

Abstract
Aim
The aim of this study was to evaluate the diagnostic utility of high‐sensitivity cardiac troponin T (hs‐cTnT) levels in discriminating cardiac amyloidosis from patients with cardiac hypertrophy caused by aetiologies other than cardiac amyloidosis.
Methods and results
Serum hs‐cTnT levels were measured in 96 patients with cardiac amyloidosis (light chain: 23, wild‐type transthyretin amyloidosis: 40, and mutated transthyretin amyloidosis: 33), and 91 patients with other causes of cardiac hypertrophy who were confirmed to have no cardiac amyloidosis by endomyocardial biopsy (control group). The diagnostic utility and cut‐off value of hs‐cTnT were evaluated by receiver operating characteristic analysis. The median hs‐cTnT levels were higher in cardiac amyloidosis than the control group [0.048 (0.029–0.073) vs. 0.016 (0.010–0.031) ng/mL; P < 0.001]. High levels of hs‐cTnT were suggestive of cardiac amyloidosis (cut‐off value: 0.0312 ng/mL, sensitivity: 0.74, specificity: 0.76, area under the curve: 0.788; 95% confidence interval: 0.723–0.854, P < 0.001), compared with brain natriuretic peptide and E/e′ ratio. The hs‐cTnT levels were also useful in differentiating each type of amyloidosis from the control group. Multivariate analysis identified log hs‐cTnT as an independent diagnostic factor for cardiac amyloidosis (odds ratio: 2.22; 95% confidence interval: 1.30–3.80; P = 0.004).
Conclusions
High serum levels of hs‐cTnT are highly suggestive of cardiac amyloidosis, allowing its differentiation from cardiac hypertrophy of other aetiologies. Further refined diagnostic approaches that include imaging modalities and histopathological examination are needed for these patients to avoid underdiagnosis of cardiac amyloidosis.
Keywords: Cardiac amyloidosis, Cardiac troponin, Diagnosis
Introduction
Cardiac amyloidosis is a progressive and infiltrative cardiomyopathy with increased ventricular wall thickness, diastolic dysfunction, and cardiac conduction system diseases.1, 2 There are three main types of cardiac amyloidosis: acquired monoclonal immunoglobulin light chain (AL amyloidosis); hereditary, mutated transthyretin amyloidosis (ATTRm); and wild‐type transthyretin amyloidosis (ATTRwt). AA amyloidosis is a complication of chronic inflammatory conditions and still the most numerous amyloidosis in non‐industrialized countries. However, clinically significant involvement of the heart is very rare. Treatment and prognosis of cardiac amyloidosis vary greatly depending on the amyloid fibril precursor. Therefore, early detection and classification are important for the selection of appropriate therapeutic strategies such as chemotherapy, liver transplantation, and novel transthyretin‐modifying therapeutics.1, 2, 3, 4
Heart failure with preserved ejection fraction (HFpEF) accounts for about half of patients with heart failure, and it is common in elderly patients with myocardial hypertrophy.5 Cardiac amyloidosis can cause HFpEF; however, the diagnosis of cardiac amyloidosis is missed or delayed because of lack of definitive finding on electrocardiography and echocardiography. It is reported that 13% of HFpEF cases were diagnosed as ATTRwt.6 Furthermore, several post‐mortem studies found cardiac amyloid deposition in up to 25% of individuals >80 years of age.7, 8 Therefore, it is important to suspect and differentiate cardiac amyloidosis from HFpEF of other aetiologies for the selection of appropriate therapy and prediction of prognosis.
Precise diagnosis of cardiac amyloidosis requires endomyocardial biopsy (EMB) to demonstrate disease‐specific deposition, but this procedure is relatively invasive and cannot be performed routinely in patients with cardiac hypertrophy. In fact, the need to perform EMB in patients with suspected cardiac amyloidosis decreased following the introduction of various non‐invasive imaging modalities and feasibility of sampling other tissues.9 Recent improvements in the diagnostic accuracy of cardiac magnetic resonance imaging using late gadolinium enhancement and T1 mapping, and bone scintigraphy, have contributed to the diagnosis of cardiac amyloidosis.6, 10, 11, 12
Persistent and modest elevation of cardiac troponin level is frequently observed and reflects ongoing subclinical myocardial damage in patients with various non‐ischaemic cardiomyopathies.13, 14, 15 It has been reported that cardiac troponin levels are higher in patients with cardiac amyloidosis compared with other forms of cardiomyopathy and high levels of cardiac troponin predict poor prognosis using conventional or high sensitive assays.6, 16, 17, 18, 19, 20 However, the utility of cardiac troponin level for the diagnosis of cardiac amyloidosis has not been fully evaluated. In the present study, we evaluated the diagnostic utility of high‐sensitivity cardiac troponin T (hs‐cTnT) level in discriminating cardiac amyloidosis, including the three types of cardiac amyloidosis, from patients with cardiac hypertrophy caused by aetiologies other than cardiac amyloidosis.
Methods
Study patients
The study was conducted in 96 patients with cardiac amyloidosis (test group) and 91 patients with non‐cardiac amyloidosis (control group). For the test group, 101 consecutive patients diagnosed with cardiac amyloidosis between February 2010 and June 2016 at Kumamoto University Hospital and National Cerebral and Cardiovascular Center were selected. Of these, we excluded three patients for renal insufficiency (estimated glomerular filtration rate <30 mL/min/1.73 m2, n = 2) and missing hs‐cTnT test values under stable condition (n = 1). Of the remaining 98 patients, 57 (AL: 16, ATTRmt: 35, and ATTRm: 6) underwent coronary angiography or computerized tomography coronary angiography, and two patients were found to have significant coronary artery stenosis requiring revascularization and were thus excluded from the study. For the control group, 592 consecutive patients underwent EMB for the diagnosis of cardiomyopathy between January 2014 and June 2016 at the aforementioned institution. We selected 106 consecutive patients with cardiac hypertrophy (interventricular septum ≥12 mm) of non‐cardiac amyloidosis aetiology as the control group. All were confirmed to be negative for amyloid deposition in the myocardium. Genetic testing was not performed among the control group, except for the patients with Fabry disease and mitochondrial cardiomyopathy to confirm the diagnosis. Among the control group, five patients had renal insufficiency, and data on hs‐cTnT level were missing in three patients with missing hs‐cTnT level and were excluded from the analysis. Of the remaining 98 patients, 94 underwent coronary angiography or computerized tomography coronary angiogram, and seven patients were found to have significant coronary artery stenosis requiring revascularization. Consequently, the final study population consisted of 96 patients with cardiac amyloidosis and 91 control patients with cardiac hypertrophy of non‐cardiac amyloidosis aetiology (Figure 1 ). The study protocol was approved by the Human Ethics Review Committee of Kumamoto University and National Cerebral and Cardiovascular Center.
Figure 1.
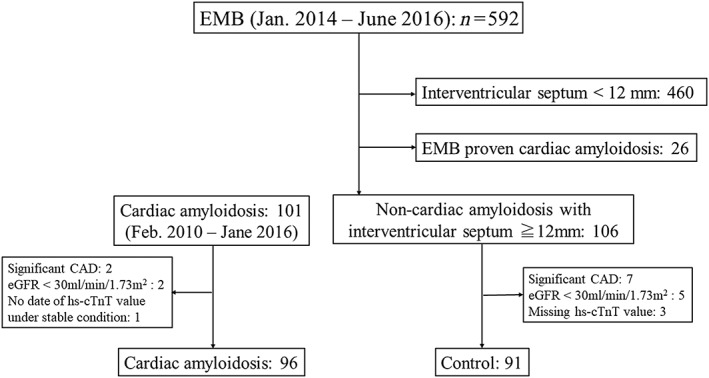
Selection of patients for the two study groups. CAD, coronary artery disease; eGFR, estimated glomerular filtration rate; EMB, endomyocardial biopsy; hs‐cTnT, high‐sensitivity cardiac troponin T.
Diagnosis of cardiac amyloidosis
The diagnosis of amyloidosis was based on Congo red staining and apple‐green birefringence examination under cross‐polarized light microscopy. Cardiac amyloidosis was diagnosed by amyloid deposition in the myocardium or increased wall thickness (interventricular septum ≥12 mm) on echocardiography in the absence of any other cause of ventricular hypertrophy, providing that a histopathological diagnosis of amyloidosis has been made in at least one involved organ, such as the abdominal subcutaneous adipose tissue, skin, or gastrointestinal tract.1 The presence of global transmural or subendocardial late gadolinium enhancement on cardiac magnetic resonance and positive finding on 99mTc‐pyrophosphate scintigraphy, if performed, added support to the diagnosis of cardiac amyloidosis.6, 10, 11, 12
Immunohistochemistry and DNA analysis were employed to determine the subtype of cardiac amyloidosis. AL amyloidosis was confirmed by the presence of monoclonal protein in the serum or urine and/or a monoclonal population of plasma cells in the bone marrow. ATTRwt amyloidosis was diagnosed by positive immunohistochemistry for transthyretin in the absence of any transthyretin mutation at DNA analysis or elderly patients without family history of ATTRm with negative findings of AL amyloidosis, if DNA analysis was not performed. ATTRm was diagnosed based on documented transthyretin mutation at DNA analysis.1
Biomarker and imaging analysis
Blood samples were collected under clinically stable condition. Serum hs‐cTnT levels were measured at diagnosis using the Elecsys 2010 Troponin T hs kit (Roche Diagnostics, Indianapolis, IN), with a lower limit of detection of 0.0005 ng/mL and a reported 99th percentile value in apparently healthy individuals of 0.0135 ng/mL with a coefficient of variation of 9%.21 Plasma brain natriuretic peptide (BNP) levels were measured using the MI02 Shionogi BNP kit (Shionogi, Osaka, Japan). The glomerular filtration rate was calculated using the Modification of Diet in Renal Disease modified for Japanese patients.
Echocardiography was performed using commercially available ultrasound equipment. Chamber size, wall thickness, left ventricular ejection fraction (LVEF), and left ventricular (LV) mass were evaluated using standard procedures.22 Peak early and late diastolic velocity of LV inflow (E and A velocity, respectively), deceleration time of E velocity, and peak early diastolic velocity on the septal corner of the mitral annulus (e′) were measured in the apical four‐chamber view, and the E/e′ ratio was calculated.23
Statistical analysis
Normally distributed parameters were expressed as mean ± SD, while data of variables with skewed distribution were expressed as medians with interquartile ranges. Differences between groups were examined by the Student's t‐test or the Mann–Whitney U test for unpaired data. Categorical values were presented as numbers (percentages) and compared by the Χ2 test or Fisher's exact test as appropriate. Receiver operating characteristic curve analysis was performed to compare the diagnostic accuracy and determine the cut‐off value of hs‐cTnT for the diagnosis of cardiac amyloidosis. Univariate logistic regression analysis was performed to identify significant parameters related to the diagnosis of cardiac amyloidosis. Then, multivariate logistic regression analysis was performed using the forced inclusion model that included the following parameters: age, gender, body mass index, log hs‐cTnT, haemoglobin, serum sodium, E/e′ ratio, and deceleration time. The Hosmer–Lemeshow statistic was applied to assess model calibration. The correlations between hs‐cTnT levels and several parameters were evaluated using Spearman's correlation. A two‐tailed value of P < 0.05 was considered statistically significant. All statistical analyses were performed with SPSS, version 19 (SPSS Inc., Chicago, IL).
Results
Diagnosis and clinical characteristics of study patients
Among 96 patients with cardiac amyloidosis, the numbers of patients with AL amyloidosis, ATTRwt, and ATTRm were 23, 40, and 33, respectively. V30M (p.V50M) was found in almost half of the ATTRm patients (n = 15: 45%). Other transthyretin mutations were found as follows: S77T: 2, G89L: 2, T60A: 2, S50I: 2, G47V: 2, T59A: 1, A45A: 1, V30A: 1, A36A: 1, T49I: 1, I107V: 1, T14C: 1, and L55P: 1. A total of 51 patients with cardiac amyloidosis (53%; AL: 13, ATTRwt: 31, and ATTRm: 7) were histopathologically found to have amyloid disposition in the myocardium. The remaining 22 patients (23%; AL: 3, ATTRwt: 9, and ATTRm: 10) had positive findings consistent with the diagnosis of cardiac amyloidosis on cardiac magnetic resonance or 99mTc‐pyrophosphate scintigraphy. DNA analysis was performed in 27 of 40 (68%) ATTRwt patients, and the findings confirmed the absence of any transthyretin mutation. Among 91 patients of the control group, hypertrophic cardiomyopathy was the most common diagnosis (n = 69: 76%), and final diagnosis was hypertensive heart disease (n = 11), severe aortic stenosis (n = 5), Fabry disease (n = 4), mitochondrial cardiomyopathy (n = 1), and dilated cardiomyopathy (n = 1).
Patient characteristics and high‐sensitivity cardiac troponin T levels
Table 1 lists the clinical characteristics, and Figure 2 shows the distribution of serum hs‐cTnT levels for the study patients. Patients with cardiac amyloidosis were more likely to be older men, especially AL amyloidosis and ATTRwt, compared with the control group. Atrial fibrillation was more common in ATTRwt amyloidosis compared with other types of cardiac amyloidosis (P < 0.05). Serum hs‐cTnT level was significantly higher in the cardiac amyloidosis group [0.048 (0.029–0.073) ng/mL] than the control group [0.016 (0.010–0.031) ng/mL; P < 0.001]. Among the three types of cardiac amyloidosis, hs‐cTnT levels were highest among patients with AL amyloidosis than those with ATTRwt (P < 0.01) and ATTRm (P < 0.01). There were no differences in plasma BNP levels and renal function between patients with cardiac amyloidosis and those of the control group. However, BNP levels were significantly higher in AL amyloidosis and lower in ATTRm than the control group. Echocardiographic analysis showed increased posterior wall thickness, higher E velocity and E/e′ ratio, and smaller LV end‐diastolic dimension and A velocity in the cardiac amyloidosis group than the control group. The use of loop diuretics and aldosterone antagonists was higher and that of β‐blockers was lower in cardiac amyloidosis than the control group.
Table 1.
Demographic and clinical characteristics of participating subjects
| Variables | Control group (n = 91) | Cardiac amyloidosis (n = 96) | AL amyloidosis (n =23) | ATTRwt amyloidosis (n = 40) | ATTRm amyloidosis (n = 33) |
|---|---|---|---|---|---|
| Age (years) | 57 ± 15 | 68 ± 12* | 64 ± 9* | 77 ± 6* | 60 ± 11 |
| Male | 48 (53%) | 83 (87%)* | 21 (91%)* | 37 (93%)* | 25 (76%) |
| BMI (kg/m2) | 24.1 ± 4.3 | 22.5 ± 3.1* | 22.7 ± 2.7 | 23.6 ± 3.1 | 21.2 ± 2.9* |
| Hypertension | 51 (56%) | 29 (30%)* | 4 (17%)* | 20 (50%) | 5 (15%)* |
| Diabetes mellitus | 13 (14%) | 13 (14%) | 1 (4%) | 7 (18%) | 5 (15%) |
| Dyslipidemia | 36 (40%) | 23 (24%)* | 5 (22%) | 10 (25%) | 8 (24%) |
| Prior HF hospitalization | 17 (19%) | 25 (26%) | 11 (48%)* | 11 (28%) | 3 (9%) |
| Atrial fibrillation | 15 (17%) | 29 (30%)* | 6 (26%) | 21 (53%)* | 2 (6%) |
| Pacemaker | 0 (0%) | 7 (7%)* | 1 (0%) | 1 (3%) | 5 (15%)* |
| ICD/CRT‐D | 5 (6%) | 7 (7%) | 3 (13%) | 3 (8%) | 1 (3%) |
| Laboratory data | |||||
| hs‐cTnT (ng/mL) | 0.016 [0.010–0.031] | 0.048 [0.029–0.073] * | 0.073 [0.055–0.142]* | 0.048 [0.036–0.080]* | 0.032 [0.020–0.050]* |
| BNP (pg/mL) | 201 [85–490] | 194 [87–489] | 843 [318–1456]* | 254 [133–455] | 73 [42–138]* |
| Haemoglobin (g/dL) | 13.6 ± 1.9 | 13.1 ± 1.7* | 12.8 ± 2.1 | 13.4 ± 1.8 | 12.8 ± 1.1* |
| Serum sodium (mEq/L) | 141 ± 2 | 139 ± 4* | 137 ± 4* | 139 ± 4* | 140 ± 2.9 |
| eGFR (mL/min/1.73m2) | 66.5 ± 21.6 | 65.8 ± 24.6 | 63.1 ± 19.2 | 53.4 ± 12.5* | 82.7 ± 29.3* |
| C‐reactive protein (mg/mL) | 0.07 [0.03–0.24] | 0.07 [0.03–0.20] | 0.18 [0.05–0.65]* | 0.10 [0.04–0.20] | 0.03 [0.02–0.11] * |
| Imaging findings | |||||
| LVDd (mm) | 46.2 ± 8.7 | 43.1 ± 6.5* | 42.9 ± 6.9 | 44.4 ± 7.6 | 41.8 ± 4.1* |
| LVDs (mm) | 29.9 ± 10.6 | 30.9 ± 7.4 | 31.0 ± 7.7 | 32.9 ± 8.5 | 28.3 ± 4.5 |
| IVS (mm) | 15.1 ± 3.7 | 15.2 ± 3.2 | 14.4 ± 3.5 | 15.6 ± 3.4 | 15.3 ± 2.5 |
| PW (mm) | 11.7 ± 2.8 | 14.3 ± 2.9* | 13.9 ± 2.9* | 14.3 ± 3.1* | 14.7 ± 2.6* |
| LV mass index (g/m2) | 152 ± 53 | 155 ± 39 | 139 ± 34 | 165 ± 43 | 154 ± 35 |
| LVEF (%) | 54.2 ± 13.0 | 51.3 ± 11.4 | 48.5 ± 9.7 | 48.2 ± 12.7* | 56.9 ± 8.6 |
| E velocity (cm/s) | 69.2 ± 25.9 | 77.7 ± 24.7* | 81.7 ± 26.8* | 77.7 ± 25.6 | 74.8 ± 22.2 |
| A velocity (cm/s) | 71.2 ± 30.8 | 51.4 ± 31.1* | 46.8 ± 28.9* | 44.5 ± 27.2* | 60.2 ± 34.2 |
| E/A ratio | 1.14 ± 0.75 | 1.44 ± 1.33* | 2.35 ± 1.87* | 1.25 ± 1.00* | 1.05 ± 0.91 |
| e' | 4.57 ± 2.01 | 4.22 ± 1.89 | 3.90 ± 1.18 | 3.98 ± 1.25 | 4.75 ± 2.71 |
| E/e′ ratio | 16.8 ± 8.4 | 21.1 ± 9.9* | 23.7 ± 13.1* | 22.1 ± 9.5* | 18.2 ± 7.2 |
| Deceleration time (ms) | 234 ± 94 | 202 ± 87* | 157 ± 51* | 204 ± 86 | 230 ± 98 |
| CMR‐LGE; (positive/n; %) | — | 54/64 (84%) | 11/13 (85%) | ]26/30 (87%) | 14/15 (93%) |
| 99mTc‐PYP scintigraphy (positive/n; %) | — | 33/47 (70%) | 2/11 (18%) | 20/20 (100%) | 11/15 (73%) |
| Medications | |||||
| Loop diuretics | 22 (24%) | 52 (54%)* | 19 (83%)* | 27 (68%)* | 6 (18%) |
| β‐blockers | 64 (70%) | 32 (33%)* | 7 (30%)* | 21 (53%)* | 4 (12%)* |
| ACE‐I or ARB | 39 (43%) | 20 (31%) | 3 (13%)* | 23 (58%) | 4 (12%)* |
| Aldosterone antagonists | 13 (14%) | 26 (27%)* | 12 (52%)* | 14 (35%)* | 0 (0%)* |
ACE‐I, angiotensin‐converting enzyme inhibitor; AL, amyloid light chain; ARB, angiotensin receptor blocker; ATTRm, mutated transthyretin amyloidosis; ATTRwt, wild‐type transthyretin amyloidosis; BMI, body mass index; BNP, brain natriuretic peptide; CMR, cardiac magnetic resonance; CRT‐D, cardiac resynchronization therapy defibrillator; eGFR, estimated glomerular filtration rate; HF, heart failure; hs‐cTnT, high‐sensitivity cardiac troponin T; ICD, implantable cardioverter defibrillator; IVS, interventricular septum; LGE, late gadolinium enhancement; LV, left ventricular; LVDd, left ventricular diameter at end diastole; LVDs, left ventricular diameter at end systole; LVEF, left ventricular ejection fraction; PW, posterior wall; PYP, pyrophosphate.
P < 0.05 vs. the control group (patients with cardiac hypertrophy of non‐amyloid aetiologies).
Figure 2.
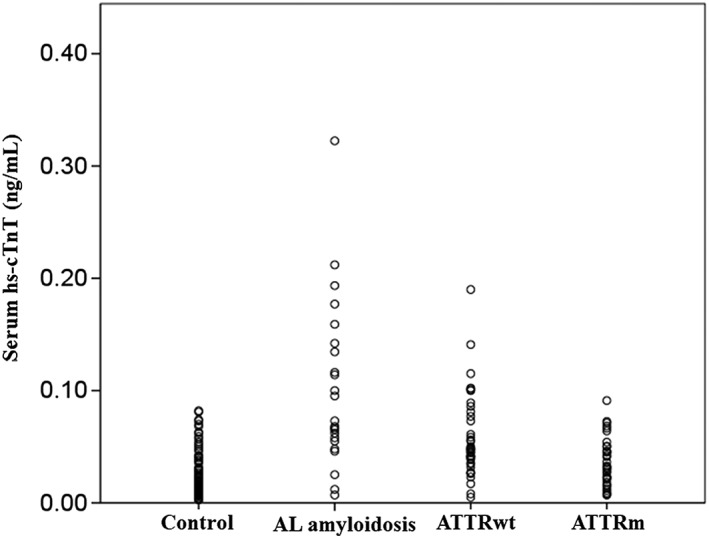
Distribution of high sensitivity cardiac troponin T levels in the study patients. AL, amyloid light chain; ATTRm, mutated transthyretin amyloidosis; ATTRwt, wild‐type transthyretin amyloidosis; hs‐cTnT, high‐sensitivity cardiac troponin T.
Diagnostic accuracy of high‐sensitivity cardiac troponin T
Figure 3 and Table 2 show the receiver operator characteristic analyses of the diagnostic accuracy of cardiac amyloidosis. hs‐cTnT showed the highest area under the curve (AUC: 0.788; 95% confidence interval: 0.723–0.854; P < 0.001, Figure 3A ) to differentiate cardiac amyloidosis from the control group, compared with BNP and E/e′ ratio, with the best hs‐cTnT cut‐off level of 0.0312 ng/mL (sensitivity: 0.74 and specificity: 0.76). In the same way, hs‐cTnT had the highest AUC to differentiate the three types of amyloidosis from the control group. The best hs‐cTnT cut‐off values were 0.0440 ng/mL (AUC: 0.881, sensitivity: 0.87, and specificity: 0.84) for AL amyloidosis (Figure 3B ), 0.0315 ng/mL (AUC: 0.820, sensitivity: 0.83, and specificity: 0.76) for ATTRwt (Figure 3C ), and 0.0286 ng/mL (AUC: 0.686, sensitivity: 0.58, and specificity: 0.68) for ATTRm (Figure 3D ).
Figure 3.
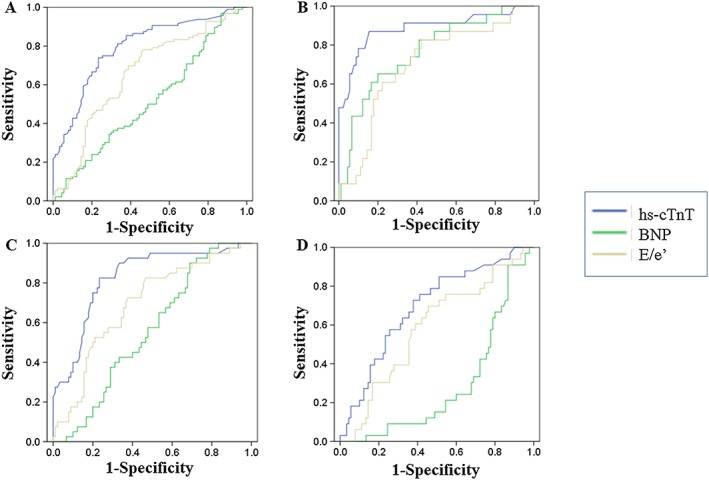
Receiver operator characteristic analyses of the diagnostic accuracy of cardiac amyloidosis in (A) cardiac amyloidosis vs. the control group, (B) amyloid light chain amyloidosis vs. the control group, (C) wild‐type transthyretin amyloidosis vs. the control group, and (D) mutated transthyretin amyloidosis vs. the control group. hs‐cTnT, high‐sensitivity cardiac troponin T.
Table 2.
Receiver operating characteristic analysis for diagnosis of cardiac amyloidosis
| AUC (95% CI) | P value | |
|---|---|---|
| A. Cardiac amyloidosis vs. control group | ||
| hs‐cTnT | 0.788 (0.723–0.854) | <0.001 |
| BNP | 0.511 (0.428–0.594) | 0.80 |
| E/e′ ratio | 0.658 (0.578–0.737) | <0.001 |
| B. AL amyloidosis vs. control group | ||
| hs‐cTnT | 0.881 (0.787–0.975) | <0.001 |
| BNP | 0.763 (0.654–0.872) | <0.001 |
| E/e′ ratio | 0.703 (0.584–0.821) | 0.003 |
| C. ATTRwt vs. control group | ||
| hs‐cTnT | 0.820 (0.741–0.898) | <0.001 |
| BNP | 0.551 (0.453–0.650) | 0.35 |
| E/e′ ratio | 0.690 (0.593–0.787) | 0.001 |
| D. ATTRm vs. control group | ||
| hs‐cTnT | 0.686 (0.583–0.789) | 0.002 |
| BNP | 0.286 (0.191–0.381) | <0.001 |
| E/e′ ratio | 0.587 (0.475–0.698) | 0.14 |
AUC, area under the curve; CI, confidence interval. For other abbreviations, see Table 1.
We used univariate and multivariate logistic regression analyses to identify the parameters associated with the diagnosis of cardiac amyloidosis. Multivariate analysis identified log serum hs‐cTnT level (odds ratio: 2.22; 95% confidence interval: 1.30–3.80; P = 0.004; Hosmer–Lemeshow Χ2 = 6.069 and P = 0.64) as an independent and significant correlate with the diagnosis of cardiac amyloidosis (Table 3).
Table 3.
Results of univariate and multivariate logistic regression analyses for the diagnosis of cardiac amyloidosis
| Variables | Univariate analysis | Multivariate analysis | ||
|---|---|---|---|---|
| OR (95% CI) | P value | OR (95% CI) | P value | |
| Age (per year) | 1.06 (1.03–1.08) | <0.001 | 1.04 (1.01–1.07) | 0.008 |
| Male | 5.72 (2.80–11.69) | <0.001 | 5.24 (2.04–13.45) | 0.001 |
| BMI (per kg/m2) | 0.89 (0.82–0.97) | 0.005 | 0.89 (0.80–0.99) | 0.04 |
| Log hs‐cTnT (per 1.0) | 3.92 (2.52–6.09) | <0.001 | 2.22 (1.30–3.80) | 0.004 |
| Log BNP (per 1.0) | 1.07 (0.85–1.34) | 0.59 | ||
| Haemoglobin (per g/dL) | 0.84 (0.71–0.99) | 0.04 | 0.87 (0.69–1.09) | 0.23 |
| Serum sodium (per mEq/L) | 0.81 (0.72–0.91) | <0.001 | 0.88 (0.76–1.01) | 0.07 |
| eGFR (per mL/min/1.73m2) | 1.00 (0.99–1.01) | 0.84 | ||
| Log C‐reactive protein (per mg/mL) | 1.01 (0.83–1.21) | 0.96 | ||
| LV mass index (per g/m2) | 1.00 (1.00–1.01) | 0.61 | ||
| LVEF (per %) | 0.98 (0.96–1.00) | 0.11 | ||
| E/e′ ratio | 1.06 (1.02–1.10) | 0.002 | 1.04 (0.99–1.09) | 0.15 |
| Deceleration time (per ms) | 1.00 (0.99–1.00) | 0.02 | 1.00 (0.99–1.00) | 0.23 |
OR, odds ratio. For other abbreviations, see Table 1.
Correlation of serum high‐sensitivity cardiac troponin T levels with other parameters
Table 4 details the results of univariate linear regression analyses for hs‐cTnT level in patients with cardiac amyloidosis. In this group of patients, hs‐cTnT levels correlated with BNP, haemoglobin, serum sodium, estimated glomerular filtration rate, C‐reactive protein, LVEF, LV mass index, and diastolic parameters of echocardiography (e.g. E/A, deceleration time, and E/e′ ratio). Notably, BNP correlated strongly with hs‐cTnT levels in each type of cardiac amyloidosis.
Table 4.
Results of univariate linear regression analyses for hs‐cTnT levels in patients with cardiac amyloidosis
| Variables | Cardiac amyloidosis | AL amyloidosis | ATTRwt amyloidosis | ATTRm amyloidosis |
|---|---|---|---|---|
| Age | 0.137 | 0.149 | 0.057 | 0.268 |
| BMI | 0.016 | −0.019 | −0.078 | −0.145 |
| BNP | 0.693* | 0.703* | 0.584* | 0.559* |
| Haemoglobin | −0.210* | −0.022 | −0.275 | −0.392* |
| Serum sodium | −0.236* | −0.387 | −0.124 | 0.064 |
| eGFR | −0.352* | −0.281 | −0.400* | −0.188 |
| C‐reactive protein | 0.447* | 0.400 | 0.294 | 0.255 |
| LVDd | −0.064 | −0.307 | 0.077 | −0.269 |
| LVDs | 0.212* | −0.057 | 0.396* | 0.029 |
| IVS | 0.184 | 0.482* | 0.130 | 0.353* |
| PW | 0.221* | 0.235 | 0.269 | 0.411* |
| LV mass index | 0.215* | 0.279 | 0.205 | 0.413* |
| LVEF | −0.358* | −0.107 | −0.486* | −0.122 |
| E velocity | 0.143 | 0.190 | 0.024 | 0.111 |
| A velocity | −0.291* | −0.316 | −0.364 | −0.076 |
| E/A ratio | 0.369* | 0.408 | 0.007 | 0.289 |
| e′ | −0.330* | −0.014 | −0.467* | −0.239 |
| E/e′ ratio | 0.373* | 0.151 | 0.396* | 0.320 |
| Deceleration time | −0.300* | −0.438* | −0.237 | −0.009 |
For abbreviations, see Table 1.
P < 0.05.
Discussion
The major findings of the present study were as follows: (i) serum hs‐cTnT levels were significantly higher in patients with cardiac amyloidosis than those with cardiac hypertrophy free of cardiac amyloidosis. (ii) High levels of serum hs‐cTnT (hs‐cTnT ≥ 0.0312 ng/mL) were highly suggestive of cardiac amyloidosis in patients with cardiac hypertrophy. (iii) Compared with BNP and E/e′ ratio, the AUC of hs‐cTnT was the best that allowed differentiation of three types of amyloidosis from the control group. Together, these findings indicate that serum hs‐cTnT is a helpful biomarker to suspect and differentiate cardiac amyloidosis from cardiac hypertrophy of non‐cardiac amyloidosis aetiologies.
Several studies have investigated the prognostic value of cardiac troponin in cardiac amyloidosis.17, 18, 19, 20 However, there are only a few studies that evaluated the diagnostic utility of cardiac troponin in patients suspected of cardiac amyloidosis and compared the levels between cardiac amyloidosis and cardiac hypertrophy of non‐cardiac amyloidosis aetiologies. Kubo et al. 16 reported that hs‐cTnT levels were significantly higher in 11 patients with infiltrative cardiomyopathy including two patients with AL amyloidosis and six with ATTRwt compared with 35 patients with hypertrophic cardiomyopathy (0.083 ± 0.057 vs. 0.027 ± 0.034 ng/mL, P < 0.001) and established the diagnostic utility of hs‐cTnT in distinguishing infiltrative cardiomyopathy from LV hypertrophy. Compared with this report, our study evaluated hs‐cTnT levels in a larger number of patients who were classified into three types of cardiac amyloidosis and in EMB‐confirmed non‐cardiac amyloidosis patients with cardiac hypertrophy. Thus, our study is the first report that highlights the diagnostic utility of serum hs‐cTnT for cardiac amyloidosis in patients with cardiac hypertrophy.
The exact mechanism responsible for cardiac troponin release from the myocardium in patients with cardiac amyloidosis remains speculative. Various reasons have been proposed for the persistent hypertroponinemia, including myocardial ischaemia, increased wall stress, myocyte damage from inflammatory cytokines and/or oxidative stress, neurohormonal activation, and coronary microvascular dysfunction in heart failure.13, 24 These potential mechanisms are enhanced in cardiac amyloidosis and boost cardiac troponin release through myocardial necrosis, apoptosis, and troponin degradation compared with non‐cardiac amyloidosis.
Microvascular angina and myocardial ischaemia without overt coronary artery disease have been reported in cardiac amyloidosis.25 Dorbala et al. 26 reported that coronary microvascular dysfunction is highly prevalent in subjects with cardiac amyloidosis even in the absence of epicardial coronary artery disease compared with patients with LV hypertrophy. The mechanisms of coronary microvascular dysfunction in cardiac amyloidosis are presumed to be due to (i) amyloid deposits in the interstitium and perivascular regions, (ii) elevated LV filling pressure, and (iii) impaired endothelial dysfunction. Amyloid deposits cause luminal stenosis and extrinsic compression of the microvasculature. Increased LV filling pressure, which causes increased wall stress due to diastolic dysfunction in cardiac amyloidosis, compresses myocardial capillaries, with subsequent decrease in their lumen. Endothelial function is impaired by microvascular toxicity induced by light chain in AL amyloidosis.27, 28 These mechanisms cause functional myocardial ischaemia, subclinical impairment of LV systolic function, and cardiac troponin release. Additionally, it has been reported that circulating amyloid light chain seems to have a direct cardiotoxic effect causing cardiac dysfunction independent of extracellular fibril deposition.29 This point may be the reason for the higher serum hs‐cTnT levels and poor prognosis in AL amyloidosis in spite of the smaller LV hypertrophy compared with ATTR amyloidosis.18, 30 It is reported that cardiac high‐sensitivity troponin I level tended to decrease after chemotherapy in patients with AL amyloidosis.31 Therefore, disease‐specific therapy could reduce cardiac troponin levels, though this needs to be confirmed by accumulation of more data. Further large studies are warranted to clarify whether disease‐specific therapy can reduce myocardial damage.
What is the best diagnostic approach for cardiac amyloidosis? Electrocardiography and echocardiography are low cost and valuable tests. Low voltage, even as myocardial hypertrophy, poor R progression in the precordial leads, and pseudo‐infract patterns in the electrocardiography and concentric thickening of LV wall with increased echogenicity and thickened valve leaflets in echocardiography are well‐known suspicious findings. However, these features have poor specificity and are different in each type of cardiac amyloidosis.1, 18, 30 Immunofixation and serum‐free light chains should always be measured when amyloidosis is suspected.32 Recently, the excellent diagnostic accuracy of cardiac magnetic resonance and bone scintigraphy was confirmed in several studies.6, 10, 11, 12 However, these modalities are expensive and available only in a limited number of institutions. In addition to the aforementioned electrocardiographic and echocardiographic findings, high serum levels of hs‐cTnT should increase the likely diagnosis of cardiac amyloidosis. A more refined diagnostic approach is needed that should also include imaging modalities and histopathological examination.
This study has certain limitations. First, histopathological confirmation of amyloid deposition in the heart and DNA analysis are required for definitive diagnosis of cardiac amyloidosis. In this study, amyloid disposition in the myocardium was confirmed by histopathology in 51 (53%) patients. However, positive findings of cardiac magnetic resonance or 99mTc‐pyrophosphate scintigraphy supported the diagnosis in 22 patients (23%). Notably, all ATTRwt patients were diagnosed by EMB or imaging findings. However, the diagnosis of cardiac amyloidosis might be overestimated in patients without EMB or imaging confirmation. Second, this study included a relatively small number of patients from two centres. Further multicentre studies of larger groups are needed to confirm the present results. Third, cardiac troponin levels increase with age and are higher in men than women. A larger percentage of our patients with cardiac amyloidosis were older men compared with those of the control group. This fact could significantly interfere with the interpretation of hs‐cTnT difference noted between the control and test groups. Fourth, evaluation of coronary artery disease was limited to about 58% of cardiac amyloidosis patients. Undiagnosed coronary artery disease may affect the increase in hs‐cTnT levels in patients with cardiac amyloidosis. Finally, serum hs‐cTnT levels are also influenced by various clinical factors such as renal clearance, exacerbation of heart failure, and presence of coronary artery disease.13 Therefore, we have to evaluate serum hs‐cTnT levels and probability of cardiac amyloidosis in the presence or absence of these factors in mind.
Conclusions
Serum hs‐cTnT levels were significantly higher in patients with cardiac amyloidosis than in those with cardiac hypertrophy free of cardiac amyloidosis. High levels of hs‐cTnT enhanced differentiation of cardiac amyloidosis from cardiac hypertrophy caused by other aetiologies. Further diagnostic approaches that include cardiac magnetic resonance, bone scintigraphy, and histopathological examination are needed in these patients to avoid underdiagnosis of cardiac amyloidosis.
Conflict of interest
None declared.
Funding
This work was supported by Grants‐in‐Aid for Young Scientists B from the Japan Society for the Promotion of Science (S.T., 26860574).
Takashio, S. , Yamamuro, M. , Izumiya, Y. , Hirakawa, K. , Marume, K. , Yamamoto, M. , Ueda, M. , Yamashita, T. , Ishibashi‐Ueda, H. , Yasuda, S. , Ogawa, H. , Ando, Y. , Anzai, T. , and Tsujita, K. (2018) Diagnostic utility of cardiac troponin T level in patients with cardiac amyloidosis. ESC Heart Failure, 5: 27–35. doi: 10.1002/ehf2.12203.
References
- 1. Falk RH. Diagnosis and management of the cardiac amyloidoses. Circulation 2005; 112: 2047–2060. [DOI] [PubMed] [Google Scholar]
- 2. Rapezzi C, Merlini G, Quarta CC, Riva L, Longhi S, Leone O, Salvi F, Ciliberti P, Pastorelli F, Biagini E, Coccolo F, Cooke RM, Bacchi‐Reggiani L, Sangiorgi D, Ferlini A, Cavo M, Zamagni E, Fonte ML, Palladini G, Salinaro F, Musca F, Obici L, Branzi A, Perlini S. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation 2009; 120: 1203–1212. [DOI] [PubMed] [Google Scholar]
- 3. Castaño A, Drachman BM, Judge D, Maurer MS. Natural history and therapy of TTR‐cardiac amyloidosis: emerging disease‐modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev 2015; 20: 163–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maurer MS, Grogan DR, Judge DP, Mundayat R, Packman J, Lombardo I, Quyyumi AA, Aarts J, Falk RH. Tafamidis in transthyretin amyloid cardiomyopathy: effects on transthyretin stabilization and clinical outcomes. Circ Heart Fail 2015; 8: 519–526. [DOI] [PubMed] [Google Scholar]
- 5. Borlaug BA, Paulus WJ. Heart failure with preserved ejection fraction: pathophysiology, diagnosis, and treatment. Eur Heart J 2011; 32: 670–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. González‐López E, Gallego‐Delgado M, Guzzo‐Merello G, de Haro‐Del Moral FJ, Cobo‐Marcos M, Robles C, Bornstein B, Salas C, Lara‐Pezzi E, Alonso‐Pulpon L, Garcia‐Pavia P. Wild‐type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J 2015; 36: 2585–2594. [DOI] [PubMed] [Google Scholar]
- 7. Tanskanen M, Peuralinna T, Polvikoski T, Notkola IL, Sulkava R, Hardy J, Singleton A, Kiuru‐Enari S, Paetau A, Tienari PJ, Myllykangas L. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2‐macroglobulin and tau: a population‐based autopsy study. Ann Med 2008; 40: 232–239. [DOI] [PubMed] [Google Scholar]
- 8. Mohammed SF, Mirzoyev SA, Edwards WD, Dogan A, Grogan DR, Dunlay SM, Roger VL, Gertz MA, Dispenzieri A, Zeldenrust SR, Redfield MM. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail 2014; 2: 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takashio S, Izumiya Y, Jinnin M, Yamamuro M, Kojima S, Ihn H, Ogawa H. Diagnostic and prognostic value of subcutaneous tissue biopsy in patients with cardiac amyloidosis. Am J Cardiol 2012; 110: 1507–1511. [DOI] [PubMed] [Google Scholar]
- 10. Syed IS, Glockner JF, Feng D, Araoz PA, Martinez MW, Edwards WD, Gertz MA, Dispenzieri A, Oh JK, Bellavia D, Tajik AJ, Grogan M. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovasc Imaging 2010; 3: 155–164. [DOI] [PubMed] [Google Scholar]
- 11. Karamitsos TD, Piechnik SK, Banypersad SM, Fontana M, Ntusi NB, Ferreira VM, Whelan CJ, Myerson SG, Robson MD, Hawkins PN, Neubauer S, Moon JC. Noncontrast T1 mapping for the diagnosis of cardiac amyloidosis. JACC Cardiovasc Imaging 2013; 6: 488–497. [DOI] [PubMed] [Google Scholar]
- 12. Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, Wechalekar AD, Berk JL, Quarta CC, Grogan M, Lachmann HJ, Bokhari S, Castano A, Dorbala S, Johnson GB, Glaudemans AW, Rezk T, Fontana M, Palladini G, Milani P, Guidalotti PL, Flatman K, Lane T, Vonberg FW, Whelan CJ, Moon JC, Ruberg FL, Miller EJ, Hutt DF, Hazenberg BP, Rapezzi C, Hawkins PN. Non‐biopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 2016; 133: 2404–2412. [DOI] [PubMed] [Google Scholar]
- 13. Kociol RD, Pang PS, Gheorghiade M, Fonarow GC, O'Connor CM, Felker GM. Troponin elevation in heart failure prevalence, mechanisms, and clinical implications. J Am Coll Cardiol 2010; 56: 1071–1078. [DOI] [PubMed] [Google Scholar]
- 14. Kawahara C, Tsutamoto T, Nishiyama K, Yamaji M, Sakai H, Fujii M, Yamamoto T, Horie M. Prognostic role of high‐sensitivity cardiac troponin T in patients with nonischemic dilated cardiomyopathy. Circ J 2011; 75: 656–661. [DOI] [PubMed] [Google Scholar]
- 15. Kubo T, Kitaoka H, Yamanaka S, Hirota T, Baba Y, Hayashi K, Iiyama T, Kumagai N, Tanioka K, Yamasaki N, Matsumura Y, Furuno T, Sugiura T, Doi YL. Significance of high‐sensitivity cardiac troponin T in hypertrophic cardiomyopathy. J Am Coll Cardiol 2013; 62: 1252–1259. [DOI] [PubMed] [Google Scholar]
- 16. Kubo T, Baba Y, Hirota T, Tanioka K, Yamasaki N, Yamanaka S, Iiyama T, Kumagai N, Furuno T, Sugiura T, Kitaoka H. Differentiation of infiltrative cardiomyopathy from hypertrophic cardiomyopathy using high‐sensitivity cardiac troponin T: a case–control study. BMC Cardiovasc Disord 2015; 15: 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dispenzieri A, Kyle RA, Gertz MA, Therneau TM, Miller WL, Chandrasekaran K, McConnell JP, Burritt MF, Jaffe AS. Survival in patients with primary systemic amyloidosis and raised serum cardiac troponins. Lancet 2003; 361: 1787–1789. [DOI] [PubMed] [Google Scholar]
- 18. Pinney JH, Whelan CJ, Petrie A, Dungu J, Banypersad SM, Sattianayagam P, Wechalekar A, Gibbs SD, Venner CP, Wassef N, McCarthy CA, Gilbertson JA, Rowczenio D, Hawkins PN, Gillmore JD, Lachmann HJ. Senile systemic amyloidosis: clinical features at presentation and outcome. J Am Heart Assoc 2013; 2 e000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dispenzieri A, Gertz MA, Kumar SK, Lacy MQ, Kyle RA, Saenger AK, Grogan M, Zeldenrust SR, Hayman SR, Buadi F, Greipp PR, Leung N, Russell SR, Dingli D, Lust JA, Rajkumar SV, Jaffe AS. High sensitivity cardiac troponin T in patients with immunoglobulin light chain amyloidosis. Heart 2014; 100: 383–388. [DOI] [PubMed] [Google Scholar]
- 20. Kristen AV, Giannitsis E, Lehrke S, Hegenbart U, Konstandin M, Lindenmaier D, Merkle C, Hardt S, Schnabel PA, Röcken C, Schonland SO, Ho AD, Dengler TJ, Katus HA. Assessment of disease severity and outcome in patients with systemic light‐chain amyloidosis by the high‐sensitivity troponin T assay. Blood 2010; 116: 2455–2461. [DOI] [PubMed] [Google Scholar]
- 21. Giannitsis E, Kurz K, Hallermayer K, Jarausch J, Jaffe AS, Katus HA. Analytical validation of a high‐sensitivity cardiac troponin T assay. Clin Chem 2010; 56: 254–261. [DOI] [PubMed] [Google Scholar]
- 22. Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward J, Shanewise JS, Solomon SD, Spencer KT, Sutton MS, Stewart WJ. Recommendations for chamber quantification: a report from the American Society of Echocardiography's Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr 2005; 18: 1440–1463. [DOI] [PubMed] [Google Scholar]
- 23. Nagueh SF, Appleton CP, Gillebert TC, Marino PN, Oh JK, Smiseth OA, Waggoner AD, Flachskampf FA, Pellikka PA, Evangelista A. Recommendations for the evaluation of left ventricular diastolic function by echocardiography. J Am Soc Echocardiogr 2009; 22: 107–133. [DOI] [PubMed] [Google Scholar]
- 24. Takashio S, Yamamuro M, Izumiya Y, Sugiyama S, Kojima S, Yamamoto E, Tsujita K, Tanaka T, Tayama S, Kaikita K, Hokimoto S, Ogawa H. Coronary microvascular dysfunction and diastolic load correlate with cardiac troponin T release measured by a highly sensitive assay in patients with nonischemic heart failure. J Am Coll Cardiol 2013; 62: 632–640. [DOI] [PubMed] [Google Scholar]
- 25. Ogawa H, Mizuno Y, Ohkawara S, Tsujita K, Ando Y, Yoshinaga M, Yasue H. Cardiac amyloidosis presenting as microvascular angina–a case report. Angiology 2001; 52: 273–278. [DOI] [PubMed] [Google Scholar]
- 26. Dorbala S, Vangala D, Bruyere J Jr, Quarta C, Kruger J, Padera R, Foster C, Hanley M, Di Carli MF, Falk R. Coronary microvascular dysfunction is related to abnormalities in myocardial structure and function in cardiac amyloidosis. JACC Heart Fail 2014; 2: 358–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Migrino RQ, Truran S, Gutterman DD, Franco DA, Bright M, Schlundt B, Timmons M, Motta A, Phillips SA, Hari P. Human microvascular dysfunction and apoptotic injury induced by AL amyloidosis light chain proteins. Am J Physiol Heart Circ Physiol 2011; 301: H2305–H2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Modesto KM, Dispenzieri A, Gertz M, Cauduro SA, Khandheria BK, Seward JB, Kyle R, Wood CM, Bailey KR, Tajik AJ, Miller FA, Pellikka PA, Abraham TP. Vascular abnormalities in primary amyloidosis. Eur Heart J 2007; 28: 1019–1024. [DOI] [PubMed] [Google Scholar]
- 29. Liao R, Jain M, Teller P, Connors LH, Ngoy S, Skinner M, Falk RH, Apstein CS. Infusion of light chains from patients with cardiac amyloidosis causes diastolic dysfunction in isolated mouse hearts. Circulation 2001; 104: 1594–1597. [PubMed] [Google Scholar]
- 30. Ng B, Connors LH, Davidoff R, Skinner M, Falk RH. Senile systemic amyloidosis presenting with heart failure: a comparison with light chain‐associated amyloidosis. Arch Intern Med 2005; 165: 1425–1429. [DOI] [PubMed] [Google Scholar]
- 31. Salinaro F, Meier‐Ewert HK, Miller EJ, Pandey S, Sanchorawala V, Berk JL, Seldin DC, Ruberg FL. Longitudinal systolic strain, cardiac function improvement, and survival following treatment of light‐chain (AL) cardiac amyloidosis. Eur Heart J Cardiovasc Imaging 2016. in press. [DOI] [PubMed] [Google Scholar]
- 32. Falk RH, Alexander KM, Liao R, Dorbala S. AL (light‐chain) cardiac amyloidosis: a review of diagnosis and therapy. J Am Coll Cardiol 2016; 68: 1323–1341. [DOI] [PubMed] [Google Scholar]