


Abstract
Terpene cyclases catalyze the highly stereospecific molding of polyisoprenes into terpenes, which are precursors to most known natural compounds. The isoprenoids are formed via intricate chemical cascades employing rich, yet highly erratic, carbocation chemistry. It is currently not well understood how these biocatalysts achieve chemical control. Here, we illustrate the catalytic control exerted by trichodiene synthase, and in particular, we discover two features that could be general catalytic tools adopted by other terpenoid cyclases. First, to avoid formation of byproducts, the enzyme raises the energy of bisabolyl carbocation, which is a general mechanistic branching point in many sesquiterpene cyclases, resulting in an essentially concerted cyclization cascade. Second, we identify a sulfur–carbocation dative bonding interaction that anchors the bisabolyl cation in a reactive conformation, avoiding tumbling and premature deprotonation. Specifically, Met73 acts as a chameleon, shifting from an initial sulfur–π interaction in the Michaelis complex to a sulfur–carbocation complex during catalysis.
Keywords: terpene cyclases, enzyme catalysis, QM/MM simulations, sulfur–π interaction, sulfur–cation interaction
Graphical abstract
INTRODUCTION
Enzymes are the most potent catalysts known, producing remarkable rate accelerations as much as 20 orders of magnitude over the uncatalyzed reactions in aqueous solution.1–4 However, enzymes are also extremely precise bioreactors, with unparalleled regio- and stereoselectivity.5,6 A fascinating example of chemical control is the biosynthesis of terpenoid compounds by a large family of enzymes called terpenoid synthases or cyclases.7,8 Approximately 60% of all known natural compounds are terpenoids,9 which are all derived from simple acyclic precursors composed of a hydrocarbon chain and a pyrophosphate moiety (PP), such as geranyl diphosphate (GPP; C10), farnesyl diphosphate (FPP; C15), and geranylgeranyl diphosphate (C20). Indeed, a most remarkable feature of these enzymes is that they all employ the same family of acyclic precursors, to produce a huge variety of carbon-skeleton architectures that have marveled organic chemists for over a century.7,10,11 Yet, these chemical transformations follow a common reaction mechanism, involving a sequence of electrophilic cycloadditions and rearrangements via highly reactive carbocation intermediates. These intermediates must be guided in the correct reaction direction, as well as be protected from nucleophilic attacks and premature quenching in the active site, thereby avoiding or minimizing formation of side products. Evidently, each enzyme offers a unique character that prefolds the acyclic substrate in a conformation analogous to its final target.7,12 Previous work has suggested that the PP moiety plays an important role both in ligand binding and in forming a controlled electrostatic environment.13–18 Undoubtedly, there is a delicate balance of kinetic, thermodynamic, and dynamic control exerted along the carbocation cyclization cascade;19,20 however, little is known about the details of the chemical regulation in terpenoid cyclases at atomic resolution.
Trichodiene synthase (TDS) catalyzes the conversion of FPP to the sesquiterpene trichodiene (TD), the parent compound of the trichothecane family of antibiotics and mycotoxins.10,21 TDS has been studied extensively by a variety of methods, including enzymology,10,22 mutagenesis,23–29 and X-ray crystallography,23,27–31 and is one of the best characterized terpene cyclases. Experimental studies, including the use of isotope-labeled substrates,10 have established the detailed chemical steps taking place in the active site of TDS (Scheme 1). The reaction commences with initial ionization and recombination of FPP via nerolidyl PP (NPP), 1, to yield TD, 9. Kinetic studies have shown that the overall rate limiting step in TDS is product release, while the rate limiting chemical step is the cleavage of the carbon–oxygen bond yielding farnesyl carbocation.22 However, the ultimate catalytic challenge for TDS, as it is for all terpenoid synthases, is to control regio- and stereoselectivity while handling erratic carbocation intermediates. Terpene cyclases rarely produce a single product, and indeed, biochemical studies of the promiscuous wild-type (WT) TDS have shown that TD is produced with 84% yield, while numerous side products are formed (Figure 1), such as α-barbatene (3.2%), β-farnesene (0.5%), β-acoradiene (0.5%), isochamigrene (3%), α-bisabolene (1%), β-bisabolene (0.9%), cuprenene (2.4%), and at least six additional unknown compounds (5%).29 Site-directed mutations of TDS have shown a reduction in the formation of TD, with a concomitant increase in one or more specific side-products.23–29
Scheme 1.
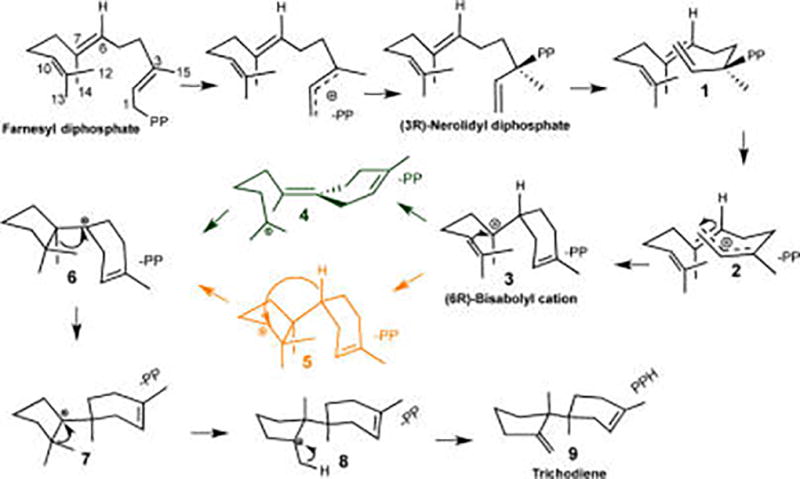
Biosynthesis of Trichodiene Catalyzed by Trichodiene Synthase, Including Possible Pathways from Bisabolyl Cation to Trichodienyl Cation
Figure 1.
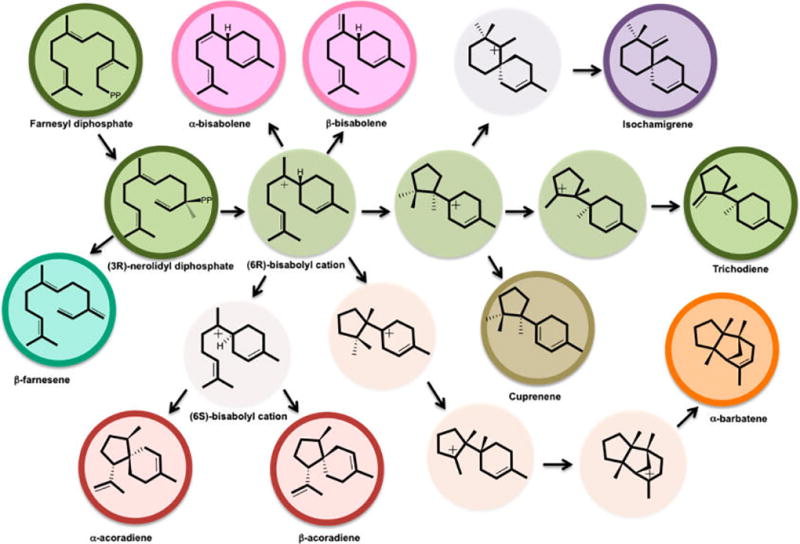
Possible side-products formed along the biosynthetic pathway in wild-type and mutant forms of trichodiene synthase. The main pathway is marked in green.
In spite of the significant amount of research conducted in the area of natural product biosynthesis, the enzymatic toolbox that enables chemical control in the face of tremendous catalytic challenges is not well understood. Herein, we carry out multiscale simulations to tackle the complex reaction mechanism in TDS, while deriving general principles of catalytic control. We present a detailed mechanism that is in agreement with the available experimental data, and address the question of the catalytic role of the enzyme. Specifically, we identify key control elements that allow the enzyme to direct the reaction flux toward TD, 9, while minimizing byproduct formation.
COMPUTATIONAL DETAILS
Gas-Phase Model Electronic Structure Calculations
Previously we have performed extensive quantum mechanics (QM) benchmark calculations to arrive at an optimal method and basis set for modeling carbocation reactions.15 Based on this work, we study the gas-phase reaction employing the M06-2X functional32 with a 6-31+G(d,p) basis set.
Enzyme Modeling and Simulations
The TDS dimeric structure30 is used in the present molecular dynamics (MD) simulations, employing the CHARMM program.33 The enzyme system was embedded in a water sphere described by stochastic boundary conditions. A crucial step in the study of enzymatic terpenoid cyclization reactions is to correctly model the initial substrate configuration and its interactions within the active site.20 Herein, we adopt a stepwise docking procedure, which generates a large number (100 000) of possible configurations of the intermediate, 6, employing molecular mechanics (MM) rigid active site docking, followed by semiempirical QM/MM Monte Carlo simulations to produce 10 000 best poses, and finally semiempirical QM/MM energy minimization to obtain the 100 best candidates. The final configuration to be used in the mechanistic studies is chosen by considering both the lowest energy structures and the requirement that the C3 and C13 atoms of 6 are in reasonable proximity to one of the PP oxygens, as a result of the initial carbocation generation and final deprotonation, respectively. We note that O’Brien et al. have suggested an alternative approach to docking the substrate and intermediates in terpene synthases.17
We adopt a computational protocol similar to that employed in our multiscale modeling study of the monoterpene system bornyl diphosphate synthase (BPPS).15,16,20 Specifically, we employ a combined QM/MM potential to model the trichodiene carbocation cyclization cascade.34,35 Similarly to our previous study,16 the substrate hydrocarbon framework, as well as the metal–pyrophosphate cluster PP-(Mg2+)3, is treated quantum mechanically, while the remaining enzyme–solvent system is represented by the CHARMM22/27 MM force field.36 The QM region is treated by density functional theory (DFT), using the M06-2X functional.15,16,20,32 The three-point charge TIP3P model is used for water.37
The potential of mean force (PMF)38 was defined as a multi-dimensional function of the reaction coordinates depending on the specific reaction step under investigation.39 The total QM(DFT)/MM simulation time was ca. 1 ns.
RESULTS
Since carbocations are highly reactive species that do not persist in aqueous solution, the reaction mechanism in the gas-phase constitutes a natural reference for understanding chemical selectivity in terpene synthases.15,16,20,40–42 Hence, we initially performed extensive model gas-phase reactions of plausible carbocation steps along the reaction pathway from FPP to TD.3,40,43 Figures 2 and S1 present the gas-phase free energy profile for stationary points for key carbocation species and transition states along the TD synthetic pathway (Table S1). The TD carbocation cascade commences with farnesyl cation, 2, via the equatorial conformation of bisabolyl cation, 3eq, and ends with trichodienyl cation, 8. The initial ring closure from farnesyl to bisabolyl cation, 3eq, occurs without a free energy barrier and is exergonic by −18 kcal/mol. As can be seen in Figure 1, numerous side products produced by TDS originate from cation 3, which may be attributed to its relative stability with longer half-life and hence time to undergo side reactions. In fact, bisabolyl cation represents a common mechanistic branching point found in many sesquiterpene synthases,43 where each enzyme channels the product flux in a unique direction. The next major mechanistic step is the second cycloaddition reaction, leading to the bicyclic cuprenyl cation, 6 (Scheme 1), which may occur via either a hydride or proton transfer pathway.41,43 Based on gas-phase calculations, Hong and Tantillo suggested that the proton-transfer pathway is preferred, thereby avoiding an anti-Markovnikov secondary cation 5,41 and this is confirmed in the present study. The proton transfer takes place via a [1,5]-shift from C6 to C10 in cation 3eq, to yield iso-bisabolyl cation 4, with a free energy barrier of 5.8 kcal/mol. Iso-bisabolyl cation undergoes an electrophilic attack at the C7 position to form cuprenyl cation, 6, which is stabilized by 8.7 kcal/mol relative to 3eq.
Figure 2.
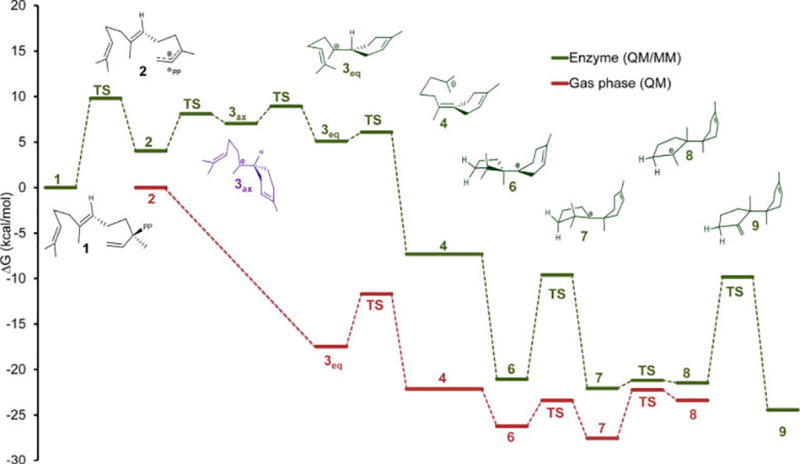
Free energy profiles (kcal/mol) for the biosynthesis of trichodiene by trichodiene synthase compared with carbocation reaction steps leading to trichodienyl in the gas-phase.
Subsequently, 6 undergoes sequential methyl migrations, yielding trichodienyl cation, 8. The final product TD may then be formed upon deprotonation, which was not modeled in the gas-phase.
The active site in TDS is divided into two primary regions:30 The PP binding region and the carbocation binding pocket (Figure 3). The PP group is bound by a multitude of polar interactions involving three magnesium ions, several amino acid residues, and water molecules. The binding site of the hydrocarbon is located below the PP moiety in a hydrophobic pocket (Figure 3), which is sequestered from water by the presence of numerous hydrophobic residues as well as the PP group. Of particular notice is a sulfur–π interaction between Trp298 and Met73. Interestingly, such sulfur–aryl interactions are abundant in proteins44,45 and are stabilizing.46 Based on site-directed mutagenesis, Vedula et al. found that the Tyr295Phe active-site mutation does not affect the reaction rate but resulted in ca. 17% increase in the formation of β-bisabolene, at the expense of TD.29
Figure 3.
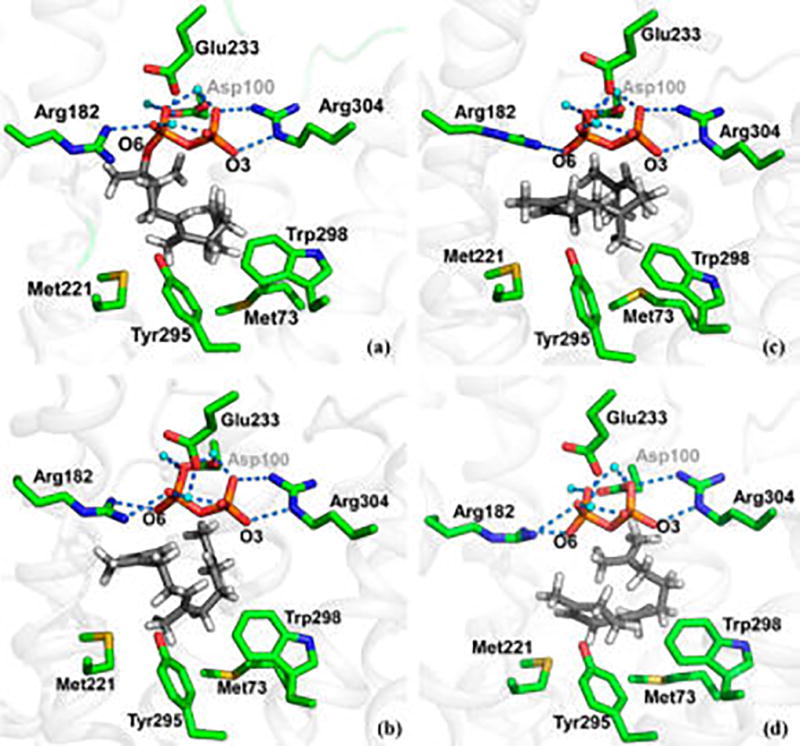
Snapshots from molecular dynamics simulations of the (a) initially folded state of the NPP substrate, (b) farnesyl cation, (c) bisabolyl cation, and (d) isobisabolyl cation in trichodiene synthase.
The current multiscale simulations of TD biosynthesis commenced with enzyme bound (3R)-NPP, 1, which is essential for producing the required cis conformation of farnesyl allyl cation, 2 (Figure 3a).47 Key features of the free energy profile for the enzymatic reaction are provided in Figure 2, with specific details of individual free energy surfaces given as Supporting Information (Figure S2–S7, Table S1). Initial heterolytic cleavage of the C3–O6 bond to form 2 and PP proceeds with a free energy barrier of 9.8 kcal/mol. The resulting ion pair is somewhat higher in free energy by 4.0 kcal/mol relative to NPP (Figure S2). Importantly, 2 is prefolded in a conformation such that the C15 methyl group is buried inside the hydrophobic pocket (Figure 3b) and is not expected to undergo premature quenching to yield β-farnesene (Figure 1).
The cis conformation of the allylic carbocation 2 is perfectly positioned for an electrophilic attack at the C6–C7 double bond to produce (6R)-bisabolyl cation, 3, in the axial conformation, which is readily relaxed to the equatorial form, 3eq (Figures 2 and 3c). The overall free energy barrier for the cyclization step is 4.9 kcal/mol, and the bisabolyl cation, 3eq, is 1.1 kcal/mol less stable than the allyl cation, in spite of the formation of a C–C bond (Figure S3). Critically, we find that the enzyme active site raises the free energy of 3eq, relative to that of 2, by ca. 20 kcal/mol relative to that in the gas phase (Figure 2). We attribute this destabilization to reduced electrostatic interactions in going from the initial allyl cation, 2, which forms an ion-pair with the pyrophosphate anion and Asp100, to the newly formed 3eq, which has a localized cation at C7, which is deeply embedded in the hydrophobic binding pocket. This is the single most remarkable difference between the enzymatic and gas-phase free energy reaction profiles (Figure 2).
However, the C7 cation in 3 is not formed without any form of stabilization; in fact we discovered a novel dative S73 → C7+ bonding interaction from the sulfur atom of Met73 (Figure 4a). To further examine the nature of this interaction, we included Met73 in the QM region in QM(DFT)/MM dynamics simulations. We found that a dative bond with an average distance of ca. 2 Å was formed within a few hundred femtoseconds (Figure 4b). This interaction compensates for the loss of electrostatic interaction with the PP moiety, as well as providing an anchoring point that prevents tumbling of the bisabolyl cation in the hydrophobic cavern. Moreover, the S73 → C7+ interaction greatly reduces the reactivity of the carbocation, allowing it to relax and fine-tune its conformation for the next chemical step. To the best of our knowledge, this is the first example of such an interaction in an enzyme, and this may be utilized in other terpenoid synthases as well. We note that the identification of a sulfur–cation interaction is supported by the D100E crystal structure with (R)-azabisabolene complexed, in which the sulfur atom of Met221 is in near proximity to the presumably protonated aza-nitrogen.28 Such an interaction may prevent formation of the side products α/β-bisabolenes and α-barbatene (Figure 1). The former side-products are formed by premature deprotonation, while the latter is produced from an incorrect conformation around the C6–C7 bond followed by several cyclization and migration steps.29
Figure 4.
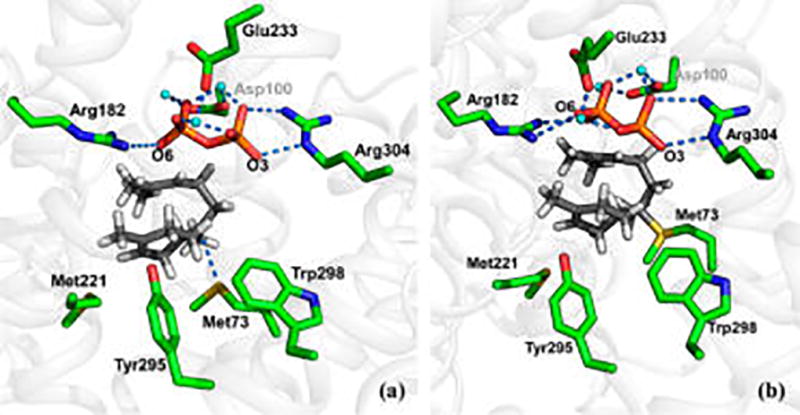
Snapshot from molecular dynamics simulations showing Met73 interacting with the intermediate bisabolyl cation in trichodiene synthase. (a) Correctly folded cation prior to dative bond formation; (b) dative S73 → C7+ bonded state.
The present QM(DFT)/MM simulations reveal that, similarly to the gas-phase reaction, bisabolyl cation, 3, undergoes a [1,5]-H+ transfer, followed by ring-closure to give 6, whereas the anti-Markovnikov route, via a secondary cation and a concomitant hydride transfer, is considerably slower and is not described here. However, this preferred mechanistic pathway is not without obstacles. First, following proton transfer in cation 3eq to yield iso-bisabolyl cation, 4, a C6–C7 double bond is formed. To be consistent with 14C labeling experiments, the resulting electrophilic addition must occur on the si face of the C7 sp2 carbocation center, where C12 is positioned in an anti-conformation relative to C14 (Figure 1).48 Second, following proton transfer, there is a free rotation around the C10–C11 bond, which could result in scrambling of the two methyl groups, again contradicting 14C labeling experiments.48 Moreover, premature deprotonation of iso-bisabolyl cation results in iso-bisabolene (Figure 1). Finally, once iso-bisabolyl cation has been formed, α- or β-chamigrene could in principle be formed via C6–C11 ring formation. Thus, the enzyme needs to prevent the formation of a stable intermediate 4, which would have sufficient time to generate unwanted side-products.
In our QM(DFT)/MM simulations, the equatorial conformation of 3eq is folded in a conformation conducive to proton transfer from C6 to C10 (Figures 3 and 4). The conformation of the folded bisabolyl cation is similar to that of (R)-azabisabolene found in the D100E and R304K mutant crystal structures.23,28 In these crystal structures, the cyclohexene ring is in the C7-equatorial conformation and the C6–C10 distance is 3.3 Å, in a conformation that is stereochemically compatible with the required proton transfer. Figure 2 shows that the formation of iso-bisabolyl cation, 4, has a free energy barrier of only 1.0 kcal/mol, which is followed by a barrierless ring closure, to yield cuprenyl cation, 6. These two steps may best be characterized as an asynchronous concerted process (Figure S4). This is in marked contrast to the gas-phase process. First, the barrier for the [1,5]-proton transfer in TDS is noticeably lower than that in the absence of the enzyme, hence kinetically avoiding accumulation of 3eq. Second, the exergonic nature of the 3eq–6 transformation provides a thermodynamic driving force (ca. 26 kcal/mol), again avoiding accumulation of 3eq. Finally, in the gas-phase several higher energy conformers of 4 are involved in structural interconversions before 6 is finally produced (Figure S1), whereas in the enzyme such conformational transitions are not needed. Hence, the enzyme active site has already prefolded the substrate in a conformation suitable for direct conversion of 3eq to 6, bypassing 4 as an intermediate altogether.
Because of the concerted nature of the process from 3eq to 6, we can rule out the possibilities of free rotation about the C10–C11 bond and premature deprotonation at C12 or C13. Thus, iso-bisabolene is not observed as a side product in TDS. The concerted [1,5]-proton transfer and cycloaddition to the C6–C7 double bond to form 6 could in principle produce an alternative tertiary carbocation, the spiro-bicyclic chamigrenyl cation (Figure 1). However, steric congestion due to the equatorial conformation adopted in the enzyme operates against the C6–C11 bond formation. Thus, the formation of chamigrenyl cation is not favored relative to 6 in TDS. In contrast, in the gas-phase model calculations, chamigrenyl cation formation was observed in the axial conformation but not in the equatorial conformer, which is adopted in the enzyme active site. Hence, the enzyme selects the equatorial conformer to avoid the chamigrenyl side product. However, isochamigrene is found in wild-type TDS with 3% yield as a result of carbocation rearrangement of the carbon skeleton of 6 to yield the tertiary isochamigrenyl cation at the C7 position (Figure 1).28 This process competes with methyl group migration, also yielding a tertiary cation on the C7 carbon. The latter is preferred due to less ring strain energy compared with the spiro bicyclic structure, emphasizing thermodynamic control.
The final steps in the TDS-catalyzed carbocation reaction cascade involve a pair of methyl group migrations (Figures S5–S6). These steps incorporate a gradual inversion of configuration at the C6 and C7 centers, via sp2 cation hybridizations. These reactions occur in a stepwise fashion with relatively small free energy barriers, and the reaction is completed by a final deprotonation by an active site base, here modeled as the PP anion, to produce trichodiene, 9 (Figure S7).
DISCUSSION
An intriguing question in enzymology is the origin of the extraordinary rate enhancement observed in enzymes and the accompanying catalytic power. In this regard, terpene synthases seem to be an exception to the rule that enzymes work mainly by accelerating the reaction rate. The main catalytic effect in terpene synthases, in its traditional sense of rate enhancement, occurs during the first ionization step.20 However, terpene synthases seemingly excel mainly at performing highly complex chemistry, and they achieve this by controlling the reaction pathways and stereochemistry of extremely reactive carbocation intermediates.7,15,16,20 For instance, biosynthesis of cholesterol, which is performed by oxidosqualene synthase, provides a vivid illustration of this concept. In that reaction, five rings and nine stereocenters are formed in essentially a single reaction step via precise enzymatic control.19 Results obtained herein seem to confirm the proposition that chemical control is no less important than rate acceleration. In particular, gas-phase QM calculations of key intermediate carbocation steps in the TD biosynthesis cascade reveal that the free energy barriers for these processes are generally low. Indeed, the highest computed free energy barrier along the TD biosynthetic pathway in the gas-phase is about 6 kcal/mol, and that in TDS is just 11 kcal/mol, lower than that corresponding to product release.22 Thus, the role of TDS is not to lower the free energy barrier for these carbocation steps, but rather one of chemical control. The question we would like to address is how such control is obtained.
In TDS, such control is partly realized by carrying out a highly asynchronous, yet nearly concerted, cascade of cyclizations to form 6 from 2 (Figure 2). First, TDS assures initial substrate folding by virtue of the active site contour, which ensures rapid formation of bisabolyl cation, 3, followed by an equally facile concerted proton transfer and cycloaddition to produce cuprenyl cation, 6. TDS facilitates such a catalytic pathway by raising the free energy of the intermediates 2 and 3, reducing the lifetime of iso-bisabolyl cation, 4, as an intermediate, hence eliminating the kinetic bottleneck prior to conversion to 6. In the absence of an enzyme, the carbocation intermediates 3 and 4 are critical branching points prone to side-product formation (Figure 1). In TDS, the lifetime of these intermediates is significantly reduced, leaving little time for side reactions to occur.
Another important finding in the present study is the precise positioning of Met73, and possibly also Met221, which can stabilize selected conformations of the reactive bisabolyl cation. Prior to formation of bisabolyl cation, 3eq, Met73 interacts with Trp298 via a weak sulfur–aryl contact,44,45 which is shifted to form a sulfur–cation dative bond as 3eq is formed (Figure 4). Such a novel sulfur–carbocation interaction plays several important roles. First, it delocalizes the cationic charge at the C7 position, reducing the likelihood of formation of the side products α/β-bisabolenes and α-barbatene. This is consistent with the observation that the Tyr295Phe mutation results in increased β-bisabolene formation, at the expense of TD. In the present simulations, the Tyr295 hydroxyl group is in close proximity to the cyclohexenyl cation. The mutation likely increases the tumbling of the bisabolyl cation, thereby exposing the C14 methyl group to a basic site (e.g., PP or Asp100). Second, following the subsequent proton transfer, the Met73 sulfur atom may stabilize the C6–C7 π-bond in iso-bisabolyl cation. The polarizable sulfur atom of Met73 behaves like a chameleon, adapting to the changing electronic requirements during catalysis. We stress that the ability of Met residues to stabilize cations, such as carbocations or metal ions, in organic systems,49,50 as well as proteins, is well-known.50–52 Furthermore, sequence alignment of sesquiterpene synthases presumed to involve a bisabolyl cation intermediate reveals that the active sites of these enzymes are decorated with Met residues (Figure S8). In TDS, the Met sulfur–carbocation interaction serves as an anchor, preventing internal rotation around the C6–C7 bond and global tumbling of the intermediate bisabolyl cation, which could lead to incorrect cyclization products, such as α-barbatene. Intermediate tumbling is likely a general problem for terpene synthases, as the carbocation intermediates become increasingly more compact as the cyclization reactions proceed and there are no hydrogen bonds to anchor intermediates in correct catalytic orientations.20,27,30 This is highlighted by biochemical studies and crystal structures of aza-bisabolyl cation analogues showing considerable binding variability in various forms of the enzyme.28,31 Hence, active site amino acids, such as Met, Cys, and also Trp,53 could stabilize intermediate carbocations.
CONCLUSIONS
According to the current simulations, TDS employs a highly evolved catalytic toolbox to achieve specificity in the face of extremely challenging chemistry. We identify two features that could be general catalytic strategies adopted by other terpenoid cyclases. First, to avoid byproducts, the enzyme raises the energy of a key intermediate that is a mechanistic branching point, resulting in an essentially concerted cyclization cascade, which greatly reduces the chances for side-reactions to occur. Second, we identify a novel sulfur–carbocation dative bond that anchors the bisabolyl cation in a reactive conformation, avoiding substrate tumbling in the active site and premature deprotonation.
Supplementary Material
Acknowledgments
This work has been supported by the Israel Science Foundation (Grant No. 2146/15) and the National Institutes of Health (Grant GM46736).
Footnotes
ASSOCIATED CONTENT
- Complete methods section, gas phase free energy profile, individual PMFs for different steps in the NPP to TD transformation in TDS, sequence alignment of different sesquiterpenes, gas-phase structures, energies, and coordinates, intrinsic reaction coordinates, and gas-phase calculations of dimethyl sulfide with bisabolyl cation (PDF)
The authors declare no competing financial interest.
References
- 1.Warshel A. Proc. Natl. Acad. Sci. U. S. A. 1978;75:5250–5254. doi: 10.1073/pnas.75.11.5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolfenden R, Snider MJ. Acc. Chem. Res. 2001;34:938–945. doi: 10.1021/ar000058i. [DOI] [PubMed] [Google Scholar]
- 3.Gao J, Ma S, Major DT, Nam K, Pu J, Truhlar DG. Chem. Rev. 2006;106:3188–3209. doi: 10.1021/cr050293k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Warshel A, Sharma PK, Kato M, Xiang Y, Liu H, Olsson MHM. Chem. Rev. 2006;106:3210–3235. doi: 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- 5.Clardy J, Walsh C. Nature. 2004;432:829–837. doi: 10.1038/nature03194. [DOI] [PubMed] [Google Scholar]
- 6.Weng J-K, Philippe RN, Noel JP. Science. 2012;336:1667–1670. doi: 10.1126/science.1217411. [DOI] [PubMed] [Google Scholar]
- 7.Christianson DW. Chem. Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 8.Salmon M, Laurendon C, Vardakou M, Cheema J, Defernez M, Green S, Faraldos JA, O’Maille PE. Nat. Commun. 2015;6:6143. doi: 10.1038/ncomms7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Firn R. Nature’s Chemicals - the Natural Products that shaped our world. Oxford University Press; Oxford, UK: 2009. [Google Scholar]
- 10.Cane DE. Chem. Rev. 1990;90:1089–1103. [Google Scholar]
- 11.Davis EM, Croteau R. Top. Curr. Chem. 2000;209:53–95. [Google Scholar]
- 12.Christianson DW. Science. 2007;316:60–61. doi: 10.1126/science.1141630. [DOI] [PubMed] [Google Scholar]
- 13.Zhou K, Peters RJ. Chem. Commun. 2011;47:4074–4080. doi: 10.1039/c0cc02960b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frick S, Nagel R, Schmidt A, Bodemann RR, Rahfeld P, Pauls G, Brandt W, Gershenzon J, Boland W, Burse A. Proc. Natl. Acad. Sci. U. S. A. 2013;110:4194–4199. doi: 10.1073/pnas.1221489110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weitman M, Major DT. J. Am. Chem. Soc. 2010;132:6349–6360. doi: 10.1021/ja910134x. [DOI] [PubMed] [Google Scholar]
- 16.Major DT, Weitman M. J. Am. Chem. Soc. 2012;134:19454–19462. doi: 10.1021/ja308295p. [DOI] [PubMed] [Google Scholar]
- 17.O’Brien TE, Bertolani SJ, Tantillo DJ, Siegel JB. Chem. Sci. 2016;7:4009–4015. doi: 10.1039/c6sc00635c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hong YJ, Tantillo DJ. Org. Biomol. Chem. 2010;8:4589–4600. doi: 10.1039/c0ob00167h. [DOI] [PubMed] [Google Scholar]
- 19.Rajamani R, Gao J. J. Am. Chem. Soc. 2003;125:12768–12781. doi: 10.1021/ja0371799. [DOI] [PubMed] [Google Scholar]
- 20.Major DT, Freud Y, Weitman M. Curr. Opin. Chem. Biol. 2014;21:25–33. doi: 10.1016/j.cbpa.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 21.Cane DE, Swanson S, Murthy PPN. J. Am. Chem. Soc. 1981;103:2136–2138. [Google Scholar]
- 22.Cane DE, Chiu HT, Liang PH, Anderson KS. Biochemistry. 1997;36:8332–8339. doi: 10.1021/bi963018o. [DOI] [PubMed] [Google Scholar]
- 23.Vedula LS, Cane DE, Christianson DW. Biochemistry. 2005;44:12719–12727. doi: 10.1021/bi0510476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cane DE, Xue Q. J. Am. Chem. Soc. 1996;118:1563–1564. [Google Scholar]
- 25.Cane DE, Xue Q, Fitzsimons BC. Biochemistry. 1996;35:12369–12376. doi: 10.1021/bi961344y. [DOI] [PubMed] [Google Scholar]
- 26.Cane DE, Xue Q, Van Epp JE, Tsantrizos YS. J. Am. Chem. Soc. 1996;118:8499–8500. [Google Scholar]
- 27.Rynkiewicz MJ, Cane DE, Christianson DW. Biochemistry. 2002;41:1732–1741. doi: 10.1021/bi011960g. [DOI] [PubMed] [Google Scholar]
- 28.Vedula LS, Rynkiewicz MJ, Pyun H-J, Coates RM, Cane DE, Christianson DW. Biochemistry. 2005;44:6153–6163. doi: 10.1021/bi050059o. [DOI] [PubMed] [Google Scholar]
- 29.Vedula LS, Jiang J, Zakharian T, Cane DE, Christianson DW. Arch. Biochem. Biophys. 2008;469:184–94. doi: 10.1016/j.abb.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rynkiewicz MJ, Cane DE, Christianson DW. Proc. Natl. Acad. Sci. U. S. A. 2001;98:13543–13548. doi: 10.1073/pnas.231313098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vedula LS, Zhao Y, Coates RM, Koyama T, Cane DE, Christianson DW. Arch. Biochem. Biophys. 2007;466:260–266. doi: 10.1016/j.abb.2007.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao Y, Truhlar DG. Theor. Chem. Acc. 2008;120:215–241. [Google Scholar]
- 33.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 34.Warshel A, Levitt M. J. Mol. Biol. 1976;103:227–249. doi: 10.1016/0022-2836(76)90311-9. [DOI] [PubMed] [Google Scholar]
- 35.Gao J. Methods and Applications of Combined Quantum Mechanical and Molecular Mechanical Potentials. Vol. 7 VCH; New York: 1995. [Google Scholar]
- 36.MacKerell AD, Jr, Bashford D, Bellott RL, Dunbrack RL, Jr, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, III, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 37.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 38.Northrup SH, Pear MR, Lee CY, McCammon JA, Karplus M. Proc. Natl. Acad. Sci. U. S. A. 1982;79:4035–4039. doi: 10.1073/pnas.79.13.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doron D, Kohen A, Major DT. J. Chem. Theory Comput. 2012;8:2484–2496. doi: 10.1021/ct300235k. [DOI] [PubMed] [Google Scholar]
- 40.Tantillo DJ. Nat. Prod. Rep. 2011;28:1035–1053. doi: 10.1039/c1np00006c. [DOI] [PubMed] [Google Scholar]
- 41.Hong YJ, Tantillo DJ. Org. Lett. 2006;8:4601–4604. doi: 10.1021/ol061884f. [DOI] [PubMed] [Google Scholar]
- 42.Allemann RK, Young NJ, Ma S, Truhlar DG, Gao J. J. Am. Chem. Soc. 2007;129:13008–13013. doi: 10.1021/ja0722067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hong YJ, Tantillo DJ. J. Am. Chem. Soc. 2014;136:2450–2463. doi: 10.1021/ja4106489. [DOI] [PubMed] [Google Scholar]
- 44.Salonen LM, Ellermann M, Diederich F. Angew. Chem. Int. Ed. 2011;50:4808–4842. doi: 10.1002/anie.201007560. [DOI] [PubMed] [Google Scholar]
- 45.Valley CC, Cembran A, Perlmutter JD, Lewis AK, Labello NP, Gao J, Sachs JN. J. Biol. Chem. 2012;287:34979–34991. doi: 10.1074/jbc.M112.374504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tatko CD, Waters ML. Protein Sci. 2004;13:2515–2522. doi: 10.1110/ps.04820104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cane DE, Ha HJ. J. Am. Chem. Soc. 1988;110:6865–6870. [Google Scholar]
- 48.Jones ERH, Lowe G. J. Chem. Soc. 1960;0:3959–3962. [Google Scholar]
- 49.Hojo M, Ichi T, Shibato K. J. Org. Chem. 1985;50:1478–1482. [Google Scholar]
- 50.Hong YJ, Giner JL, Tantillo DJ. J. Org. Chem. 2013;78:935–941. doi: 10.1021/jo3024208. [DOI] [PubMed] [Google Scholar]
- 51.Xue Y, Davis AV, Balakrishnan G, Stasser JP, Staehlin BM, Focia P, Spiro TG, Penner-Hahn JE, O’Halloran TV. Nat. Chem. Biol. 2008;4:107–109. doi: 10.1038/nchembio.2007.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ranaghan KE, Hung JE, Bartlett GJ, Mooibroek TJ, Harvey JN, Woolfson DN, van der Donk WA, Mulholland AJ. Chem. Sci. 2014;5:2191–2199. [Google Scholar]
- 53.Gebler JC, Woodside AB, Poulter CD. J. Am. Chem. Soc. 1992;114:7354–7360. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.