) Schematic of MOLM-13 xenograft mouse model for mitochondrial TMRE assay experiment. (O) TMRE staining of mitochondria from human leukemia cells selected by human CD45 and CD15 antibodies from bone marrow of vehicle- and BTSA1-treated cohorts (n=5). Data represent mean ± SD at indicated n points. See also Figure S6.
We generated a second AML xenograft model using the MOLM-13 cell line. Infiltration of human leukemia cells was assessed after 21 days of engraftment during which mice were treated with either vehicle or 10 mg/Kg BTSA1 every 48 hr (Figure 6G). Treatment with BTSA1 reduced significantly the infiltration of human cells in both bone marrow and peripheral blood samples (Figure 6H, 6I). To confirm that decrease in infiltration of human cells was associated with apoptotic cell death, bone marrow samples were analyzed for caspase cleavage and TUNEL stain by immunohistochemistry. Indeed, densitometric analysis of immunohistochemistry for caspase cleavage and TUNEL stain demonstrated that BTSA1 promotes apoptotic cell death in bone marrow infiltrates consistent with an on-target effect of BTSA1 in vivo. (Figure 6J–6M). Alternatively, to directly demonstrate the pharmacodynamic effect of BTSA1 on BAX, we developed an in vivo mitochondrial membrane potential assay. As shown above, an immediate effect of BTSA-induced BAX activation in cultured cells is a decreased in mitochondrial membrane potential (Figure 3F). MOLM-13 mice xenografts were treated 15-days post-transplantation for two days with BTSA1 or vehicle (Figure 6N). After two days treatment, bone marrow samples from sacrificed mice were isolated and stained with TMRE to label active mitochondria. As expected, reduced infiltration of human leukemia cells from BTSA1 treatment was associated with a decrease in mitochondrial membrane potential from bone marrow samples (Figure 6O).
To evaluate tolerance of BTSA in vivo, a toxicology study was performed by treating mice for 30 days with a higher dose (15 mg/kg) of BTSA1 than that used in our efficacy studies. Mice tolerated BTSA1 for the entire duration of treatment exhibiting normal behavior and no detectable toxicity was observed. Specifically, body weights before and after treatment showed no loss of weight (Figure 7A). Blood counts of white blood cells, red blood cells and platelets were maintained at normal range and comparable to levels of vehicle-treated mice (Figure 7B–7D), and histological evaluation of several tissues showed no evidence of toxicity (Figure 7E). Additionally, bone marrow samples of BTSA1-treated mice were analyzed by histopathology analysis (Figure 7E, S7A) and determination of frequency of immature hematopoietic stem and progenitor cell populations by FACS. BTSA1 had no toxic effects on healthy hematopoiesis, including healthy stem cell-enriched (LSK) cells, common myeloid progenitors (CMP), granulocyte-monocyte progenitors (GMP), and megakaryocyte-erythrocyte progenitors (MEP) (Figure S7B). Thus, the data demonstrate that BTSA1 is orally bioavailable with excellent pharmacokinetics, has significant anti-tumor activity in leukemia xenografts by promoting apoptosis and at therapeutically effective doses it does not show any detectable toxicity in the hematopoietic system or other tissues.
Figure 7. BTSA1 is well tolerated without toxicity to normal cells in vivo.

(A) Body weight measurements of NSG mice on day 1 and after treatment for 30 days with 15 mg/kg BTSA1 per body weight or vehicle. (B–D) Counts of peripheral red blood cells (B), white blood cells (C), and platelets (D) in NSG mice treated with vehicle or 15 mg/kg BTSA1 per body weight for 30-days. Data in (A–D) represent mean ± SD (n=3). (E) Representative tissue sections of liver, spleen, kidney, lung, heart, brain and bone marrow using Hematoxylin & Eosin staining from mice after treatment of vehicle and 15 mg/Kg BTSA1. Scale bars, 100 μm. See also Figure S7.
DISCUSSION
Since the discovery of pro-apoptotic BAX more than two decades ago (Olitvai et al., 1993), numerous studies have provided evidence that BAX is a critical protein that controls the gateway to mitochondrial outer membrane permeabilization and apoptosis (Luna-Vargas and Chipuk, 2016; Walensky and Gavathiotis, 2011; Wei et al., 2001). Direct BAX activation by select BH3-only proteins such as BIM, BID and PUMA has been established as a physiological mechanism that initiates the mitochondrial apoptotic pathway during development and homeostasis (Ren et al., 2010). Moreover, cancer cells neutralize BAX activation by deregulation or suppression of BH3-only proteins, and/or overexpression of anti-apoptotic BCL-2 proteins (Letai, 2008; Juin et al., 2013; Leverson et al., 2015). We previously provided structural, biochemical and cellular data for the identification and characterization of a BH3-binding site of inactive BAX that regulates activation of cytosolic BAX (Gavathiotis et al., 2008; Gavathiotis et al., 2010). We reasoned that these studies would enable a therapeutic strategy in cancer cells to provide an effective drug that would directly activate BAX and lead to mitochondrial permeabilization and apoptosis. Essentially, such a drug would replace the function of the BH3-only proteins that are kept suppressed either by anti-apoptotic BCL-2 proteins or other mechanisms in cancer (Hata et al., 2015; Letai, 2008).
Here, we described the discovery and characterization of BTSA1 that binds potently and specifically to the BAX trigger site and initiates BAX conformational changes resulting in BAX mitochondrial translocation, oligomerization and mitochondrial dysfunction (Gavathiotis et al., 2008; Kim et al., 2009). BTSA1 induces activation of soluble (cytosolic) BAX leading to prompt and robust mitochondrial apoptosis in leukemia cell lines and AML samples. The efficacy of BTSA1 is regulated by the availability of anti-apoptotic BCL-2 proteins to inhibit activated BAX and therefore, higher levels of cytosolic BAX monomer correlated with higher efficacy of BAX-mediated mitochondrial dysfunction. Our data suggest that AML or other cancer cells that have anti-apoptotic BCL-2 family proteins neutralized by pro-apoptotic members would be sensitive to direct BAX activation and BTSA1, while cells that keep the expression levels of BAX suppressed or tip the balance of BCL-2 family proteins towards overexpression of anti-apoptotic proteins, would be less sensitive. Moreover, we previously reported the formation of the autoinhibited BAX dimer, which can provide a mechanism to reduce sensitivity to apoptosis induction (Garner et al., 2016). Here we found that BAX in leukemia cell lines is in monomeric conformation and primed for BAX activation by BTSA1, whereas the formation of the autoinhibited BAX dimer can provide significant resistance to BAX activation by BTSA1. These regulatory mechanisms to BAX activation may be more effective in blocking direct BAX activation and decreasing the activity of BTSA1 in normal cells compared to AML cells providing an applicable therapeutic index as demonstrated in our in vivo studies. Consistent with our data, a recent report demonstrated the insensitivity of most adult healthy somatic tissues to BAX activation by BIM BH3 (Sarosiek et al., 2017).
AML remains a challenging cancer to treat and current chemotherapy regimens result in unsatisfying 5-year survival (< 25 %), which is even worse in the elderly individuals who represent the majority of AML patients. Unfavorable outcomes have been associated with the existence of leukemic and pre-leukemic stem cells that are resistant to current treatments and drive AML relapse (Huff et al., 2006; Jan et al., 2012; Slush et al., 2014, Steidl et al., 2006, Will et al. 2015). Our findings demonstrate that BTSA1-induced BAX activation is effective in primary AML samples and human AML xenograft models. It is noteworthy that BTSA1 is also effective in leukemic stem cell-enriched fractions (CD34+CD38−). Surprisingly, we found that not only do AML blasts display higher expression of BAX but also highly purified stem and progenitor cell populations from AML patients when compared to healthy counterparts. This may be a critical factor for the overall sensitivity of AML blasts and pre-leukemic stem cells to direct BAX activation. Overall, the data strongly support direct BAX activation as effective monotherapy treatment for AML and suggest that agents such BTSA1 should be also useful in combinatorial treatments with other targeted agents such as BCL-2 inhibitors.
In summary, this study provides preclinical proof-of-concept for BAX as a druggable target and the therapeutic potential of direct BAX activation as an effective treatment strategy in AML. We present the discovery of BTSA1, a preclinical lead candidate with drug-like properties such as high potency, well-characterized mechanism of action, selectivity, excellent pharmacokinetic profile, oral bioavailability and synthetic ease that warrants further evaluation of its therapeutic potential in cancer.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Evripidis Gavathiotis (evripidis.gavathiotis@einstein.yu.edu).
Experimental Model and Subject Details
Patient Samples
Primary human AML and healthy control samples were obtained with informed consent and studies were approved by IRB of Albert Einstein College of Medicine. Patient characteristics are described in figure S5A. Samples were maintained in Iscove’s modified Dulbecco’s medium with 10% fetal bovine serum at 37°C.
Mice
All animal experiments were approved by and performed in compliance with the guidelines and regulations approved by the Institutional Animal Care and Use Committee of the Albert Einstein College of Medicine (protocols 20160613, 20161008). For xenograft studies, toxicity and pharmacodynamics analysis experiments utilized 6–8 weeks old NOD-SCID IL2Rγ null (NSG) male and female mice purchased from Jackson Laboratory. For pharmacokinetics studies, 6–8 weeks old ICR (CD-1) male mice were used from Charles River. All mice were kept under standard conditions and diet and had a weight > 20 grs.
Primary Cells
Murine bone marrow were aspirated or isolated from cleaned tibias and femurs of mice. Red blood cells were lysed by incubation with ACK lysis buffer. Cells were resuspended in RPMI 1640 media (Life Technologies) supplemented with 10% FBS (ThermoFisher Scientific), 100 U ml–1 penicillin/streptomycin (ThermoFisher Scientific) and 2 mM l-glutamine (ThermoFisher Scientific).
Cell lines
Cell lines were purchased from ATCC and DSMZ. Leukemia cells were also provided by Ulrich Steidl’s laboratory and MEF cells were provided by Emily Cheng’s laboratory. Leukemia cells were maintained in RPMI 1640 media (Life Technologies) supplemented with 10% FBS, 100 U ml–1 penicillin/streptomycin, 2 mM l-glutamine, and 50 μM β-mercaptoethanol. OCI-AML3 was maintained in MEM α (Life Technologies) supplemented with 10% FBS, 100 U ml–1 penicillin/streptomycin, 2 mM l-glutamine and 50 μM β-mercaptoethanol. WT MEFs, BAX/BAK DKO MEFs, BAX KO MEFs and DKO BAX mutant MEFs were maintained in DMEM (Life Technologies) supplemented with 10% FBS, 100 U ml–1 penicillin/streptomycin, 2 mM l-glutamine, 0.1 mM MEM nonessential amino acids, and 50 μM β-mercaptoethanol. H9C2 myocytes and 3T3 fibroblasts were maintained in DMEM (Life Technologies) supplemented with 10% FBS, 100 U ml–1 penicillin/streptomycin and 2 mM l-glutamine.
Microbe strains
For recombinant protein production, E. coli strain was grown in standard Luria Broth media at 37°C and cells were typically induced at optical density of 0.6–1 at 30°C for 4 hr (Gavathiotis et al, 2012; Uchime et al., 2016).
Methods Details
Compounds
Hydrocarbon-stapled peptides corresponding to the BH3 domain of BIM, Ac-BIM SAHBA2: N-acetylated EIWIAQELRS5IGDS5FNAYYA-CONH2 and FITC-BIM SAHBA2: FITC-βAla-EIWIAQELRS5IGDS5FNAYYA-CONH2, where S5 represents the non-natural amino acid inserted for olefin metathesis, were synthesized, purified at >95% purity by CPC Scientific Inc. and characterized as previously described (Gavathiotis et al., 2012). BTSA2 compound was obtained from Scientific Exchange, Inc. (cat. # X-013360). BAX channel blocker was obtained from EMD Millipore (cat. # 196805). Venetoclax was purchased from MedCheM Express (cat. # HY-15531) and Navitoclax was obtained from SelleckChem (cat. # S1001). Compounds were >95% pure, reconstituted in 100% DMSO and diluted in aqueous buffers or cell culture medium for assays.
Chemical Syntheses
All chemical reagents and solvents were obtained from commercial sources (Aldrich, Acros, Fisher) and used without further purification unless otherwise noted. Anhydrous solvents (tetrahydrofurane, toluene, dichloromethane, diethyl ether) were obtained using a Pure Solv™ AL-258 solvent purification system. Ethanol was dried over activated 4 Å molecular sieves. Microwave reactions were performed on an Anton Paar Monowave 300. Chromatography was performed on a Teledyne ISCO CombiFlash Rf 200i using disposable silica cartridges (4, 12, and 24 g). Analytical thin layer chromatography (TLC) was performed on aluminum-backed Silicycle silica gel plates (250 μm film thickness, indicator F254). Compounds were visualized using a dual wavelength (254 and 365 nm) UV lamp and/or staining with CAM (cerium ammonium molybdate) or KMnO4 stains. NMR spectra were recorded on Bruker DRX 300 and DRX 600 spectrometers. 1H and 13C chemical shifts (δ) are reported relative to tetramethyl silane (TMS, 0.00/0.00 ppm) as internal standard or to residual solvent (CD3OD: 3.31/49.00 ppm; CDCl3: 7.26/77.16 ppm; dmso-d6: 2.50/39.52 ppm). Mass spectra were recorded on a Shimadzu LCMS 2010EV (direct injection unless otherwise noted). High-resolution electrospray ionization mass spectra (ESI-MS) were obtained at the Albert Einstein College of Medicine’s Laboratory for Macromolecular Analysis and Proteomics. Elemental analyses were obtained from Intertek USA, Inc. (Whitehouse, NJ; http://www.intertek.com/pharmaceutical/analysis/whitehouse-nj/). N-(3-(2-(2-(3-aminopropoxy)ethoxy)ethoxy)propyl)-5-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanamide (S3) and 1-(3-(2-(2-(3-aminopropoxy)ethoxy)ethoxy)propyl)-3(3′,6′dihydroxy-3-oxo-3H-spiro[isobenzofuran-1,9′-xanthen]-5-yl)thiourea were synthesized according to literature procedures (Hang et al., 2004; Meijer et al., 2005). See also Method S1 for chemical synthesis and analytical characterization for BTSA1, biotin-labeled BTSA1 and fluorescein-labeled BTSA1.
Production of recombinant proteins
Human, recombinant and tagless BAX, BAKΔC, BCL-XLΔC, MCL-1ΔNΔC, BFL-1/A1ΔC were expressed in Escherichia coli and purified as previously reported (Gavathiotis et al, 2012; Uchime et al., 2016). BAX wild type and mutant monomers were purified by size exclusion chromatography in a buffer containing 20 mM HEPES pH 7.2, 150 mM KCl, 1 mM DTT. BCL-XLΔC, MCL-1ΔNΔC, and BFL-1/A1ΔC and BAKΔC monomeric proteins were purified by size exclusion chromatography in a buffer containing 50 mM Tris pH 7.4, 150 mM NaCl, 1 mM DTT. Superdex 75 10/300 GL and 200 10/300 GL (GE Healthcare) columns were used.
Fluorescence polarization binding assays
Fluorescence polarization assays (FPA) were performed as previously described (Gavathiotis et al., 2012). Firstly, direct binding isotherms were generated by incubating FITC-BIM SAHBA2 (50 nM) with serial dilutions of full-length BAX, BCL-XLΔC, MCL-1ΔNΔC, BFL-1/A1ΔC and fluorescence polarization was measured at 20 min on a F200 PRO microplate reader (TECAN). Subsequently, in competition assays, a serial dilution of small molecule or acetylated BIM SAHBA2 (Ac-BIM SAHB) was combined with FITC-BIM SAHBA2 (50 nM), followed by the addition of recombinant protein at ~EC75 concentration, as determined by the direct binding assay (BAX: 500 nM; BCL-XLΔC, MCL-1ΔNΔC, BFL-1/A1ΔC: 200 nM). For direct binding of FITC-BTSA1 (25 nM) to BAX, serial dilutions of full-length BAX were incubated and fluorescence polarization was measured at 20 min on a F200 PRO microplate reader. EC50 or IC50 values were calculated by nonlinear regression analysis of competitive binding curves using Graphpad Prism software.
NMR samples and spectroscopy
The uniformly 15N-labelled protein samples were prepared by growing the bacteria in minimal medium containing 15N-labeled NH4Cl followed by the same purification procedure as previously described (Uchime et al., 2016). Protein samples were prepared in 25 mM sodium phosphate, 50 mM NaCl solution at pH 6.0 in 5% D2O. Correlation 1H-15N-HSQC spectra were recorded on 15N-labelled BAX at 50 μM and titrations up to 100 mM of BTSA1 were performed. NMR spectra were acquired at 25 °C on a Bruker 600 MHz spectrometer equipped with a cryoprobe, processed using Topsin and analyzed with CCPNMR. BAX wild type cross-peak assignments were applied as previously reported (Gavathiotis et al., 2008). The weighted average chemical shift difference Δ(CSP) was calculated as √(ΔδH1)2+(ΔδN15/5)2 in p.p.m. The absence of a bar indicates no chemical shift difference, or the presence of a proline or a residue that is overlapped and not used in the analysis. The significance threshold for backbone amide chemical shift changes (0.01 p.p.m.) was calculated based on the average chemical shift across all residues plus the standard deviation, in accordance with standard methods. Mapping of chemical shifts on BAX structure and structural analysis was performed with PYMOL (Schrodinger, LLC). BAX activation and oligomerization were monitored by 1H-15N-HSQC spectra using BAX at 50 μM and BTSA1 at 100 μM or BIM SAHBA2 at 100 μM in the presence of NMR buffer plus 0.3% CHAPS to stabilize the oligomeric BAX in solution.
Molecular docking and molecular dynamics
NMR-guided docking of BTSA1 into the NMR structure of BAX (PDB ID: 2KW7) was performed using GLIDE (Glide, version 6.5, Schrödinger, LLC, New York, NY, 2014) with or without constraints based on residues undergoing significant chemical shifts. BTSA1 was converted to 3D all atom structure with LIGPREP (LigPrep, version 3.2, Schrödinger, LLC, New York, NY, 2014) and assigned partial charges with EPIK (Epik, version 3.0, Schrödinger, LLC, New York, NY, 2014). BTSA1 was docked using the extra precision (XP) docking mode. The lowest-energy docking pose is consistent with the observed NMR-chemical shift perturbation data. The lowest energy structure pose from XP docking was selected for further analysis and subjected to a 10 ns molecular dynamics (MD) simulation using DESMOND (DESMOND, version 3, Schrödinger, LLC, New York, NY, 2014). The lowest energy BAX structure from the NMR ensemble (PDB ID: 1F16) was subjected to a 10 ns MD simulation using DESMOND for comparison. MD runs were performed in truncated octahedron SPC water box using OPLS_2005 force field, 300 K and constant pressure of 1.0325 bar. Clustering and analysis of the trajectory was performed with MAESTRO tools (Maestro, version 10.0, Schrödinger, LLC, New York, NY, 2014). PYMOL (The PyMOL Molecular Graphics System. Version 1.7; Schrödinger, LLC: New York, 2014) was used for preparing the highlighted poses.
BAX oligomerization assay
The NMR samples started from mixtures of BTSA1-BAX monomer and BIM SAHBA2-BAX monomer as well as a BAX monomer sample were subjected to analysis by size-exclusion chromatography using a SD75 column in 25 mM sodium phosphate and 50 mM NaCl solution at pH 6.0 running buffer. The monomeric and oligomeric BAX fractions elute at ~13.0 min and ~8.5 min, respectively. Protein standards (GE Healthcare) were used to calibrate the molecular weights of gel filtration peaks. Replicates were performed using independent preparations of freshly SEC-purified monomeric BAX protein.
BAX conformational change assay
The N-terminal conformational change exposing the 6A7 epitope on activated BAX (1 μM) was measured by immunoprecipitation assay in 20 mM Hepes pH 7.2, 150 mM KCl buffer containing increasing doses of BTSA1 for 15 min at room temperature. The mixture was added to 280 μl 3% BSA in PBS and 30 μl of the resulting mixture (10%) was kept for input analysis. The remaining mixture was mixed with 3 μl 6A7 antibody (sc-23959, Santa Cruz) and pre-washed protein A/G beads (Santa Cruz) and incubated for 2h at 4°C with rotation. Beads were collected by brief spin, washed three times with 1 mL of 3% BSA buffer, and then solubilized with 25 μl LDS/DTT loading buffer. Samples were resolved by SDS-PAGE electrophoresis and western blot analysis using an anti-BAX antibody (Cell Signaling Cat. 2772).
BAX “BH3-Domain” Immunoprecipitation Assay
Exposure of the BH3 domain of BAX was assessed by immunoprecipitation with a BAX BH3-Domain specific antibody purchased from Abgent (cat. # AP1302a). Recombinant full-length BAX (200 nM) was incubated with increasing concentrations of BTSA1 in assay buffer (10 mM HEPES, 200 mM KCl, 0.250% CHAPS, 0.2 mM EDTA, 5 mM MgCl2,, pH 7.2) for 30 min. After incubation, 1 μl of BAX BH3-domain antibody was added to every sample and mixed pre-washed protein A/G beads (Santa Cruz) and incubated for 2 hr at 4°C with rotation. Beads were collected by brief spin, washed three times with 1 mL of assay buffer, and then solubilized with 25 μl LDS/DTT loading buffer. Samples were resolved by SDS-PAGE electrophoresis and western blot analysis an anti-BAX antibody (Cell Signaling Cat. 2772)
Liposomal Release Assay
Liposomes were prepared and release assays performed as previously described (Uchime et al. 2016). Liposomes of the following lipid composition (Avanti Polar Lipids): phosphatidylcholine, 48%; phosphatidylethanolamine, 28%; phosphatidylinositol, 10%; dioleoyl phosphatidylserine, 10%; and tetraoleoyl cardiolipin, 4% are mixed (1 mg total) and resuspended in liposome assay buffer (10 mM HEPES, 200 mM KCl, 0.2 mM EDTA, 5 mM MgCl2,, pH 7.2) containing 12.5 mM of the fluorescent dye ANTS (8-aminonaphthalene-1,3,6-trisulfonic acid, disodium salt) and 45 mM of the quencher DPX (p-xylene-bis-pyridinium bromide). The resulting slurry is vortexed for 10 min and freeze-thawed five times in liquid nitrogen and a 38°C water bath, respectively. The solution is then passed through an Avanti Mini-Extruder Set (#610000) equipped with a 100 nm filter, followed by passage through a Sepharose column (GE Healthcare) to remove residual ANTS/DPX. The liposomes are brought up to a volume of 3 mL to produce a final liposome stock. For the liposomal release assay, recombinant BAX (20 μM) is pre-incubated with increasing concentrations of BTSA1 in liposome assay buffer containing 0.1250% CHAPS for 1 hr. Following pre-incubation, mixture is then diluted 10 fold (final BAX concentration 200 nM) and transfer into a 96-well opaque plate (Costar) containing 10 μL liposomes in a total volume of 100 μL. ANTS/DPX release was quantified based on the increase in fluorescence intensity that occurs when the ANTS fluorophore is separated from the DPX quencher upon release from the liposomes into the supernatant. Fluorescence (λex = 355 nm and λem = 520 nm) was measured over time at 30°C using a Tecan Infinite M1000 plate reader. The percentage release of ANTS/DPX at 90 min was calculated as percentage release = ((F − F0)/(F100 − F0)) × 100, where F0 and F100 are baseline and maximal fluorescence, respectively. 1% Triton treatment is used to determine the maximum amount of liposomal release per assay, and this value sets the 100% value.
Liposomal translocation assay
Liposomes of the following lipid composition (Avanti Polar Lipids): phosphatidylcholine, 47%; phosphatidylethanolamine, 27%; phosphatidylinositol, 10%; dioleoyl phosphatidylserine, 10%; tetraoleoyl cardiolipin, 4%; and biotiny-phosphatidylethanolamine, 2% were mixed (1 mg total) and resuspended in liposome assay buffer (10 mM HEPES, 200 mM KCl, 0.2 mM EDTA, 5 mM MgCl2, pH 7.2). The resulting slurry was vortexed for 10 min and sonicated in a sonicating water bath at 4°C for 10 min. The solution was then passed through an Avanti Mini-Extruder Set (#610000) equipped with a 100 nm filter, followed by passage through a Sepharose column (GE Healthcare). For the liposomal translocation assay, recombinant BAX (20 μM) is pre-incubated with increasing concentrations of BTSA1 or 60 nM tBID in liposome assay buffer containing 0.1250% CHAPS for 1 hr. Mixture was then diluted 10 times (final BAX concentration 200 nM) and transferred to a 96-well low binding plate (Costar) containing 10 μL liposomes in a total volume of 100 μL. Reaction mixtures were incubated for 45 min at room temperature with shaking. Streptavidin was immobilized onto 96-well EIA/RIA plates (Corning Inc.) at 5 μg per well (incubated at 4°C over night in PBS) and blocked for 2 hr at room temperature with 3% BSA in PBS. Liposome translocation reaction mixtures were mixed with BSA (final concentration 0.6%) and loaded on to streptavidin coated 96-well EIA/RIA plate and incubated for 15 min at room temperature, after which 50 μg of biotin was added to each reaction and incubated for 5 min. Non-translocated BAX was removed by 4 washes with PBS. BAX translocation was monitored using horseradish peroxidase (HRP)-N20-anti-BAX antibody conjugate (Santa Cruz sc-493 HRP) incubated at 1/1000 dilution in 3% BSA for 1 hr at room temperature and washed 4 times with PBS. Followed by incubation with 3,3,5,5 -tetramethylbenzidine HRP substrate quenched with 0.5 M sulphuric acid and measured at 450 nm. Each well was normalized to 100% BAX translocation induced by BAX incubated with 1% triton X and liposomes.
Immunoprecipitation and Immunoblotting
Protein lysates were obtained by cell lysis in Triton X-100 buffer (50 mM Tris-HCL pH 7.40, 150 mM NaCl, 5 mM MgCl2, 1 mM EGTA, 10% Glycerol, 1% Triton X-100. Immunoprecipitation was performed in 600 μl of 500 μg of proteins, which was precleared by centrifugation followed by exposure to 12 μl (50% slurry) protein A/G beads (Santa Cruz) at 4°C for 1 hr. Cleared extracts were incubated overnight with 1 μl of anti-BAX antibody (Cell Signaling Cat. 2772). Samples were then exposed to 20 μl (50% slurry) protein A/G beads (Santa Cruz) at 4°C for 2 hr and later centrifuged and washed three times with Triton X-100 buffer and boiled in loading buffer (Life Technologies). Protein samples were electrophoretically separated on 4–12% NuPage (Life Technologies) gels, transferred to mobilon-FL PVDF membranes (Millipore) and subjected to immunoblotting. For visualization of proteins with Odyssey Infrared Imaging System (LI-COR Biosciences) membranes were blocked in PBS containing 5% dry milk. Primary antibodies were incubated overnight at 4°C in a 1:1,000 dilution. After washes, membranes were incubated with an IRdye800-conjugated goat anti-rabbit IgG or IRdye800-conjugated goat anti-mouse IgG secondary antibodies (LI-COR Biosciences) in a 1:5,000 dilution. Antibodies were used to detect the following proteins on membrane: BCL-XL (BD Cat. 610747), MCL-1 (Cell Signaling Cat. 4572), BAX (Cell Signaling Cat. 2772), BCL-2 (Cell Signaling Cat. 4223), BAK (Millipore Cat. 06-536), Cytochrome c (BD Cat. 556433), VDAC1/Porin (Abcam Cat. 15895), β-Actin (Sigma Cat. A1978).
Western blotting
Protein lysates were obtained by cell lysis in Triton X-100 buffer (50 mM Tris-HCL pH 7.40, 150 mM NaCl, 5 mM MgCl2, 1 mM EGTA, 10% Glycerol, 1% Triton X-100 [Sigma]). Protein samples were electrophoretically separated on 4–12% NuPage (Life Technologies) gels, transferred to mobilon-FL PVDF membranes (Millipore) and subjected to immunoblotting. For visualization of proteins with Odyssey Infrared Imaging System (LI-COR Biosciences) membranes were blocked in PBS containing 5% dry milk. Primary antibodies were incubated overnight at 4°C in a 1:1,000 dilution. After washing, membranes were incubated with an IRdye800-conjugated goat anti-rabbit IgG or IRdye800-conjugated goat anti-mouse IgG secondary antibodies (LI-COR Biosciences) in a 1:5,000 dilution. Proteins were detected with Odyssey Infrared Imaging System. Antibodies were used to detect the following proteins on membrane: BCL-XL (BD Cat. 610747), MCL-1 (Cell Signaling Cat. 4572), BAX (Cell Signaling Cat. 2772), BCL-2 (Cell Signaling Cat. 4223), BAK (Millipore Cat. 06-536), Cytochrome c (BD Cat. 556433), VDAC1/Porin (Abcam Cat. 15895), β-Actin (Sigma Cat. A1978).
Western blot protein quantification
Densitometry of protein bands were acquired using a LI-COR Odyssey® scanner. Quantification and analysis was performed using the Western Analysis tool from the Image Studio 3.1 software. Relative expression levels were quantified based on protein expression of β-Actin.
BAX crosslinking
BAX oligomerization was detected using a crosslinking approach by incubating isolated mitochondria from liver and recombinant BAX protein (200 nM) at indicated doses of BTSA1 with 20 x Bismaleimidohexane (BMH, Pierce) for 30 min at room temperature followed by quenching with 1 mM DTT. Samples were denatured at 95°C and analyzed by 4–12% NuPage (Life Technologies) gel electrophoresis followed by immunoblotting with anti-BAX antibody (Cell Signaling Cat. 2772).
Pull Down Assay
OCI-AML3 cytosolic and mitochondrial lysates were generated by Dounce Homogenization. Cells were lysed by Dounce homogenizer in lysis buffer (LB) containing 10 mM Tris, pH 7.5, 1 mM EGTA, 200 mM sucrose plus Complete Protease Inhibitors. The cell lysates were centrifuged at 700 x g for 10 min to remove unbroken cells and nuclei. The supernatants were centrifuged at 12000 x g for 10 min at 4 °C and the resulting pellet was collected as the mitochondrial fraction and supernatant as the cytosolic fraction. The mitochondrial fraction was resuspended in LB with 1% CHAPS to solubilize proteins. In summary, 500 ug of lysates were incubated for 2 hr at room temperature with biotin-labeled BTSA1 or vehicle. Biotin capture was achieved by incubation with high capacity streptavidin agarose (50 μL 50% slurry/reaction - Pierce) for 1 hr at room temperature. The streptavidin beads were washed (3x) at room temperature with 1X PBS. Cellular BAX bound to the biotin-labeled BTSA1 was eluted by boiling for 20 min in LDS buffer and then subjected to BAX (2772S, Cell Signaling), BCL-2 (BD Biosciences Cat. 610539), BCL-XL (Cell Signaling Cat. 2764) and MCL-1 (Cell Signaling Cat. 4572) western blot analysis.
Cell viability assay
AML cells (2 × 104 cells/well) were seeded in 96-well white plates and incubated with serial dilutions of BTSA1 or BTSA2 or vehicle (0.15% DMSO) in no FBS media for 2.5 hr, followed by 10% FBS replacement to a final volume of 100 μl. Cell viability was assayed at 24 hr by addition of CellTiter-Glo or ApoTox-Glo™ Triplex Assay reagents according to the manufacturer’s protocol (Promega), and luminescence measured using a F200 PRO microplate reader (TECAN). For the combination experiment with Venetoclax, AML cells were seeded as described above and co-treated with Venetoclax and BTSA1 at the indicated doses. Viability assays were performed in at least triplicate and the data normalized to vehicle-treated control wells. IC50 values were determined by nonlinear regression analysis using Prism software (Graphpad). For healthy cells, WT MEFs (1 × 104 cells/well), BAX/BAK DKO MEFs (1 × 104 cells/well), BAX KO MEFs (1 × 104 cells/well), H9C2 (1 × 104 cells/well) and 3T3 (1 × 104 cells/well) cells (~75%–80% cellular confluence) were seeded in 96-well white plates for 18 hr, then cells were treated with serial dilutions of BTSA1 or vehicle (0.15% DMSO) in serum-free growth media for 2 hr followed by serum replacement for a final volume of 100 μl containing 10% FBS. Cell viability was measured at 24 hr or at the indicated time point. For the combination experiments with Navitoclax, WT MEFs (2.5 × 103 cells/well) and DKO BAX mutant MEFs (2.5 × 103 cells/well) were seeded in a 384-well white plate for 18 hr, then cells were pre-treated with serial dilution concentrations of Navitoclax in complete media (10% FBS). Following 48 hr pre-treatment, media from cells was removed, and cells were treated with BTSA1 for 24 hr at the indicated concentration in serum-free growth media for 2.5 hr followed by serum replacement. Dilutions of compounds was performed using a TECAN D300e Digital Dispenser from 10 mM stocks.
Mitochondria isolation
Liver from Bak−/− mouse was homogenized in mitochondria isolation buffer [MIB: 250 μM Sucrose, 10 mM Tris-HCl, 0.1 mM EGTA, complete protease inhibitors (Roche Applied Science) by using a Teflon Dounce homogenizer and mitochondria recovered by differential centrifugation at 7000 x g for 10 min at 4°C.
In cell mitochondrial BAX translocation assay
Cells (1 × 106 cells/well) were seeded in 6-well clear bottom plate and incubated with serial dilutions of BTSA1 or vehicle (0.2% DMSO) in media with no FBS in a final volume of 2000 μL. After 2 hr, FBS was supplemented to a final concentration of 10%. Following 4 hr treatment, cells were lysed in 100 μL of digitonin buffer [20 mM Hepes, pH 7.2, 10 mM KCl, 5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 250 mM sucrose, 0.025% Digitonin (from 5% w/v stock) and complete protease inhibitors (Roche Applied Science)] and incubated on ice for 10 min. The supernatants were isolated by centrifugation at 15,000 x g for 10 min and the mitochondrial pellets solubilized in 1% Triton X-100/PBS for 1 hr at 4 °C. Pellets were solubilized, subjected to a 15,000 x rpm spin for 10 min, and 50 ng of protein was mixed with 25 μl LDS/DTT loading buffer. The equivalent fractional volume of the corresponding supernatant samples was mixed with 25 μl LDS/DTT loading buffer. The mitochondrial supernatant and pellet fractions were then separated by 4–12% NuPage (Life Technologies) gels, and analyzed by immunoblotting with anti-BAX antibody (2772S, Cell Signaling). VDAC1/Porin (Abcam Cat. 15895) and β-Actin (Sigma Cat. A1978) are used for loading control of mitochondrial and supernatant fractions respectively.
In vitro mitochondrial BAX translocation assay
Mitochondria from liver of Bak−/− mice (1 mg/ml) were resuspended in experimental buffer (125 mM KCl, 10 mM Tris-MOPS [pH 7.4], 5 mM glutamate, 2.5 mM malate, 1 mM K3PO4, 0.1mM EGTA-Tris [pH 7.4]) and treated with the indicated concentrations of BTSA1 and recombinant BAX protein (200 nM), singly and in combination, and incubated at room temperature for 60 min. The supernatant fractions were isolated by centrifugation at 5500 x g for 10 min and the mitochondrial pellets resuspended and washed with 0.1 M sodium carbonate (pH 11.5) for 30 min, centrifuged at 13,000 x g for 10 min at 4°C, and then solubilized in 1% Triton X-100/PBS for 1 h at 4°C. Mitochondrial supernatant and pellet fractions were separated by 4–12% NuPage (Life Technologies) gels and analyzed by immunoblotting with anti-BAX antibody (Cell Signaling Cat. 2772).
In cell cytochrome c release assay
Cells (1×106 cells/well) were seeded in 6-well clear bottom plate and incubated with serial dilutions of BTSA1 or vehicle (0.2% DMSO) in RPMI media with no FBS in a final volume of 2000 μL. After 2 hr FBS was supplemented to a final concentration of 10%. Following 4hr treatment, cells were lysed in 100 μL of digitonin buffer [20 mM Hepes, pH 7.2, 10 mM KCl, 5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 250 mM sucrose, 0.025% Digitonin (from 5% w/v stock) and complete protease inhibitors (Roche Applied Science)] and incubated on ice for 10 min. The supernatants were isolated by centrifugation at 15,000 x g for 10 min and the mitochondrial pellets solubilized in 1% Triton X-100/PBS for 1 h at 4 °C. Pellets were solubilized, subjected to a 15,000 x g rpm spin for 10 min, and 50 ng of protein was mixed with 25 μl LDS/DTT loading buffer. The equivalent fractional volume of the corresponding supernatant samples was mixed with 25 μl LDS/DTT loading buffer. The mitochondrial supernatant and pellet fractions were then separated by 4–12% NuPage (Life Technologies) gels, and analyzed by immunoblotting with anti-cytochrome c antibody (BD Biosciences Cat. 556433).
In vitro cytochrome c release assay
Mitochondria from liver of WT mice (1mg/ml) were resuspended in experimental buffer (125 mM KCl, 10 mM Tris-MOPS [pH 7.4], 5 mM glutamate, 2.5 mM malate, 1 mM K3PO4, 0.1 mM EGTA-Tris [pH 7.4]) and treated with the indicated concentrations of BTSA1 and recombinant BAX protein (200 nM), singly and in combination, and incubated at room temperature for 90 min. The supernatants were isolated by centrifugation at 5500 x g for 10 min and the mitochondrial pellets solubilized in 1% Triton X-100/PBS. Mitochondrial supernatant and pellet fractions were separated by 4–12% NuPage (Life Technologies) gels and analyzed by immunoblotting with anti-cytochrome c antibody (BD Biosciences Cat. 556433). VDAC1/Porin (Abcam Cat. 15895) and β-Actin (Sigma Cat. A1978) are used for loading control of mitochondrial and supernatant fractions respectively.
Caspase-3/7 activation assay
Cells were treated with BTSA1 alone or concurrently with the BAX channel inhibitor or in combination with Venetoclax as described in cell viability assays, and caspase-3/7 activation was measured at 6 hr or at the indicated time points by addition of the Caspase-Glo 3/7 chemiluminescence reagent in accordance with the manufacturer’s protocol (Promega). Luminescence was detected by a F200 PRO microplate reader (TECAN).
Mitochondrial depolarization assay (mtΔΨ)
Cells (3× 105 cells/well) were seeded in 6-well clear bottom plate and incubated with serial dilutions of BTSA1 or vehicle (0.2% DMSO) as described for cell viability assays. Following BTSA1 treatment, cells were stained with 100 nM TMRE (Sigma Cat. 87917) for 30 min at 37 °C. Subsequently, cells were pelleted by centrifugation and resuspended in 1xPBS to eliminate background fluorescence and transfer to a black 96-well plate. Fluorescence intensity was detected by a M100 microplate reader (TECAN, Ex: 540 nm, Em: 579 nm).
Cellular transfections
Anti-Apoptotic BCL-2 proteins over-expression: NB4 cells were transfected with human BCL-2, BCL-XL or MCL-1using the Amaxa Nucleofector Kit V according to the manufacturer’s protocol. Briefly, 3 × 106 cells were resuspended into Amaxa N solution V with 2 μg human BCL-2 pCDNA3, human MCL-1 pCDNA3 or human BCL-XL Kozark start-stop pCDNA3 followed by electroporation using program X-001 of Nucleofector Device. Nucleofected cells (3 × 106) were incubated for 48 hr before protein extraction and 30 μg protein/sample were separated by 4–12% NuPage (Life Technologies) gels and analyzed by immunoblotting with anti-BCL2 (BD Biosciences Cat. 610539), anti-MCL-1 (Cell Signaling Cat. 4572) or anti-BCL-XL (Cell Signaling Cat. 2764) to evaluate transfection efficiency. For viability experiments, 48 hr post-nucleofection, NB4 cells were treated with BTSA1 as described above for cell viability assays. Bax silencing: NB4 cells were transfected with siBAX using the Amaxa Nucleofector Kit V according to the manufacturer’s protocol. Briefly, 3 × 106 cells were resuspended into Amaxa N solution V with 300 nM siBAX s1890 (Life Technologies Cat. 4390824) or 300 nM Select Negative Control No. 2 siRNA (Life Technologies Cat. 4390846) followed by electroporation using program X-001 of Nucleofector Device. Nucleofected cells (3 × 106) were incubated for 24 hr before protein extraction and 30 μg protein/sample were separated by 4–12% NuPage (Life Technologies) gels and analyzed by immunoblotting with anti-BAX antibody (Cell Signaling Cat. 2772) to evaluate transfection efficiency. For viability experiments, 24 hr post-nucleofection, NB4 cells were treated with BTSA1 as described above for cell viability assays.
Evaluation of apoptosis in primary human cells
Samples were incubated with 5 and 10 μM of BTSA1 in Iscove’s modified Dulbecco’s medium with 10% fetal bovine serum at 37 °C. After 48 hr, cells were rinsed with PBS and stained with antibodies against CD34 (8G12), CD38 (HIT2) or CD4. After washing with PBS, cells were mixed with prediluted fluorescein isothiocyanate-conjugated Annexin V (Life Technologies), incubated at room temperature for 15 min, and resuspended in 0.5 mL of Annexin binding buffer (Life Technologies). Just before flow cytometric analysis, DAPI (Acros Organics) was added to the cells at a final concentration of 1 μg/mL. Viability, apoptosis, and necrosis of CD34+, CD4+, CD34+CD38− stem cells were analyzed by flow cytometry using a FACSAria II Special Order System (BD Biosciences) as previously performed (Schinke et al., 2015).
Gene expression data
Data on 200 AML samples was obtained from TCGA. Gene expression data from 183 MDS CD34+ samples and 17 controls was obtained from GEO (GSE19429). Gene expression data on sorted LT-HSCs, ST-HSCs, and GMPs from AML/MDS patients and healthy controls is deposited in the GEO database (GSE35008 and GSE35010).
Pharmacokinetic analysis
ICR (CD-1) male mice were fasted at least three hr and water was available ad libitum before the study. Animals were housed in a controlled environment, target conditions: temperature 18 to 29°C, relative humidity 30 to 70%. Temperature and relative humidity was monitored daily. An electronic time controlled lighting system was used to provide a 12 hr light/12 hr dark cycle. 3 mice for each indicated time point were administered 10 mg/Kg BTSA1 in 1% DMSO, 30% PEG-400, 65% D5W (5% dextrose in water), 4% Tween-80 either by an oral gavage or intravenous injection. Mice were sacrificed, and plasma samples were harvested at 0 hr, 0.25 hr, 0.5 hr, 1 hr, 2 hr, 4 hr, 8 hr, 24 hr, 48 hr and analyzed for BTSA1 levels using LC-MS/MS. Pharmacokinetics parameters were calculated using Phoenix WinNonlin 6.3. Experiments performed at SIMM-SERVIER joint Biopharmacy Laboratory.
Human leukemia xenografts studies
For survival experiments, 6–8 weeks old NOD-SCID IL2Rγ null (NSG) mice (Jackson Laboratory) were subjected to sublethal (200 rad) total body irradiation and 3 hr later injected with 5 x105 THP-1 cells by tail vein. Ten days after inoculation, mice with AML disease, were divided into two groups (n = 7 per arm), and treated with vehicle and 10 mg/kg BTSA1 by IP route every two days. The vehicle for BTSA1 was 1% DMSO, 30% PEG-400, 65% D5W, 4% Tween-80. The survival distributions of BTSA1- and vehicle-treated mice were determined using the Kaplan-Meier method and compared using the log-rank test. Body weight and blood counts of animals were monitored during disease progression and treatment. For measurement of THP-1 infiltration, 6–8 weeks old NOD-SCID IL2Rγ null (NSG) mice were subjected to sublethal (200 rad) total body irradiation and 3 hr later injected with 3 × 105 THP-1 cells by tail vein. Experiments were repeated as above with two groups (n= 4 per arm) and mice sacrificed at 3 weeks post treatment initiation and tissues collected for FACS analysis. Peripheral blood from NOD-SCID IL2Rγ null (NSG) mice was obtained by facial vein puncture and collected in EDTA-coated tubes (BD Cat. 365973). Blood counts were determined on a Forcyte Veterinary Hematology Analyzer (Oxford Science Inc.) For measurement of MOLM-13 infiltration, 6–8 weeks old NOD-SCID IL2Rγ null (NSG) mice (Jackson Laboratory) were subjected to sublethal (200 rad) total body irradiation and 3 hr later injected with 5 x105 MOLM-13 cells by tail vein. Three days after inoculation, mice with AML disease, were divided into two groups (n = 5 per arm), and treated with vehicle, 10 mg/kg BTSA1 by IP every two days. For MOLM-13 transplanted NOD-SCID IL2Rγ null (NSG) mice, infiltration was assayed two weeks after inoculation by bone marrow aspirate extraction of blood samples followed by FACS analysis. Additionally, infiltration was also quantified from sacrificed animals three weeks after inoculation by FACS analysis from spleen, bone marrow, peripheral blood and liver samples.
Analysis of engraftment and tumor burden
To determine human cells in THP-1-transplanted NOD-SCID IL2Rγ null (NSG) mice, cells from blood, bone marrow, liver and spleen were isolated. Briefly, mononuclear cells were purified by lysis of erythrocytes. To distinguish donor from host cells in transplanted mice, cells were stained with murine CD45 Ly-5 30-F11 PE (eBioscience Cat. 12-0451-83) human CD45 2D1 APC (eBioscience Cat. 17-9459-42) and human CD15 HI98 FITC (eBioscience Cat. 11-0159-42) conjugated antibodies. To distinguish donor from MOLM-13 cells in transplanted mice, cells were stained with murine CD45.1 FITC Monoclonal Antibody (A20) (Affymetrix Cat. 11045382), human CD45 PE (BD Cat. 560975) and human CD15 PB (Thermo Fisher Cat. MHCD1528) conjugated antibodies. Analysis and sorting were performed using a FACSAria II Special Order System (BD Biosciences, San Jose, CA).
In vivo mitochondrial depolarization assay
NOD-SCID IL2Rγ null (NSG) mice (Jackson Laboratory) were subjected to sublethal (200 rad) total body irradiation and 3 hr later injected with 5 x105 MOLM-13 cells by tail vein. 15 days after transplantation, mice were treated with 10 mg/kg BTSA1 or vehicle via IP for two consecutive days. Following treatment, mice were sacrificed and peripheral blood and bone marrow samples harvested. Briefly, erythrocytes from bone marrow samples were lysed using an ACK buffer. Immediately, samples were resuspended in RPMI 1640 media (Life Technologies) supplemented with 10% FBS, 100 U ml–1 penicillin/streptomycin, and 2 mM l-glutamine. Samples were then stained with 100nM TMRE (Sigma Cat. 87917) for 30 min at 37°C. Subsequently, cells were pelleted by centrifugation and washed two times in 1xPBS/0.5% BSA. Cells were then stained with murine CD45.1 FITC Monoclonal Antibody (A20) (Affymetrix Cat. 11045382), human CD45 PE (BD Cat. 560975) and human CD15 PB (Thermo Fisher Cat. MHCD1528) for 30 min on ice. After incubation, cells were pelleted by centrifugation followed by two washes with 1xPBS/0.5% BSA. Cells where then resuspended in 300 μL of 1xPBS/0.5% BSA and analyzed by FACS. Analysis and sorting were performed using a FACSAria II Special Order System (BD Biosciences, San Jose, CA).
Immunohistochemistry analysis
Femur from MOLM-13 xenografts of mice treated with 10 mg/kg BTSA1 or vehicle for 3 weeks were dissected and fixed in 10% buffered formalin (Fisher Scientific) for 24 hr followed by decalcification of bone. Paraffin-embedded sections were then stained with cleaved caspase-3 antibody (Cell Signaling Cat. 9661) or with the ApopTag Peroxidase in situ Apoptosis Detection Kit (Millipore Cat. S7100), which detects apoptotic cells in situ by labeling and detecting DNA strand breaks by the TUNEL method. Stained slides were imaged using the PerkinElmer P250 High Capacity Slide Scanner under brightfield using a 20X objective. Quantification and analysis of densitometry of immunohistochemistry (IHC) was performed using Panoramic Viewer. Using Panoramic Viewer, bone marrow sections from slides were selected followed by analysis using the “Densito Quant Module” This module, measures the density of immunostain on the digital slides by distributing pixels to negative and 3 grades of positive classes by their RGB values. For caspase cleavage stain, the percentage of total positive signals of BTSA1 or vehicle treated samples was obtained using the Densito Quant Module and plotted as bar-graphs. For TUNEL stain, the percentage the of total medium and strong positive signals of BTSA1 or Vehicle treated samples was obtained using the Densito Quant Module and plotted as bar-graphs.
Toxicity studies in vivo
6–8 weeks old NOD-SCID IL2Rγ null (NSG) mice (Jackson Laboratory) were divided into three groups (n = 3 per arm), and treated with vehicle and 15 mg/Kg BTSA1 by IP route every other day. Mice were monitored daily and body weight was monitored every two days. After 30 days of vehicle and drug treatments mice were subjected to euthanasia and necropsy (Animal Pathology Core, Albert Einstein College of Medicine) and tissues (e.g. bone morrow, spleen, liver, kidney, lung heart, brain) were harvested for fixation in 10% buffered formalin (Fisher Scientific). Paraffin-embedded sections (5 μm) were stained with H&E. Peripheral blood from NOD-SCID IL2Rγ null (NSG) mice was obtained by facial vein puncture and collected in EDTA-coated tubes (BD cat. 365973). Blood counts were determined on a Forcyte Veterinary Hematology Analyzer (Oxford Science Inc.).
Analysis of hematopoiesis by FACS
Mice were treated daily via IP injection with 15 mg/kg of BTSA1 or vehicle for 5 days. After treatment, mice were sacrificed and femurs dissected and fixed with formalin for 24 hr for bone marrow biopsy analysis by H&E staining. Alternatively, bone marrow samples were also isolated for FACS analysis. Briefly, isolation of whole bone marrow samples were obtained using a bone crushing technique. Cleaned tibias and femurs from mice were dissected and transferred into a sterile mortar containing 2 ml of 1X PBS supplemented with 2 mM EDTA and 0.5% BSA. Bones were crushed into bone fragments, releasing marrow within using a pestle. Supernatant from mortar were collected into a 50 ml canonical tube using a 40 micron cell strainer and samples centrifuged for 5 min at 300 g. Red blood cells were lysed by incubation with ACK lysis buffer. Samples were then stained with the following antibodies to assess more immature hematopoietic compartments: CD4-PE-Cy5 (Affymetrix Cat. 15-0041-82), CD8a–PE-Cy5 (Affymetrix Cat. 15-0081-82), CD19-PE-Cy5 (Affymetrix Cat. 15-0193-82), Ter119-PE-Cy5 (Affymetrix Cat. 15-5921-82), CD11b-PE-Cy5 (Affymetrix Cat. 15-0112-82), Gr-1-PE-Cy5 (Affymetrix Cat. 15-5931-83), B220-PE-Cy5 (Affymetrix Cat. 15-0452-82), c-Kit(CD117)-APC (Affymetrix Cat. 17-1172-82), Sca1-BV410 (Biolegend Cat. 108127), CD34-FITC (Affymetrix Cat. 11-0341-82) and FcgammaR II/III PE-Cy7 (Affymetrix Cat. 25-0161-82). Analysis and sorting was performed using a FACSAria II Special Order System (BD Biosciences, San Jose, CA).
Quantification And Statistical Analysis
Statistical significance for pair-wise comparison of groups was determined by 2-tailed Student’s T test using GraphPad PRISM software (Graph Pad Inc., CA). Statistics for mouse survival studies using Kaplan-Meier analysis were assessed with the log-rank test using GraphPad PRISM software. p values of less than 0.05 were considered significant. Additional statistical details are provided in figure legends and methods details.
Supplementary Material
SIGNIFICANCE.
Resistance to apoptosis is a hallmark of cancer. The BCL-2 protein family contains anti- and pro-apoptotic members that regulate mitochondrial apoptosis. Cancer cells ensure their survival and resistance to treatments by overexpression of anti-apoptotic BCL-2 proteins to maintain BAX and other pro-apoptotic members suppressed. Therapeutic strategies to selectively activate apoptosis in cancer have focused on inhibiting anti-apoptotic BCL-2 proteins. Here, we present the development of BTSA1, a lead compound with high affinity and specificity for the BAX N-terminal activation site. Our results demonstrate that direct BAX activation by BTSA1 promotes selective apoptosis and efficacious anti-tumor activity without adversely affecting healthy cells. This study provides insights for the therapeutic potential of direct BAX activation in AML and potentially other cancers.
Highlights.
Small molecule BTSA1 interacts potently and specifically with the BAX trigger site
BTSA1 triggers BAX activation leading to mitochondrial membrane permeabilization
BTSA1 induces promptly apoptosis in AML cell lines and patient samples
BTSA1 suppresses leukemia growth in vivo without toxicity to normal cells
Acknowledgments
Studies were supported by an NCI grant 1R01CA178394 to E.G. and awards from the Sidney Kimmel Foundation for Cancer Research, the Gabrielle’s Angels Foundation for Cancer Research, the Alexandrine and Alexander L. Sinsheimer Foundation and the Pershing Square Sohn Cancer Research Alliance to E.G. Partial support was also provided by NCI grant 5R35CA197583, R01CA217092, a William Lawrence and Blanche Hughes Foundation leukemia grant. U.S., A.V, and L.D.W. are supported by Leukemia and Lymphoma Scholar awards. NMR data were collected with support from NIH awards 1S10OD016305, P30 CA013330 a grant from NYSTAR. This work was supported by the Stem Cell Isolation and Xenotransplantation Facility of the Gottesman Institute for Stem Cell Research and Regenerative Medicine (NYSTEM grant #C029154). L.D.W. is a scientific advisory board member and consultant for Aileron Therapeutics
Footnotes
AUTHOR CONTRIBUTIONS
D.E.R. designed and performed biochemical, cellular and in vivo studies, T.P.G performed NMR, biochemical and molecular modeling studies, A.L performed cellular studies, F.K. performed synthetic chemistry and analytical characterization. A.V, G.S.C, A.S, B.D designed and performed experiments with primary cells. U.S, S.R.S and K.M. designed and performed in vivo studies. B.A.B performed gene expression analysis. L.D.W. contributed to the research design. E.G. conceived the research study, designed and performed experiments and wrote the manuscript, which was edited by all authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barreyro L, Will B, Bartholdy B, Zhou L, Todorova TI, Stanley RF, Ben-Neriah S, Montagna C, Parekh S, Pellagatti A, et al. Overexpression of IL-1 receptor accessory protein in stem and progenitor cells and outcome correlation in AML and MDS. Blood. 2012;120:1290–1298. doi: 10.1182/blood-2012-01-404699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmar J, Fesik SW. Small molecule Mcl-1 inhibitors for the treatment of cancer. Pharmacol & Ther. 2015;145:76–84. doi: 10.1016/j.pharmthera.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleicken S, Jeschke G, Stegmueller C, Salvador-Gallego R, Garcia-Saez AJ, Bordignon E. Structural model of active Bax at the membrane. Mol Cell. 2014;56:496–505. doi: 10.1016/j.molcel.2014.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bombrun A, Gerber P, Casi G, Terradillos O, Antonsson B, Halazy S. 3,6-dibromocarbazole piperazine derivatives of 2-propanol as first inhibitors of cytochrome c release via Bax channel modulation. J Med Chem. 2003;46:4365–4368. doi: 10.1021/jm034107j. [DOI] [PubMed] [Google Scholar]
- Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ, et al. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell. 2013;152:519–531. doi: 10.1016/j.cell.2012.12.031. [DOI] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, Tu HC, Kim H, Cheng EH, Tjandra N, et al. BAX activation is initiated at a novel interaction site. Nature. 2008;455:1076–1081. doi: 10.1038/nature07396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner TP, Reyna DE, Priyadarshi A, Chen HC, Li S, Wu Y, Ganesan YT, Malashkevich VN, Almo SS, Cheng EH, et al. An Autoinhibited Dimeric Form of BAX Regulates the BAX Activation Pathway. Mol Cell. 2016;63:485–497. doi: 10.1016/j.molcel.2016.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavathiotis E, Reyna DE, Davis ML, Bird GH, Walensky LD. BH3-triggered structural reorganization drives the activation of proapoptotic BAX. Mol Cell. 2010;40:481–492. doi: 10.1016/j.molcel.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavathiotis E, Reyna DE, Bellairs JA, Leshchiner ES, Walensky LD. Direct and selective small-molecule activation of proapoptotic BAX. Nat Chem Biol. 2012;8:639–645. doi: 10.1038/nchembio.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff DJ, Court Recart A, Sadarangani A, Chun HJ, Barrett CL, Krajewska M, Leu H, Low-Marchelli J, Ma W, Shih AY, et al. A Pan-BCL2 inhibitor renders bone-marrow-resident human leukemia stem cells sensitive to tyrosine kinase inhibition. Cell Stem Cell. 2013;12:316–328. doi: 10.1016/j.stem.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hang HC, Yu C, Pratt MR, Bertozzi CR. Probing Glycosyltransferase Activities with the Staudinger Ligation. J Am Chem Soc. 2004;126:6–7. doi: 10.1021/ja037692m. [DOI] [PubMed] [Google Scholar]
- Hata AN, Engelman JA, Faber AC. The BCL2 Family: Key Mediators of the Apoptotic Response to Targeted Anticancer Therapeutics. Cancer Discov. 2015;5:475–487. doi: 10.1158/2159-8290.CD-15-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff CA, Matsui W, Smith BD, Jones RJ. The paradox of response and survival in cancer therapeutics. Blood. 2006;107:431–434. doi: 10.1182/blood-2005-06-2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer S, Anwari K, Alsop AE, Yuen WS, Huang DC, Carroll J, Smith NA, Smith BJ, Dewson G, Kluck RM. Identification of an activation site in Bak and mitochondrial Bax triggered by antibodies. Nature Comm. 2016;7:11734. doi: 10.1038/ncomms11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR, Majeti R. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4:149ra118. doi: 10.1126/scitranslmed.3004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juin P, Geneste O, Gautier F, Depil S, Campone M. Decoding and unlocking the BCL-2 dependency of cancer cells. Nat Rev Cancer. 2013;13:455–465. doi: 10.1038/nrc3538. [DOI] [PubMed] [Google Scholar]
- Kim H, Tu HC, Ren D, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell. 2009;36:487–499. doi: 10.1016/j.molcel.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopleva M, Contractor R, Tsao T, Samudio I, Ruvalo PP, Kitada S, Deng XM, Zhai DY, Shi YX, Sneed T, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–388. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T, McKeegan E, Salem AH, Zhu M, Ricker JL, et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discovv. 2016;6:1106–1117. doi: 10.1158/2159-8290.CD-16-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, Chanrion M, Kelly GL, Gong JN, Moujalled DM, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538:477–482. doi: 10.1038/nature19830. [DOI] [PubMed] [Google Scholar]
- Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, Ashton JM, Pei S, Grose V, O’Dwyer KM, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12:329–341. doi: 10.1016/j.stem.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letai AG. Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat Rev Cancer. 2008;8:121–132. doi: 10.1038/nrc2297. [DOI] [PubMed] [Google Scholar]
- Leshchiner ES, Braun CR, Bird GH, Walensky LD. Direct activation of full-length proapoptotic BAK. PNAS USA. 2013;110:E986–995. doi: 10.1073/pnas.1214313110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leverson JD, Phillips DC, Mitten MJ, Boghaert ER, Diaz D, Tahir SK, Belmont LD, Nimmer P, Xiao Y, Ma XM, et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci Transl Med. 2015;7:279ra240. doi: 10.1126/scitranslmed.aaa4642. [DOI] [PubMed] [Google Scholar]
- Llambi F, Green DR. Apoptosis and oncogenesis: give and take in the BCL-2 family. Curr Opin Gen & Dev. 2011;21:12–20. doi: 10.1016/j.gde.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llambi F, Moldoveanu T, Tait SW, Bouchier-Hayes L, Temirov J, McCormick LL, Dillon CP, Green DR. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell. 2011;44:517–531. doi: 10.1016/j.molcel.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber B, Andrews DW. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell. 2008;135:1074–1084. doi: 10.1016/j.cell.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Luna-Vargas MP, Chipuk JE. Physiological and Pharmacological Control of BAK, BAX, and Beyond. Trends Cell Biol. 2016;26:906–917. doi: 10.1016/j.tcb.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijler MM, Kaufmann GF, Qi L, Mee JM, Coyle AR, Moss JA, Wirsching P, Matsushita M, Janda KD. Fluorescent Cocaine Probes: A Tool for the Selection and Engineering of Therapeutic Antibodies. J Am Chem Soc. 2005;127:2477–2484. doi: 10.1021/ja043935e. [DOI] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, Cortes J, DeAngelo DJ, Debose L, Mu H, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014;4:362–375. doi: 10.1158/2159-8290.CD-13-0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perciavalle RM, Opferman JT. Delving deeper: MCL-1’s contributions to normal and cancer biology. Trends Cell Biol. 2013;23:22–29. doi: 10.1016/j.tcb.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren D, Tu HC, Kim H, Wang GX, Bean GR, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science. 2010;330:1390–1393. doi: 10.1126/science.1190217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, Kipps TJ, Anderson MA, Brown JR, Gressick L, et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. New Eng J Med. 2016;374:311–322. doi: 10.1056/NEJMoa1513257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JC. Bcl-2-family proteins and hematologic malignancies: history and future prospects. Blood. 2008;111:3322–3330. doi: 10.1182/blood-2007-09-078162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, Hann CL, Garon EB, Ribeiro de Oliveira M, Bonomi PD, Camidge DR, Chu Q, Giaccone G, Khaira D, Ramalingam SS, et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res. 2012;18:3163–3169. doi: 10.1158/1078-0432.CCR-11-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarosiek KA, Fraser C, Muthalagu N, Bhola PD, Chang W, McBrayer SK, Cantlon A, Fisch S, Golomb-Mello G, Ryan JA, et al. Developmental Regulation of Mitochondrial Apoptosis by c-Myc Governs Age- and Tissue-Specific Sensitivity to Cancer Therapeutics. Cancer Cell. 2017;31:142–156. doi: 10.1016/j.ccell.2016.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinke C, Giricz O, Li W, Shastri A, Gordon S, Barreyro L, Bhagat T, Bhattacharyya S, Ramachandra N, Bartenstein M, et al. IL8-CXCR2 pathway inhibition as a therapeutic strategy against MDS and AML stem cells. Blood. 2015;125:3144–3152. doi: 10.1182/blood-2015-01-621631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, Kennedy JA, Schimmer AD, Schuh AC, Yee KW, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506:328–333. doi: 10.1038/nature13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- Uchime O, Dai Z, Biris N, Lee D, Sidhu SS, Li S, Lai JR, Gavathiotis E. Synthetic Antibodies Inhibit Bcl-2-associated X Protein (BAX) through Blockade of the N-terminal Activation Site. J Biol Chem. 2016;291:89–102. doi: 10.1074/jbc.M115.680918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2004;59:687–96. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- Walensky LD, Gavathiotis E. BAX unleashed: the biochemical transformation of an inactive cytosolic monomer into a toxic mitochondrial pore. Trends Biochem Sci. 2011;36:642–652. doi: 10.1016/j.tibs.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B. Role of BAX in the apoptotic response to anticancer agents. Science. 2000;290:989–992. doi: 10.1126/science.290.5493.989. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Subramaniam S, Kale J, Liao C, Huang B, Brahmbhatt H, Condon SG, Lapolla SM, Hays FA, Ding J, et al. BH3-in-groove dimerization initiates and helix 9 dimerization expands Bax pore assembly in membranes. EMBO J. 2016;35:208–236. doi: 10.15252/embj.201591552. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.