

Abstract
Among the complex pathophysiological events following spinal cord injury (SCI), one of the most important molecular level consequences is a dramatic reduction in neuronal cyclic adenosine monophosphate (cAMP) levels. Many studies shown that rolipram (Rm), a phosphodiesterase IV inhibitor, can protect against secondary cell death, reduce inflammatory cytokine levels and immune cell infiltration, and increase white matter sparing and functional improvement. Previously, we developed a polymeric micelle nanoparticle, poly(lactide-co-glycolide)-graft-polyethylenimine (PgP), for combinatorial delivery of therapeutic nucleic acids and drugs for SCI repair. In this study, we evaluated PgP as an Rm delivery carrier for SCI repair. Rolipram's water solubility was increased ∼6.8 times in the presence of PgP, indicating drug solubilization in the micelle hydrophobic core. Using hypoxia as an in vitro SCI model, Rm-loaded PgP (Rm-PgP) restored cAMP levels and increased neuronal cell survival of cerebellar granular neurons. The potential efficacy of Rm-PgP was evaluated in a rat compression SCI model. After intraspinal injection, 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl indotricarbocyanine Iodide–loaded PgP micelles were retained at the injection site for up to 5 days. Finally, we show that a single injection of Rm-PgP nanoparticles restored cAMP in the SCI lesion site and reduced apoptosis and the inflammatory response. These results suggest that PgP may offer an efficient and translational approach to delivering Rm as a neuroprotectant following SCI.
Keywords: : cAMP, compression spinal cord injury, polymeric micelle nanoparticle, rolipram, secondary injury
Introduction
Spinal cord injury (SCI) disrupts axonal pathways, leading to permanent motor, sensory, and autonomic dysfunction, as well as chronic pain, respiratory impairment, and loss of bowel or bladder control.1,2 Several complex pathophysiological mechanisms limit spontaneous recovery following SCI. First, damage from the initial trauma progressively expands through a process of secondary injury characterized by free radical production, excitotoxicity, ischemia, and inflammation that lead to further axonal degeneration, demyelination, and apoptosis.1,3–5 Secondly, although some damaged axons form new growth cones, their capacity for regeneration and target re-innervation is severely limited by growth inhibitory molecules present in degenerating myelin and the glial scar and injury-induced and age-related changes in neuronal biochemistry. The only clinical treatments available for SCI are spinal decompression and the steroidal anti-inflammatory drug methylprednisolone, which has largely been discontinued due to complications and extensive controversy over its efficacy.6,7 Currently, a wide range of treatments targeting neuroprotection or axonal regeneration are in pre-clinical studies,1,3,8 with the most promising results commonly obtained using combinatorial therapies simultaneously targeting two or more mechanisms of SCI pathophysiology.
Among the molecular consequences of SCI, one of the most important is a reduction in cyclic adenosine monophosphate (cAMP), a cytoplasmic second messenger produced by adenylyl cyclase in response to numerous extracellular stimuli. cAMP has been implicated in both neuroprotection and axonal regeneration, making it a particularly attractive therapeutic target. Cai and colleagues9 first identified that a developmental transition in the neuronal response to myelin from growth promotion to growth inhibition coincided with a decline in cAMP levels and that exogenous elevation of cAMP could overcome myelin-mediated growth inhibition. Elevation of cAMP also was found to be an important mechanism underlying the growth response elicited by the peripheral conditioning lesion model.10 Although not completely elucidated, cAMP signaling appears to act through independent activation of protein kinase A (PKA) and EPAC2. PKA signaling through cAMP response element-binding protein leads to transcriptional activation of multiple targets including arginase I that support axonal growth,11 while EPAC2 activates the growth-promoting b-Raf pathway.12 Interestingly, both PKA and EPAC2 can inhibit Rho activation, a critical convergence point in the signaling of multiple extracellular growth inhibitors.13
In addition to exogenous stimuli that activate adenyl cyclase, cAMP levels are also controlled by a family of degradative enzymes, phosphodiesterases (PDEs).14,15 PDE4 is the most predominantly expressed cAMP-specific PDE in neural tissue16 as well as in immune cells.17 Therapeutic modulation of cAMP levels has been primarily pursued through use of the drug rolipram (Rm), a PDE4 inhibitor that can cross the blood–brain barrier after systemic administration. Rm was initially used primarily in combination with cell and tissue transplants and shown to contribute to improved functional recovery in animal injury models. Recently, studies using Rm alone have reported reduced oligodendrocyte death,18 reduced inflammatory cytokine levels and immune cell infiltration,19 increased white matter sparing,20 and functional improvement.21 Reduction in caspase 3 activity has been proposed as a mechanism for the anti-apoptotic activity of Rm.22 Despite its efficacy in pre-clinical models, there are several challenges with systemic administration of Rm, including the need for organic solvents to increase its solubility and the potential for side effects when targeting such a ubiquitous signaling molecule as cAMP.13, 23–25 Therefore, the development of carriers for efficient delivery of Rm to the injured spinal cord is required.
The application of nanotechnology in medicine (called “nanomedicine”) has considerable potential to improve healthcare. Recently, polymeric micellar nanoparticles26–32 have proven to be useful in drug and gene delivery. Amphiphilic block copolymers spontaneously form micellar nanoparticles in aqueous solutions composed of a hydrophobic core and hydrophilic shell, capable of increasing the solubility of hydrophobic drugs. Additionally, incorporation of a cationic polymer for the hydrophilic shell allows the formation of polyelectrolyte complexes with anionic therapeutic nucleic acids and can enhance particle stability, increase association with the cell membrane, and enhance endosomal escape.33–36 Therefore, polymeric micelle nanoparticles offer an attractive platform for combinatorial drug and gene delivery.
Our goal is to develop multi-functional polymeric micelle nanocarriers for combinatorial therapy of multiple therapeutic agents to promote axonal regeneration and plasticity. To achieve this goal, we developed a cationic, amphiphilic block copolymer, poly(lactide-co-glycolide)-graft-polyethylenimine (PgP) that offers three important capabilities: 1) electrostatic complexation of therapeutic nucleic acids with the cationic hydrophilic shell; 2) loading of hydrophobic drug in the hydrophobic core; and 3) neuron-targeting through surface conjugation of cell-type specific targeting moiety such as ligands or antibodies (Fig. 1). In our previous work, we synthesized PgP and demonstrated its ability to efficiently transfect pGFP and small interfering RNA (siRNA) in various neural cell lines and primary chick forebrain neurons in 10% serum condition in vitro, as well as in the normal rat spinal cord.37 We also have reported that PgP can deliver siRNA targeting RhoA to the injured spinal cord and maintain RhoA knockdown for up to 4 weeks post-injection, reduce astrogliosis and cavitation, and increase axonal regeneration.38 Here, we demonstrate that PgP can efficiently load Rm in its hydrophobic core and Rm-loaded PgP (Rm-PgP) restores cAMP levels, and reduces apoptosis and inflammation in the injured spinal cord after local injection in a rat compression spinal cord injury (SCI) model.
FIG. 1.

Proposed target-specific poly(lactide-co-glycolide)-graft-polyethylenimine (PgP) micelle nanotherapeutics. Color image is available online at www.liebertpub.com/neu
Methods
Materials
Poly(lactide-co-glycolide) (PLGA; 25kDa, 50:50) was purchased from Durect Corporation (Cupertino, CA). Anhydrous dimethylformamide, N-hydroxysuccinimide, N,N′-dicyclohexylcarbodiimide (DCC), branched polyethylenimine 25kDa (PEI), deuterium oxide (D2O), cytosine arabinoside (AraC), laminin, and poly-L-lysine hydrobromide were obtained from Sigma-Aldrich (St. Louis, MO). Dialysis tubing (MWCO; 50 kDa) was obtained from Spectrum labs (Rancho Dominguez, CA). Rm was purchased from LC Laboratories (Woburn, MA) and 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl indotricarbocyanine Iodide (DiR) from PromoCell GmbH (Germany). Lebowitz (L-15) and Basal Medium Eagle (BME) media were obtained from ATCC and Gibco (Life Technologies, Carlsbad, CA), respectively. DyLight® 488-conjugated goat anti-mouse IgG (H+L) secondary antibodies were obtained from Fisher Scientific (Waltham, MA). Mouse anti-beta III tubulin (2G10) and goat anti-mouse Cy3 conjugated IgG antibodies were purchased from Abcam (Cambridge, MA). Mouse monoclonal anti-macrophage/monocyte clone ED1 antibodies and the ApopTag® Fluorescein In Situ Apoptosis Detection Kit were obtained from EMD Millipore (Darmstadt, Germany). Mouse monoclonal anti-Bax and rabbit polyclonal anti-Bcl-2 primary antibodies were obtained from Santa Cruz Biotechnology (Dallas, TX). The cAMP Parameter Assay ELISA kit was purchased from R&D Systems (Minneapolis, MN).
Synthesis and characterization of PgP
PgP was synthesized as previously described.37 Briefly, the carboxylic end group of PLGA (25 kDa, 60 μmole) was activated by (72 μmole) and DCC (72 μmole), and the resulting dicyclohexyl urea byproduct was removed by filtration. The activated PLGA solution was added dropwise to the branched polyethylenimine (bPEI; 25kDa, 50 μmole) solution over 30 min and allowed to react for 24 h at room temperature with stirring. PgP was purified by dialysis against deionized water using a membrane filter (MWCO; 50,000), centrifuged at 5000 rpm for 10 min to remove unreacted PLGA precipitate, and lyophilized. The structure of PgP was determined by 1H- NMR (300 MHz, Bruker) using D2O as a solvent.
Rm loading in PgP and stability of Rm-loaded PgP nanoparticles
Rm was loaded into PgP micelle nanoparticles by the solvent evaporation method. Briefly, various concentrations of Rm were prepared in ethanol and 100 μL of each Rm solution was added into 1 mL of PgP solution (1 mg PgP/mL water) solution. The mixture was sealed and incubated for 4 h at room temperature, then the ethanol was allowed to evaporate overnight. To remove unloaded Rm, the solutions were filtered using a 0.2 μm syringe filter. The amount of loaded Rm in Rm-PgP solution was measured by high-performance liquid chromatography (HPLC; Waters, Milford, MA) using Rm standard curve prepared in dimethylsulfoxide (DMSO). The HPLC system was composed of Waters 1525 binary HPLC pump, Waters 2998 photodiode array detector, and a Shodex C18 column (Showa Denko America, New York, NY). The mobile phase was water:acetonitrile (60:40). The injection volume was 20 μL and run time was 6 min. The loading efficiency was calculated as:
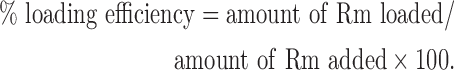 |
We also evaluated the stability of Rm-PgP solution. Rm was loaded into PgP micelle solution as described above and incubated at 37°C for 6 weeks. Each week, solutions containing Rm-PgP were sampled and filtered with 0.2 μm syringe filter to remove released precipitated Rm and then Rm amount was measured by HPLC as described above. The % Rm retained was calculated as:
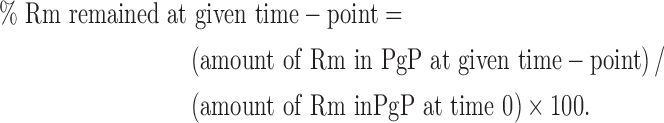 |
Particle size of Rm-PgP nanoparticles
To evaluate the effect of Rm loading, the particle size of PgP micellar nanoparticles and Rm-PgP (1 mg Rm loaded in 1 mg/mL PgP) micellar nanoparticles was measured by dynamic light scattering using a 90 Plus Particle Size Analyzer (Brookhaven Instruments Corporation, Holtzville, NY).
Rat cerebellar granule neurons isolation and culture
Primary cerebellar granule neurons (CGNs) were isolated from 3-day post-natal (P3) Sprague Dawley rat pups. The pups were first euthanized by decapitation and the cerebella excised and placed into L-15 dissection medium. The meninges of each cerebella were removed, the tissue minced, and digested in 0.25% Trypsin in Hank's Balanced Salt Solution for 10 min. Full culture medium (BME with 33 mM D-glucose, potassium chloride 20 mM, glutamine 2 mM, penicillin/streptomycin 50 μg/mL, and 10% fetal bovine serum) with 0.1% DNase was added and the digested tissue centrifuged at 1100 g for 7 min. After removal of the supernatant and addition of fresh culture medium, the tissue was triturated with a fire polished glass Pasteur pipet. The dissociated cells were plated at 5 × 105 cells/mL in 12 well-plates that were previously coated with 0.1% poly-L-lysine overnight at 37°C followed by 10 μg/mL laminin solution for 2 h at 37°C. To prevent glial cell contamination and maintain purity of the CGN culture, half the media volume was exchanged after 2 days and 1 μM AraC (Sigma Aldrich) added. Subsequently, half of the media volume was replaced every 2 days using full media without AraC. After 5 days incubation, the CGN cultures were used to evaluate the effect of Rm-PgP.
Effect of Rm-PgP on cAMP and neurite length of CGNs cultured in hypoxia condition in vitro
To evaluate the effect of Rm-PgP on cAMP level and neuronal survival in vitro, P3 rat CGNs were cultured in hypoxia condition as an in vitro spinal injury model. CGNs were cultured under normoxia condition (normal atmosphere with 5% added CO2) for 5 days, then transferred to a hypoxia chamber (StemCell Technologies) with an atmosphere of 95% N2 and 5% CO2. After 24 h incubation, experimental wells were treated with Rm-PgP (10 μg Rm/well). Free Rm dissolved in DMSO (Rm-DMSO, 10 μg Rm/well); PgP without Rm (10 μg PgP/well) was used for comparison, and untreated CGNs were used as a negative control. The cells were incubated an additional 24 h in hypoxia condition and then lysed for measurement of cAMP level or fixed for neurite length evaluation. CGNs maintained through the culture period under normoxia condition were used as a positive control.
cAMP measurement
To evaluate the effect of Rm-PgP treatment on the cAMP level of CGN cells cultured in hypoxia condition, a Mouse/Rat cAMP Parameter Assay Kit (R&D Systems) was used to evaluate the cAMP concentration of collected cell lysates according to manufacturer's instructions. Culture medium was replaced with phosphate-buffered saline (PBS) and CGNs were removed by scraping on ice, collected, and centrifuged. The PBS supernatant was removed and the cells re-suspended in 0.1N HCl/cell lysis buffer 5 at approximately 1 × 107 cells/100 uL. Following lysis, the samples were centrifuged at 600 g for 10 min to remove cell debris. The supernatant was collected and neutralized using 1N NaOH prior to 2-fold dilution with Calibrator Diluent RD5-55. Streptavidin-coated plates were incubated with biotinylated mouse monoclonal antibodies to cAMP, washed, and incubated with cAMP conjugate (cAMP conjugated to horse radish peroxidase) and sampled for 2 h at room temperature. After washing thoroughly, substrate solution was added and incubated for 30 min. The reaction was halted using an acidic stop solution and then the absorbance was measured at 450 nm and 570 nm. Optical density values at 570 nm were subtracted from the values at 450 nm to correct for background. The cAMP levels from three separate wells were averaged for each biological replicate (n = 3) and data presented as mean ± standard error of the mean (SEM).
Neurite length measurement
CGNs were fixed with ice cold methanol for 5 min and then rinsed with staining media (PBS with 5% bovine growth serum) and permeabilized with 0.1% Triton X solution. The cells were incubated with monoclonal anti-beta-III tubulin (1:250) for 2 h at room temperature, washed, and incubated for 1 h with DyLight® 488-conjugated secondary antibody (1:250). The wells were then digitally imaged using an Axiovert 200 inverted epifluorescent microscope (Zeiss). After imaging, the neurite lengths of CGN cells for each group (Rm-PgP, Rm-DMSO, PgP, untreated in hypoxia, and normoxia) were measured using ImageJ software for three biological replicates (n = 3/experiment) and data presented as mean ± SEM.
Generation of rat compression SCI model
All surgical procedures and post-operative care were conducted according to National Institutes of Health (NIH) guidelines for the care and use of laboratory animal (NIH publication No. 86-23, revised 1996) and under the supervision of the Clemson University Animal Research Committee (approved animal protocol no. AUP2014-012). The rat clip comprssion SCI model was generated as previously described by Gwak and colleagues.38 Sprague Dawley rats (male; 200 g) were anesthetized under isoflurane gas, their backs shaved, and the surgical sites cleaned with chlorohexidine, betadine solution, and sterile water. To produce the injury, a 4-cm long longitudinal incision was made on the dorsal side over the mid-thoracic region revealing the spinal column. The T9 spinous process was identified and removed using orthopedic bone cutters, then the ligamentum flavum was removed to expose the intervertebral space. A vascular clip was placed on the dorsal T9 spinal cord and compressed for 10 min. Following injury, the paraspinal muscles were closed with 4-0 vicryl suture and the skin was closed with surgical clip. After surgery, animals were warmed by heating blanket for recovery. Animals received cefazolin (40 mg/kg; Hikma Pharmaceuticals) and buprenorphine (0.01 mg/kg; Hospira Inc.) for 2 weeks after surgery and bladders were manually expressed three times daily.
Retention of locally injected DiR-loaded PgP in the SCI lesion site
We first evaluated whether the drug-loaded PgP nanoparticles would remain in the spinal cord injury lesion after local injection or diffuse away from the injury site. The hydrophobic fluorescent dye DiR was loaded in the PgP micelle nanoparticle solution for visualization. DiR was dissolved in acetone and then added into a PgP (1 mg/mL; water) solution (final concentration of 25 μg DiR/mL). The acetone was evaporated overnight and then the solution was filtered with a 0.2 μm syringe filter to remove precipitated DiR. After compression SCI, 10 μL of DiR-PgP solution was injected into the injury site using a 26 G Hamilton syringe (Hamilton®, Reno, NV). At 2, 4, 6, 24, 72, and 120 h post-injection, the animals were anesthetized under isoflurane gas and imaged by a live animal fluorescence imaging system (IVIS Luminar XR; Caliper Life Sciences). At 2, 6, 24, 72, and 120 h post-imaging, animals were euthanized using carbon dioxide and the spinal cord was excised (1.5 cm-long piece from the center of the injury) for ex vivo imaging using an IVIS Luminar XR.
Effect of Rm-PgP on cAMP level in rat compression SCI model
After spinal cord compression injury, 10 μL Rm-PgP (10 μg Rm) was injected immediately after injury into the injured dorsal T9 spinal cord using a 26-gauge Hamilton syringe. Untreated SCI and sham animal groups were used as controls. At 1, 2, 3, and 7 days post-injury, animals were sacrificed with CO2 overdose and spinal cords (0.5 cm-long piece from the center of the injury) were harvested, frozen in liquid nitrogen, and stored at −80°C. For cAMP analysis, the tissue samples were weighed individually, then homogenized in 0.1N HCl/lysis buffer 5 solution at a 1/5 (w/v) ratio. The samples were centrifuged at 10,000 g for 10 min at 4°C and the supernatant was removed, neutralized with 0.1N NaOH, and then diluted 2-fold with Diluent RD5-55. Measurement of cAMP concentration was performed in the manner previously described for in vitro samples.
Effect of Rm-PgP on secondary injury in rat compression SCI model
To evaluate the effect of Rm-PgP treatment on apoptosis in SCI lesion site, terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining was performed using an ApopTag® Plus Fluorescein In Situ Apoptosis Detection Kit. At 3 days after injury and Rm-PgP injection, the rats were anesthetized by isoflurane gas and sacrificed by cardiac perfusion with 4% paraformaldehyde in PBS (pH 7.4). The spinal cords were excised (0.5 cm on either side of the injury site) and fixed with 4% paraformaldehyde solution. The fixed tissue was embedded in O.C.T. compound, sectioned longitudinally at 10 μm thickness, and mounted onto positively charged glass slides. Sections approximately 30 μm and 60 μm lateral from the lesion epicenter (total n = 18 sections/group; three rats/group, six sections/rat) were stained using the ApopTag® Kit and nuclei were counterstained by 4',6-diamidino-2-phenylindole (DAPI). Images of the entire stained sample were captured at 50 × using an AZ100 fluorescent microscope (Nikon Instruments) and the images were stitched together using Photoshop software. Additional images were obtained at 400 × from the lesion epicenter and 2 mm and 4 mm rostral and caudal from the epicenter. The number of TUNEL and DAPI+ cells were counted for each image area using Photoshop software, and the percentage of TUNEL+ cells per DAPI+ cells were calculated. Sham animals and untreated SCI animals were used for comparison.
To further characterize the effect of Rm-PgP treatment on secondary injury related and apoptosis, immunohistochemical staining was performed for Bax, a pro-apoptotic marker related to permeability of the mitochondrial membrane and release of cytochrome-c, and Bcl-2, an anti-apoptotic marker associated with homeostasis of the mitochondrial membrane. Sections approximately 30 μm and 60 μm lateral from the lesion epicenter (total n = nine sections/group; three rats/group, three sections/rat) were incubated with mouse monoclonal anti-Bax (1:200; sc-23959) and rabbit polyclonal anti-Bcl-2 (1:200; sc-492), washed, and incubated with anti-mouse Cy3-conjugated (1:200; ab97035; Abcam) and AlexaFluor 488 (1:2000, A11008, Life Technologies). Sections were digitally imaged and the numbers of Bax+ and Bcl-2+ cells were counted and a ratio of Bcl-2+/Bax+ cells was calculated. Sham animals and untreated SCI animals were used for comparison.
To evaluate effect of Rm-PgP treatment on the inflammatory response in the SCI lesion, immunohistochemical staining for ED1 was performed on spinal cord sections from 3-day post-injury samples. Sections approximately 30 μm and 60 μm lateral from the lesion epicenter (total n = 18 sections/group; three rats/group, six sections/rat) were incubated with mouse monoclonal anti-ED1 antibodies (1:200; MAB1435) for active microglia/macrophages, washed, and incubated with Cy3-conjugated secondary (1:200; ab97035; Abcam). Sections were digitally imaged as described above and the number of ED1 and DAPI+ cells were counted using Photoshop software. Cells were determined as ED1+ when the DAPI stained nucleus was completely surrounded by ED1 specific staining.
Statistical analysis
Quantitative data are presented as the mean ± SEM. The statistical significance was analyzed between groups by a Student's t-test. A p value of less than 0.05 was considered significant.
Results
Synthesis and characterization of PgP
The amphiphilic copolymer PgP was synthesized by conjugating the carboxyl groups of PLGA to the amine groups of bPEI through amide bonds. Following synthesis and purification, the structure of PgP was confirmed by 1H-NMR using D2O as a solvent. The peaks of bPEI backbone -CH2-CH2 were observed at δ2.4∼3.5 (m), the peaks of methyl (-CH3), methylene (–CH2), and methine (-CH) of PLGA was observed at δ1.4∼1.6 (d, 3H), δ3.9 (s, 2H), and δ4.3 (q, 1H), respectively.
Rm loading in PgP micelle nanoparticles
Like many drugs, Rm is relatively hydrophobic and has limited aqueous solubility (solubility in water: 0.2 mg/mL). We first evaluated the ability of Rm to be loaded into the hydrophobic core of PgP micelles, increasing its water solubility. As presented in Figure 2, the amount of Rm loaded in the PgP increased with the amount of Rm added, ultimately reaching a peak and then decreasing due to precipitation by supersaturation. The amount of Rm loaded in PgP solution was approximately 1.36 ± 0.042 mg as determined from three individual loading replicates. This represents about 6.8 times higher than Rm solubility in water (0.2 mg/mL) indicated by the dashed red line (Fig. 2A). The Rm loading efficiency was calculated as approximately 90%. Figure 2B shows the stability of Rm-PgP solution incubated at 37°C and the Rm-PgP was stable up to 5 weeks and then started to decrease reaching 63.3% at 6 weeks.
FIG. 2.

(A) Rolipram (Rm) loading into poly(lactide-co-glycolide)-graft-polyethylenimine (PgP) micelle nanoparticles by the solvent evaporation method. 100 μL containing various concentrations of Rm (0–2 mg/mL in ethanol) was added into PgP (1 mg/mL in water) solution, incubated to allow ethanol evaporation, and filtered to remove free Rm, and the concentration of loaded Rm measured by high-performance liquid chromatography (HPLC). The maximum water solubility of free Rm is indicated by dashed red line (0.2 mg/mL). (B) The long-term stability of Rm-PgP incubated at 37°C for 6 weeks. Each week, solutions containing Rm-PgP were sampled and filtered with 0.2 μm syringe filter to remove released precipitated Rm and then Rm amount was measured by HPLC. The % Rm remained was calculated as: % Rm remained at given time-point = (amount of Rm in PgP at given time-point)/ (amount of Rm in PgP at time 0) × 100.
Particle size of Rm-PgP micelle nanoparticles
To evaluate whether Rm loading into PgP affects micelle particle size, we measured the particle size of PgP and Rm-PgP nanoparticles. Rm loading did not change the PgP micellar nanoparticle size and the mean particle sizes of PgP and Rm-PgP were 123.5 ± 3.5 nm with polydispersity 0.215 ± 0.021 and 121.8 ± 1.9 nm with polydispersity 0.279 ± 0.315, respectively.
Rm-PgP increases cAMP levels of CGNs cultured in hypoxia condition
The cAMP level of CGNs cultured under hypoxia was significantly lower than that of CGNs cultured under normoxia conditions (Fig. 3). In both Rm-PgP and Rm-DMSO treated groups, the cAMP levels of CGNs were restored to levels not significantly different from cells cultured in the normoxia condition. We used PgP without Rm as a mock control to evaluate the effect of PgP alone. Interestingly, PgP alone did not indicate any cytotoxicity, but increased cAMP level slightly, compared with the untreated hypoxia group.
FIG. 3.

Rolipram-loaded poly(lactide-co-glycolide)-graft-polyethylenimine (Rm-PgP) increases cyclic adenosine monophosphate (cAMP) levels of cerebellar granular neurons (CGNs) cultured in hypoxia condition. Rat CGNs were cultured in hypoxia for 24 h and then treated with Rm-PgP (10 μg Rm/well), Rm dissolved in dimethylsulfoxide (Rm-DMSO, 10 μg Rm/well) and PgP only (10 μg PgP/well), and cultured an additional 24 h in hypoxia. Untreated CGNs cultured in hypoxia and CGNs cultured in normoxia was used as negative and positive control, respectively. cAMP levels were measured by enzyme-linked immunosorbent assay. Data presented as mean ± standard error of the mean (n = 9). *p < 0.05, compared with normoxia.
Rm-PgP increases neurite length of CGNs cultured in hypoxia condition
Figure 4A shows representative images of CGNs cultured under normoxia (i) and hypoxia conditions either untreated (ii) or treated with PgP alone (iii), Rm-PgP (iv), or Rm-DMSO (v). Quantitative image analysis showed that neurite length was significantly lower in hypoxia condition relative to normoxia condition, while neurite length of CGNs treated with Rm-PgP and Rm-DMSO under hypoxia was not significantly different from the normoxia control (Fig. 4B).
FIG. 4.

Rolipram-loaded poly(lactide-co-glycolide)-graft-polyethylenimine (Rm-PgP) increases neurite length of cerebellar granular neurons (CGNs) cultured in hypoxia condition. Rat CGNs were cultured in hypoxia for 24 h and then treated with Rm-PgP (10 μg Rm/well), Rm dissolved in dimethylsulfoxide (Rm-DMSO, 10 μg Rm/well) and PgP only (10 μg PgP/well) and cultured an additional 24 h in hypoxia. Untreated CGNs cultured in hypoxia and CGNs cultured in normoxia was used as negative and positive control, respectively. CGNs were immunostained for beta-III tubulin and imaged. (A) Representative images for β-3 tubulin stained CGNs cultured in (i) normoxia, and (ii) untreated, (iii) PgP-only treated, (iv) Rm-PgP treated, and (v) Rm-DMSO treated CGNs after culture in hypoxia condition. Scale bars represent 100 μm. (B) Neurite length of CGNs cultured in various conditions. Data presented as mean ± standard error of the mean (n = 9). *p < 0.05, compared with normoxia. Color image is available online at www.liebertpub.com/neu
DiR-PgP is retained at SCI lesion site after local injection
To evaluate the duration of retention of locally injected PgP micellar nanoparticles in the spinal cord lesion site, the hydrophobic dye DiR was loaded in PgP nanoparticle solution using the solvent evaporation method. Figure 5A shows that DiR was soluble in the PgP micelle core, while DiR was filtered out in water after filtering with 0.2 μm membrane filter due to its low solubility. Figure 5B shows that injected DiR-PgP nanoparticles were retained at the injection site up to 5 days post-injection. Figure 5C shows that the injected DiR-PgP nanoparticles retained in excised spinal cord at each time-point.
FIG. 5.

Rolipram-loaded poly(lactide-co-glycolide)-graft-polyethylenimine (Rm-PgP) is retained at spinal cord injury lesion site after local injection. (A) 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl indotricarbocyanine Iodide (DiR) loading in PgP solution and water. (B) Visualization of DiR-PgP nanoparticles by live animal imaging system at 2, 4, 6, 24, 72, and 120 h post-injection. Left: untreated control animal. Right: DiR-PgP injected animal. (C) Images of spinal cord isolated at 2, 4, 6, 24, 72, and 120 h post-injection. Color image is available online at www.liebertpub.com/neu
Rm-PgP restores cAMP levels after compression SCI
After SCI, cAMP levels measured at all time-points were significantly reduced relative to the sham control (Fig. 6). Injection of Rm-PgP (10 μg Rm/rat) restored cAMP to levels not significantly different from the sham animal group at 1 and 2 days post-injection. cAMP levels in animals receiving Rm-PgP decreased at Day 3 and were significantly lower than the sham control, but still significantly higher than untreated animals. At Day 7, cAMP levels in untreated and Rm-PgP-treated animals were not significantly different.
FIG. 6.

Rolipram-loaded poly(lactide-co-glycolide)-graft-polyethylenimine (Rm-PgP) restores cyclic adenosine monophosphate (cAMP) levels after compression spinal cord injury. Rm-PgP (10 μg Rm) was injected immediately after clip compression injury and the spinal cord was harvested at 1, 2, 3, and 7 days post-injection. cAMP levels were measured by enzyme-linked immunosorbent assay assay. Sham and untreated spinal cord injury animal groups were used for comparison. Data presented as mean ± standard error of the mean (n = 6). *p < 0.05, compared with the sham group, indicated by the dashed red line. Color image is available online at www.liebertpub.com/neu
Rm-PgP reduces apoptosis and inflammatory response after compression SCI
Apoptosis was measured by the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay at the 3-day post-injury/Rm-PgP injection. Figure 7A shows representative images of TUNEL+ cells in the entire stained spinal cord (0.5 cm on either side of the injury site) captured at 50 × . We observed much less TUNEL + (apoptotic) cells in the spinal cord of animals treated with Rm-PgP than in the spinal cord of animals in the untreated group. Figure 7B shows representative images of TUNEL + cells in the spinal cord centered on the lesion epicenter and spanning 2 and 4 mm towards the rostral and caudal ends with 400 × magnification. The % TUNEL+ cells was significantly decreased in Rm-PgP treated animals relative to untreated SCI animals at all positions (Fig. 7C).
FIG. 7.

Rolipram-loaded poly(lactide-co-glycolide)-graft-polyethylenimine (Rm-PgP) reduces apoptosis after compression spinal cord injury (SCI). Rm-PgP (10 μg Rm) was injected immediately after clip compression injury. At 3 days post-injection, spinal cords were explanted, embedded, sectioned, and stained for terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) + cells (green) and nuclei (blue). Sham and untreated SCI animal groups were used for comparison. (A) Representative images of TUNEL stained longitudinally sectioned spinal cord (0.5 cm-long piece from the center of the injury). Scale bar indicates 500 μm. Top: sham. Middle: untreated SCI. Bottom: Rm-PgP treated SCI rats. (B) Representative images of TUNEL+ cells in the spinal cord at the lesion epicenter and 2 and 4 mm rostral and caudal. Scale bar indicates 50 μm, Top: sham. Middle: untreated SCI. Bottom: Rm-PgP treated SCI animals. (C) The % TUNEL+ cells quantified from total 18 different sections of spinal cords from each group (six sections/rat, three rats/group). *p < 0.05, compared with untreated SCI. Color image is available online at www.liebertpub.com/neu
Figure 8A and 8B show representative images of Bax+ and Bcl-2+ cells, respectively, in the the spinal cord centered on the lesion epicenter and spanning 2 and 4 mm towards the rostral and caudal ends with 400 × magnification. Figure 8C shows the ratio of Bcl-2+/Bax+ cells calculated by number of Bcl-2+ and Bax+ cells quantified. We observed that the ratios of Bcl-2+/Bax+ in treated animal group were significantly higher than those in the untreated SCI animal group at all positions.
FIG. 8.

Rolipram-loaded poly(lactide-co-glycolide)-graft-polyethylenimine (Rm-PgP) increases the ratio of Bcl-2+/Bax+ cells after compression spinal cord injury (SCI). Rm-PgP (10 μg Rm) was injected immediately after clip compression injury. At 3 days post-injection, spinal cords were explanted, embedded, sectioned, and stained for Bax, pro-apototic protein and Bcl-2, anti-apoptotic protein. Sham and untreated SCI animal groups were used for comparison. Representative images of Bcl-2+ cells (A) and Bax+ cells (B) in the spinal cord at the lesion epicenter, 2, and 4 mm rostral and caudal. Scale bar indicates 50 μm, Top: sham. Middle: untreated SCI. Bottom: Rm-PgP treated SCI animals. (C) The ratio of Bcl-2+/Bax+ cells were calculated from a total of nine different sections of spinal cords from each group (three sections/rat, three rats/group). Sections were digitally imaged and the numbers of Bax+ and Bcl-2 + cells were counted. *p < 0.05, compared with untreated SCI. Color image is available online at www.liebertpub.com/neu
We also evaluated the effect of Rm-PgP administration on the inflammatory response. Figure 9A shows representative images of ED1+ cells in the entire stained spinal cord (0.5 cm on either side of the injury site) captured at 50 × . We observed fewer ED1+ activated microglia and/or infiltrated macrophages in spinal cords from the Rm-PgP treated group than from the untreated animal group. Figure 9B shows representative images of ED1+ cells in the spinal cord centered on the lesion epicenter and spanning 4 mm towards the rostral and caudal ends captured at 400 × . The % ED1+ cells was significantly decreased in Rm-PgP treated animals at 4 mm rostral and caudal of the epicenter relative to untreated SCI animals (Fig. 9C).
FIG. 9.

Rolipram-loaded poly(lactide-co-glycolide)-graft-polyethylenimine (Rm-PgP) reduces inflammatory cell accumulation after compression spinal cord injury (SCI). Rm-PgP (10 μg Rm) was injected immediately after clip compression injury. At 3 days post-injection, spinal cords were explanted, embedded, sectioned, and stained for ED1+ microglia/macrophages (red) and nuclei (blue). Sham and untreated SCI animal groups were used for comparison. (A) Representative images of immunostaining for ED1 in longitudinally sectioned spinal cord (0.5 cm-long piece from the center of the injury). Scale bar indicates 500 μm. Top: sham. Middle: untreated SCI. Bottom: Rm-PgP–treated SCI rats. (B) Representative images of ED1+ cells in the spinal cord at the lesion epicenter, 2 and 4 mm rostral and caudal. Scale bar indicates 50 μm. Top: sham. Middle: untreated SCI. Bottom: Rm-PgP–treated SCI animals. (C) The % ED1+ cells was quantified from a total of 18 different sections of spinal cords from each group (six sections/rat, three rats/group). *p < 0.05, compared with untreated SCI. Color image is available online at www.liebertpub.com/neu
Discussion
After traumatic injury, further neuronal cell loss occurs due to ischemia, inflammation, calcium influx, free radical formation, and glutamate-mediated excitotoxicity. Further, the regenerative capacity of surviving neurons is limited by extracellular growth inhibitors and deficiencies in biochemical signaling and gene transcription. To overcome this complex pathophysiology and develop an effective treatment for SCI, we developed multi-functional polymeric nanocarrier composed of a cationic, amphiphilic graft copolymer, poly (lactide-co-glycolide)-graft-polyethylenimine (PgP) for combinatorial drug/gene delivery. Previously, we reported the synthesis and characterization of PgP as a carrier for siRNA and pDNA in vitro in the presence of serum and pDNA to the native spinal cord.37 We also have reported that PgP is capable of efficiently delivering siRNA to knockdown RhoA gene expression in the rat SCI lesion site for up to 4 weeks and increase axonal regeneration.38 Here, we report the ability of PgP to serve as a rolipram (Rm) delivery carrier in cerebellar granular neurons (CGNs) cultured in hypoxia condition in vitro, as well as in a rat compression spinal cord injury model.
We first examined the loading efficiency of Rm in the hydrophobic core of PgP nanoparticles by solvent evaporation method and the amount of Rm loaded in PgP (1 mg/mL) was about 7 times higher than Rm's solubility in water (0.2 mg/mL). The success of loading Rm within PgP is consistent with the demonstrated performance of amphiphilic micelles for loading of drugs with poor aqueous solubility.39–41 We believe that this increased water solubility of Rm by PgP may increase the opportunity to translate Rm as a neuroprotectant for SCI without unwanted side effects by organic solvents, such as DMSO or ethanol.42 In our long-term stability study, we observed that Rm-PgP was stable up to 5 weeks at 37°C. We believe that the increased water solubility without the need for organic solvent and stability of Rm-PgP are important features for commercial and clinical application.
We next examined the effect of Rm-PgP on cAMP levels and neurite length in rat primary cerebellar granular neurons (CGNs) cultured in hypoxia condition as an in vitro SCI model. We found that Rm-PgP restored cAMP levels in CGNs cultured in hypoxia to that of CGNs cultured in normoxia condition and was not significantly different from levels attained using free Rm dissolved in DMSO. We also found that Rm-PgP could restore neurite outgrowth in hypoxia condition to levels not significantly different than cells cultured under normoxia. We used the PgP-only group as a mock control to evaluate the effect of polymer alone. Interestingly, PgP alone did not show any cytotoxicity but showed slight increase of cAMP, as well as neurite length, compared with the untreated hypoxia group. These results help to confirm the cytocompatibility of our PgP as a delivery carrier.
Before evaluating the efficacy of Rm-PgP effect in in vivo SCI animal model, we questioned whether the injected nanoparticle will remain in the injection site or diffuse away. Using fluorescent dye-loaded PgP (DiR-PgP) and live animal imaging, we show that DiR-PgP are retained at the injected lesion site for up to 5 days after a single injection. After verifying retention of injected nanoparticle, Rm-PgP (10 μg Rm/rat, 10 μL Rm-PgP) was injected in injured spinal cord and a single injection of Rm-PgP was able to restore cAMP to levels not significantly different from sham control for up to 2 days before beginning to decline at 3 days post-injection. These data show for the first time that locally injected Rm-loaded nanoparticles in the injured spinal cord can provide prolonged therapeutic effect with sustained release of the drug. In many other studies, Rm was administrated systemically by intravenous or intraperitoneal injection, or locally by subcutaneously implanted osmotic pumps for at least 1 or 2 weeks after dissolving in DMSO or ethanol. Therefore, we believe that PgP nanocarrier may increase therapeutic effect of Rm, as well as patient compliance, because only one single injection of Rm-PgP (10 μg Rm) can significantly increase cAMP level up to 5 days, compared with the untreated SCI animal group.
We further evaluated the effect of restored cAMP level in the injured spinal cord by Rm-PgP on apoptosis and inflammatory response. We found that Rm-PgP was able to reduce apoptosis relative to the untreated SCI control group at all positions and the anti-apopototic activity of Rm-PgP was further confirmed by the increased ratios of Bcl-2+/Bax+ in the treated group relative to the untreated group at all positions. We also observed reduced numbers of ED1+ cells indicative of lower immune cell infiltration/activation. These results indicate that Rm-PgP treatment reduces important aspects of secondary injury by significantly reducing cell death, as well as inflammation, within the injury lesion.
Further studies will include more detailed evaluation of inflammatory signaling effects, in addition to demonstrating long-term functional recovery of treated animals through use of the Basso, Beattie, and Bresnahan scoring.43 These studies may clarify whether PgP can be used in the development of an effective therapeutic for SCI through multi-modal modulation of the injury micro-environment. The ultimate strength of this polymeric micelle nanotechnology will be its ability to deliver a loaded drug cargo alongside conjugated therapeutic nucleic acids, and therefore future application towards treatment of SCI will be approached through incorporation of Rm and small interfering RNAs (siRNAs) against inhibitory molecules such as RhoA38 for the combinatorial therapy.
Conclusion
In this study, we demonstrate that PgP is a promising carrier for the hydrophobic drug, rolipram (Rm) to mitigate secondary injury following SCI. We show that PgP micelle nanoparticle can increase water solubility of rolipram up to ∼6.8 times by dissolving rolipram in the hydrophobic core of PgP micelle. We show that Rm loaded PgP (Rm-PgP) can restore cAMP levels and increase neurite outgrowth of cerebellar granular neurons (CGNs) cultured in hypoxia condition in vitro. The potential efficacy of Rm-PgP was evaluated in a rat compression SCI model. We found that hydrophobic dye (DiR) loaded PgP (DiR-PgP) nanoparticles were retained at the injection site up to 5 days. We also show that a single injection of Rm-PgP in rat spinal cord lesion site restores cAMP levels and reduces apoptosis and the inflammatory response in rat SCI compression injury. These results suggest that PgP may offer an efficient and translational approach to delivering Rm as a neuroprotectant following SCI.
Acknowledgments
We thank Dr. Naren Vyavahare, Director of SC BioCRAFT, for his support for the project, Dr. Guzeliya Korneva from COBRE Bioengineering and Bioimaging Core for assistance in performing HPLC, and Dr. Terri Bruce from COBRE Bioengineering and Bioimaging Core and CLIF center for her technical support and access to the AZ100 microscope. We thank Dr. John Parrish and Godley-Snell animal facility staff, especially Tina Parker and Travis Pruitt, for their assistance with the animal study. We gratefully acknowledge Dr. Bruce Gao in Bioengineering Department for sharing neural tissue for isolation of primary cerebellar granule neuron from rat pups euthanized in his lab. We thank Dr. Ken Webb in the Bioengineering Department, Clemson University, for assistance in editing the manuscript. Research reported in this publication was supported by NIGMS of the National Institutes of Health under award number 5P20GM103444-07 and South Carolina Spinal Cord Injury Research Foundation under award number SCIRF # 2014 I-02.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Mothe A. and Tator C. (2013). Review of transplantation of neural stem/progenitor cells for spinal cord injury. Int. J. Dev. Neurosci. 31, 701–713 [DOI] [PubMed] [Google Scholar]
- 2.Colello R., Chow W., Bigbee J., Lin C., Dalton D., Brown D., Jha B., Mathern B., Lee K., and Simpson D. (2016). The incorporation of growth factor and chondroitinase ABC into an electrospun scaffold to promote regrowth following spinal cord injury. J. Tissue Eng. Regen. Med. 10, 656–668 [DOI] [PubMed] [Google Scholar]
- 3.Kabu S., Gao Y., Kwon B., and Labhasetwar V. (2015). Drug delivery, cell-based therapies, and tissue engineering approaches for spinal cord injury. J. Control. Release 219, 141–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Onishi K., Hollis E., and Zou Y. (2014) Axon guidance and injury-lessons from Wnts and Wnt signaling. Curr. Opin. Neurobiol. 27, 232–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giger R., Hollis E., 2nd, and Tuszynski M. (2010) Guidance molecules in axon regeneration, Cold Spring Harb. Perspect. Biol. 2, a001867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bydon M., Lin J., Macki M., Gokaslan Z., and Bydon A. (2014) The current role of steroids in acute spinal cord injury. World Neurosurg. 82, 848–854 [DOI] [PubMed] [Google Scholar]
- 7.Borgens R., Shi R., and Bohnert D. (2001) Behavioral recovery from spinal cord injury following delayed application of polyethylene glycol. J. Exp. Biol. 205, 1–12 [DOI] [PubMed] [Google Scholar]
- 8.Hurlbert R., Hadley M., Walters B., Aarabi B., Dhall S., Gelb D., Rozzelle C., Ryken T., and Theodore N. (2013). Pharmacological therapy for acute spinal cord injury. Neurosurgery 72, 93–105 [DOI] [PubMed] [Google Scholar]
- 9.Cai D., Qiu J., Cao Z., McAtee M., Bregman B., and Filbin M. (2001). Neuronal cyclic amp controls the developmental loss in ability of axons to regenerate. J. Neurosci. 21, 4731–4739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qiu J., Cai D., Dai H., McAtee M., Hoffman P., Bregman B., and Filbin M. (2002). Spinal axon regeneration induced by elevation of cyclic amp. Neuron 34, 895–903 [DOI] [PubMed] [Google Scholar]
- 11.Cai D., Deng K., Mellado W., Lee J., Ratan R., and Filbin M. (2002). Arginase I and polyamines act downstream from cyclic amp in overcoming inhibition of axonal growth mag and myelini vitro. Neuron 35, 711–719 [DOI] [PubMed] [Google Scholar]
- 12.O'Donovan K., Ma K., Guo H., Wang C., Sun F., Han S., Kim H., Wong J., Charron J., Zou H., Son Y., He Z., and Zhong J. (2014). B-raf kinase drives developmental axon growth and promotes axon regeneration in the injured matured CNS. J. Exp. Med. 211, 801–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Batty N., Fenrich K., and Fouad K. (2016). The role of camp and its downstream targets in neurte growth in the adult nervous system. Neurosci. Lett. 652, 56–63 [DOI] [PubMed] [Google Scholar]
- 14.Beavo J. (1995). Cyclic nucleotide phosphodiesterases: functional implications of multiple isoforms. Physiol. Rev. 76, 725–748 [DOI] [PubMed] [Google Scholar]
- 15.Souness J. and Rao S. (1997). Proposal for pharmacologically distinct conformers of pde4 cyclic amp phosphodiesterases. Cell Signal 9, 227–236 [DOI] [PubMed] [Google Scholar]
- 16.Iona S., Cuomo M., Bushnik T., Naro F., Sette C., Hess M., Shelton E., and Conti M. (1998). Characterization of the rolipram-sensitive, cyclic amp-specific phosphodiesterases: identification and differential expression of immunologically distinct forms in the rat brain. Mol. Pharmacol. 53, 23–32 [DOI] [PubMed] [Google Scholar]
- 17.Verghese M., McConnell R., Lenhard J., Hamacher L., and Jin C. (1996). Regulation of distinct cyclic amp-specific phosphodiesterase (phosphodiesterase type 4) isozymes in human monocytic cells. Mol. Pharmacol. 47, 1164–1171 [PubMed] [Google Scholar]
- 18.Whitaker C., Beaumont E., Wells M., Magnuson D., Hetman M., and Onifer S. (2008). Rolipram attenuates acute oligodendrocyte death in the adult rat ventrolateral funiculus following contusive cervical spinal cord injury. Neurosci. Lett. 438, 200–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schaal S., Garg M., Ghosh M., Lovera L., Lopez M., Patel M., Louro J., Patel S., Tuesta L., Chan W., and Pearse D. (2012). The therapeutic profile of rolipram, pde target and mechanism of action as a neuroprotectant following spinal cord injury. PLOS One 7, e43634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Costa L., Pereira J., Filipe V., Magalhȁes L., Cuoto P., Gonzales-Orden J., Raimondo S., Geuna S., Mauricio A., and Nikulina E. (2013). Rolpram promotes functional recovery after contusive thoracic spinal cord injury in rats. Behav. Brain Res. 243, 66–73 [DOI] [PubMed] [Google Scholar]
- 21.Nikulina E., Tidwell J., Dal H., Bregman B., and Filbin M. (2004). The phosphodiesterase inhibitor rolipram delivered after a spinal cord injury lesion promotes axonal regeneration and functional recovery. PNAS 101, 8786–8790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen R., Williams A., Liao Z., Yao C., Tortella F., and Dave J. (2007). Broad spectrum neuroprotection profile of phosphodiesterase inhibitors as related to modulation of cell-cycle elements and caspase-3 activation. Neurosci. Lett. 418, 165–169 [DOI] [PubMed] [Google Scholar]
- 23.Kim C., Shah B., Subramaniam P., and Lee K. (2011). Synergistic induction of apoptosis in brain cancer cells by targeted codelivey of sirna and anticancer drugs. Mol. Pharm. 8, 1955–1961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nout Y., Culp E., Schmidt M., Tovar C., Pröschel C., Mayer-Pröschel M., Noble M., Beattie M., and Breshahan J. (2011). Glial restricted precursor cell transplant with cyclic adenosine monophosphate improved some autonomic functions but resulted in reduced graft size after spinal cord contusion injury in rats. Exp. Neurol. 227, 159–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murray A., Tucker S., and Shewan D. (2009). cAMP-dependent axon guidance Is distincly regulated by epac and protein kinase a. J. Neurosci. 29, 15434–15444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bae Y., Jang W., Nishiyama N., Fukushima S., and Kataoka K. (2005). Multifunctional polymeric micelles with folate-mediated cancer cell targeting and ph-triggered drug releasing properties for active intracellular drug delivery. Mol. Biosyst. 1, 242–250 [DOI] [PubMed] [Google Scholar]
- 27.Itaka K., Kanayama N., Nishiyama N., Jang W., Yamasaki Y., Nakamura K., Kawaguchi H., and Kataoka K. (2004). Supramolecular nanocarrier of sirna from peg-based block catiomer carrying diamine side chain with distinctive pKa directed to enhance intracellular gene silencing. J. Am. Chem. Soc. 126, 13612–13613 [DOI] [PubMed] [Google Scholar]
- 28.Nishiyama N. and Kataoka K. (2006). Current state, acheivements and future prospects of polymeric micelles as nanocarriers for drug and gene delivery. Pharmacol. Ther. 112, 630–648 [DOI] [PubMed] [Google Scholar]
- 29.Torchilin V., Lukyanov A., Gao Z., and Papahadjopoulos-Sternberg B. (2003). Immunomicelles: targeted pharmaceutical carriers for poorly soluble drugs. Proc. Natl. Acad. Sci. U.S.A. 100, 6039–6044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishiyama N., Matsumra Y., and Kataoka K. (2016). Development of polymeric micelles for targeting intractable cancers. Cancer Sci. 107, 867–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mochida Y., Cabral H., and Kataoka K. (2017). Polymeric micelles for targeted tumor therapy of platinum anticancer drugs. Expert Opin. Drug Deliv. 1–16 [DOI] [PubMed] [Google Scholar]
- 32.Yousefpour M. and Yari A. (2017). Polymeric micelles as mighty nanocarriers for cancer gene therapy: a review. Cancer Chemother. Pharmacol. 79, 637–649 [DOI] [PubMed] [Google Scholar]
- 33.Danhier F., Ansorena E., Silva J., Coco R., Breton A., and Préat V. (2012). Plga-based nanoparticles: an overview of biomedical applications. J. Control. Release 161, 505–522 [DOI] [PubMed] [Google Scholar]
- 34.Cabral H. and Kataoka K. (2014). Progress of drug-loaded polymeric micelles into clinical studies. J. Control. Release 190, 465–476 [DOI] [PubMed] [Google Scholar]
- 35.Bala I., Hariharan S., and Kumar M. (2004). Plga nanoparticles in drug delivery the state of the art. Crit. Rev. Ther. Drug Carrier Syst. 21, 387–422 [DOI] [PubMed] [Google Scholar]
- 36.Honary S. and Zahir F. (2013). Effect of zeta potential on the properties of nano-drug delivery systems—A Review (Part 2). Trop. J. Pharm. Res. 12, 265–273 [Google Scholar]
- 37.Gwak S., Nice J., Zhang J., Green B., Macks C., Bae S., Webb K., and Lee J. (2016). Cationic, amphiphilic copolymer micelles as nucleic acid carriers for enhanced transfection in rat spinal cord. Acta Biomater. 35, 98–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gwak S., Macks C., Jeong D., Kindy M., Lynn M., Webb K., and Lee J. (2017). Rhoa knockdown by cationic amphiphilic copolymer/siRhoA polyplexes enhances axonal regeneration in rat spinal cord injury model. Biomaterials 121, 155–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kataoka K., Harada A., and Nagasaki Y. (2001). Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv. Drug Deliv. Rev. 47, 113–131 [DOI] [PubMed] [Google Scholar]
- 40.Lee J., Cho E., and Cho K. (2004). Incorportation and release behavior of hydrophobic drug in functionalized poly(D,L-lactide)-block-poly(ethylene oxide) micelles. J. Control. Release 94, 323–335 [DOI] [PubMed] [Google Scholar]
- 41.Tang Y., Liu S., Armes S., and Billingham N. (2003). Solubilization and controlled release of a hydrophobic drug using novel micelle-forming abc triblock copolymers. Biomacromolecules 4, 1636–1645 [DOI] [PubMed] [Google Scholar]
- 42.Galvao J., Davis B., Tilley M., Normando E., Duchen M., and Cordeiro M. (2014). Unexpected low-dose toxicity of the universal solvent dmso. FASEB J. 28, 1317–1330 [DOI] [PubMed] [Google Scholar]
- 43.Basso D., Beattie M., and Bresnahan J. (1995). A sensitive and reliable locomotor rating scale for open field testing in rats. J. Neurotrauma 12, 1–21 [DOI] [PubMed] [Google Scholar]